

Abstract
Activation of macrophages by Toll-like receptors (TLRs) and functionally related proteins is essential for host defense and innate immunity. TLRs recognize a wide variety of pathogen-associated molecules. Here, we demonstrate that the meningococcal outer membrane protein NhhA has immunostimulatory functions and triggers release of proinflammatory cytokines from macrophages. NhhA-induced cytokine release was found to proceed via two distinct pathways in RAW 264.7 macrophages. Interleukin-6 (IL-6) secretion was dependent on activation of TLR4 and required the TLR signaling adaptor protein MyD88. In contrast, release of tumor necrosis factor (TNF) was TLR4 and MyD88 independent. Both pathways involved NF-κB-dependent gene regulation. Using a PCR-based screen, we could identify additional targets of NhhA-dependent gene activation such as the cytokines and growth factors IL-1α, IL-1β, granulocyte colony-stimulating factor (G-CSF), and granulocyte-macrophage colony-stimulating factor (GM-CSF). In human monocyte-derived macrophages, G-CSF, GM-CSF, and IL-6 were found to be major targets of NhhA-dependent gene regulation. NhhA induced transcription of IL-6 and G-CSF mRNA via TLR4-dependent pathways, whereas GM-CSF transcription was induced via TLR4-independent pathways. These data provide new insights into the role of NhhA in host-pathogen interaction.
INTRODUCTION
The Gram-negative bacterium Neisseria meningitidis is a leading cause of bacterial meningitis and septicemia. Disease progression is rapid, and meningococcal infections can be fatal as a result of acute inflammatory responses. Individual bacterial components can be recognized by the innate immune system and trigger host defense mechanisms. The meningococcal endotoxin lipooligosaccharide (LOS) is a potent proinflammatory factor that triggers release of cytokines in human macrophages by binding to and activating TLR4 (30). LOS-dependent macrophage activation also requires CD14 and MD-2, which bind TLR4 and thereby function as coreceptors (3). In addition, meningococci may cause LOS-independent induction of proinflammatory responses (24). LOS-deficient meningococci cause fatal sepsis in an animal model of meningococcal disease (20). N. meningitidis capsular polysaccharides (CPS) trigger innate immunity responses by activating TLR2 and TLR4 (29). In addition, neisserial porins have immunostimulatory activities that depend on TLR2 and the intracellular adaptor protein myeloid differentiation primary response gene 88 (MyD88) (15). TLR receptors frequently undergo heterodimerization, and meningococcal PorB was reported to activate the TLR2/TLR1 complex by binding directly to TLR2 (16).
Meningococcal LOS-induced TLR4 signaling results in MyD88-dependent release of the cytokines tumor necrosis factor (TNF), interleukin-1β (IL-1β), monocyte chemoattractant protein 1 (MCP-1), and macrophage inflammatory protein 3α (MIP-3α) (31). MyD88-independent TLR4 signaling promotes synthesis of beta interferon (IFN-β), nitric oxide (NO), and IFN-γ inducible protein 10 (IP-10) (31). MyD88-independent TLR signaling has been linked only to activation of TLR3 or TLR4, whereas all other members of the TLR family appear to exclusively activate MyD88-dependent signaling pathways. TLR signaling induces transcription of genes related to infection and inflammation by activating mitogen-activated protein kinases (MAPKs), phosphoinositide 3-kinase (PI3K), and nuclear factor kappa B (NF-κB) (3).
The Neisseria Hia/Hsf homologue (NhhA) is a meningococcal protein related to the Hia and Hsf adhesins of Haemophilus influenzae (18). The NhhA gene is conserved in different N. meningitidis strains. Screening for vaccine candidates against serogroup B meningococci revealed that NhhA is a surface-exposed antigen that can produce an antibactericidal antibody response in mice (19, 27). Direct interaction between purified NhhA and the extracellular matrix components laminin and heparan sulfate was demonstrated (21). NhhA promotes the interaction between capsulated, wild-type meningococci and macrophages (22). In addition, meningococci lacking NhhA are more sensitive to complement-mediated bacterial killing in human serum (22). NhhA-dependent serum resistance was shown to depend on the interaction between NhhA and vitronectin (7). Using a mouse model of meningococcal disease, NhhA was found to play a role in colonization of nasal mucosa. Challenge with NhhA-deleted mutant bacteria leads to enhanced survival and less proinflammatory reactions in mice (22).
Here we demonstrate that NhhA activates macrophages and triggers NF-κB-dependent cytokine release via two distinct pathways. In human monocyte-derived macrophages, IL-6 and granulocyte colony-stimulating factory (G-CSF) were the major targets of NhhA-dependent gene regulation via TLR4, whereas granulocyte-macrophage colony-stimulating factor (GM-CSF) transcription was induced through TLR4-independent pathways.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
FAM20, an N. meningitidis serogroup C strain, was grown at 37°C and 5% CO2 on GC medium base (Acumedia).
Cloning and purification of recombinant NhhA.
A sequence encoding amino acids 51 to 510 of NhhA was amplified by PCR using genomic DNA from FAM20 as the template using the following primers: 5′-GCGAATGGATCCGATACCGATGAAGATG-3′ and 5′-CCTTTAAGCTTTGCGACGTTTGTAACATC-3′. After purification and digestion, the PCR product was cloned into the vector pET-21a (Novagen) using restriction enzymes BamHI and HindIII. Recombinant NhhA was expressed in Escherichia coli in fusion with a C-terminal hexahistidine tag. NhhA was purified by affinity chromatography using talon resin (Clontech). Recombinant NhhA was then further purified by fast protein liquid chromatography (FPLC) gel filtration. Purification was monitored by SDS-PAGE with Coomassie brilliant blue staining. The meningococcal protein NMC0101 was expressed and purified in the same way to serve as a control.
Cell culture and stimulation.
All cell lines were obtained from American Type Culture Collection except for the HEK 293-hTLR4A-MD2-CD14 cells, which were from InvivoGen. The murine macrophage cell line RAW 264.7 was cultured in Dulbecco's modified Eagle's medium (DMEM) plus 10% heat-inactivated fetal calf serum at 37°C and 5% CO2. The human monocytic cell line THP-1 was cultured in RPMI 1640 and 10% heat-inactivated fetal calf serum at 37°C and 5% CO2. After seeding RAW 264.7 cells (500,000/ml) into 6-well cell culture plates, cells were allowed to attach for 6 h. THP-1 cells were differentiated to macrophages using 100 nM phorbol 12-myristate 13-acetate (PMA) (Sigma) for 3 days. Thereafter, PMA was removed and fresh medium added. Cells were maintained in serum-containing growth medium during differentiation and stimulation.
HEK 293 cells were cultured in DMEM with 10% heat-inactivated fetal calf serum, 50 u/ml penicillin, 50 μg/ml streptomycin, and 2 mM l-glutamine. HEK 293-hTLR4A-MD2-CD14 cells were cultured in the same growth medium supplemented with 10 μg/ml Blasticidin (InvivoGen) and 50 μg/ml HygroGold (InvivoGen). Cells (500,000/ml) were seeded into 6-well cell culture plates. After attachment, the supernatant was removed and fresh medium added.
Human CD14+ monocytes were isolated from freshly prepared buffy coat (Karolinska Institute Biobank) using the pluriBead cell separation kit (pluriSelect GmbH). Isolated monocytes (1,000,000/ml) were seeded into 6-well cell culture plates and incubated for 2 h at 37°C in RPMI 1640 medium supplemented by 10% fetal bovine serum (FBS), 100 IU/ml penicillin, and 100 μg/ml streptomycin. After nonadherent cells were removed, monocytes were differentiated into macrophages in fresh medium with 50 ng/ml rhM-CSF (ImmunoTools) for 7 days. Medium was replaced two times during the differentiation period. Subsequently, M-CSF was removed and fresh medium added.
If indicated, cells were pretreated with 1 μM polymyxin B (InvivoGen), 1 μM CLI-095 (InvivoGen), 5 μM Celastrol (InvivoGen), or 10 μM chloroquine (InvivoGen). After 1 h of pretreatment, cells were exposed to 0 to 400 nM NhhA for 5 to 20 h or 500 ng/ml E. coli LPS (Sigma-Aldrich) for 20 h. Before cells were stimulated, NhhA was pretreated with polymyxin B at 10 μg/ml to remove possible LPS contamination.
Measurement of cytokine release.
Cell supernatants were collected from stimulated RAW 264.7 cells, and IL-6 and TNF concentrations were determined by enzyme-linked immunosorbent assay (ELISA) using IL-6 and TNF antibody pairs (Invitrogen) according to the manufacturer's instructions. Standard curves were constructed using ELISA MAXTM standard sets (BioLegend).
siRNA transfection.
The predesigned FlexiTube small interfering RNAs (siRNAs) CD14-1, MyD88-4, Tlr1-1, and Tlr2-2 targeting mouse CD14, MyD88, Tlr1, and Tlr2, respectively, were obtained from Qiagen. The AllStar control siRNA (Qiagen) was used to monitor unspecific effects. RAW 264.7 cells were transfected using HiPerFect transfection reagent (Qiagen) according to the manufacturer's instructions. In brief, aliquots of 20,000 cells in 30 μl of growth medium were seeded in 96-well plates. siRNAs were suspended in 30 μl growth medium without serum at 50 nM and 1 μl HiPerFect transfection reagent was added. After 10 min of incubation, the mixture was added dropwise to the cells. After 6 h, 140 μl complete growth medium was added. Twenty-four hours after transfection, cells were either subjected to RNA extraction or stimulated with 50 ng/ml Pam3CSK4 (InvivoGen), 0.5 μg/ml E. coli LPS (Sigma), or 400 nM NhhA. After stimulation for 20 h, cell supernatants were collected and cytokine release was determined by ELISA.
RNA isolation and gene expression analysis.
RNA was isolated using the RNeasy minikit (Qiagen). RNA quality was assessed by agarose gel electrophoresis, and the concentration was determined by measuring absorbance at 260 nm. Three hundred nanograms of total RNA was used for cDNA synthesis in a final reaction volume of 20 μl. cDNA for conventional real-time PCR was synthesized using the RevertAid H minus first-strand cDNA synthesis kit (Fermentas). cDNA for PCR array analysis was synthesized using the RT2 first-strand kit (Qiagen). Real-time PCR was performed using the LightCycler 480 SYBR green I master (Roche). PCR primers are specified in Tables 1 and 2. PCR array analysis was performed using mouse Toll-like receptor signaling pathway PCR arrays (Qiagen). All PCR amplifications were performed using a 480 LightCycler (Roche). Primer efficiencies were determined by PCR analysis of serially diluted cDNA. Advanced relative quantifications corrected for primer efficiency were performed using the LightCycler software. For PCR array, a set of five housekeeping genes were used as reference genes. For conventional real-time PCR, Gapdh (mouse cells) or RPL37A (human cells) were used as reference genes. RPL37A was chosen on the basis of superior expression stability during PMA-induced THP-1 differentiation (13).
Table 1.
Mouse real-time PCR primers
| Gene product | Primer sequence |
|---|---|
| Gapdh | Forward, CAACTTTGTCAAGCTCATTTCCTG |
| Reverse, CCTCTCTTGCTCAGTGTCCTT | |
| Csf3 | Forward, TCCAGGCCAGCGGCTCGGTG |
| Reverse, TGTCTGCTGCAGGGCCTGGC | |
| Il1b | Forward, GCCTCGTGCTGTCGGACCCATA |
| Reverse, TGCAGGGTGGGTGTGCCGTCTT | |
| Il6 | Forward, AGCTGGAGTCACAGAAGGAGTG |
| Reverse, ATAACGCACTAGGTTTGCCGAG | |
| Csf2 | Forward, GCCATCAAAGAAGCCCTGAA |
| Reverse, GCGGGTCTGCACACATGTTA | |
| Il1a | Forward, GGAGAAGACCAGCCCGTGTTGCT |
| Reverse, CCGTGCCAGGTGCACCCGACTT | |
| Ptgs2 | Forward, GCCAGCTCCACCGCCACCACT |
| Reverse, GAGCCCCAGGGCAGCGCAGA | |
| Il10 | Forward, TGCACCCACTTCCCAGTCGGCCA |
| Reverse, TGGCTGAAGGCAGTCCGCAGCTC | |
| Ccl2 | Forward, TTGCCGGCTGGAGCATCCACGT |
| Reverse, AGTAGCAGCAGGTGAGTGGGGCG | |
| Ifnb1 | Forward, GCCTGGATGGTGGTCCGAGCA |
| Reverse, TACCAGTCCCAGAGTCCGCCTCT | |
| Cxcl10 | Forward, TCCGGAAGCCTCCCCATCAGCACC |
| Reverse, TGCAGCGGACCGTCCTTGCGA | |
| Tnf | Forward, TCCCAGGTTCTCTTCAAGGGA |
| Reverse, GGTGAGGAGCACGTAGTCGG |
Table 2.
Human real-time PCR primers
| Gene product | Primer sequence |
|---|---|
| RPL37A | Forward, ATTGAAATCAGCCAGCACGC |
| Reverse, AGGAACCACAGTGCCAGATCC | |
| CSF3 | Forward, GCTTGAGCCAACTCCATAGC |
| Reverse, TTCCCAGTTCTTCCATCTGC | |
| IL1B | Forward, TCCCCAGCCCTTTTGTTGA |
| Reverse, TTAGAACCAAATGTGGCCGTG | |
| IL-6 | Forward, GGCACTGGCAGAAAACAACC |
| Reverse, GCAAGTCTCCTCATTGAATCC | |
| CSF2 | Forward, AGAAATGTTTGACCTCCAGGA |
| Reverse, TTGCACAGGAAGTTTCCG | |
| IL1A | Forward, CGCCAATGACTCAGAGGAAGA |
| Reverse, AGGGCGTCATTCAGGATGAA | |
| TNF | Forward, CCTGCCCCAATCCCTTTATT |
| Reverse, CCCTAAGCCCCCAATTCTCT |
Statistical analysis.
Student's t test for paired samples was used for statistical analysis. Differences between means were considered significant if P values were ≤0.05.
RESULTS
NhhA triggers cytokine production in macrophages.
In order to perform functional studies of NhhA, we expressed NhhA in soluble form by excluding the C-terminal membrane anchoring domain. A sequence encoding amino acids 51 to 510 of NhhA from FAM20, an N. meningitidis serogroup C strain, was cloned into the vector pET-21a and expressed in E. coli in fusion with a histidine tag. The N terminus of NhhA contains a predicted leader peptide that was excluded from the NhhA construct. NhhA was purified by affinity chromatography followed by gel chromatography to avoid endotoxin contamination. The capacity of purified recombinant NhhA to induce proinflammatory responses was investigated by exposing RAW 264.7 mouse macrophages to various concentrations of NhhA. After 20 h of incubation, the presence of the cytokines IL-6 and TNF in the extracellular medium was determined. NhhA was found to trigger a dose-dependent release of both these cytokines, and stimulation of cytokine production could be observed at the lowest dose tested, 3 nM NhhA (Fig. 1A and B). In unstimulated cells, the extracellular IL-6 level was 14.9 ± 3.7 pg/ml (mean ± standard deviation [SD]; n = 3 samples), whereas TNF was undetectable. The release of IL-6 at different time points after NhhA stimulation was determined (Fig. 1C). After 8 h, significant increases in IL-6 levels were observed (P = 0.003 versus untreated cells). IL-6 accumulated at later time points, indicating an ongoing production of the cytokine. To confirm the specificity of NhhA-induced cytokine production, we cloned, expressed, and purified an unrelated His-tagged meningococcal protein (NMC0101). NMC0101 failed to induce IL-6 or TNF production in RAW 264.7 cells (results not shown).
Fig 1.
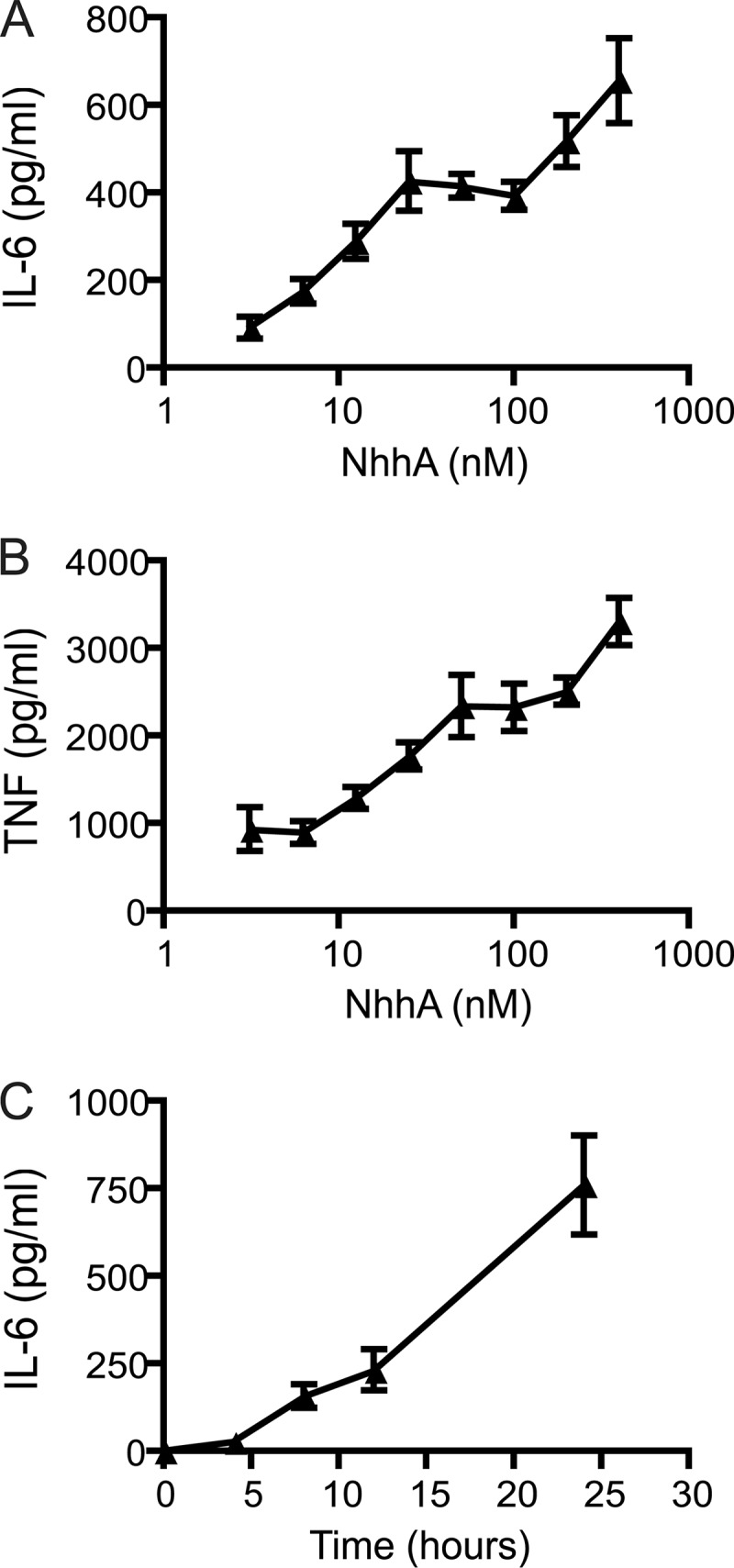
NhhA induces cytokine production in macrophages. (A and B) RAW 264.7 macrophages were exposed to various concentrations of NhhA for 20 h, and the release of IL-6 and TNF was determined by ELISA. (C) RAW 264.7 mouse macrophages were exposed to 400 nM NhhA for various time points and the release of IL-6 was determined. Values indicate means ± SD (n = 3 samples) of one representative experiment out of three.
NhhA-induced cytokine release occurs via multiple pathways.
The mechanisms involved in NhhA-stimulated cytokine release were investigated by using a set of chemical inhibitors (Fig. 2A). CLI-095/TAK242 is an inhibitor of TLR4 that prevents TLR4 signaling by binding its intracellular domain (10). As expected, CLI-095 inhibited LPS-induced cytokine release (Fig. 2B). In NhhA-stimulated cells, CLI-095 prevented IL-6 release but not TNF release (Fig. 2A). Celastrol, an inhibitor of the transcription factor NF-κB, was able to block NhhA-induced IL-6 and TNF release (Fig. 2A). The LPS binding compound polymyxin B did not prevent NhhA-induced cytokine release, confirming that our preparations of purified recombinant NhhA were not contaminated with LPS. In order to further investigate the possibility of LPS contamination, NhhA was heated at 95°C for 10 min before treatment of RAW 264.7 cells. Heat treatment reduced the capacity of NhhA (400 nM) to induce release of IL-6 by 93.7 ± 5.9% (mean ± SD; n = 3 experiments). In contrast, heat treatment of LPS only reduced the capacity of LPS (500 ng/ml) to induce release of IL-6 by 2.7 ± 7.0% (mean ± SD; n = 3 experiments). Since LPS is highly resistant to heat inactivation (14), these data confirm that NhhA-induced cytokine release did not result from LPS contamination.
Fig 2.

Role of TLR4 and NF-κB in NhhA-induced cytokine release. (A) RAW 264.7 macrophages were preincubated with or without (control) 1 μM CLI-095 (TLR4 inhibitor), 5 μM Celastrol (NF-κB inhibitor), or 1 μM polymyxin B (PMB) for 1 h. Thereafter, cells were stimulated with NhhA (400 nM, 20 h), and the release of IL-6 and TNF was determined by ELISA. *, P < 0.05 versus NhhA-treated control. (B) RAW 264.7 macrophages were preincubated with or without (control) 1 μM CLI-095 or 1 μM polymyxin B for 1 h. Thereafter, cells were stimulated with 500 ng/ml E. coli LPS for 20 h, and the release of IL-6 and TNF was determined by ELISA. Values indicate means ± SD from three independent experiments. *, P < 0.05 versus LPS-treated control.
Pretreatment of RAW 264.7 cells with chloroquine, an inhibitor of endosomal acidification and TLR9 signaling, did not alter the release of IL-6 or TNF (results not shown). In order to verify the inhibitory functions of CLI-095 and polymyxin B under our experimental conditions, the capacity of these drugs to inhibit LPS-induced cytokine release was verified (Fig. 2B). These results suggested that NhhA-induced cytokine release was NF-κB dependent and could occur via TLR4-dependent as well as TLR4-independent pathways.
NhhA-induced IL-6 release requires MyD88.
The role of additional factors related to TLR signaling was investigated using siRNA-mediated downregulation. RAW 264.7 cells were exposed to siRNAs specifically targeting TLR1, TLR2, CD14, or MyD88. Specific downregulation of the corresponding mRNA was verified by quantitative real-time reverse transcription (RT)-PCR (results not shown). To verify that siRNA treatment induced functional changes, cells were stimulated with Pam3CSK4 or LPS (Fig. 3A and B). In line with previous reports, TLR1 or TLR2 knockdown reduced Pam3CSK4-induced cytokine release, and CD14 or MyD88 knockdown reduced LPS-induced cytokine release.
Fig 3.

Role of TLR1, TLR2, CD14, and MyD88 in NhhA-induced cytokine release. (A) RAW 264.7 macrophages were treated with siRNA against TLR1 or TLR2. Alternatively, a scrambled control siRNA (AllStar) was used. Thereafter, cells were stimulated with 50 ng/ml Pam3CSK4 for 20 h. Extracellular IL-6 levels were determined by ELISA. (B) RAW 264.7 macrophages were treated with siRNA against CD14 or MyD88. Thereafter, cells were stimulated with 500 ng/ml E. coli LPS for 20 h, and IL-6 release was determined. (C) RAW 264.7 macrophages were treated with siRNA against TLR1, TLR2, CD14, or MyD88. Thereafter, cells were stimulated with 400 nM NhhA for 20 h. Extracellular IL-6 and TNF levels were determined. Values indicate means ± SD from three independent experiments. *, P < 0.05 versus AllStar control.
Next, the role of these proteins in NhhA-induced cytokine release was investigated. None of these factors could significantly alter release of TNF after NhhA stimulation. However, MyD88 siRNA treatment almost completely abolished IL-6 release. These data pointed to an essential role of MyD88 in NhhA-induced release of IL-6.
Identification of NhhA-regulated genes.
The capacity of NhhA to regulate genes involved in inflammatory responses was investigated further using a PCR array-based screening. RNA was isolated from unstimulated and NhhA-stimulated RAW 264.7 macrophages and reverse transcribed. The cDNA was applied to commercially available 96-well plates preloaded with primers recognizing 84 genes involved in TLR signaling. A set of five housekeeping genes were used as a reference. A group of 10 mRNAs that were 100-fold or more upregulated after NhhA exposure could be identified (Fig. 4A). These are described in Table 3 and consist mainly of chemokines and cytokines, including IL-6. In addition, the enzyme prostaglandin synthase 2 (Ptgs2), which is involved in arachidonic acid metabolism, was also identified as a target of NhhA-induced gene regulation. TNF mRNA displayed an 11-fold upregulation after NhhA exposure. The result of the PCR array screen was validated by conventional real-time RT-PCR (Fig. 4B) using Gapdh as the reference gene. The mRNAs encoding GM-CSF/CSF2, G-CSF/CSF3, IL-1α, IL-1β, and IL-6 were upregulated more than 10,000-fold after NhhA stimulation. Since meningococci are human-specific pathogens, it was important to confirm that NhhA-mediated gene regulation could occur also in human macrophages. THP-1 monocytes were differentiated to macrophage-like cells by treatment with phorbol 12-myristate 13-acetate (PMA). The expression of mRNAs encoding GM-CSF, G-CSF, IL-1α, IL-1β, and IL-6 was investigated by real-time RT-PCR (Fig. 5A) using RPL37A as a reference gene. All investigated transcripts were clearly induced, ranging from more than 1,000-fold upregulation of the IL-6 transcript to around 10-fold upregulation of IL-1β and TNF mRNA. In addition, NhhA-induced cytokine mRNA induction was examined in human monocyte-derived macrophages. In these cells, transcription of mRNA encoding GM-CSF, G-CSF, and IL-6 was increased around 100-fold, whereas the other mRNAs investigated were induced to a lesser extent (Fig. 5C). These results demonstrate potent gene regulatory actions induced by NhhA stimulation.
Fig 4.

Identification of NhhA-regulated genes. (A) RAW 264.7 macrophages were stimulated with 400 nM NhhA for 6 h followed by RNA isolation and cDNA synthesis. PCR array experiments analyzing 84 genes involved in TLR signaling were performed. Unstimulated cells served as the control. Relative changes in gene expression were determined by quantitative real-time PCR. A scatter plot was generated to visualize up- or downregulated genes. Red dots indicate genes upregulated 100-fold or more after NhhA exposure. (B) Genes upregulated 100-fold or more after NhhA exposure were selected for validation experiments. RAW 264.7 macrophages were stimulated as described above, and conventional real-time RT-PCR experiments were performed using primer sets unrelated to the PCR array. Unstimulated cells served as the control. Values indicate means from two independent experiments (PCR array) or means ± SD from three independent experiments (validation). For comparison, PCR array results for the selected genes were plotted also in panel B.
Table 3.
NhhA target gene products in RAW 264.7 macrophages
| Gene product | Description |
|---|---|
| Csf3 | Granulocyte colony-stimulating factor (cytokine, growth factor) |
| Il1b | Interleukin-1β (proinflammatory cytokine) |
| Il6 | Interleukin-6 (proinflammatory cytokine) |
| Csf2 | Granulocyte macrophage colony-stimulating factor (cytokine, growth factor) |
| Il1a | Interleukin-1α (proinflammatory cytokine) |
| Ptgs2 | Prostaglandin synthase 2 (enzyme involved in arachidonic acid metabolism) |
| Il10 | Interleukin-10 (anti-inflammatory cytokine) |
| Ccl2 | Chemokine ligand 2 (chemokine) |
| Ifnb1 | Interferon-β (cytokine, antiviral) |
| Cxcl10 | C-X-C motif chemokine 10 (chemokine) |
Fig 5.

TLR4 mediates cytokine mRNA induction by NhhA in macrophages. (A) Human THP-1 cells differentiated to macrophages by PMA treatment were exposed to 400 nM NhhA for 6 h. RNA was isolated and real-time RT-PCR experiments were performed. Relative changes in cytokine mRNA expression were determined using unstimulated cells as the control. (B) THP-1-derived and RAW 264.7 macrophages were pretreated with 1 μM CLI-095 (TLR4 inhibitor) for 1 h before exposure to 400 nM NhhA for 6 h. RNA was isolated and real-time RT-PCR experiments were performed. Relative changes in cytokine mRNA expression were determined. NhhA-exposed cells not pretreated with CLI-095 were used as the control to visualize the effect of CLI-095 pretreatment. (C) Human monocyte-derived macrophages were exposed to 400 nM NhhA for 6 h. RNA was isolated and real-time RT-PCR experiments were performed. Relative changes in cytokine mRNA expression were determined using unstimulated cells as the control. (D) Human monocyte-derived macrophages were pretreated with 1 μM CLI-095 for 1 h before exposure to 400 nM NhhA for 6 h. RNA was isolated and real-time RT-PCR experiments were performed. Relative changes in cytokine mRNA expression were determined. NhhA-exposed cells not pretreated with CLI-095 were used as a control to visualize the effect of CLI-095 pretreatment. Values indicate means ± SD from three independent experiments. *, P < 0.05 versus NhhA-treated control.
TLR4 mediates NhhA-dependent IL-6 gene regulation.
The role of TLR4 in NhhA-induced expression of cytokine mRNAs was investigated using the TLR4 inhibitor CLI-095 (Fig. 5B). Preincubation of RAW 264.7 or PMA-treated THP-1 cells with the TLR4 inhibitor did not prevent induction of TNF mRNA upon NhhA exposure. However, the TLR4 inhibitor efficiently blocked NhhA-induced induction of IL-6 mRNA in both cell types. In differentiated THP-1 cells, the induction of G-CSF mRNA was also substantially reduced, whereas IL-1β, GM-CSF, and IL-1α mRNA levels remained largely unaffected after TLR4 inhibition. In RAW 264.7 cells, the expression of all investigated mRNAs, except TNF mRNA, was lowered after CLI-095 pretreatment. In monocyte-derived macrophages, IL-6 and G-CSF induction was found to be TLR4 dependent, whereas induction of GM-CSF did not require TLR4 (Fig. 5D). These results indicate TLR4-dependent as well as TLR4-independent upregulation of cytokine gene expression upon exposure of macrophages to NhhA. IL-6 mRNA induction was consistently found to require TLR4 activation in monocyte-derived macrophages and macrophage cell lines.
In order to confirm a role of TLR4 in NhhA-induced cytokine mRNA induction, we stimulated TLR4-expressing HEK 293 cells with NhhA for 6 h (Fig. 6). Nontransfected cells did not respond to NhhA stimulation. In TLR4-expressing HEK 293 cells, increased expression of mRNA encoding TNF was detected. In contrast, IL-6 transcript levels were nearly unaffected by NhhA stimulation. These data confirm the capacity of TLR4 to mediate NhhA-dependent cytokine mRNA induction.
Fig 6.
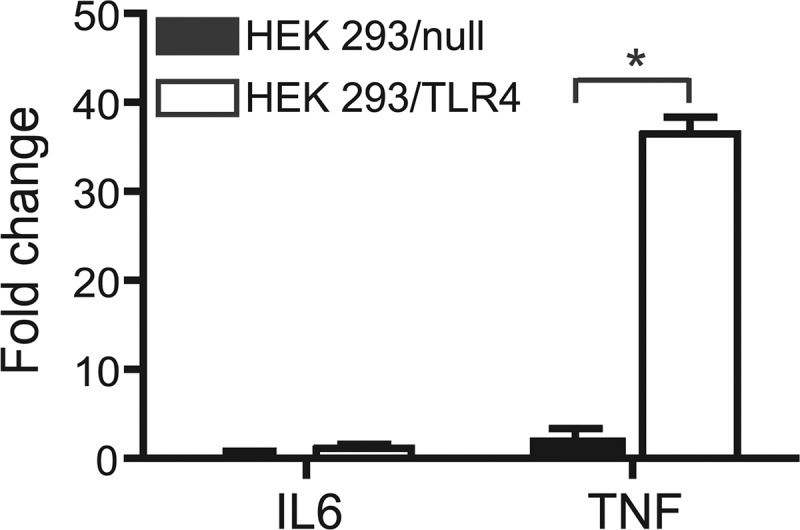
TLR4 mediates cytokine mRNA induction by NhhA in HEK 293 cells. Nontransfected HEK 293 control cells (HEK 293/null) and stably transfected HEK 293 cells expressing TLR4 and its coreceptors MD2 and CD14 (HEK 293/TLR4) were exposed to 400 nM NhhA for 6 h. RNA was isolated and real-time RT-PCR experiments were performed. Relative changes in cytokine mRNA expression were determined using unstimulated cells as the control. Values indicate means ± SD from three independent experiments. *, P < 0.05.
DISCUSSION
In addition to the well-studied proinflammatory effects of carbohydrate-based molecules such as meningococcal LOS and CPS, proteins present on the bacterial surface may also contribute to innate immunity responses. Meningococcal porin B triggers TLR2/TLR1-dependent cytokine release (16). In addition, the neisserial adhesin NadA provokes cytokine production and stimulates differentiation of monocytes to macrophages (5). NadA-dependent monocyte stimulation was recently shown to be TLR4 dependent and to involve interaction between NadA and extracellular Hsp 90 (2).
We found that exposure of macrophages to purified recombinant NhhA triggered release of the proinflammatory cytokines IL-6 and TNF in a time- and dose-dependent manner. Already at 3 nM NhhA, stimulation of IL-6 and TNF secretion was observed. The specific TLR4 inhibitor CLI-095 (10) blocked NhhA-induced IL-6 release but not NhhA-induced TNF release. However, both IL-6 and TNF productions were found to be NF-κB dependent. The involvement of additional factors related to TLR signaling was investigated using siRNA-based silencing. MyD88 was identified as an additional factor required for NhhA-induced IL-6 release in RAW 264.7 macrophages. TLR4 and MyD88 signaling is more closely linked in certain cell types (9). Therefore, NhhA-induced IL-6 release might not be MyD88 dependent in all cell types. NhhA-induced TNF production was not affected by depletion of TLR1, TLR2, CD14, or MyD88.
Using a PCR array-based screening, the upregulation of mRNA encoding IL-6 and TNF was confirmed. In addition, a number of cytokines and chemokines were identified as targets of NhhA-dependent gene regulation, and striking upregulation of mRNA encoding, for example, GM-CSF, G-CSF, IL-1α, and IL-1β was observed. In line with our biochemical findings, we observed that TLR4 inhibition blocked NhhA-induced IL-6 mRNA induction but not TNF mRNA induction in murine and human macrophage cell lines. In human monocyte-derived macrophages, IL-6, G-CSF, and GM-CSF were identified as major targets of NhhA-dependent gene regulation. These data point to important differences in NhhA and LPS stimulation of macrophages since LPS has only minor effects on G-CSF and GM-CSF transcription (1). In monocyte-derived macrophages, NhhA induced transcription of IL-6 and G-CSF mRNA via TLR4-dependent pathways, whereas GM-CSF transcription was induced via TLR4-independent pathways. The ability of NhhA to activate TLR4 and induce cytokine mRNA expression was confirmed in TLR4-expressing HEK 293 cells, where TNF mRNA was found to be upregulated. In contrast to macrophages, NhhA failed to induce IL-6 transcription in HEK 293 cells. TLR4 is emerging as a potential target for treatment of sepsis (11). The data presented here might contribute to the understanding of how TLR4 inhibition could influence meningococcal sepsis.
The ability of an individual bacterial component to stimulate cytokine release via two apparently independent pathways is unusual. Therefore, the identification of mechanisms involved in mediating NhhA-dependent TNF release is an interesting target of future studies. Live meningococci stimulate TLR signaling via TLR2, TLR4, and TLR9 (17). We could not link NhhA-induced TNF release to TLR2 or TLR4. Since TLR9 signals via MyD88, it is an unlikely mediator of NhhA-induced TNF release. Furthermore, chloroquine, an inhibitor of endosomal acidification and TLR9 signaling (8), did not prevent TNF release after NhhA stimulation (results not shown). Instead, NhhA-induced TNF release might involve other pattern recognition receptors (PRRs) such as members of the scavenger receptor (SR) family or NOD-like proteins. NhhA-induced TNF release was NF-κB dependent, and both SR and NOD-like proteins have been reported to activate NF-κB (6, 25, 26). Screening for human proteins interacting with NhhA by using biochemical or yeast two-hybrid-based methods might be helpful to identify additional NhhA receptors.
NhhA was recently shown to stimulate apoptosis in macrophages (23). Since TNF has been linked to proapoptotic signaling (28), NhhA-induced TNF production might play a role in inducing macrophage apoptosis. However, antibody-mediated inhibition of TNF signaling failed to prevent NhhA-induced apoptosis (23). Another function of NhhA related to host defense was highlighted in a recent study. It was shown that NhhA can bind to vitronectin and thereby increase serum resistance by regulating complement activity (7). Since vitronectin is abundant in serum, the vitronectin-NhhA interaction could potentially regulate other actions of NhhA, such as its cytokine production-stimulating activity described here. Bovine vitronectin binds to NhhA with an affinity similar to that of human vitronectin (7). Since our experiments were carried out in the presence of bovine serum, we draw the conclusion that NhhA-induced cytokine production is not prevented by vitronectin. Further studies will be needed to elucidate to what extent the different functions of NhhA influence each other.
NhhA has been shown to trigger antibactericidal antibody responses in mice and has been suggested to be a candidate for vaccine development (19, 27). Immunostimulatory adjuvants may enhance vaccine responses (12), and therefore the immunostimulatory functions of NhhA described here may be of importance for successful development of NhhA-based vaccines. Also, the need for nonantibiotic or adjunctive therapies to complement antibiotic-based treatment of sepsis and septic shock is now being increasingly recognized (4). Continued studies on how membrane-bound factors, such as NhhA, stimulate host responses will be important for supporting complementary approaches to potentially decrease inflammatory reactions to meningococci. In summary, we demonstrate that NhhA has immunostimulatory functions that involve potent gene regulatory events. These findings will contribute to understanding the role of NhhA in host-pathogen interaction.
ACKNOWLEDGMENTS
This work was supported by the Swedish Research Council (grant no. 2008-2572, 2008-3367), the Swedish Society of Medicine, the Magnus Bergvalls Foundation, the Harald Jeanssons Foundation, the Tore Nilsons Foundation, the Henrik Granholms Foundation, the Åke Wiberg Foundation, and Stockholm University.
Footnotes
Published ahead of print 4 September 2012
REFERENCES
- 1. Bjorkbacka H, et al. 2004. The induction of macrophage gene expression by LPS predominantly utilizes Myd88-independent signaling cascades. Physiol. Genomics 19:319–330 [DOI] [PubMed] [Google Scholar]
- 2. Cecchini P, et al. 2011. The soluble recombinant Neisseria meningitidis adhesin NadA(Delta351-405) stimulates human monocytes by binding to extracellular Hsp90. PLoS One 6:e25089 doi:10.1371/journal.pone.0025089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen K, et al. 2007. Toll-like receptors in inflammation, infection and cancer. Int. Immunopharmacol. 7:1271–1285 [DOI] [PubMed] [Google Scholar]
- 4. Cohen J. 2009. Non-antibiotic strategies for sepsis. Clin. Microbiol. Infect. 15:302–307 [DOI] [PubMed] [Google Scholar]
- 5. Franzoso S, et al. 2008. Human monocytes/macrophages are a target of Neisseria meningitidis adhesin A (NadA). J. Leukoc. Biol. 83:1100–1110 [DOI] [PubMed] [Google Scholar]
- 6. Goh JW, Tan YS, Dodds AW, Reid KB, Lu J. 2010. The class A macrophage scavenger receptor type I (SR-AI) recognizes complement iC3b and mediates NF-kappaB activation. Protein Cell 1:174–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Griffiths NJ, et al. 2011. Meningococcal surface fibril (Msf) binds to activated vitronectin and inhibits the terminal complement pathway to increase serum resistance. Mol. Microbiol. 82:1129–1149 [DOI] [PubMed] [Google Scholar]
- 8. Ivory CP, Prystajecky M, Jobin C, Chadee K. 2008. Toll-like receptor 9-dependent macrophage activation by Entamoeba histolytica DNA. Infect. Immun. 76:289–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kawai T, et al. 2001. Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J. Immunol. 167:5887–5894 [DOI] [PubMed] [Google Scholar]
- 10. Kawamoto T, Ii M, Kitazaki T, Iizawa Y, Kimura H. 2008. TAK-242 selectively suppresses Toll-like receptor 4-signaling mediated by the intracellular domain. Eur. J. Pharmacol. 584:40–48 [DOI] [PubMed] [Google Scholar]
- 11. Leon CG, Tory R, Jia J, Sivak O, Wasan KM. 2008. Discovery and development of Toll-like receptor 4 (TLR4) antagonists: a new paradigm for treating sepsis and other diseases. Pharm. Res. 25:1751–1761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu HM, et al. 1998. Immunostimulatory CpG oligodeoxynucleotides enhance the immune response to vaccine strategies involving granulocyte-macrophage colony-stimulating factor. Blood 92:3730–3736 [PubMed] [Google Scholar]
- 13. Maess MB, Sendelbach S, Lorkowski S. 2010. Selection of reliable reference genes during THP-1 monocyte differentiation into macrophages. BMC Mol. Biol. 11:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Majde JA. 1993. Microbial cell-wall contaminants in peptides: a potential source of physiological artifacts. Peptides 14:629–632 [DOI] [PubMed] [Google Scholar]
- 15. Massari P, et al. 2002. Cutting edge: immune stimulation by neisserial porins is Toll-like receptor 2 and MyD88 dependent. J. Immunol. 168:1533–1537 [DOI] [PubMed] [Google Scholar]
- 16. Massari P, et al. 2006. Meningococcal porin PorB binds to TLR2 and requires TLR1 for signaling. J. Immunol. 176:2373–2380 [DOI] [PubMed] [Google Scholar]
- 17. Mogensen TH, Paludan SR, Kilian M, Ostergaard L. 2006. Live Streptococcus pneumoniae, Haemophilus influenzae, and Neisseria meningitidis activate the inflammatory response through Toll-like receptors 2, 4, and 9 in species-specific patterns. J. Leukoc. Biol. 80:267–277 [DOI] [PubMed] [Google Scholar]
- 18. Peak IR, Srikhanta Y, Dieckelmann M, Moxon ER, Jennings MP. 2000. Identification and characterisation of a novel conserved outer membrane protein from Neisseria meningitidis. FEMS Immunol. Med. Microbiol. 28:329–334 [DOI] [PubMed] [Google Scholar]
- 19. Pizza M, et al. 2000. Identification of vaccine candidates against serogroup B meningococcus by whole-genome sequencing. Science 287:1816–1820 [DOI] [PubMed] [Google Scholar]
- 20. Plant L, Wan H, Jonsson AB. 2007. Non-lipooligosaccharide-mediated signalling via Toll-like receptor 4 causes fatal meningococcal sepsis in a mouse model. Cell Microbiol. 9:657–669 [DOI] [PubMed] [Google Scholar]
- 21. Scarselli M, et al. 2006. Neisseria meningitidis NhhA is a multifunctional trimeric autotransporter adhesin. Mol. Microbiol. 61:631–644 [DOI] [PubMed] [Google Scholar]
- 22. Sjolinder H, Eriksson J, Maudsdotter L, Aro H, Jonsson AB. 2008. Meningococcal outer membrane protein NhhA is essential for colonization and disease by preventing phagocytosis and complement attack. Infect. Immun. 76:5412–5420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sjolinder M, et al. 2012. Meningococcal outer membrane protein NhhA triggers apoptosis in macrophages. PLoS One 7:e29586 doi:10.1371/journal.pone.0029586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sprong T, et al. 2002. Neisseria meningitidis can induce pro-inflammatory cytokine production via pathways independent from CD14 and Toll-like receptor 4. Eur. Cytokine Netw. 13:411–417 [PubMed] [Google Scholar]
- 25. Stewart CR, et al. 2010. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 11:155–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tukhvatulin AI, et al. 2011. A in vitro and in vivo study of the ability of NOD1 ligands to activate the transcriptional factor NF-κB. Acta Naturae 3:77–84 [PMC free article] [PubMed] [Google Scholar]
- 27. Weynants VE, et al. 2007. Additive and synergistic bactericidal activity of antibodies directed against minor outer membrane proteins of Neisseria meningitidis. Infect. Immun. 75:5434–5442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xaus J, et al. 2000. LPS induces apoptosis in macrophages mostly through the autocrine production of TNF-alpha. Blood 95:3823–3831 [PubMed] [Google Scholar]
- 29. Zughaier SM. 2011. Neisseria meningitidis capsular polysaccharides induce inflammatory responses via TLR2 and TLR4-MD-2. J. Leukoc. Biol. 89:469–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zughaier SM, et al. 2004. Neisseria meningitidis lipooligosaccharide structure-dependent activation of the macrophage CD14/Toll-like receptor 4 pathway. Infect. Immun. 72:371–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zughaier SM, Zimmer SM, Datta A, Carlson RW, Stephens DS. 2005. Differential induction of the Toll-like receptor 4-MyD88-dependent and -independent signaling pathways by endotoxins. Infect. Immun. 73:2940–2950 [DOI] [PMC free article] [PubMed] [Google Scholar]