

Abstract
Our immune system has to constantly strike a balance between activation and inhibition of an inflammatory response to combat invading pathogens and avoid inflammation-induced collateral tissue damage. Toll interleukin-1 receptor 8 (IL-1R-8)/single Ig domain IL-1R-related molecule (TIR8/SIGIRR) is an inhibitor of Toll-like receptor (TLR)/IL-1R signaling, which is predominantly expressed in the kidney. The biological role of renal TIR8 during infection is, however, unknown. We therefore evaluated renal TIR8 expression during Escherichia coli pyelonephritis and explored its role in host defense using TIR8−/− versus TIR8+/+ mice. We found that TIR8 protein is abundantly present in the majority of cortical tubular epithelial cells. Pyelonephritis resulted in a significant downregulation of TIR8 mRNA in kidneys of TIR8+/+ mice. TIR8 inhibited an effective host response against E. coli, as indicated by diminished renal bacterial outgrowth and dysfunction in TIR8−/− mice. This correlated with increased amounts of circulating and intrarenal neutrophils at the early phase of infection. TIR8−/− tubular epithelial cells had increased cytokine/chemokine production when stimulated with lipopolysaccharide (LPS) or heat-killed E. coli, suggesting that TIR8 played an anti-inflammatory role during pathogen stimulation by inhibiting LPS signaling. These data suggest that TIR8 is an important negative regulator of an LPS-mediated inflammatory response in tubular epithelial cells and dampens an effective antibacterial host response during pyelonephritis caused by uropathogenic E. coli.
INTRODUCTION
The innate immune system employs Toll-like receptors (TLRs) to sense a wide range of pathogen-associated motifs and to trigger innate immune responses that eventually lead to the clearance of pathogens from the host and the establishment of adaptive immunity to control any subsequent infection (6). However, an excessive and prolonged immune response may result in severe collateral organ damage and even the risk of multiple-organ failure, shock, and death. This implies that the immune system has to continuously find a balance between activation and inhibition of TLRs to battle pathogens and to avoid harmful inflammation. To achieve immune homeostasis, negative-feedback regulation of TLR signaling is crucial. More than a decade ago, a negative regulator of TLR/interleukin-1 receptor 8 (IL-1R-8) signaling, named TIR8 (Toll IL-1R-8) or SIGIRR (single immunoglobulin domain IL-1R-related molecule) was discovered (35) and shown to sequester the formation of TLR signaling complexes, leading to dampening of an inflammatory response (28). TIR8 is highly conserved among vertebrates (29), and its mRNA is predominantly expressed in the kidney (14, 26, 38), especially in tubular epithelial cells (TECs) (21, 26). This conservation and specific renal epithelial expression pattern suggest that TIR8 is under strong evolutionary pressure and, as such, is an important molecule in the kidney. The biological role of renal TIR8 during renal infection, however, remains unknown.
Renal epithelial cells are a crucial component of the innate immune system as they are the first to encounter ascending pathogens and respond to microbial challenge by initiating an inflammatory response that leads to the recruitment and activation of neutrophils. In addition to TIR8, renal epithelial cells constitutively express TLR4 (36, 39). TLR4 signaling is negatively regulated by TIR8 (28) and is essential for the innate resistance of the urinary tract to invasion with uropathogenic Escherichia coli (4, 11, 15, 34). Mice deficient or mutant in TLR4 are highly susceptible to experimental urinary tract infection (UTI) and unable to properly clear E. coli from the urinary tract (11, 15, 34). The susceptibility of these mice to UTI was related to an impaired influx of neutrophils (31), and TLR4 on renal cells appeared to be involved in the recruitment of these inflammatory cells to the primary site of infection (25). Children with and without anatomic abnormalities who have a TLR4 Asp(299)Gly mutation are furthermore at increased risk of UTI (19). Considering that TIR8 is able to specifically inhibit TLR4-mediated signaling and inflammation (28), this receptor could be crucial for host defense against UTI and avoidance of harmful inflammation. UTIs are considered one of the most common infections affecting mankind (10), with a major health and economic burden (10). Therefore, novel insights into the factors that contribute to the battle against UTIs are warranted.
In the present project we investigated the role of TIR8 during experimental UTI as TIR8 may be an important determinant of whether renal host defense or injury prevails and as it might unravel the reason for the conserved and high, organ-specific expression of this receptor.
MATERIALS AND METHODS
Bacteria.
E. coli 1677 was isolated from a patient with UTI and donated by W. J. Hopkins (University of Wisconsin Medical School, Madison, WI). Virulence characteristics include type 1 and P fimbriae, hemolysin, aerobactin, and the O6 serotype (8). The strain was cultured for 16 h at 37°C in Trypticase soy broth (TSB). After dilution, the suspension was grown to mid-logarithmic phase, washed, and resuspended in sterile phosphate-buffered saline (PBS).
Mice.
Pathogen-free, 8- to 10-week-old female C57BL/6 wild type ([wt] TIR8+/+) mice were purchased from Charles River Breeding Laboratories. TIR8−/− mice were a kind gift of Alberto Mantovani and Cecilia Garlanda (Istituto Clinico Humanitas, IRCCS, Rozzano, and University of Milan, Italy); they were generated as described previously (14), backcrossed to a C57BL/6 background six times, and bred in the animal facility of the Academic Medical Center in Amsterdam, The Netherlands. Age- and sex-matched mice were used in all experiments. The Animal Care and Use Committee of the University of Amsterdam approved all experiments.
Pyelonephritis.
Pyelonephritis (upper UTI) was induced as described before (7, 24). Under general anesthesia (0.07 ml/10 g of body weight of FFM mixture, containing 1.25 mg/ml midazolam [Roche], 0.08 mg/ml fentanyl citrate, and 2.5 mg/ml fluanisone [Janssen Pharmaceutica]) TIR8+/+ and TIR8−/− mice (n = 7 to 11 per group) were administered via the urethra 5 × 108 CFU per mouse in a volume of 100 μl, as assessed by plating 10-fold serial dilutions of the suspension on blood agar plates. Administering this volume did not lead to vesicoureteral reflux of bacteria, as determined by plating kidney homogenate of 3 animals 10 min after bacterial injection. Animals were sacrificed 4, 8, 24, and 48 h after inoculation. Sham control mice underwent the same procedure with administration of sterile PBS instead of bacterial suspension. At the time of sacrifice, blood was collected by heart puncture in heparin-containing tubes, and kidneys were harvested for further analysis.
Bacterial outgrowth.
Kidneys were homogenized in 4 volumes of sterile saline with a tissue homogenizer (Polytron PT1300D homogenizer; Kinematica AG), and 10-fold serial dilutions were plated onto blood agar plates to determine bacterial loads. Colonies were counted 16 h after incubation at 37°C.
Renal function.
Renal function was determined by measuring creatinine in plasma samples by enzyme reactions involving creatinase and using standard autoanalyzer methods by our hospital research services. Systemic creatinine is used as a traditional biomarker of renal tubular injury and renal dysfunction.
Oxidative burst.
Oxidative burst of neutrophils was analyzed by flow cytometry according to Kampen et al. (18). Whole blood (2.5 × 105 white blood cells) of TIR8+/+ and TIR8−/− mice was loaded with 1.5 mg/ml dihydrorhodamine 123 (DHR; Sigma) for 30 min. Neutrophils in whole blood were stimulated by the addition of 1.6 × 105 CFU of E. coli 1677 for 30 min, followed by staining with anti-mouse Ly6G-allophycocyanin (APC) (BD Pharmingen) and red blood cell lysis for 10 min at 4°C with 160 mM NH4Cl, 10 mM KHCO3, and 0.1 mM EDTA (pH 7.4). Cells were analyzed by fluorescence-activated cell sorting (FACS) (Becton Dickinson). Conversion of dihydrorhodamine in neutrophils was determined by gating the Ly6G-positive (Ly6G+) population.
White blood cell count and differentiation.
White blood cell (WBC) counts in peripheral blood were determined using a Coulter counter (Beckmann Coulter, Fullerton, CA). For differentiation, 2 μl of whole blood was used for blood smear and stained with Giemsa (Diff-Quick; Baxter, McGraw Park, Il). A total of 100 WBCs were counted in a high-magnification (×400) field, (after which percentages of different cell types were determined.
MPO ELISA.
As the influx of granulocytes during pyelonephritis is very focal, we used a myeloperoxidase (MPO) enzyme-linked immunosorbent assay (ELISA) for quantification. For MPO measurements, snap-frozen kidneys were diluted in lysis buffer containing 75 mM NaCl, 7.5 mM Tris, 0.5 mM MgCl2, 0.5 mM CaCl2, 0.5% Triton X-100, and 1% protease inhibitor cocktail II (Sigma Chemical Co.); samples were homogenized and incubated at 4°C for 30 min. Homogenates were subsequently centrifuged at 10,000 × g at 4°C for 10 min, and MPO in the supernatant was measured by ELISA according to the manufacturer's protocol (Hycult Biotechnology).
Primary culture of mouse renal TECs.
Primary mouse renal TECs were isolated as described by Wuthrich et al. (41). Briefly, the renal capsule was removed, and tissue from the outer cortex was cut into small pieces. Then, single cortical tubular cell suspensions of freshly dissected kidney cortices from TIR8+/+ and TIR8−/− mice (5 mice per group) were prepared by collagenase type 1A (Sigma Chemical Co.) dispersion at 37°C for 1 h and washed in RPMI 1640 medium (Gibco-BRL). TECs were subsequently grown in duplicates to 80% confluence on six-well plates in Dulbecco's modification of Eagle's medium (DMEM)-Ham's F12 50/50 mix supplemented with 10% fetal calf serum (FCS; Integro), 5 μg/ml insulin, 5 μg/ml transferrin, 5 ng/ml sodium selenite, 20 ng/ml triiodothyronine, 50 ng/ml hydrocortisone, 5 ng/ml prostaglandin E1 (all from Sigma), 100 IU/ml penicillin, and 100 μg/ml streptomycin (both from Life Technologies, Inc.). Cells exhibited the cobblestone morphology of epithelial cells as seen by phase-contrast microscopy and were screened positive for ZO-1 mRNA as assessed by reverse transcription-PCR (RT-PCR). TECs were subsequently incubated for 24 h with either conditioned medium, lipopolysaccharide (LPS; 10 ng/ml), or heat-killed E. coli 1677 (108 microorganisms/ml). Supernatants were subsequently collected and stored at −80°C until ELISAs were performed. The number of viable cells present in the wells after stimulation was determined by using trypan blue, after which cells were counted. Possible contamination of leukocytes was ruled out by RT-PCR for CD45 and CD11b with primary murine macrophages as a positive control.
ELISA.
Cytokines and chemokines (tumor necrosis factor alpha [TNF-α], IL-1β, IL-6, keratinocyte chemoattractant [KC], macrophage inflammatory protein 2 [MIP-2] or CXCL2, and monocyte chemoattractant protein 1 [MCP-1] or CCL2) were measured in supernatants or renal homogenate using specific ELISAs (R&D Systems) according to the manufacturer's instructions. The detection limits were 31 pg/ml for TNF-α, 8 pg/ml for IL-1, 16 pg/ml for IL-6, 12 pg/ml for KC, 94 pg/ml for CXCL2, and 7 pg/ml for CCL2. Cytokine levels were corrected for the amount of protein present using a Bio-Rad protein assay (Bio-Rad Laboratories) with IgG as the standard.
Detection of TIR8 by immunohistochemistry.
Kidneys were snap-frozen in liquid nitrogen and stored at −80°C prior to use. For histological detection of TIR8, kidneys were cut into 5-μm sections and fixed for 10 min with ice-cold acetone and air dried. The sections were incubated in 5% normal swine serum (Dako) for 10 min and then exposed overnight to goat anti-mouse SIGIRR/TIR8 antibody (Ab) (R&D Systems) in PBS. Endogenous peroxidase activity was quenched by a solution of 0.1% NaN3/0.3% H2O2 (Merck) in PBS. Slides were incubated with swine anti-goat-horseradish peroxidase (HRP) Ab (Biosource) with 5% normal mouse serum, rinsed again, and developed using 0.015% H2O2 and diaminobenzidine (DAB; Sigma) in 0.05 M Tris-HCl (pH 7.9).
RT-PCR.
Total RNA was extracted from renal tissue sections or stimulated TECs with TRIzol reagent (Invitrogen) and converted to cDNA. TIR8 and TLR4 gene expression was analyzed by RT-PCR using SYBR green PCR master mix. Specific gene expression was normalized to expression of the household gene hypoxanthine phosphoribosyltransferase (HPRT). SYBR green dye intensity was analyzed with linear regression analysis. The following primer sequences were used: TLR4, 5′-GGACTCTGATCATGGCACTG-3′ (forward) and 5′-CTGATCCATGCATTGGTAGGT-3′ (reverse); TIR8, 5′-GTGGCTGAAAGATGGTCTGGCATTG-3′ (forward) and 5′-CAGGTGAAGGTTCCATAGTCCTCTGC-3′ (reverse); MyD88, 5′-TGGCCTTGTTAGACCGTGA-3′ (forward) and 5′-AAGTATTTCTGGCAGTCCTCCTC-3′ (reverse); IRAK1, 5′-CACAAGGTGCAAAGACCAAGT-3′ (forward) and 5′-CACAGAGTAGGCTGGGTGCT-3′ (reverse); TRAF6, 5′-AAGCCTGCATCATCAAATCC-3′ (forward) and 5′-GATTTTCCAGCAGTATTTCATTGTC-3′ (reverse); TAK1, 5′-CTGGGACAGAACCTGGTCA-3′ (forward) and 5′-GTCCTGAGGTAGTGATCATTCTGA-3′ (reverse); ICAM-1, 5′-CTTCTGAGCGGCGTCGAGCC-3′ (forward) and 5′-GCCGAGGACCATACAGCACGT-3′ (reverse); CXCR2, 5′-CAATTACAGGGAGAAAGAGGTCA-3′ (forward) and 5′-TCAGTAGGCATACCAAGATGGA-3′ (reverse); CCR2, 5′-ACCTGTAAATGCCATGCAAGT-3′ (forward) and 5′-TGTCTTCCATTTCCTTTGATTTG-3′ (reverse); HPRT, 5′-TCCTCCTCAGACCGCTTTT-3′ (forward) and 5′-CCTGGTTCATCATCGCTAATC-3′ (reverse).
Immunohistochemical detection of renal inflammatory infiltrate.
Sections of 5 μm were deparaffinized, and antigen retrieval was performed by 0.25% pepsin digestion (Sigma) in 0.1 M HCl for granulocyte detection. Slides were subsequently exposed to fluorescein isothiocyanate (FITC)-labeled anti-mouse Ly6G monoclonal antibody (MAb; BD-Pharmingen) and rabbit anti-FITC antibody (Dako Cytomation), which was followed by a further incubation with HRP-conjugated goat anti-rabbit IgG (Immunovision Technologies Co.). The slides were developed using 1% H2O2 and DAB (Sigma-Aldrich) in 0.05 M Tris-HCl (pH 7.9) and counterstained with methyl green (Sigma-Aldrich). The number of positive cells during renal ischemia-reperfusion was counted within 10 nonoverlapping fields (magnification, ×400).
Plasma biochemical analysis.
Renal function was determined by measuring creatinine and urea in plasma samples by enzyme reactions involving creatinase and urease and using standard autoanalyzer methods by our hospital research services.
Statistics.
All data are expressed as means± standard errors of the means (SEM). Serial data were analyzed using a Kruskal-Wallis test. Differences between TIR8+/+ and TIR8−/− mice were analyzed by a Mann-Whitney U test. Differences in survival between the groups of mice were compared by a chi-square test. A P value of <0.05 was considered to represent a statistically significant difference.
RESULTS
TIR8 expression in the kidney during experimental upper UTI.
To determine whether TIR8 is modulated during pyelonephritis, TIR8+/+ mice were inoculated with uropathogenic E. coli and killed at different time points after infection. Real-time quantitative PCR revealed that TIR8 mRNA was constitutively present in the kidneys and was altered during the course of infection (Fig. 1a). Eight hours after bacterial inoculation, renal TIR8 mRNA was significantly downregulated while the mRNA levels started to return to baseline levels after 24 h. Immunohistochemical analysis of TIR8 protein demonstrated extensive TIR8 staining in kidneys of normal, healthy mice (Fig. 1b and c). TIR8 was abundantly present in the majority of tubular epithelial cells of the cortex. Expression was predominantly located at the apical side of renal proximal tubules and extended into the cytoplasm (Fig. 1d). Medulla, glomeruli (Fig. 1e, g), vessels (Fig. 1e, V), renal pelvis, papilla, and ureter were all negative for TIR8 staining (Fig. 1b and c). Analysis of kidneys obtained from TIR8−/− mice, used as negative controls, confirmed the specificity of the TIR8 staining (Fig. 1f). Kidneys from infected mice displayed the same patterns of TIR8 positivity as kidneys from healthy TIR8+/+ mice; i.e., TECs stained positive for TIR8 (data not shown).
Fig 1.

TIR8 expression and localization in kidneys from TIR8+/+ mice. (a) Renal TIR8 mRNA expression from TIR8+/+ mice with upper urinary tract infection as determined by semiquantitative PCR. Data are means and SEM of 6 to 8 mice per group. *, P < 0.05. (b to e) Immunohistochemical analysis of TIR8 protein in kidney from TIR8+/+ mice (G, glomerulus; V, vessel). (f) Analysis of kidneys obtained from TIR8−/− mice, used as negative controls, confirmed the specificity of the TIR8 staining. Magnifications, ×20 (b), ×100 (c and f), ×400 (d), and ×200 (e); scale bars, 5 μm (b), 100 μm (c and f), 20 μm (d), and 50 μm (e).
TIR8 impairs bacterial clearance and renal function during experimental upper UTI.
To investigate whether the profound expression of renal TIR8 had an effect on the outcome of infection, we examined bacterial loads in kidneys from TIR8+/+ and TIR8−/− mice at 24 and 48 h after induction of UTI. One day after inoculation, TIR8−/− mice had 4,200 times less bacterial load in their kidney homogenates than TIR8+/+ mice (P < 0.01) (Fig. 2a). At 48 h after induction of UTI, no differences could be found. Sham control animals did not have bacteria in their kidneys. Mice with UTI had impaired renal function 24 h after infection, as reflected by a modest elevation of serum creatinine levels (Fig. 2b). Renal dysfunction, however, was significantly reduced in TIR8−/− mice compared to TIR8+/+ mice at 24 h after infection. After 48 h UTI starts to resolve, as reflected by the complete recovery of baseline renal function. These data demonstrate that TIR8 conveys early sensitivity to UTI.
Fig 2.
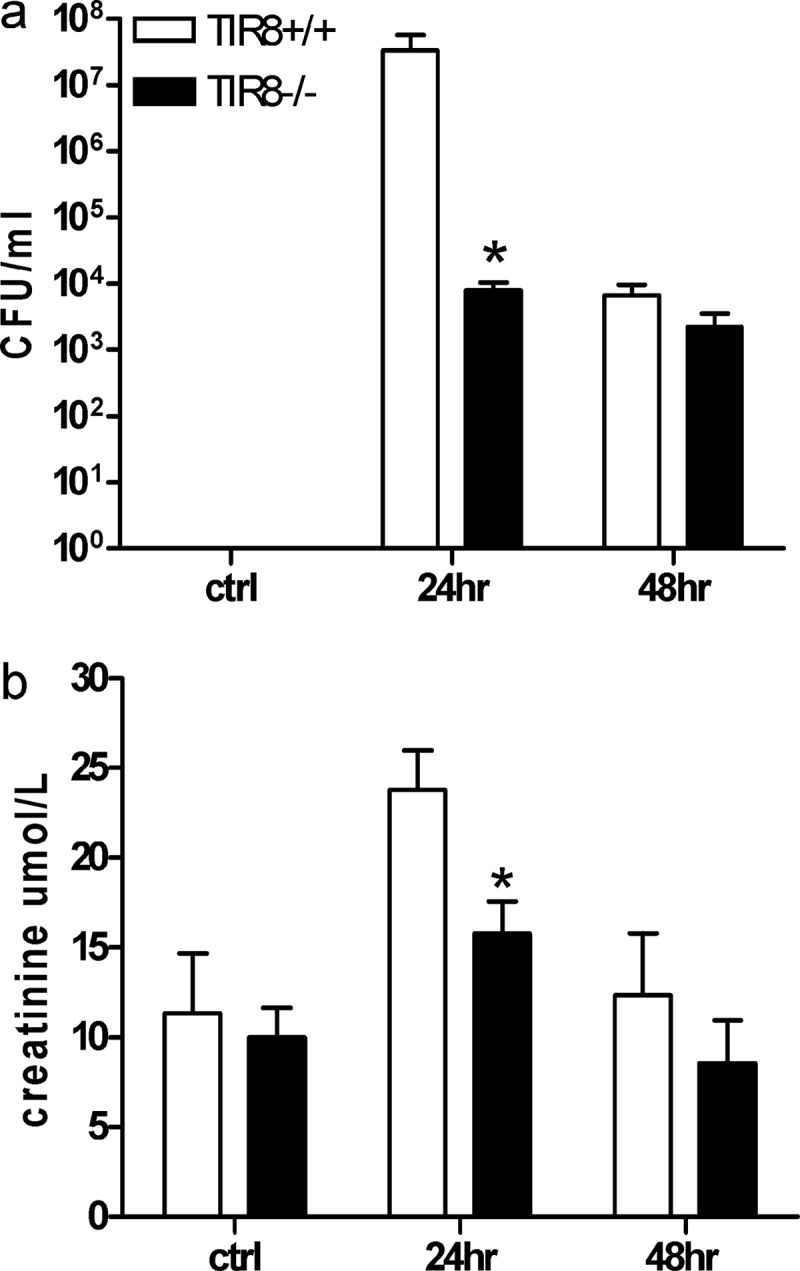
Bacterial outgrowth and renal function. The effect of TIR8 deficiency on outgrowth of uropathogenic E. coli in kidney homogenate (a) and on renal function (b) at 24 and 48 h after transurethral bacterial challenge in TIR8+/+ and TIR8−/− mice. Data represent means ± SEM of 8 mice per group. *, P < 0.05. Ctrl, control.
TIR8 is not essential for neutrophil oxidative burst in response to uropathogenic E. coli.
Neutrophils are a requisite component of the host defense against uropathogenic E. coli and express low levels of TIR8 mRNA (26). The improved bacterial clearance from the urinary tract of TIR8−/− mice may, therefore, be intrinsic to the neutrophil itself or may result from changes in the number of available neutrophils. To establish if TIR8 is critical for the antimicrobial mechanism of neutrophils, blood cells from TIR8−/− and TIR8+/+ mice were stimulated with uropathogenic E. coli, and oxidative burst activity of neutrophils was analyzed using a dihydrorhodamine-based flow cytometry assay. Neutrophils from both TIR8−/− and TIR8+/+ mice had increased respiratory burst responses when mice were challenged with E. coli (Fig. 3a and b). However, no differences were observed in the total numbers of neutrophils undergoing burst activity in TIR8−/− and TIR8+/+ neutrophils (Fig. 3a). In addition, the levels of mean fluorescence intensity (MFI), corresponding to the mean oxidative burst activity per cell, were similar in stimulated TIR8−/− and TIR8+/+ neutrophils. These results suggest that the improved bacterial clearance in TIR8−/− mice is not due to an abnormal E. coli-stimulated oxidative burst response of neutrophils.
Fig 3.
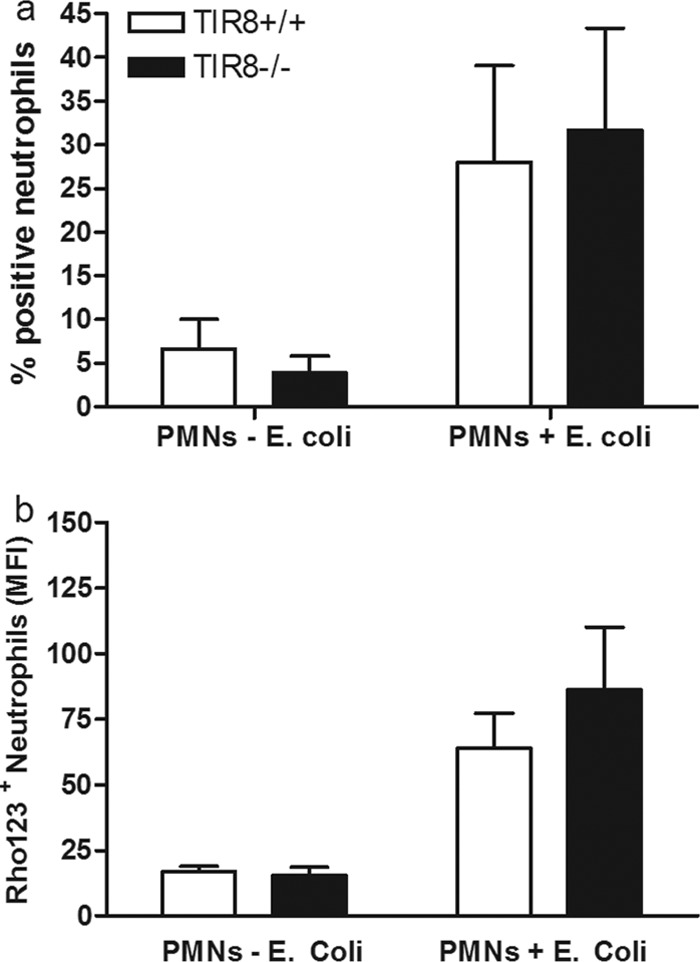
Neutrophil respiratory burst activity in response to uropathogenic E. coli. Oxidative burst of TIR8+/+ and TIR8−/− granulocytes after infection with E. coli. With the use of a dihydrorhodamine 123 (Rho123)-based flow cytometry assay, the percentage of stimulated neutrophils undergoing oxidative burst (a) and the mean fluorescence intensity (MFI) per cell (b) were measured in neutrophils from blood of TIR8+/+ and TIR8−/− mice. Data are means and SEM (n = 4 per group).
TIR8 impairs an early increase in circulating and intrarenal recruitment of neutrophils during experimental upper UTI.
We next determined the impact of TIR8 deficiency on the number of available neutrophils. For this, we infected TIR8+/+ and TIR8−/− mice with E. coli and sacrificed them 4 and 8 h thereafter. At these early time points, the bacterial loads did not differ between the mouse strains (data not shown), indicating that the improved bacterial clearance in TIR8−/− mice 24 h after infection might occur due to an impact of TIR8 deficiency on the immune response in the kidneys at this early phase. We first assessed whole-blood samples from infected TIR8+/+ and TIR8−/− mice and found that TIR8 deficiency was associated with an increased number of neutrophils in blood 4 h after infection (Fig. 4a), whereas no significant differences were found in the amounts of lymphocytes (Fig. 4b). In addition, a moderate increase in circulating monocyte numbers was seen in TIR8−/− mice compared to levels in TIR8+/+ mice at 4 h after infection (Fig. 4c). We next investigated whether the enhanced number of neutrophils in the circulation corresponded with an increase in tissue-associated neutrophils. Indeed, we found that renal neutrophil influx, as measured by tissue MPO activity, was more profound in TIR8−/− mice than in TIR8+/+ mice at 8 h after infection (Fig. 4d). In line with this observation, we found that TIR8−/− mice had more granulocytes in the pyelum region after infection than TIR8+/+ mice (Fig. 4e). At the later phases of UTI (24 and 48 h), TIR8−/− mice had lower MPO concentrations (ng/mg of protein) in their kidneys than TIR8+/+ mice (at time t = 24 h, 32.9 ± 9.5 for TIR8+/+ mice and 13.0 ± 3.2 for TIR8−/− mice, P = 0.087; at t = 48 h, 42.0 ± 10.9 for TIR8+/+ mice and 13.5 ± 3.0 for TIR8−/− mice, P = 0.042), probably reflecting the reduced bacterial load in these mice after 24 h. Together, these data indicate that TIR8 deficiency is associated with enhanced neutrophil mobilization and recruitment to the kidneys in the early phase of the infection.
Fig 4.

Leukocyte dynamics in response to infection: neutrophil (a), lymphocyte (b), and monocyte (c) cell counts in blood, renal MPO levels (d), and neutrophil influx in the renal pyelum region (e) after transurethral bacterial challenge in TIR8+/+ and TIR8−/− mice. Data in panels a to d represent means ± SEM of 6 to 8 mice per group. *, P < 0.05.
TIR8 expressed in renal epithelial cells functions as a negative regulator of a TLR4-mediated inflammatory response.
To further elucidate the mechanism underlying the enhanced intrarenal neutrophil recruitment in TIR8-deficient mice, we analyzed the inflammatory response of tubular epithelial cells to the TLR4 ligand LPS and heat-killed uropathogenic E. coli. We found that LPS and heat-killed E. coli led to a significant increase of the proinflammatory molecules KC, CXCL2 (MIP-2), CCL2 (MCP-1), and TNF-α in TIR8+/+ TECs (Fig. 5b) and increased mRNA levels of MyD88, IRAK1, IRAK4, and TAK1 (see Fig. S1 in the supplemental material) that together indicate the activation of the MyD88-dependent TLR4 signaling pathway. To study whether TIR8 would modulate the MyD88-dependent inflammatory cytokine response, we also stimulated TIR8−/− TECs, which revealed that LPS-stimulated primary TECs of TIR8−/− mice produce significantly more of the proinflammatory mediators KC, CXCL2, CCL2, and TNF-α than TECs from TIR8+/+ mice (Fig. 5b). In line with this, we found increased levels of KC, CCL2, and TNF-α in supernatants of TIR8−/− TECs when they were stimulated with heat-killed E. coli compared with levels in TIR8+/+ TECs (Fig. 5b). No difference could be observed in IL-6 production levels between TIR8−/− and TIR8+/+ TECs when they were stimulated with either LPS or heat-killed bacteria (data not shown). Hence, TEC-associated TIR8 functions as a negative regulator for the TLR4-mediated inflammatory response against uropathogenic E. coli.
Fig 5.

Inflammatory response of tubular epithelial cells. Graphs show levels of cytokines and chemokines in supernatants obtained from TIR8+/+ and TIR8−/− TECs after stimulation with LPS or heat-killed (HK) uropathogenic E. coli. Data are means ± SEM (n = 5 per group). *, P < 0.05.
In support of this notion, we found increased levels of KC and MCP-1 in TIR8−/− kidneys compared to levels in TIR8+/+ kidneys at 8 h after infection (for KC, 27.1 ± 0.7 pg/mg of protein in TIR8+/+ mice versus 76.4 ± 49.9 pg/mg of protein in TIR8−/− mice; for MCP-1, 1.6 ± 0.2 pg/mg of protein in TIR8−/− mice versus 3.1 ± 1.8 pg/mg of protein in TIR8+/+ mice). Unfortunately, this difference did not reach statistical significance due to high variations in the TIR8−/− group. No differences were found between the control groups and the other groups at 4-h postinfection (data not shown). Moreover, we analyzed the expression of intercellular adhesion molecule 1 (ICAM-1), which is a crucial adhesion molecule for the migration of polymorphonuclear leukocytes (PMNs) to the kidney during ascending pyelonephritis (1). This revealed a trend toward increased ICAM-1/HPRT mRNA expression in TIR8−/− kidneys at t = 8 compared to TIR8+/+ kidneys (TIR8+/+, 0.95 ± 0.06 for TIR8+/+ versus TIR8−/− 1.78 ± 0.74, ICAM-1/HPRT), while values for sham-operated animals and animals at 4 h postinfection were similar (data not shown). The expression levels of chemokine receptors CXCR2 and CCR2 were not statistically different in all tested groups and could not, therefore, explain the difference in PMN influx levels (data not shown).
To study whether differences in bacterial clearance and inflammatory response can also be explained by a modulatory effect of TIR8 on TLR4 expression, we next analyzed the expression of TIR8 and TLR4 in stimulated TECs and kidneys from infected mice. This revealed that TIR8 does not modulate the mRNA expression of TLR4 either in vitro (Fig. 6a) or in vivo (Fig. 6b).
Fig 6.

TLR4 mRNA expression. (a) Expression of TLR4 mRNA by TECs from TIR8+/+ and TIR8−/− mice stimulated with LPS or heat-killed (HK) uropathogenic bacteria. Data are means ± SEM (n = 5 per group). (b) TLR4 mRNA expression in kidneys from TIR8+/+ and TIR8−/− mice with upper urinary tract infection as determined by semiquantitative PCR. Data are means and SEM of 6 to 8 mice per group.
DISCUSSION
To ensure that the host survives infections without inducing inflammation-driven collateral tissue damage and loss of vital organ function, the immune system has to achieve and maintain a delicate equilibrium between proinflammatory and anti-inflammatory processes. TIR8 is a key regulator in inflammation, cancer-related inflammation, and autoimmunity as it has the ability to inhibit TLR signaling by trapping TLR signaling complexes, which leads to the diminishing of an inflammatory response (12). TIR8 mRNA is predominantly expressed in the kidney, in particular, in TECs. As TLRs and renal epithelium control innate immunity during UTI, we evaluated the expression of TIR8 during upper UTI and explored its role in host defense by comparing TIR8−/− to TIR8+/+ mice. We found that renal epithelium-associated TIR8 inhibits an efficient host response against uropathogenic E. coli during UTI. TIR8 deficiency caused a decreased outgrowth of bacteria in the kidney, which was associated with an enhanced LPS-mediated inflammatory response of renal TECs, an early increase of circulating and tissue-associated neutrophils, and reduced renal dysfunction during infection.
Data of the role of TIR8 in host defense against infection are highly limited. In a murine model of pulmonary tuberculosis and acute Pseudomonas aeruginosa lung infection, TIR8−/− mice were shown to have increased mortality (13, 37). Huang et al. (16) reported that mice treated with anti-TIR8 Ab had increased stromal damage and bacterial load in the cornea in a model of Pseudomonas aeruginosa keratitis. Moreover, it was reported that TIR8-deficient mice had a decreased survival and increased fungal burden in response to candidiasis (2). To the best of our knowledge, other investigations on the role of TIR8 in infection have not been reported. The role of the predominant expression of TIR8 in the kidney during infection remained unknown. The results of the present study demonstrate a crucial role of TIR8 in the early host defense against upper UTI. The lack of TIR8 led to an increase in the amount of circulating and renal tissue-specific neutrophils within a few hours after infection. Consequently, early host defense against uropathogenic bacteria was strongly improved in TIR8−/− mice, leading to an increase in bacterial clearance and a more preserved renal function. Although TIR8+/+ mice eventually recovered and overcame infection with uropathogenic E. coli at almost the same level as TIR8−/− mice, early clearance of the bacteria from the kidneys was highly inefficient. Conceivably, the unfavorable effect of TIR8 during UTI depends on the stage of the infection, becoming less important as the clearance of the bacteria is efficient. Alternatively, the absence of TIR8 in TIR8−/− mice may be compensated for by other mechanisms at later stages of infection.
What could be the mechanism by which TIR8 dampens an inflammatory response during infection with uropathogenic E. coli? We found that endogenous TIR8 on TECs plays an anti-inflammatory role during pathogen stimulation by inhibiting LPS signaling. This is reflected by increased cytokine (TNF-α) and chemokine (KC, CCL2, and CCL3) production in TIR8−/− TECs compared to TIR8+/+ TECs when they are stimulated with LPS or heat-killed uropathogenic E. coli. As these chemokines and cytokines are well-known MyD88-dependent pathway molecules and as other investigators have shown that TIR8 is involved in the negative regulation of the MyD88-dependent TLR signaling (38), it seems reasonable to suggest that the MyD88-dependent TLR4 signaling pathway was affected in TIR8−/− TECs after pathogenic stimulation. These in vitro observations correspond to our in vivo data, in which we found enhanced circulating leukocytes and intrarenal granulocytes in TIR8-deficient mice and a mild increase, although not statistically different, in local chemokines and adhesion molecule ICAM-1. Apparently, the small differences in chemokines and ICAM-1 are large enough to lead to differences in PMN influx levels in kidneys between TIR8−/− and TIR8+/+ mice. Alternatively, differences in other unidentified factors might play an additional role in the difference in PMN influx levels. Together, these results show that renal cells can modulate local immune responses that are induced by the TLR4 signaling pathway via TIR8. This ascribes a much more active role to epithelial cells in the regulation of local immune responses than anticipated so far. The Anders group previously showed that TIR8 suppresses chemokine and cytokine production in renal resident myeloid cells or CD45-positive cells (21, 33). It could therefore be that some of the differences we found in inflammation and bacterial clearance are mediated as well by leukocyte-associated TIR8. In contrast to our results, the Anders group did not find a difference in cytokine/chemokine responses in primary TECs, with or without TIR8, that are exposed to LPS in vitro (21, 33). One plausible explanation could be that in these studies the amounts of cytokines and chemokine are not normalized to the amounts of (viable) cells that were stimulated. Since TIR8 is involved in cell survival, proliferation, and apoptosis of epithelial cells (9, 42) and since LPS can induce cell death in tubular epithelial cells (3), we chose to correct the level of produced chemokines for the amount of viable TECs. In line with our results, Wald et al. found that TIR8-deficient primary renal epithelial cells had an increase in their NF-κB DNA binding activities in response to IL-1 and LPS (38). Accordingly, it was shown that the activation of NF-κB of LPS-stimulated human alveolar epithelial cells was inhibited by TIR8 overexpression (9) and that LPS-mediated IL-8 production was significantly greater in intestinal epithelial cells transfected with TIR8 small interfering RNA (siRNA) (17). However, one has to keep in mind that using LPS and heat-killed E. coli as a cell stimulus is a simplification of the system. Using living bacteria carrying a heterogeneous and dynamic set of molecules might be more suitable to dissect the response-inducing agonists or mechanisms, and this possibility warrants further investigation.
The differences that were found in microbial outgrowth between different infection models indicate that TIR8 has a pathogen- and organ-specific role in host defense against infection. Studies on the expression of TIR8 demonstrate a highly conserved pattern among different animal species from humans to ungulates, birds, carnivores, and rodents (29). In a wide panel of organs and tissues of these animal species, TIR8 mRNA is, overall, most abundantly expressed in the kidney (14, 26, 29, 38), especially on tubular epithelial cells (21, 26; also this study). This conservation and organ-specific expression in different animal species suggest that TIR8 is a crucial receptor in the kidney. Indeed, we found that the level of TIR8 mRNA is altered in the kidney during infection, suggesting that this receptor is important for regulating inflammatory responses in this organ. TIR8 mRNA was significantly downregulated in the kidney at early time points and started to increase toward baseline levels at later time points after UTI. It can be speculated that this downregulation would enhance an inflammatory response directed against renal infection or elimination of debris during tissue injury. Indeed, TIR8−/− mice with infected or lethally injured kidneys had increased inflammatory responses compared to TIR8+/+ mice. The subsequent upregulation of TIR8 later during the infection could, in its turn, avoid the induction of collateral damage due to an exaggerated or prolonged inflammatory response. This corresponds with studies demonstrating that TIR8 deficiency renders mice more susceptible to acute renal ischemic injury (20), acute graft rejection, and tubular necrosis after kidney allotransplantation (23), while mice with TLR4 and TLR2 deficiencies are less susceptible to acute renal ischemic injury (22, 27, 32, 40). The high level of constitutive TIR8 expression in the kidney during homeostasis could be a mechanism by which organisms try to avoid the induction of a disproportional and detrimental inflammatory response when only few pathogens are present. Although ascending bacteria (4, 5) and TIR8 expression (this study) in the kidney seem to be located in different compartments in the kidney, it might be envisioned that residual bacterial antigens or tissue injury debris that persists subsequent to bacterial killing is rapidly spread through the renal tissue and in this way can reach and activate TLR4- and TIR8-positive tubular cells.
UTI is one of the most common infections afflicting mankind, with significant medical and financial implications. The acute inflammatory response during UTI is decisive for an effective host defense but also for the development of renal damage and scarring (30). Our findings imply an important role for tubular epithelial cell-associated TIR8 in the regulation of microbial-mediated inflammatory responses and the host defense against upper UTI.
Supplementary Material
ACKNOWLEDGMENTS
TIR8−/− mice were a kind gift of Alberto Mantovani (Istituto Clinico Humanitas, IRCCS, Rozzano, Italy, and University of Milan, Milan, Italy) and Cecilia Garlanda (Istituto Clinico Humanitas, IRCCS, Rozzano, Italy).
This study was supported by a grant from the Dutch Kidney foundation to L.M.B. (C10-2350), I.S. (C08.2277), and W.P.P. (IP10.20) and from The Netherlands Organization for Scientific Research to J.C.L. (016.126.386).
We have no financial and commercial conflicts of interest.
Footnotes
Published ahead of print 13 August 2012
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1. Agace WW, Patarroyo M, Svensson M, Carlemalm E, Svanborg C. 1995. Escherichia coli induces transuroepithelial neutrophil migration by an intercellular adhesion molecule-1-dependent mechanism. Infect. Immun. 63:4054–4062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bozza S, et al. 2008. Lack of Toll IL-1R8 exacerbates Th17 cell responses in fungal infection. J. Immunol. 180:4022–4031 [DOI] [PubMed] [Google Scholar]
- 3. Bussolati B, David S, Cambi V, Tobias PS, Camussi G. 2002. Urinary soluble CD14 mediates human proximal tubular epithelial cell injury induced by LPS. Int. J. Mol. Med. 10:441–449 [PubMed] [Google Scholar]
- 4. Chassin C, et al. 2006. Renal collecting duct epithelial cells react to pyelonephritis-associated Escherichia coli by activating distinct TLR4-dependent and -independent inflammatory pathways. J. Immunol. 177:4773–4784 [DOI] [PubMed] [Google Scholar]
- 5. Chassin C, Goujon JM, Le BC, Buzoni-Gatel D, Vandewalle A. 2007. A novel function for renal collecting duct intercalated cells: defense against uropathogenic Escherichia coli. Med. Sci. (Paris) 23:32–34 (In French.) [DOI] [PubMed] [Google Scholar]
- 6. Creagh EM, O'Neill LA. 2006. TLRs, NLRs and RLRs: a trinity of pathogen sensors that co-operate in innate immunity. Trends Immunol. 27:352–357 [DOI] [PubMed] [Google Scholar]
- 7. Dessing MC, et al. 2010. S100A8/A9 is not involved in host defense against murine urinary tract infection. PLoS One 5:e13394 doi:10.1371/journal.pone.0013394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Elkahwaji JE, Ott CJ, Janda LM, Hopkins WJ. 2005. Mouse model for acute bacterial prostatitis in genetically distinct inbred strains. Urology 66:883–887 [DOI] [PubMed] [Google Scholar]
- 9. Feng T, et al. 2010. Single immunoglobulin IL-1 receptor-related protein attenuates the lipopolysaccharide-induced inflammatory response in A549 cells. Chem. Biol. Interact 183:442–449 [DOI] [PubMed] [Google Scholar]
- 10. Foxman B. 2002. Epidemiology of urinary tract infections: incidence, morbidity, and economic costs. Am. J. Med. 113(Suppl 1A):5S–13S [DOI] [PubMed] [Google Scholar]
- 11. Frendeus B, et al. 2001. Escherichia coli P fimbriae utilize the Toll-like receptor 4 pathway for cell activation. Mol. Microbiol. 40:37–51 [DOI] [PubMed] [Google Scholar]
- 12. Garlanda C, Anders HJ, Mantovani A. 2009. TIR8/SIGIRR: an IL-1R/TLR family member with regulatory functions in inflammation and T cell polarization. Trends Immunol. 30:439–446 [DOI] [PubMed] [Google Scholar]
- 13. Garlanda C, et al. 2007. Damping excessive inflammation and tissue damage in Mycobacterium tuberculosis infection by Toll IL-1 receptor 8/single Ig IL-1-related receptor, a negative regulator of IL-1/TLR signaling. J. Immunol. 179:3119–3125 [DOI] [PubMed] [Google Scholar]
- 14. Garlanda C, et al. 2004. Intestinal inflammation in mice deficient in Tir8, an inhibitory member of the IL-1 receptor family. Proc. Natl. Acad. Sci. U. S. A. 101:3522–3526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hagberg L, et al. 1984. Difference in susceptibility to gram-negative urinary tract infection between C3H/HeJ and C3H/HeN mice. Infect. Immun. 46:839–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang X, Hazlett LD, Du W, Barrett RP. 2006. SIGIRR promotes resistance against Pseudomonas aeruginosa keratitis by down-regulating type-1 immunity and IL-1R1 and TLR4 signaling. J. Immunol. 177:548–556 [DOI] [PubMed] [Google Scholar]
- 17. Kadota C, et al. 2010. Down-regulation of single immunoglobulin interleukin-1R-related molecule (SIGIRR)/TIR8 expression in intestinal epithelial cells during inflammation. Clin. Exp. Immunol. 162:348–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kampen AH, Tollersrud T, Lund A. 2004. Flow cytometric measurement of neutrophil respiratory burst in whole bovine blood using live Staphylococcus aureus. J. Immunol. Methods 289:47–55 [DOI] [PubMed] [Google Scholar]
- 19. Karoly E, et al. 2007. Heat shock protein 72 (HSPA1B) gene polymorphism and Toll-like receptor (TLR) 4 mutation are associated with increased risk of urinary tract infection in children. Pediatr. Res. 61:371–374 [DOI] [PubMed] [Google Scholar]
- 20. Lech M, et al. 2009. Resident dendritic cells prevent postischemic acute renal failure by help of single Ig IL-1 receptor-related protein. J. Immunol. 183:4109–4118 [DOI] [PubMed] [Google Scholar]
- 21. Lech M, et al. 2007. Different roles of TiR8/Sigirr on Toll-like receptor signaling in intrarenal antigen-presenting cells and tubular epithelial cells. Kidney Int. 72:182–192 [DOI] [PubMed] [Google Scholar]
- 22. Leemans JC, et al. 2005. Renal-associated TLR2 mediates ischemia/reperfusion injury in the kidney. J. Clin. Invest. 115:2894–2903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Noris M, et al. 2009. The Toll-IL-1R member Tir8/SIGIRR negatively regulates adaptive immunity against kidney grafts. J. Immunol. 183:4249–4260 [DOI] [PubMed] [Google Scholar]
- 24. Olszyna DP, et al. 2001. CXC chemokine receptor 2 contributes to host defense in murine urinary tract infection. J. Infect. Dis. 184:301–307 [DOI] [PubMed] [Google Scholar]
- 25. Patole PS, et al. 2005. Toll-like receptor-4: renal cells and bone marrow cells signal for neutrophil recruitment during pyelonephritis. Kidney Int. 68:2582–2587 [DOI] [PubMed] [Google Scholar]
- 26. Polentarutti N, et al. 2003. Unique pattern of expression and inhibition of IL-1 signaling by the IL-1 receptor family member TIR8/SIGIRR. Eur. Cytokine Netw. 14:211–218 [PubMed] [Google Scholar]
- 27. Pulskens WP, et al. 2008. Toll-like receptor-4 coordinates the innate immune response of the kidney to renal ischemia/reperfusion injury. PLoS One 3:e3596 doi:10.1371/journal.pone.0003596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Qin J, Qian Y, Yao J, Grace C, Li X. 2005. SIGIRR inhibits interleukin-1 receptor- and Toll-like receptor 4-mediated signaling through different mechanisms. J. Biol. Chem. 280:25233–25241 [DOI] [PubMed] [Google Scholar]
- 29. Riva F, et al. 2009. The expression pattern of TIR8 is conserved among vertebrates. Vet. Immunol. Immunopathol. 131:44–49 [DOI] [PubMed] [Google Scholar]
- 30. Rushton HG. 1997. Urinary tract infections in children. Epidemiology, evaluation, and management. Pediatr. Clin. North Am. 44:1133–1169 [DOI] [PubMed] [Google Scholar]
- 31. Shahin RD, Engberg I, Hagberg L, Svanborg Eden C. 1987. Neutrophil recruitment and bacterial clearance correlated with LPS responsiveness in local gram-negative infection. J. Immunol. 138:3475–3480 [PubMed] [Google Scholar]
- 32. Shigeoka AA, et al. 2007. TLR2 is constitutively expressed within the kidney and participates in ischemic renal injury through both MyD88-dependent and -independent pathways. J. Immunol. 178:3475–3480 [DOI] [PubMed] [Google Scholar]
- 33. Skuginna V, et al. 2011. Toll-like receptor signaling and SIGIRR in renal fibrosis upon unilateral ureteral obstruction. PLoS One 6:e19204 doi:10.1371/journal.pone.0019204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Svanborg Eden C, Briles D, Hagberg L, McGhee J, Michalec S. 1984. Genetic factors in host resistance to urinary tract infection. Infection 12:118–123 [DOI] [PubMed] [Google Scholar]
- 35. Thomassen E, Renshaw BR, Sims JE. 1999. Identification and characterization of SIGIRR, a molecule representing a novel subtype of the IL-1R superfamily. Cytokine 11:389–399 [DOI] [PubMed] [Google Scholar]
- 36. Tsuboi N, et al. 2002. Roles of Toll-like receptors in C-C chemokine production by renal tubular epithelial cells. J. Immunol. 169:2026–2033 [DOI] [PubMed] [Google Scholar]
- 37. Veliz Rodriguez T, et al. 2012. Role of Toll interleukin-1 receptor (IL-1R) 8, a negative regulator of IL-1R/Toll-like receptor signaling, in resistance to acute Pseudomonas aeruginosa lung infection. Infect. Immun. 80:100–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wald D, et al. 2003. SIGIRR, a negative regulator of Toll-like receptor-interleukin 1 receptor signaling. Nat. Immunol. 4:920–927 [DOI] [PubMed] [Google Scholar]
- 39. Wolfs TG, et al. 2002. In vivo expression of Toll-like receptor 2 and 4 by renal epithelial cells: IFN-gamma and TNF-alpha mediated up-regulation during inflammation. J. Immunol. 168:1286–1293 [DOI] [PubMed] [Google Scholar]
- 40. Wu H, et al. 2007. TLR4 activation mediates kidney ischemia/reperfusion injury. J. Clin. Invest. 117:2847–2859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wuthrich RP, et al. 1990. MHC class II, antigen presentation and tumor necrosis factor in renal tubular epithelial cells. Kidney Int. 37:783–792 [DOI] [PubMed] [Google Scholar]
- 42. Xiao H, et al. 2007. The Toll-interleukin-1 receptor member SIGIRR regulates colonic epithelial homeostasis, inflammation, and tumorigenesis. Immunity 26:461–475 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.