

Abstract
Pneumocystis is an opportunistic fungal pathogen that causes pneumonia in a variety of clinical settings. An early step in Pneumocystis infection involves the attachment of organisms to alveolar epithelial cells (AECs). AECs produce chemokines in response to Pneumocystis stimulation, but the upstream host-pathogen interactions that activate AEC signaling cascades are not well-defined. MyD88 is an adaptor molecule required for activation of proinflammatory signaling cascades following Toll-like receptor (TLR)-dependent recognition of conserved molecular patterns on pathogens. To determine whether the TLR/MyD88 pathway is required for the AEC chemokine response to Pneumocystis, wild-type (WT) and MyD88-deficient AECs were incubated with Pneumocystis. As expected, WT AECs produced CCL2 and CXCL2 following Pneumocystis stimulation. In contrast, MyD88-deficient AECs were severely impaired in their ability to respond to Pneumocystis. MyD88-deficient AECs did not display Pneumocystis-induced Jun N-terminal protein kinase activation and produced much less chemokine than Pneumocystis-stimulated WT AECs. Using a panel of TLR agonists, primary murine AECs were found to respond vigorously to TLR2 and TLR4 agonists. However, the AEC chemokine response to Pneumocystis did not require TLR2 or TLR4. Surprisingly, the interleukin-1 receptor (IL-1R) was required for an AEC chemokine response to Pneumocystis. The role of MyD88 in early responses during Pneumocystis infection was supported by in vivo studies demonstrating that MyD88-deficient mice showed impaired Pneumocystis-stimulated chemokine production and impaired inflammatory cell recruitment. These data indicate an important role for MyD88 in the AEC inflammatory response to Pneumocystis.
INTRODUCTION
Pneumocystis is an opportunistic pathogen that takes advantage of immunocompromised hosts, causing severe, frequently fatal pneumonia. Pneumocystis pneumonia (PCP) remains the most common AIDS-defining illness and remains a common opportunistic infection and a leading cause of death in HIV-infected patients (13, 21). PCP is also becoming more prevalent in a growing population of non-HIV-infected patients, such as patients undergoing long-term anti-tumor necrosis factor (anti-TNF) therapy for rheumatoid arthritis and Crohn's disease (14). Thus, determining the mechanisms by which Pneumocystis causes disease will provide opportunities to improve current treatment.
Although antibiotics effective against Pneumocystis are available, they do not always produce rapid clinical improvement, probably because host inflammation is a key contributor to PCP-related lung injury (19, 33). Early clinical studies of patients with PCP found that the degree of pulmonary inflammation rather than organism burden correlated with the severity of disease (19). In humans and animal models, PCP is associated with elevated numbers of T cells, alveolar macrophages (AMs), and neutrophils in the alveoli (16, 25, 33, 36). Cytokines and chemokines are also produced in the lungs in response to Pneumocystis infection. Lung levels of tumor necrosis factor alpha (TNF-α), gamma interferon (IFN-γ), CCL2, CXCL2, and CCL5 (RANTES) are all elevated during Pneumocystis infection (34, 35). CCL2 or monocyte chemoattractant protein-1 (MCP-1) is a member of the CC chemokine family that mainly recruits AMs and T cells to sites of infection and inflammation. CCL2 contributes to the pathophysiology of chronic lung disease and also modulates the pulmonary immune response to infectious agents (2, 4, 22). Our laboratory has found that CCL2 levels increase in bronchoalveolar lavage (BAL) fluid of mice after Pneumocystis infection and that alveolar epithelial cells (AECs) express CCL2 in vivo during Pneumocystis infection (30). These findings suggest that production of chemokines by AECs initiates and/or sustains inflammatory injury during Pneumocystis infection by recruiting immune cells to the alveoli.
In response to infectious agents and other stimulants, AECs can modulate the pulmonary immune/inflammatory response by secreting cytokines and chemokines. One of the earliest events following entry of Pneumocystis into the lung is the attachment to the alveolar epithelium (18, 26, 38, 39). Importantly, our laboratory has found that Pneumocystis stimulates a proinflammatory AEC response that includes secretion of the chemokines CCL2 and CXCL2 (10, 30, 31, 36). Other studies have reported that purified Pneumocystis β-glucan also induces AEC chemokine production (11). Thus, the interaction of Pneumocystis and AECs is an important early event which likely leads to subsequent immune-mediated inflammatory lung injury. CCL2 release by Pneumocystis-stimulated AECs is mediated through an NF-κB- and mitogen-activated protein kinase (MAPK)-dependent pathway (30, 31). However, the upstream receptors or adaptor proteins involved in this response have not been identified (31). Recent studies have shown that human AECs express certain pathogen recognition receptors (PRRs), including Toll-like receptors (TLRs) (3, 9). Most TLR signaling requires the adaptor molecule myeloid differentiation factor 88 (MyD88), which signals downstream molecules belonging to the NF-κB and MAPK pathways that ultimately control expression of TLR- and MyD88-dependent proinflammatory genes (15).
The current study utilizes in vitro and in vivo approaches to demonstrate that the AEC chemokine response to Pneumocystis is mediated through a MyD88-dependent pathway. These results enhance our understanding of the innate immune pathway utilized by AECs in the early inflammatory responses to Pneumocystis. Understanding the mechanisms by which early events occur during Pneumocystis infection in the lung will identify more specific therapeutic targets that might be used to improve current therapy and modulate inflammatory responses in the lung.
MATERIALS AND METHODS
Mice.
CB.17 severe combined immunodeficient (SCID) and C57BL/6 wild-type mice were bred at the University of Rochester. C57BL6 MyD88−/−, C57BL/6 TLR2−/−, C57BL/6 TLR4−/−, and C57BL/6 mice with a TIR-domain-containing adaptor protein inducing IFN-β (TRIF) knockout (TRIF−/−) were previously generated and generously provided by S. Akira (Osaka University, Japan) (1, 32). TLR2−/− and TLR4−/− mice were crossed to generate TLR2/4−/− double-knockout mice on the C57BL/6 background. All animal protocols were approved by the University Committee for Animal Research (UCAR) at the University of Rochester Medical Center.
Isolation and culture of primary murine AECs.
Primary type II AECs were isolated from mouse lungs as previously described with few modifications (30). Briefly, the lungs were perfused with 1× Hank's balanced salt solution (HBSS). A catheter was inserted into the trachea and 2 ml of dispase solution (BD Biosciences) was instilled, followed by slow instillation of 0.45 ml of low-melting-point agarose (Gibco-BRL) at 45°C. The lungs were cooled on ice for 2 min, removed, and incubated in dispase for 45 min at room temperature. The lung tissue was microdissected and incubated for 10 min in Dulbecco's modified Eagle medium (DMEM) with 0.01% DNase at room temperature. The cell suspensions were filtered through nylon monofilament screens (pore sizes, 100 and 40 μm [BD Falcon] and 25 μm [Spectrum Labs]) and centrifuged at 150 × g for 8 min at 4°C. Immune and inflammatory cells were removed from the AEC preparation by magnetic selection. Cell suspensions were incubated with biotin-conjugated antibodies against CD16/32 and CD45 to remove hematopoietic cell-derived cells and leukocytes, followed by removal of positive cells with streptavidin-conjugated magnetic beads in a magnetic separator. AECs were centrifuged at 150 × g for 8 min at 4°C and incubated in tissue culture treated plates (Corning) at 37°C for 4 to 12 h, after which nonattached AECs were centrifuged again and enumerated. With this procedure, the AEC yield was approximately 1 × 106 cells/mouse. Typically, the AECs were >85% viable and >90% pure, as assessed by Papanicolaou staining. In addition, approximately 95% of the isolated cells were typically positive for surfactant protein C (SP-C) expression. AECs were cultured on Matrigel/rat tail collagen-coated plates (ratio, 70/30, vol/vol; BD Biosciences) under conditions previously described by Rice et al. (27) or on precoated mouse alveolar primary cell culture extracellular matrix plates (Celprogen). Cells were maintained at 37°C with 5% CO2 in bronchial epithelial cell growth medium (BEGM) without hydrocortisone (Cambrex) supplemented with 5% charcoal-stripped fetal bovine serum (HyClone, Logan, UT) and 10 ng/ml keratinocyte growth factor (KGF; Calbiochem).
In vitro AEC treatments.
Primary AECs were cultured to approximately 90% confluence. Culture medium was replaced with BEGM without KGF 6 h before treatment. Cells were treated with freshly isolated Pneumocystis diluted in serum-free DMEM at the indicated Pneumocystis cyst-to-AEC ratios. Other controls used were TNF-α (10 ng/ml), lipopolysaccharide (LPS; 10 ng/ml), and Pam3CSK4 (0.5 μg/ml). In some experiments, the following Toll-like receptor agonists were used to treat the cells: TLR1/2 agonist (Pam3CSK4), TLR2 agonist (heat-killed Listeria monocytogenes), TLR2/6 agonist (FSL1), TLR3 agonist [poly(I·C)], TLR4 agonist (Escherichia coli K-12 LPS), TLR5 agonist (Salmonella enterica serovar Typhimurium flagellin), TLR7 agonist (single-stranded RNA 40 [ssRNA40]), and TLR9 agonist (ODN1826) at the concentrations shown in Table 1. After treatment, supernatants were collected and stored at −80°C until used.
Table 1.
TLR agonists used to stimulate primary murine AECsa
| Control treatmentb | Treatment designation | Value |
|---|---|---|
| TLR1/2 agonist Pam3CSK4 | A | 0.1 μg/ml |
| B | 0.5 μg/ml | |
| C | 1 μg/ml | |
| TLR2 agonist HKLM | A | 106 cells/ml |
| B | 107 cells/ml | |
| C | 108 cells/ml | |
| TLR2/6 agonist FSL1 | A | 10 ng/ml |
| B | 100 ng/ml | |
| C | 1 μg/ml | |
| TLR3 agonist poly(I·C) | A | 1 μg/ml |
| B | 10 μg/ml | |
| C | 100 μg/ml | |
| TLR4 agonist LPS | A | 1 ng/ml |
| B | 10 ng/ml | |
| C | 100 ng/ml | |
| TLR5 agonist ST-FLA | A | 10 ng/ml |
| B | 100 ng/ml | |
| C | 1 μg/ml | |
| TLR7 agonist ssRNA40 | A | 0.25 μg/ml |
| B | 2.5 μg/ml | |
| C | 10 μg/ml | |
| TLR9 agonist ODN1826 | A | 0.5 μM |
| B | 1 μM | |
| C | 5 μM | |
| TNF-α | 10 ng/ml | |
| Pneumocystis-to-AEC ratios of: | ||
| 0.5:1 | 0.75 × 5 Pneumocystis cysts/well | |
| Pc 1:1 | 1.5× 105 Pneumocystis cysts/well | |
| Pc 3:1 | 4.5 × 105 Pneumocystis cysts/well | |
| Pc depleted |
Primary murine AECs were isolated from C57BL/6 (WT) mice. Confluent monolayers were stimulated for 6 h with a panel of TLR ligands used at the indicated concentrations. CCL2 and CXCL2 levels were measured in supernatants (Fig. 5).
Pam3CSK4, Pam3CysSerLys4; HKLM, heat-killed preparation of L. monocytogenes; FSL1, Pam2CGDPKHPKSF; ST-FLA, flagellin from S. Typhimurium; ODN1826, CpG synthetic oligonucleotide; Pc depleted, supernatant of Pneumocystis-depleted preparation.
Isolation and enumeration of mouse Pneumocystis.
Pneumocystis was isolated from the lungs of heavily infected SCID mice and enumerated by Gomori's methenamine silver (GMS) staining as previously described (31). In order to ensure that the AEC response was specific to Pneumocystis and not to any non-Pneumocystis contaminant that might have been coisolated from the lung tissue, a cocktail of Pneumocystis-specific antibodies linked to magnetic beads was used to remove Pneumocystis organisms from the purified Pneumocystis preparation as previously described (31). The remaining preparation with any non-Pneumocystis contaminants that may have been present, but depleted of Pneumocystis, was then used to treat AECs as controls. This is referred to as “Pneumocystis depleted.”
Phospho-JNK ELISA.
Primary AECs from WT and MyD88−/− mice were grown to confluence and were exposed to medium only or 2 × 105 Pneumocystis cysts for 0.5, 1, and 2 h. As a control, AECs were treated with TNF-α (10 ng/ml) for 10 min. After treatment, cells were fixed with 4% formaldehyde and phosphorylation of Jun N-terminal protein kinase (JNK) was measured by a cellular activation of signaling enzyme-linked immunosorbent assay (ELISA) (CASE) kit for JNK T183/Y185, according to the manufacturer's instructions (SuperAray Bioscience Corporation, Frederick, MD).
RNA isolation and RPAs.
Primary murine alveolar epithelial cells were grown to confluence and then stimulated for 6 h. Total RNA isolation was performed using TRIzol reagent according to the manufacturer's instructions (Life Technologies, Grand Island, NY). For the RNase protection assay (RPA), a custom RPA template was used to transcribe radiolabeled, antisense riboprobes for murine CCL2, murine ribosomal protein L32, and glyceraldehyde-3-phosphate dehydrogenase as previously described (31, 35).
In vivo Pneumocystis infection and BAL.
Mice were intratracheally inoculated with 1 × 106 freshly isolated Pneumocystis cysts. After 3, 8, and 24 h, BAL fluid was recovered from experimental mice by lavaging both lungs with three, 1-ml aliquots of 1× HBSS. The recovered BAL fluid was centrifuged for 10 min at 1,110 rpm at 4°C to pellet cells. Cell-free BAL fluid was used for chemokine ELISA. Recovered cells were counted using a hemacytometer (Hausser Scientific).
Cytokine and chemokine ELISA.
Culture supernatants and BAL fluid were collected and then centrifuged at 12,000 × g for 10 min to remove cell debris. Supernatants and BAL fluid were kept at −80°C until used. CCL2 and CXCL2 chemokine concentrations were measured using a commercially available ELISA kit (R&D Systems) and utilized according to the manufacturer's instructions.
Statistical analysis.
Data are presented as the mean ± 1 standard error measurement (SEM). Differences between treatments were analyzed using one-way analysis of variance (ANOVA) and Tukey's multiple-comparison test as a posttest. Differences between treatments at different doses or at different time points were analyzed using two-way ANOVA and Bonferroni's multiple-comparison test as a posttest. Differences were considered significant at P values of <0.05. All data were analyzed using GraphPad Prism (version 5.00) software (GraphPad Software, San Diego, CA).
RESULTS
AEC chemokine responses to Pneumocystis require MyD88.
AECs produce chemokines in response to Pneumocystis in a time- and dose-dependent manner (30). To determine whether MyD88 plays a role in the signaling cascade leading to chemokine production by Pneumocystis-stimulated AECs, primary murine AECs isolated from WT and MyD88-deficient mice were exposed to Pneumocystis. Confluent monolayers were treated with freshly isolated murine Pneumocystis at cyst-to-AEC ratios of 1:1 and 3:1. CCL2 levels in the supernatants at 6 h and 12 h were measured by ELISA. CCL2 responses in WT cells after 6 h of treatment with Pneumocystis at cyst-to-AEC ratios of 1:1 or 3:1 were 13- and 25-fold higher than those in untreated controls, respectively (Fig. 1A). Similarly, CCL2 levels of Pneumocystis-treated AECs after 12 h of stimulation were 22- and 24-fold higher than those in untreated controls, respectively (Fig. 1B). In contrast, MyD88−/− AECs were severely impaired in their ability to produce CCL2 in response to Pneumocystis stimulation at both doses and both time points (Fig. 1A and B). WT and MyD88−/− AECs displayed similar increases in CCL2 production in response to treatment with TNF-α, demonstrating that MyD88−/− AECs do not have an intrinsic deficiency in CCL2 responses. Furthermore, MyD88−/− AECs were also impaired in their ability to respond to MyD88-dependent TLR agonists LPS and Pam3SCK4, confirming that these cells are MyD88 deficient.
Fig 1.

CCL2 and CXCL2 responses to Pneumocystis (Pc) in primary alveolar epithelial cells are dependent on MyD88. Primary mouse AECs were isolated and cultured from C57BL/6 mice (WT) and C57BL/6 Myd88−/− mice. Confluent monolayers were treated with freshly isolated murine Pneumocystis at cyst-to-AEC ratios of 1:1 and 3:1. AECs were also treated with either medium alone as a negative control (NT) or TNF-α (10 ng/ml) as a non-MyD88-dependent positive control. Cells were also treated with LPS (10 ng/ml) and Pam3CSK4 (0.5 μg/ml). CCL2 levels in the supernatants at 6 h (A) and 12 h (B) were measured by ELISA (*, P < 0.05 compared to the same treatment in WT). (C) CXCL2 levels in supernatants were measured at 6 h and 12 h (*, P < 0.05 compared to all other groups at each time point). Results are shown as the fold increase relative to medium-only treatment. Bars represent means ± SEMs (n = 3) from a representative experiment that was repeated four times.
Murine AECs have also been shown to produce the chemokine CXCL2 in response to Pneumocystis stimulation (31). To determine whether MyD88 is also necessary for the upregulation of this chemokine, we measured CXCL2 in the culture supernatants of stimulated AECs. As shown in Fig. 1C, Pneumocystis-stimulated WT AECs had greater than 3- and 6-fold increases in CXCL2 production after 6 h and 12 h stimulation, respectively. In contrast, Pneumocystis-stimulated MyD88−/− AECs had no upregulation of CXCL2 production and had responses comparable to those of the untreated cells. Overall, these data show that CCL2 and CXCL2 chemokine production by AECs is dependent on signaling through the adaptor molecule MyD88.
Our data demonstrated impaired production and release of CCL2 from Pneumocystis-treated MyD88−/− AECs compared to WT AECs. In order to determine if MyD88 plays a role in the transcription of Pneumocystis-mediated CCL2, we sought to determine if the steady-state levels of CCL2 RNA were different between WT and MyD88−/− Pneumocystis-stimulated AECs. RNA was isolated from WT and MyD88−/− AECs after 6 h of Pneumocystis stimulation, and RPAs were performed. CCL2 mRNA was not detectable in untreated cells from either strain of mice (Fig. 2A and B). Similar to ELISA data, CCL2 mRNA levels were increased >10-fold in Pneumocystis-stimulated WT AECs. However, the CCL2 mRNA level in Pneumocystis-stimulated MyD88−/− AECs was not elevated above untreated control levels (Fig. 2A and B). These data show that MyD88 controls CCL2 protein production by Pneumocystis-stimulated AECs through a transcriptional control mechanism.
Fig 2.
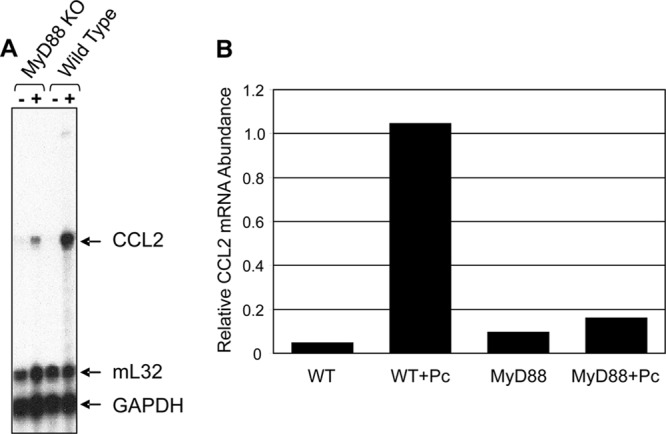
MyD88 is required for Pneumocystis-stimulated CCL2 mRNA expression in AECs. Primary murine pneumocytes were isolated from C57BL/6 WT and Myd88−/− mice. Confluent monolayers were exposed to freshly isolated murine Pneumocystis at a Pneumocystis-to-AEC ratio of 3:1. AECs were also treated with medium alone as a negative control (WT or MyD88−/−). (A) RNA was collected after 6 h of treatment and analyzed by RPA. KO, knockout. (B) MCP-1 mRNA levels were quantified and normalized to the levels of the housekeeping gene mL32.
CCL2 production by Pneumocystis-stimulated AECs is independent of the adaptor molecule TRIF.
CCL2 production by AECs in response to Pneumocystis is dependent on NF-κB and JNK. NF-κB is also known to be activated by a MyD88-independent pathway, the TRIF (37). TRIF is the main adaptor molecule utilized by TLR3 and the MyD88-independent pathway from TLR4 signaling (37). Thus, to determine if the response to Pneumocystis was specific to MyD88 or was codependent on TRIF and MyD88, confluent monolayers of primary AECs were isolated from WT or TRIF−/− mice and treated with freshly isolated Pneumocystis for 6 h. Supernatants were collected and analyzed by ELISA. Interestingly, CCL2 responses from Pneumocystis-treated TRIF−/− AECs were comparable to responses from Pneumocystis-treated WT AECs at both Pneumocystis cyst-to-AEC ratios tested (Fig. 3). CCL2 responses to TNF-α (10 ng/ml) were also comparable to those of WT AECs. These data show that the CCL2 responses to Pneumocystis are independent of the adaptor molecule TRIF and specifically dependent on MyD88.
Fig 3.
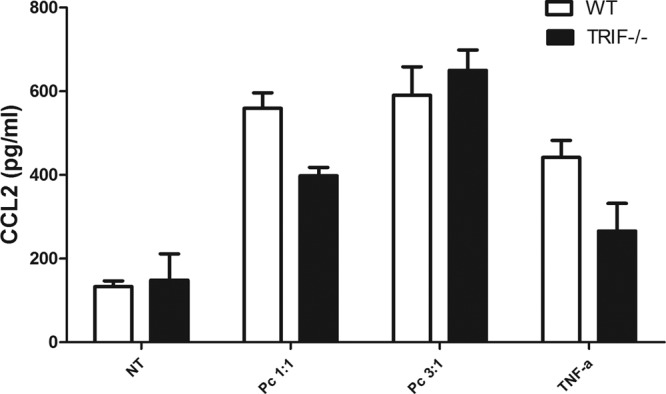
CCL2 response of Pneumocystis-stimulated AECs is independent of TRIF. Primary murine pneumocytes were isolated from C57BL/6 WT and TRIF−/− mice. Confluent monolayers were exposed to freshly isolated murine Pneumocystis at cyst-to-AEC ratios of 1:1 and 3:1. AECs were also treated with either medium alone (negative control [NT]) or TNF-α (10 ng/ml). CCL2 levels in the supernatants at 6 h were measured by ELISA (P > 0.05). Bars represent means ± SEs (n = 3) from a representative experiment that was repeated twice.
Pneumocystis-stimulated JNK activation requires MyD88-dependent signaling in alveolar epithelial cells.
Our group has previously reported that CCL2 production by Pneumocystis-stimulated AECs is dependent on JNK activation (30). To determine whether JNK activation is dependent on MyD88, primary murine pneumocytes were isolated from WT and MyD88−/− mice. Confluent monolayers were treated with Pneumocystis or with medium only. After 30 min, 1 h, and 2 h, AECs were tested for JNK phosphorylation by ELISA. As expected, WT AECs exhibited increased JNK activation at 30 min after Pneumocystis stimulation (Fig. 4A) (30). In contrast, Pneumocystis stimulation did not induce JNK signaling in AECs lacking MyD88 (Fig. 4A). This lack of JNK phosphorylation in MyD88−/− AECs was specific to the treatment with Pneumocystis, as JNK activation was observed in both WT and MyD88−/− AECs following stimulation with TNF-α, which signals through a MyD88-independent pathway (Fig. 4B). These data suggest that the MyD88-dependent mechanism through which Pneumocystis-stimulated AECs produce CCL2 involves activation of the JNK pathway, leading to increased gene transcription.
Fig 4.
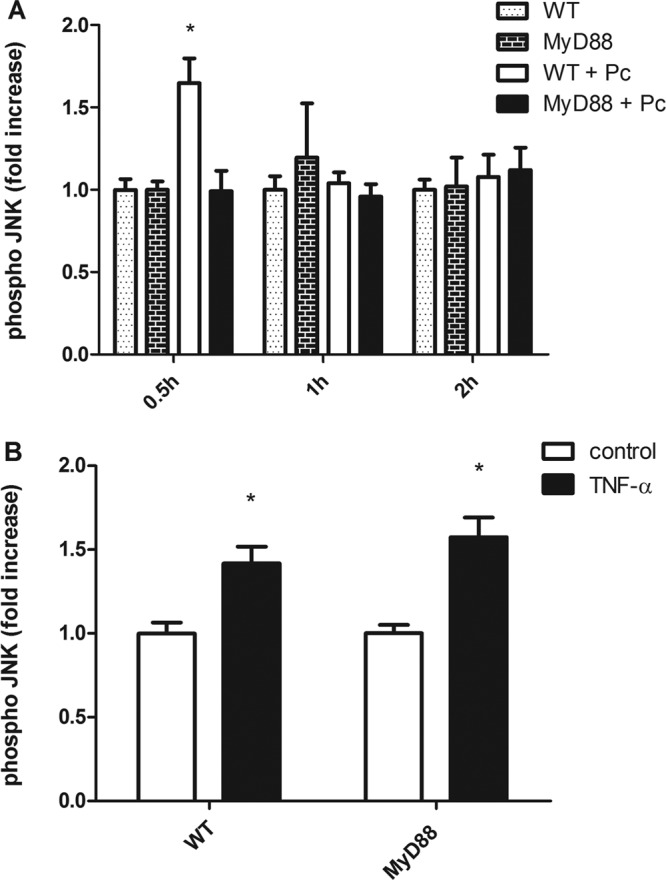
MyD88 signaling is required for Pneumocystis-stimulated JNK activation in primary murine AECs. Primary murine pneumocytes were isolated from C57BL/6 WT and Myd88−/− mice. Confluent monolayers were exposed to freshly isolated murine Pneumocystis (WT + Pc or MyD88 + Pc) (A). AECs were also treated with either medium alone as a negative control (WT or MyD88 in panel A and control in panel B) or TNF-α (10 ng/ml) (B) for 10 min. After 0.5, 1, or 2 h of stimulation, phospho-JNK was measured by ELISA in cells. *, P < 0.05 compared to each group at a specific time point (A) or compared to the control from the same strain (B). Bars represent means ± SEs from a representative experiment that was repeated two times.
Stimulation through TLR2, TLR3, TLR4, and TLR5, but not TLR7 or TLR9, induces chemokine production by AECs.
Our studies demonstrate that the AEC chemokine response to Pneumocystis requires the adaptor molecule MyD88. MyD88 is required for signaling initiated by most TLRs, with the exception of TLR3 and the MyD88-independent pathway of TLR4 (15). However, little is known about the responsiveness of primary murine AECs to various TLR ligands. Thus, we sought to elucidate which TLR ligands induce chemokine production in primary murine AECs. Confluent cultures of WT murine AECs were stimulated with a panel of TLR ligands specific for TLR1/2 (Pam3CSK4), TLR2 (heat-killed Listeria monocytogenes), TLR2/6 (FSL1), TLR3 [poly(I·C) and poly(I·C) low molecular weight (LMW)], TLR4 (E. coli K-12 LPS), TLR5 (S. enterica serovar Typhimurium flagellin), TLR7(ssRNA40), and TLR9 (ODN1826) (Table 1; Fig. 5). There was a significant increase in CCL2 production in response to the ligands for TLR1/2, TLR2/6, TLR3, TLR4, and TLR5 but not to the ligands for TLR2 only, TLR7, or TLR9 (Fig. 5A). Moreover, AEC CXCL2 production was increased by TLR2/6, TLR4, and TLR5 ligands but not TLR1/2, TLR2, TLR3, TLR5, TLR7, or TLR9 ligands (Fig. 5B). As expected, the WT AECs responded to Pneumocystis at Pneumocystis-to-AEC ratios of 1:1 and 3:1. To ensure that the AEC response was specific to Pneumocystis and not to any non-Pneumocystis contaminant that might have been coisolated from the lung tissue, a cocktail of Pneumocystis-specific antibodies was used to deplete Pneumocystis from lung homogenates. AECs were treated with the Pneumocystis-depleted preparation, and no CCL2 or CXCL2 responses were observed, confirming that the observed responses were due to Pneumocystis. These data suggest that murine primary AECs express TLRs, respond to various TLR ligands, and are able to respond to pathogen-associated molecular patterns with chemokine secretion. These results imply that AECs in the lung are capable of initiating innate immune responses to pathogens in the lung.
Fig 5.

Primary murine AECs respond to TLR agonists. Primary murine AECs were isolated from C57BL/6 WT mice, and confluent monolayers were stimulated with a panel of TLR ligands for 6 h at increasing concentrations (Table 1): TLR1/2 (Pam3CSK4), TLR2 (heat-killed Listeria monocytogenes), TLR2/6 (FSL1), TLR3 [poly(I·C) and poly(I·C) LMW], TLR4 (E. coli K-12 LPS), TLR5 (S. enterica serovar Typhimurium flagellin), TLR7(ssRNA40), and TLR9 (ODN1826). AECs were also exposed to freshly isolated murine Pneumocystis at cyst-to-AEC ratios of 1:1 (Pc 1) and 3:1 (Pc 3), antibody-depleted Pneumocystis preparations (Pc depleted), and medium alone as negative control (control). CCL2 (A) and CXCL2 (B) levels in the supernatants at 6 h were measured by ELISA. Results are shown as the fold increase relative to the medium-only treatment. Bars represent means ± SEs (n = 3) from a representative experiment that was repeated two times in triplicate. (*, P < 0.05 compared to control).
Chemokine production by Pneumocystis-stimulated AECs does not require TLR2 or TLR4.
Recent studies have suggested that alveolar macrophages respond to Pneumocystis in a proinflammatory manner, releasing cytokines and chemokines in a TLR2- and TLR4-dependent manner (7, 40). We have shown that primary murine AECs respond to several TLR ligands, including TLR2 and TLR4 agonists. Thus, we hypothesized that the MyD88-dependent AEC chemokine response to Pneumocystis was also dependent on the interaction of TLR2 or TLR4 with Pneumocystis organisms. Primary AECs were isolated from WT, TLR2−/−, TLR4−/−, and TLR2/4−/− mice on the C57BL/6 background. Cells were then exposed to Pneumocystis at cyst-to-AEC ratios of 1:1 and 3:1. TNF-α (10 ng/ml), LPS (10 ng/ml), and Pam3CSK4 (0.5 μg/ml) were used as controls. At 6 h posttreatment, cell supernatants were collected and CCL2 chemokine responses were tested by ELISA. As expected, WT AECs produced CCL2 in response to TNF-α and Pneumocystis, as well as in response to the TLR2 and TLR4 agonists Pam3CSK4 and LPS, respectively (Fig. 6). MyD88-deficient AECs produced CCL2 in response to TNF-α stimulation but were significantly impaired in their ability to respond to Pneumocystis, LPS, or Pam3CSK4. Surprisingly, TLR2- and TLR4-deficient AECs also responded to Pneumocystis stimulation with significantly elevated CCL2 production compared to that by untreated controls. In order to address whether the presence of either TLR2 or TLR4 might compensate for the absence of the other, we also generated TLR2/4 double-knockout mice and isolated primary AECs. TLR2/4 double-deficient mice did not respond to the TLR2 and TLR4 agonists Pam3CSK4 and LPS, respectively, but did produce CCL2 in response TNF-α and Pneumocystis stimulation (Fig. 6). TLR2, TLR4, and TLR2/4 double-deficient AECs also produced CXCL2 in response to Pneumocystis stimulation (data not shown). These data show that the MyD88-dependent chemokine response of Pneumocystis-stimulated AECs does not require TLR2 or TLR4.
Fig 6.
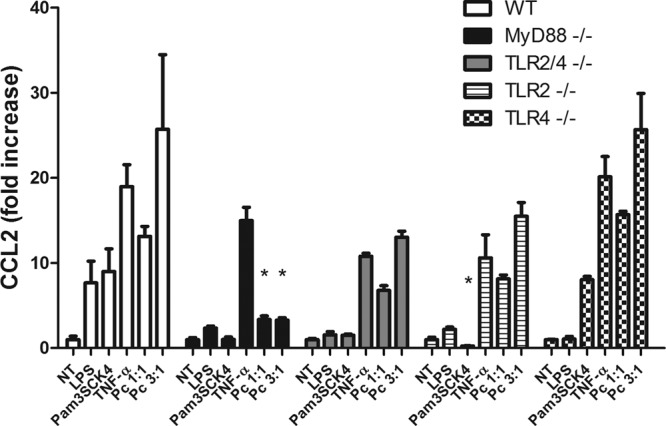
CCL2 production by Pneumocystis-stimulated AECs is independent of TLR2 and/or TLR4. Primary murine AECs were isolated from WT and MyD88−/−, TLR2−/−, TLR4−/−, and TLR2/4 double-knockout mice all on the C57BL/6 background. Confluent monolayers were exposed to Pneumocystis (at cyst-to-AECs ratios of 1:1 and 3:1), TNF-α (10 ng/ml), LPS (10 ng/ml), or Pam3CSK4 (0.5 μg/ml). At 6 h poststimulation, cell supernatants were collected and CCL2 chemokine responses were tested by ELISA. Bars represent means ± SEs (n = 3) from a representative experiment that was repeated three times (*, P < 0.05 compared to the same treatment in WT).
Chemokine production by Pneumocystis-stimulated AECs requires the IL-1 receptor.
Since the likeliest candidate TLRs (TLR2 and TLR4) were found to not be required for chemokine production by Pneumocystis-stimulated AECs, other MyD88-dependent pathways were explored. MyD88 is also required for signaling through the interleukin-1 (IL-1) receptor complex (IL-1R). Therefore, the chemokine response following Pneumocystis stimulation was evaluated in primary AECs isolated from WT and IL-1R-knockout mice. As expected, WT AECs produced CCL2 in response to stimulation with Pneumocystis, TNF, or IL-1β (Fig. 7). In contrast, IL-1R-deficient AECs produced CCL2 in response to TNF but did not respond to Pneumocystis or IL-1β stimulation (Fig. 7). These data indicate that the MyD88-dependent mechanism through which Pneumocystis stimulates chemokine production by AECs requires a functional IL-1R.
Fig 7.
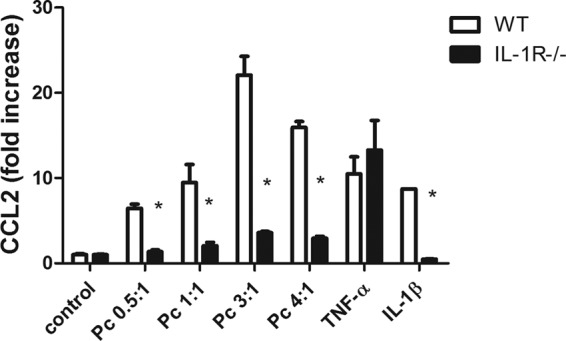
The CCL2 response to Pneumocystis in primary alveolar epithelial cells is dependent on IL-1R. Primary murine AECs were isolated from C57BL/6 WT and IL-1R−/− mice. Confluent monolayers were exposed to freshly isolated murine Pneumocystis at cyst-to-AEC ratios of 0.5:1, 1:1, 3:1, and 4:1. AECs were also treated with either medium alone (control), TNF-α (10 ng/ml), or IL-1β (10 ng/ml). CCL2 levels in the supernatants at 6 h were measured by ELISA. Bars represent means ± SEs (n = 3) from a representative experiment that was repeated twice (*, P < 0.05 compared to same treatment in WT).
MyD88 signaling is required for early chemokine release and cell recruitment following Pneumocystis infection in vivo.
AECs are the first cells in the lungs that interact with Pneumocystis and likely the cells that first detect and respond to Pneumocystis infection. MyD88-dependent signaling was found to play a major role in the AEC chemokine response to Pneumocystis in vitro (Fig. 1). To determine whether MyD88 signaling controls the chemokine response to Pneumocystis infection in vivo, WT and MyD88-deficient mice were intratracheally inoculated with 1 × 106 Pneumocystis cysts. At 3 h, 6 h, and 24 h postinfection, BAL was performed on individual mice and chemokine levels and total cells in the BAL fluid were determined. At 3 h postinfection, CCL2 and CXCL2 levels were significantly elevated in the lungs of WT mice compared to MyD88-knockout mice (Fig. 8A and B). However, by 8 h and 24 h, CCL2 levels declined in WT mice and were not significantly different from those in MyD88-knockout mice. Since AECs express CCR2, the receptor for CCL2, we speculate that the decline in CCL2 levels is at least partly due to a negative-feedback loop caused by high CCL2 levels in the lung at 3 h postinoculation. Additionally, the recruitment of CCR2-positive macrophages to the lung in response to CCL2 has been shown to downregulate local CCL2 production (20). In vivo, these mechanisms would result in a decrease in CCL2 levels in BAL fluid because of diffusion of CCL2 into the lung and circulation. CXCL2 levels remained elevated in WT mice at 8 and 24 h and were greater than those in MyD88-knockout mice. The number of cells recovered in the BAL fluid of Pneumocystis-infected WT mice was higher than that recovered from infected MyD88-knockout mice at all time points (Fig. 8C). Importantly, the number of cells in the BAL fluid of infected MyD88-knockout mice was not significantly different from that in the BAL fluid of uninfected control mice. These data demonstrate that MyD88 is important for chemokine production and early cell recruitment to the airspaces following Pneumocystis infection in vivo.
Fig 8.
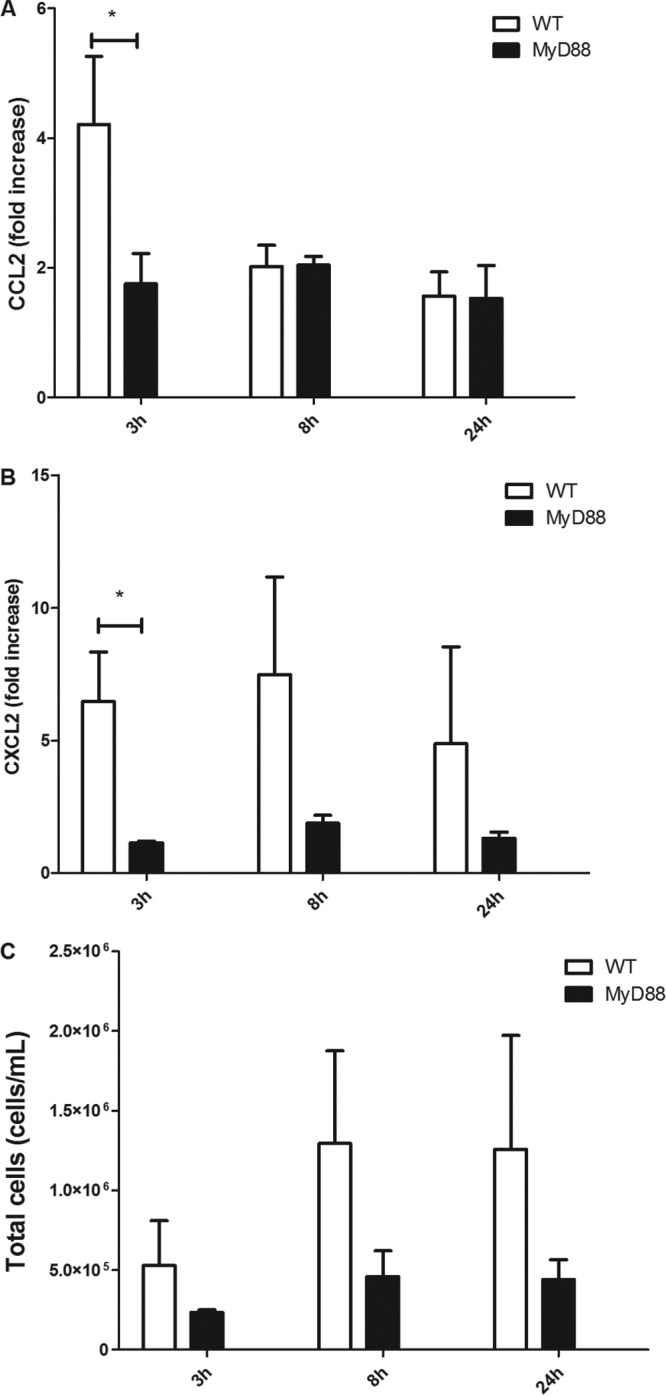
Early Pneumocystis-stimulated chemokine release and cell recruitment to the lung are dependent upon MyD88 signaling. C57BL/6 WT and MyD88−/− mice were intratracheally inoculated with 1 × 106 freshly isolated Pneumocystis cells. After 3 h, 8 h, and 24 h of Pneumocystis infection, BAL fluid from lungs were obtained to measure protein levels of CCL2 (A) and CXCL2 (B) by ELISA and for assessment of total cell recovery (C) (*, P < 0.05). Bars represent means ± SEs from pooled data from two independent experiments (n ≥ 6 mice per group).
DISCUSSION
Pneumocystis is an extracellular pathogen that interacts closely with the alveolar epithelium. Ultrastructural studies have shown interdigitation between Pneumocystis and the membrane of host AECs, and it has been suggested that attachment to AECs is required to establish infection, leading to the development of PCP (18, 26). Previously, AECs were mainly recognized as barrier cells and producers of surfactant. However, there is an increasing body of evidence suggesting an important role for AECs in host defense. Because of their barrier position in the lung, AECs are likely to mediate the initial encounter and response to inhaled pathogens. Furthermore, since AECs are uniquely situated in close proximity to the blood supply, they can affect the recruitment of inflammatory cells to the alveoli. Our laboratory has demonstrated that the direct interaction of viable Pneumocystis with lung epithelial cells induces the rapid production of proinflammatory chemokines, such as CCL2 and CXCL2, through a JNK-dependent mechanism (30, 31). In this study, we expanded upon those results by demonstrating that Pneumocystis-stimulated JNK activation and subsequent chemokine production require the adaptor molecule MyD88. Surprisingly, the AEC chemokine response was independent of TLR2 or TLR4 but did require AEC expression of IL-1R, which also signals through a MyD88-dependent mechanism. In vivo studies revealed that MyD88 is involved in the early pulmonary chemokine response to Pneumocystis infection and is also important for the early recruitment of inflammatory cells into the lung. Together, these data demonstrate a role for MyD88 in the generation of Pneumocystis-induced pulmonary inflammation and suggest that the AECs may modulate organism clearance in immunocompetent hosts and/or lung injury caused by an exacerbated inflammatory response in susceptible hosts.
It is well documented that professional phagocytes such as macrophages have important proinflammatory and host defense roles in the lung. These cells utilize TLRs and other PRRs such as dectin-1 to detect infectious agents and initiate inflammatory responses. TLRs signal through MyD88, and the alveolar macrophage response to Pneumocystis β-glucan is at least partly dependent upon MyD88 signaling (17). In addition, recent studies have suggested a role for TLR2 in the alveolar macrophage CXCL2 and TNF-α response to Pneumocystis organisms in vitro and in vivo (40). Moreover, a study by Ding et al. showed that TLR4-deficient AMs have impaired CXCL2, IL-10, and IL-12p40 responses to Pneumocystis in vitro and in vivo (7). However, they unexpectedly found that TLR4-deficient mice showed increased levels of TNF-α and IL-6 in BAL fluid, as well as higher lung injury scores and weight loss. These data suggested that other cell populations, such as AECs, may be involved in the host response to Pneumocystis infection in vivo. In the current study, we have shown that murine AECs respond to TLR2 and TLR4 agonists with CCL2 and CXCL2 release. However, we have also shown that the AEC response to Pneumocystis is independent of TLR2 or TLR4, suggesting that an alternate MyD88-dependent pathway is utilized by AECs in response to Pneumocystis.
Prior studies have identified several receptors that may be involved in the recognition of Pneumocystis and subsequent activation of proinflammatory pathways. Dectin-1 recognizes β-glucans found in cell walls of fungi and has been implicated in the AM response to Pneumocystis β-glucan (29). However, AECs express neither dectin-1 nor the integrin CD11b/CD18 (Mac-1, CR3) β-glucan receptor. Lactosylceramide (CDw17) has been identified as the alternate glucan receptor on AECs (8, 11). Interestingly, our group found that CCL2 production by Pneumocystis-stimulated murine primary AECs is not mediated by β-glucan (12), suggesting that other mechanisms that mediate the AEC response to viable Pneumocystis organisms exist. Benfield et al. reported that a nonglucan component of Pneumocystis, surface glycoprotein A, induces the production of IL-8 and CCL2 by the human alveolar epithelial cell line A549 (5). In our experiments, we stimulated AECs with freshly isolated Pneumocystis, with the aim of mimicking the interactions that occur in vivo. Our studies further enhance our understanding of the pathways that AECs utilize to recognize Pneumocystis and signal proinflammatory chemokine release.
It has been suggested that IL-1/IL-1 receptor signaling plays an important role in Pneumocystis clearance (6, 34), but the role of IL-1R in the AEC response to Pneumocystis has not been determined. We have now shown that IL-1R is required for the AEC chemokine response to Pneumocystis (Fig. 7), and there are several possible mechanisms through which IL-1R may be involved in the Pneumocystis inflammatory response. It is possible that Pneumocystis or a Pneumocystis product could directly interact with IL-1R to stimulate MyD88-dependent signaling. Alternatively, Pneumocystis may activate the inflammasome to induce production and maturation of IL-1, which then stimulates AECs in an autocrine manner. Some cell types require two signals for IL-1β to be secreted: (i) an initial step that is activated through TLRs, C-type lectins, or other signaling pathways to produce intracellular pro-IL-1β and (ii) a second step where caspase-1 is activated to cleave pro-IL-β into mature IL-1β that can be secreted (23, 24, 28). In respiratory syncytial virus and Francisella novicida infection models, TLR2 activation precedes the second step of inflammasome activation for IL-1β maturation and release (12, 28). Based on previous results that suggest a role for TLR2 in the host response to Pneumocystis and our results that show a slightly reduced chemokine response in TLR2-deficient AECs compared to WT AECs, we speculate that TLR2 plays a role in the initial step required for pro-IL-1 production. Thus, perhaps such a dramatic reduction in Pneumocystis-stimulated AEC chemokine release is observed in MyD88-deficient AECs because MyD88 is required for both the initial TLR2 signaling and the subsequent IL-1R-dependent signaling cascades.
In order to prevent and treat inflammation-mediated lung injury caused by Pneumocystis infection, it is necessary to understand how different lung cell populations interact with and respond to the organism. Understanding the contribution of AECs to the host's inflammatory response will aid in the development of improved treatments by targeting anti-inflammatory therapy to specific cell types and specific pathways utilized by the host. Further studies exploring the specific mechanisms by which the MyD88 and IL-1R pathway is exploited by Pneumocystis in AECs will help target appropriate treatment.
ACKNOWLEDGMENTS
We thank Nelissa Pérez-Nazario, Nabilah Khan, and Jane Malone for expert technical assistance.
This work was supported by Public Health Service grant R01 HL083761.
Footnotes
Published ahead of print 27 August 2012
REFERENCES
- 1. Adachi O, et al. 1998. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity 9:143–150 [DOI] [PubMed] [Google Scholar]
- 2. Arias MA, et al. 2007. Mycobacterium tuberculosis antigens specifically modulate CCR2 and MCP-1/CCL2 on lymphoid cells from human pulmonary hilar lymph nodes. J. Immunol. 179:8381–8391 [DOI] [PubMed] [Google Scholar]
- 3. Armstrong L, et al. 2004. Expression of functional Toll-like receptor-2 and -4 on alveolar epithelial cells. Am. J. Respir. Cell Mol. Biol. 31:241–245 [DOI] [PubMed] [Google Scholar]
- 4. Balamayooran G, Batra S, Balamayooran T, Cai S, Jeyaseelan S. 2011. Monocyte chemoattractant protein 1 regulates pulmonary host defense via neutrophil recruitment during Escherichia coli infection. Infect. Immun. 79:2567–2577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Benfield TL, Lundgren B, Shelhamer JH, Lundgren JD. 1999. Pneumocystis carinii major surface glycoprotein induces interleukin-8 and monocyte chemoattractant protein-1 release from a human alveolar epithelial cell line. Eur. J. Clin. Invest. 29:717–722 [DOI] [PubMed] [Google Scholar]
- 6. Chen W, et al. 1992. Interleukin 1: an important mediator of host resistance against Pneumocystis carinii. J. Exp. Med. 176:713–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ding K, et al. 2005. Impaired recognition by Toll-like receptor 4 is responsible for exacerbated murine Pneumocystis pneumonia. Microbes Infect. 7:195–203 [DOI] [PubMed] [Google Scholar]
- 8. Evans SE, et al. 2005. Pneumocystis cell wall beta-glucans stimulate alveolar epithelial cell chemokine generation through nuclear factor-kappaB-dependent mechanisms. Am. J. Respir. Cell Mol. Biol. 32:490–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gentry M, et al. 2007. Role of primary human alveolar epithelial cells in host defense against Francisella tularensis infection. Infect. Immun. 75:3969–3978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gigliotti F, Crow EL, Bhagwat SP, Wright TW. 2006. Sensitized CD8+ T cells fail to control organism burden but accelerate the onset of lung injury during Pneumocystis carinii pneumonia. Infect. Immun. 74:6310–6316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hahn PY, et al. 2003. Pneumocystis carinii cell wall beta-glucan induces release of macrophage inflammatory protein-2 from alveolar epithelial cells via a lactosylceramide-mediated mechanism. J. Biol. Chem. 278:2043–2050 [DOI] [PubMed] [Google Scholar]
- 12. Jones CL, Weiss DS. 2011. TLR2 signaling contributes to rapid inflammasome activation during F. novicida infection. PLoS One 6:e20609 doi:10.1371/journal.pone.0020609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kaplan JE, et al. 2000. Epidemiology of human immunodeficiency virus-associated opportunistic infections in the United States in the era of highly active antiretroviral therapy. Clin. Infect. Dis. 30(Suppl 1):S5–S14 [DOI] [PubMed] [Google Scholar]
- 14. Kaur N, Mahl TC. 2007. Pneumocystis jirovecii (carinii) pneumonia after infliximab therapy: a review of 84 cases. Dig. Dis. Sci. 52:1481–1484 [DOI] [PubMed] [Google Scholar]
- 15. Kawai T, Akira S. 2007. TLR signaling. Semin. Immunol. 19:24–32 [DOI] [PubMed] [Google Scholar]
- 16. Kroe DM, Kirsch CM, Jensen WA. 1997. Diagnostic strategies for Pneumocystis carinii pneumonia. Semin. Respir. Infect. 12:70–78 [PubMed] [Google Scholar]
- 17. Lebron F, Vassallo R, Puri V, Limper AH. 2003. Pneumocystis carinii cell wall beta-glucans initiate macrophage inflammatory responses through NF-kappaB activation. J. Biol. Chem. 278:25001–25008 [DOI] [PubMed] [Google Scholar]
- 18. Limper AH. 1991. Parasitic adherence and host responses in the development of Pneumocystis carinii pneumonia. Semin. Respir. Infect. 6:19–26 [PubMed] [Google Scholar]
- 19. Limper AH, Offord KP, Smith TF, Martin WJ., II 1989. Pneumocystis carinii pneumonia. Differences in lung parasite number and inflammation in patients with and without AIDS. Am. Rev. Respir. Dis. 140:1204–1209 [DOI] [PubMed] [Google Scholar]
- 20. Maus UA, et al. 2005. CCR2-positive monocytes recruited to inflamed lungs downregulate local CCL2 chemokine levels. Am. J. Physiol. Lung Cell. Mol. Physiol. 288:L350–L358 [DOI] [PubMed] [Google Scholar]
- 21. Morris A, et al. 2004. Current epidemiology of Pneumocystis pneumonia. Emerg. Infect. Dis. 10:1713–1720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Morrison BE, Park SJ, Mooney JM, Mehrad B. 2003. Chemokine-mediated recruitment of NK cells is a critical host defense mechanism in invasive aspergillosis. J. Clin. Invest. 112:1862–1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Netea MG, et al. 2009. Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood 113:2324–2335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Netea MG, et al. 2010. IL-1beta processing in host defense: beyond the inflammasomes. PLoS Pathog. 6:e1000661 doi:10.1371/journal.ppat.1000661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Norris KA, Morris A, Patil S, Fernandes E. 2006. Pneumocystis colonization, airway inflammation, and pulmonary function decline in acquired immunodeficiency syndrome. Immunol. Res. 36:175–187 [DOI] [PubMed] [Google Scholar]
- 26. Pottratz ST. 1998. Pneumocystis carinii interactions with respiratory epithelium. Semin. Respir. Infect. 13:323–329 [PubMed] [Google Scholar]
- 27. Rice WR, et al. 2002. Maintenance of the mouse type II cell phenotype in vitro. Am. J. Physiol. Lung Cell. Mol. Physiol. 283:L256–L264 [DOI] [PubMed] [Google Scholar]
- 28. Segovia J, et al. 2012. TLR2/MyD88/NF-kappaB pathway, reactive oxygen species, potassium efflux activates NLRP3/ASC inflammasome during respiratory syncytial virus infection. PLoS One 7:e29695 doi:10.1371/journal.pone.0029695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vassallo R, Standing J, Limper AH. 1999. Beta-glucan from Pneumocystis carinii stimulates TNF alpha release from alveolar macrophages. J. Eukaryot. Microbiol. 46:145S. [PubMed] [Google Scholar]
- 30. Wang J, Gigliotti F, Bhagwat SP, Maggirwar SB, Wright TW. 2007. Pneumocystis stimulates MCP-1 production by alveolar epithelial cells through a JNK-dependent mechanism. Am. J. Physiol. Lung Cell. Mol. Physiol. 292:L1495–L1505 [DOI] [PubMed] [Google Scholar]
- 31. Wang J, et al. 2005. Pneumocystis carinii activates the NF-kappaB signaling pathway in alveolar epithelial cells. Infect. Immun. 73:2766–2777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Werts C, et al. 2001. Leptospiral lipopolysaccharide activates cells through a TLR2-dependent mechanism. Nat. Immunol. 2:346–352 [DOI] [PubMed] [Google Scholar]
- 33. Wright TW, et al. 1999. Immune-mediated inflammation directly impairs pulmonary function, contributing to the pathogenesis of Pneumocystis carinii pneumonia. J. Clin. Invest. 104:1307–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wright TW, Johnston CJ, Harmsen AG, Finkelstein JN. 1997. Analysis of cytokine mRNA profiles in the lungs of Pneumocystis carinii-infected mice. Am. J. Respir. Cell Mol. Biol. 17:491–500 [DOI] [PubMed] [Google Scholar]
- 35. Wright TW, Johnston CJ, Harmsen AG, Finkelstein JN. 1999. Chemokine gene expression during Pneumocystis carinii-driven pulmonary inflammation. Infect. Immun. 67:3452–3460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wright TW, et al. 2004. TNF receptor signaling contributes to chemokine secretion, inflammation, and respiratory deficits during Pneumocystis pneumonia. J. Immunol. 172:2511–2521 [DOI] [PubMed] [Google Scholar]
- 37. Yamamoto M, et al. 2003. Role of adaptor TRIF in the MyD88-independent Toll-like receptor signaling pathway. Science 301:640–643 [DOI] [PubMed] [Google Scholar]
- 38. Yoneda K, Walzer PD. 1983. Attachment of Pneumocystis carinii to type I alveolar cells studied by freeze-fracture electron microscopy. Infect. Immun. 40:812–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yoneda K, Walzer PD. 1980. Interaction of Pneumocystis carinii with host lungs: an ultrastructural study. Infect. Immun. 29:692–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang C, et al. 2006. Toll-like receptor 2 mediates alveolar macrophage response to Pneumocystis murina. Infect. Immun. 74:1857–1864 [DOI] [PMC free article] [PubMed] [Google Scholar]