

Abstract
Anaplasma phagocytophilum is the tick-transmitted obligate intracellular bacterium that causes human granulocytic anaplasmosis (HGA). A. phagocytophilum binding to sialyl Lewis x (sLex) and other sialylated glycans that decorate P selectin glycoprotein 1 (PSGL-1) and other glycoproteins is critical for infection of mammalian host cells. Here, we demonstrate the importance of A. phagocytophilum outer membrane protein A (OmpA) APH_0338 in infection of mammalian host cells. OmpA is transcriptionally induced during transmission feeding of A. phagocytophilum-infected ticks on mice and is upregulated during invasion of HL-60 cells. OmpA is presented on the pathogen's surface. Sera from HGA patients and experimentally infected mice recognize recombinant OmpA. Pretreatment of A. phagocytophilum organisms with OmpA antiserum reduces their abilities to infect HL-60 cells. The OmpA N-terminal region is predicted to contain the protein's extracellular domain. Glutathione S-transferase (GST)-tagged versions of OmpA and OmpA amino acids 19 to 74 (OmpA19-74) but not OmpA75-205 bind to, and competitively inhibit A. phagocytophilum infection of, host cells. Pretreatment of host cells with sialidase or trypsin reduces or nearly eliminates, respectively, GST-OmpA adhesion. Therefore, OmpA interacts with sialylated glycoproteins. This study identifies the first A. phagocytophilum adhesin-receptor pair and delineates the region of OmpA that is critical for infection.
INTRODUCTION
Anaplasma phagocytophilum is a tick-transmitted bacterium in the order Rickettsiales and family Anaplasmataceae that colonizes peripherally circulating neutrophils, causing human granulocytic anaplasmosis (HGA). Presentation of this febrile illness ranges from subclinical to severe or even fatal infection (57, 64). Since HGA became a reportable disease in the United States 13 years ago (22), the number of HGA cases has risen annually (57). HGA is increasingly recognized in Europe and Asia (57, 64), and A. phagocytophilum infection is the most prevalent tick-transmitted disease of animals in Europe (14).
Promyelocytic and endothelial cell lines are useful in vitro models for studying A. phagocytophilum-host cell interactions (24, 28–30, 46, 62, 63, 77). A. phagocytophilum undergoes a biphasic developmental cycle (45, 46, 52, 68), the kinetics of which have been tracked in promyelocytic HL-60 cells. The cycle begins with attachment and entry of an infectious dense-cored (DC) organism. Once intracellular, the DC organism differentiates to the noninfectious reticulate cell (RC) form and replicates by binary fission to produce a bacterium-filled organelle called a morula. Later, the RCs transition back to DC organisms, which initiate the next round of infection (68).
Sialic acids are usually the terminal monosaccharide units on glycan chains of glycoproteins and glycolipids that cover mammalian cell surfaces. Given their outermost location on glycans, it is unsurprising that many bacterial and viral proteins bind sialic acids to promote infection (71). A. phagocytophilum's ability to infect human neutrophils and HL-60 cells is largely predicated on its interactions with the sialylated glycoprotein, P-selectin glycoprotein ligand 1 (PSGL-1) (20, 25). The ectodomain of PSGL-1 is decorated with sialylated O-glycans. The tetrasaccharide sialyl Lewis x (sLex; NeuAcα2,3Galβ1,4[Fucα1,3]GlcNac) is the terminal portion of a core-2 O-glycan that caps the PSGL-1 N terminus (41). A. phagocytophilum cooperatively binds to α2,3-sialic acid and α1,3-fucose of sLex and an amino acid sequence in the human PSGL-1 N terminus (5, 20, 25, 78). Interaction with α2,3-sialic acid of sLex is critical for the bacterium to invade human myeloid cells (20). Pretreatment of myeloid cells with the CSLEX1 monoclonal antibody (MAb), which recognizes the α2,3-linked sialic acid determinant of sLex (13), or enzymatic removal of sialic acid residues results in inefficient A. phagocytophilum binding to sLex-capped PSGL-1 and markedly inhibits infection (5, 20). The PSGL-1 N-terminal peptide determinant is important for A. phagocytophilum to infect human neutrophils but not murine neutrophils (5, 78), whereas sialic acid residues are crucial for the organism to interact with human and murine neutrophils (5). Therefore, binding to sialic acid is critical for A. phagocytophilum to infect neutrophils of both its natural murine and incidental human hosts. An A. phagocytophilum invasin that targets sialic acid or any other known determinant required for infection has yet to be identified.
Outer membrane protein A (OmpA), also known as peptidoglycan-associated lipoprotein, is conserved among most Gram-negative bacteria and interacts with peptidoglycan to maintain outer membrane integrity (7, 19). It is also important for the virulence of several Gram-negative pathogens (19, 53, 54). A. phagocytophilum and Ehrlichia chaffeensis, which is an Anaplasmataceae member that infects monocytes (32), encode OmpA but lack most peptidoglycan synthesis genes (26). E. chaffeensis OmpA contributes to infection, as pretreating bacteria with OmpA antiserum inhibits infection of monocytes (10). Accordingly, we hypothesized that A. phagocytophilum OmpA may be important for the pathogen to infect mammalian host cells.
MATERIALS AND METHODS
Cell lines and cultivation of uninfected and A. phagocytophilum-infected HL-60 cells.
Chinese hamster ovary (CHO) cells transfected to express sLex-capped PSGL-1 (PSGL-1 CHO cells) (35, 76), untransfected CHO cells, and RF/6A rhesus monkey choroidal endothelial cells (ATCC CRL-1780; American Type Culture Collection [ATCC], Manassas, VA) were cultivated as described previously (28, 68). Uninfected HL-60 cells (ATCC CCL-240) and HL-60 cells infected with the A. phagocytophilum NCH-1 strain or a transgenic A. phagocytophilum HGE1 strain expressing green fluorescent protein (GFP) (12) were cultivated as described previously (56). Spectinomycin (100 μg/ml; Sigma-Aldrich, St. Louis, MO) was added to HL-60 cultures harboring transgenic HGE1 bacteria.
Analyses of ompA expression over the course of infection.
HL-60 cells were synchronously infected with A. phagocytophilum DC organisms (68). Indirect immunofluorescence microscopic examination of aliquots recovered at 24 h confirmed that ≥60% of HL-60 cells contained morulae and that the mean number of morulae per cell was 2.8 ± 0.6. The infection time course proceeded for 36 h at 37°C in a humidified atmosphere of 5% CO2. The length of the time course enabled the bacteria to complete their biphasic developmental cycle and initiate a second round of infection (68). Every 4 h, aliquots were removed and processed for RNA isolation. Reverse transcriptase quantitative PCR (RT-qPCR) was performed as described previously (69). Gene-specific primers used for RT-qPCR are listed in Table 1. Relative transcript levels for each target were normalized to the transcript levels of the A. phagocytophilum 16S rRNA gene (aph_1000) using the 2−ΔΔCT method (38). To present the ompA transcript levels in the context of the A. phagocytophilum biphasic developmental cycle, normalized transcript levels were calculated as the fold change in expression relative to expression at 16 h, a time point at which the bacterial population consists exclusively of RC organisms (68).
Table 1.
Oligonucleotides used in this study
| Designation | Sequence (5′ to 3′)a | Targeted nucleotidesb |
|---|---|---|
| Ap 16S-527F | TGTAGGCGGTTCGGTAAGTTAAAG | 527–550 (+) |
| Ap 16S-753R | GCACTCATCGTTTACAGCGTG | 753–773 (−) |
| aph_0338-026F | GTCTACTGGCACTGCTCAGTGTTAC | 26–50 (+) |
| aph_0338-188R | CCGGGACCCTTTAGATCGTACTTCC | 164–188 (−) |
| aph_0338-055F-ENTR | CACCTGTGGGACTCTTCTTCCAGATAGTAACG | 55–82 (+) |
| aph_0338-618R | CTAGTTAGCGATTGCGCTAGAGAATTC | 592–618 (−) |
| aph_0338-222R | CTAGAGCTGCTCAACAAGCTCCAGA | 201–222 (−) |
| aph_0338-223F-ENTR | CACCAGACAGGATGACAGCATGT | 223–241 (+) |
| ott_1320-EcoRI-64F | TGCTGAATTCTGTTTATGGCAAAGATCTAAACATAGTAAC | 64–94 (+) |
| ott_1320-XhoI-615R | GATCCTCGAGCTATGCTATATTACTTTTAATAATTGTGACAGACC | 581–615 (−) |
Nucleotides in bold text correspond to a Gateway entry vector-compatible sequence; underlined nucleotides correspond to an added stop codon; double-underlined nucleotides correspond to restriction sites preceded by spacer nucleotides.
(+), positive strand; (−), negative strand.
Transmission feeding of A. phagocytophilum-infected Ixodes scapularis nymphs.
Transmission feeding of A. phagocytophilum-infected I. scapularis nymphs on C3H/HeJ mice and RNA extraction from salivary glands obtained from transmission-fed and uninfected control nymphs were performed as described previously (40). RT-qPCR was performed as described above.
Recombinant protein and antiserum production.
A. phagocytophilum genes of interest and Orientia tsutsugamushi (Ikeda strain) OmpA were amplified using primers listed in Table 1 and Platinum Pfx DNA polymerase (Invitrogen, Carlsbad, CA). Amplicons were cloned into pENTR/TEV/D-TOPO (Invitrogen) as described previously (30) to yield pENTR-candidate gene entry plasmids containing the genes of interest. Plasmid inserts were verified, and recombination of the candidate gene insert downstream of and in frame with the gene encoding GST was achieved using the pDest-15 vector (Invitrogen) as described previously (30). Expression and purification of GST-OmpA, GST-tagged OmpA residues 19 to 74 (GST-OmpA19-74), GST-OmpA75-205, GST-APH_1387112-579, GST-major surface protein 5 (Msp5), GST-tagged O. tsutsugamushi OmpA (GST-OtOmpA), and GST alone were performed as described previously (69). Generation of murine polyclonal antisera against each protein was performed as described previously (69).
Western blot analyses and spinning-disk confocal microscopy.
Antisera generated in this study and previous studies targeted OmpA, Msp5, APH_0032 (29), Asp55 (55-kDa A. phagocytophilum surface protein), and Asp62 (62-kDa A. phagocytophilum surface protein) (16, 50, 59). Western blot analyses were performed as described previously (69). A. phagocytophilum-infected HL-60 cells were processed and analyzed via indirect immunofluorescence using spinning-disk confocal microscopy as described previously (3, 27).
Surface trypsin digestion of intact A. phagocytophilum DC organisms.
The surface trypsin digestion of intact A. phagocytophilum DC organisms was performed as described by Wang and colleagues (74). Intact DC bacteria were incubated at a 10:1 ratio of total protein to trypsin (Thermo Scientific, Waltham, MA) in 1× phosphate-buffered saline (PBS) or vehicle alone at 37°C. After 30 min, phenylmethanesulfonyl fluoride (Sigma-Aldrich) was added to a final concentration of 2 mM to terminate the digestion. Bacteria were pelleted at 5,000 × g for 10 min, after which pellets were resuspended in urea lysis buffer and processed as described previously (69). Lysates of trypsin- and vehicle-treated A. phagocytophilum organisms were fractionated by SDS-PAGE, subjected to Western blot analysis, and screened with antibodies targeting OmpA, Asp55 (16), Msp5, and APH_0032 (29).
Flow cytometry.
HL-60 cells (1 × 107) infected with transgenic HGE1 organisms expressing GFP were sonicated followed by differential centrifugation to pellet host cellular debris (68). GFP-positive A. phagocytophilum DC bacteria and remaining host cellular debris were pelleted (68) and resuspended in PBS containing preimmune mouse serum, serum from a mouse that had been experimentally infected with A. phagocytophilum (72), mouse anti-OmpA, or a secondary antibody control (rabbit anti-mouse IgG conjugated to Alexa Fluor 594; BD Biosciences, San Jose, CA). Antibody incubations and wash steps were performed (55). GFP-positive DC bacteria were assessed for Alexa Fluor 594 signal by analyzing samples on a FACSCanto II flow cytometer (Becton, Dickinson, Franklin Lakes, NJ). A total of 1 × 108 events were collected in the Virginia Commonwealth University (VCU) Flow Cytometry and Imaging Shared Resource Facility. Post-data-acquisition analyses were performed using the FCS Express 4 flow cytometry software package (De Novo Software, Los Angeles, CA).
Anti-OmpA serum inhibition of A. phagocytophilum infection.
A. phagocytophilum DC organisms were incubated with heat-inactivated mouse polyclonal antiserum targeting GST or GST-OmpA (1.5 mg/ml) for 30 min. The bacteria were added to HL-60 cells in the presence of antiserum for 1 h. Unbound bacteria were removed, and aliquots of host cells were examined for bound A. phagocytophilum organisms using indirect immunofluorescence microscopy (55). The remainders of the samples were incubated for 48 h, after which host cells were examined for morulae (55).
In silico analyses.
The MEMSAT-SVM algorithm (bioinf.cs.ucl.ac.uk/psipred) was used to predict the membrane topology of A. phagocytophilum OmpA. Predicted signal sequences for Anaplasma sp., Ehrlichia sp., and O. tsutsugamushi OmpA proteins were determined using TMPred (www.ch.embnet.org/software/TMPRED_form). Alignments of OmpA sequences (minus the predicted signal sequences) were generated using CLUSTAL W (66). The tertiary structure for A. phagocytophilum OmpA was predicted using the PHYRE2 (Protein Homology/analogy Recognition Engine, version 2.0) server (www.sbg.bio.ic.ac.uk/phyre2) (33).
Binding of GST-OmpA to mammalian host cells and competitive inhibition of A. phagocytophilum infection.
For recombinant protein binding studies, RF/6A, CHO, or PSGL-1 CHO cells were incubated with 4 μM GST, GST-OmpA, GST-OmpA19-74, GST-OmpA75-205, GST-OtOmpA, or GST-APH_1387112-579 at 37°C for 1 h. Host cells were washed with PBS to remove unbound proteins, fixed with 4% (vol/vol) paraformaldehyde in PBS for 1 h, and permeabilized with ice-cold methanol for 30 s. Bound GST-fusion protein was detected using a rabbit anti-GST antibody (Invitrogen) followed by Alexa Fluor 488-conjugated goat anti-rabbit IgG (Invitrogen) and indirect immunofluorescence microscopy or flow cytometry. In some cases, RF/6A cells were treated with trypsin (10 μg/ml; Sigma) at 37°C for 30 min or a sialidase cocktail at 37°C for 1 h prior to flow cytometric analyses (5). To monitor the efficacy of enzymatic removal of sialic acid residues, mock- and sialidase-treated RF/6A cells were incubated with either biotinylated Maackia amurensis lectin II (MAL-II) followed by streptavidin-conjugated Alexa Fluor 488 or fluorescein-conjugated Sambucus nigra agglutinin (SNA). MAL-II and SNA binding was measured by flow cytometry as described by Heimburg-Molinaro and colleagues (23). For blocking of A. phagocytophilum infection, HL-60 or RF/6A cells were incubated with recombinant proteins (4 μM) for 1 h, after which A. phagocytophilum organisms were added, and incubation of bacteria with the host cells continued in the presence of recombinant protein for 2 h. Unbound bacteria were removed, and the samples were either assessed for surface-associated bacteria or incubated for 48 h followed by immunofluorescence microscopy analysis for the presence of morulae. In some cases, host cells were treated with 10 μg/ml of MAb CSLEX1 or mouse IgM isotype control (BD Biosciences) on ice for 1 h prior to the addition of bacteria.
Statistical analyses.
Statistical analyses were performed using the Prism 5.0 software package (Graphpad, San Diego, CA). If one-way analysis of variance (ANOVA) indicated a group difference (P < 0.05), then Dunnett's post hoc test was used to test for a significant difference among groups. In some instances, the Student t test was used to assess statistical significance. Statistical significance was set at P values of <0.05.
RESULTS
A. phagocytophilum upregulates ompA expression during invasion of myeloid cells and during transmission feeding of infected I. scapularis nymphs.
We determined the ompA transcriptional profile over the course of A. phagocytophilum infection in HL-60 cells. ompA expression increased during the first 8 h of infection relative to the DC inoculum, after which it subsided until it reached its lowest level at 28 h (Fig. 1A). Between 28 and 36 h, a time period that corresponds to RC-to-DC organism differentiation, DC organism exit, and initiation of the second round of infection (68), ompA expression increased. It takes up to 4 h for the majority of bound A. phagocytophilum organisms to enter and reside within nascent host cell-derived vacuoles (2, 4, 31). Thus, genes that are upregulated between 0 and 4 h and in the initial hours following bacterial entry may encode products that are important for infection. ompA expression steadily increased during the initial 4 h of infection of HL-60 cells but remained relatively stagnant during the first 4 h of infection of RF/6A endothelial cells (Fig. 1B and C).
Fig 1.

A. phagocytophilum ompA is upregulated during bacterial binding and invasion of HL-60 cells and during infected I. scapularis transmission feeding. (A) HL-60 cells were synchronously infected with A. phagocytophilum DC organisms. The infection proceeded for 36 h, a time period that allows for the bacteria to complete their biphasic developmental cycle and reinitiate infection. Total RNA was isolated from the DC inoculum and from infected host cells at several postinfection time points. RT-qPCR was performed using gene-specific primers. Relative transcript levels for each target were normalized to A. phagocytophilum 16S rRNA gene transcript levels using the 2−ΔΔCT method. To determine relative ompA transcription between RC and DC organisms, normalized transcript levels of each gene per time point were calculated as the fold change in expression relative to expression at 16 h, a time point at which the A. phagocytophilum population consists exclusively of RC organisms. The data are the means and standard deviations of results for triplicate samples and are representative of two independent experiments that yielded similar results. (B through D) DC organisms were incubated with HL-60 (B), RF/6A (C), and PSGL-1 CHO (D) cells for 4 h, a period that is required for bacterial adherence and for ≥90% of bound bacteria to invade host cells. A. phagocytophilum cannot invade PSGL-1 CHO cells. Total RNA was isolated from the DC inoculum and from host cells at 1, 2, 3, and 4 h following bacterial addition. (E) A. phagocytophilum-infected I. scapularis nymphs were allowed to feed on mice for 72 h. Total RNA was isolated from the salivary glands of uninfected and transmission-fed ticks that had been removed at 24, 48, and 72 h postattachment. Total RNA was isolated from combined salivary glands and midguts from unfed ticks. (B through E) RT-qPCR was performed using gene-specific primers. Relative transcript levels for ompA were normalized to A. phagocytophilum 16S rRNA gene transcript levels. The normalized values in panels B through D are presented relative to ompA transcript levels of the DC inoculum. Data are the means and standard deviations of results for triplicate samples and are representative of two independent experiments that yielded similar results.
We investigated whether bacterial engagement of PSGL-1 upregulates ompA transcription. Chinese hamster ovary cells transfected to express sLex-capped PSGL-1 are ideal models for studying A. phagocytophilum interactions with PSGL-1 and sLex (5, 35, 68, 76, 78). PSGL-1 CHO cells support A. phagocytophilum binding but not infection, while untransfected CHO cells lacking PSGL-1 expression do not support bacterial binding (5, 70, 78). A. phagocytophilum binding to PSGL-1 CHO cells occurs through bacterial engagement of sLex-capped PSGL-1 and excludes interactions with other undefined receptors that facilitate PSGL-1/sLex-independent adherence (5, 55, 56, 58, 70, 78). DC bacterial binding to PSGL-1 CHO cells did not increase ompA transcription (Fig. 1D).
A. phagocytophilum genes that are induced during transmission feeding of infected I. scapularis ticks are presumably important for establishing infection in mammals. We examined ompA expression in A. phagocytophilum-infected I. scapularis nymphs during transmission feeding on naïve mice. Transcripts for ompA were not detected in unfed A. phagocytophilum-infected nymphs (Fig. 1E). However, ompA expression was induced during transmission feeding, being first detected at 48 h.
A. phagocytophilum expresses OmpA during infection of HL-60 cells, humans, and mice.
The ompA coding region (19.9 kDa, excluding the signal sequence) was cloned and expressed in E. coli, resulting in an N-terminal GST-tagged fusion protein (GST-OmpA) (Fig. 2A). After glutathione-Sepharose affinity chromatography, purified GST-OmpA appeared as a 46.0-kDa band upon SDS-PAGE. The fusion protein was used to immunize mice. Polyclonal OmpA antisera recognized proteins of 22.1 kDa and 19.9 kDa, which correspond to the anticipated sizes for OmpA preprotein and mature OmpA, respectively, in a whole-cell lysate of A. phagocytophilum organisms derived from infected HL-60 cells but not an uninfected HL-60 cell lysate (Fig. 2B). HGA patient serum and A. phagocytophilum-infected mouse serum each recognized GST-OmpA (Fig. 2C), signifying that A. phagocytophilum expresses OmpA during infection of humans and mice and that OmpA stimulates the humoral immune response. Two additional HGA patient serum samples also recognized GST-OmpA (data not shown).
Fig 2.
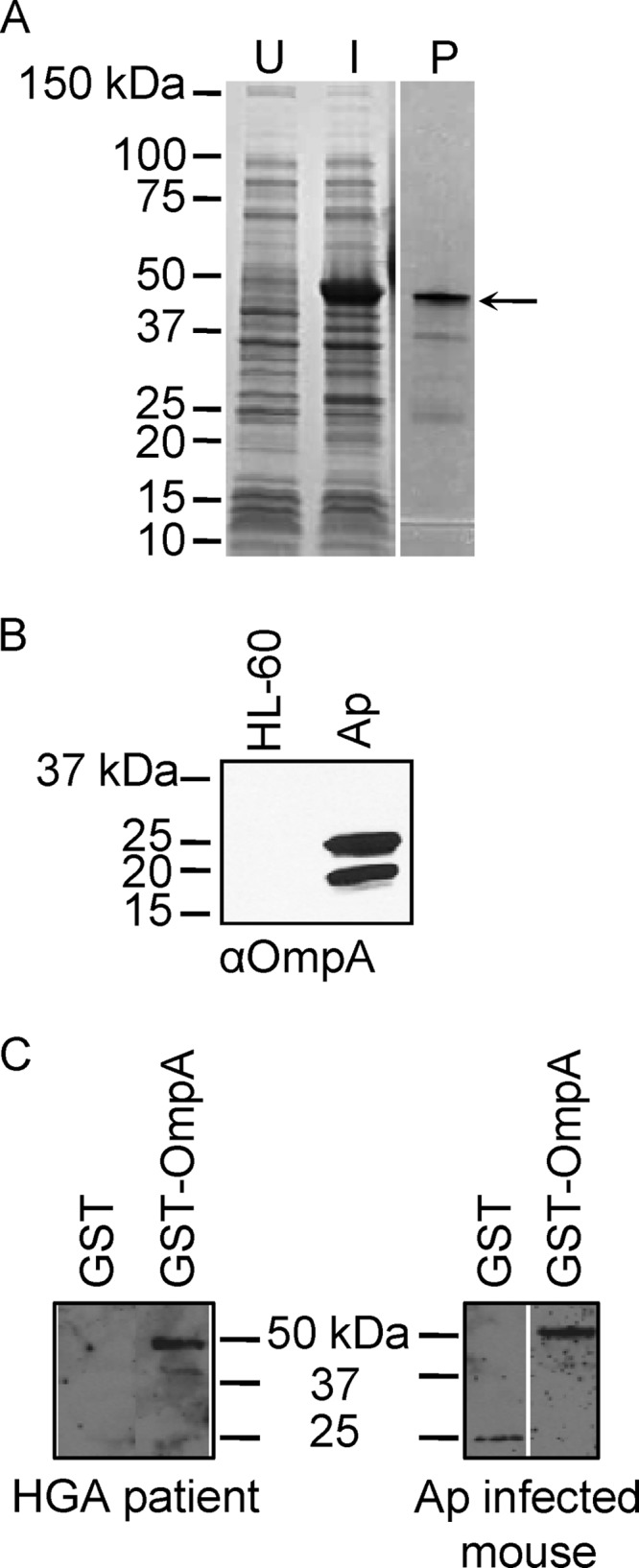
A. phagocytophilum (Ap) expresses OmpA during in vitro and in vivo infection. (A) Whole-cell lysates of uninduced E. coli (U) and of E. coli induced to express GST-OmpA (I) and GST-OmpA purified by glutathione Sepharose affinity chromatography (P) were separated by SDS-PAGE and stained with Coomassie blue. The arrow denotes the anticipated size for GST-OmpA. (B) Western blot analyses in which mouse anti-OmpA (αOmpA; raised against GST-OmpA) was used to screen whole-cell lysates of uninfected HL-60 cells and A. phagocytophilum organisms derived from infected HL-60 cells. (C) Western blots of GST-OmpA and GST screened with sera from an HGA patient and an experimentally infected mouse.
A. phagocytophilum differentially expresses OmpA during infection of mammalian versus tick cells.
Because A. phagocytophilum infects myeloid cells, endothelial cells, and I. scapularis cells (24, 57, 63, 75), we examined OmpA expression in infected HL-60 cells, RF/6A cells, and ISE6 cells, respectively. Screening with antibodies targeting Msp2 (P44; used to identify the bacteria) and OmpA during asynchronous infection revealed that 100.0% ± 0.0% and 48.6% ± 15.9% of morulae in HL-60 and RF/6A cells, respectively, harbored organisms that expressed OmpA (Fig. 3). Examination of morulae at 16, 20, 24, 32, and 36 h following a synchronous infection of HL-60 cells revealed that 100% were OmpA positive for all time points (data not shown). Only 7.0% ± 3.5% of morulae in ISE6 cells contained OmpA-positive bacteria (Fig. 3). Similar results were observed upon examining morulae within ISE6 cells each day over a 7-day period following a synchronous infection (data not shown).
Fig 3.
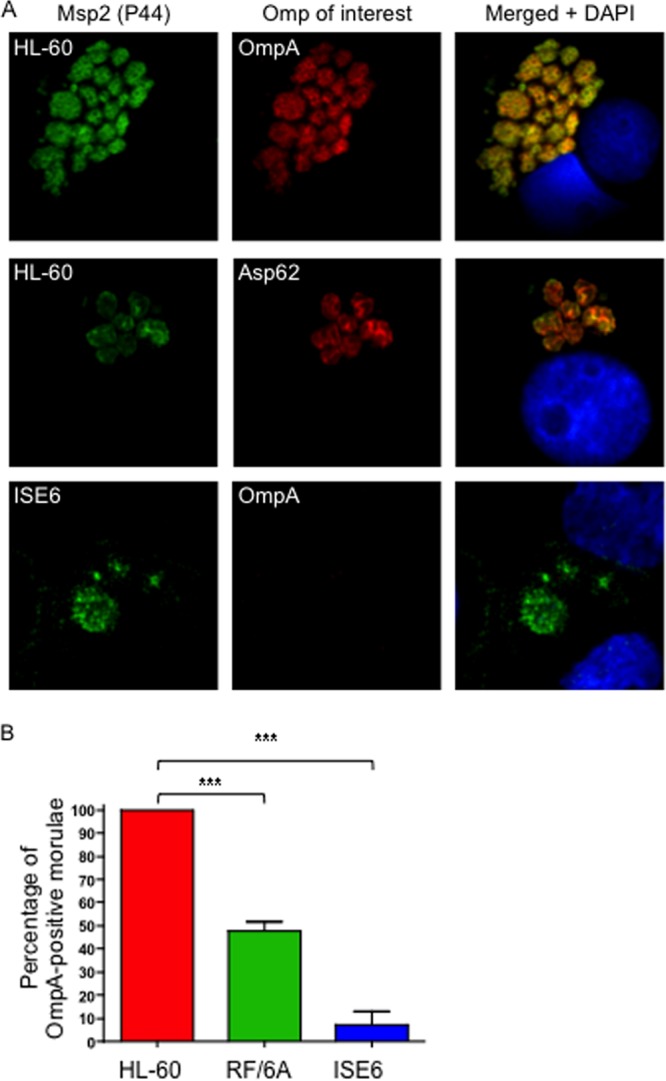
A. phagocytophilum differentially expresses OmpA during infection of mammalian and tick cells. (A) A. phagocytophilum-infected HL-60, RF/6A, and ISE6 cells were fixed and viewed by indirect immunofluorescence confocal microscopy to determine immunoreactivity with antibodies against Msp2 (P44) (major surface protein; used to identify bacteria) and OmpA or Asp62 (confirmed surface protein). Host cell nuclei are stained with DAPI (4′,6-diamidino-2-phenylindole). Note that staining of OmpA and staining of Asp62 yield comparable ring-like bacterial surface staining patterns. (B) Percentages of morulae [based on the presence of Msp2 (P44)-positive A. phagocytophilum organisms] that are positive for OmpA in infected HL-60, RF/6A, and ISE6 cells. The data are the means and standard deviations of results of at least two separate experiments. At least 200 Msp2 (P44)-positive morulae were scored for OmpA per condition. Statistically significant (***, P < 0.001) values are indicated.
OmpA is presented on the A. phagocytophilum surface.
Anti-OmpA recognition of intracellular A. phagocytophilum organisms yielded a ring-like staining pattern on the periphery of each bacterium that overlapped with signal corresponding to Msp2 (P44) and was similar to that of Asp62, both of which are confirmed surface proteins (16, 57) (Fig. 3A). To assess surface presentation of OmpA, we employed a method that has been used to verify the surface localization of Chlamydia trachomatis major outer membrane protein (74). Intact A. phagocytophilum DC organisms were incubated with trypsin followed by solubilization, Western blotting, and screening with anti-OmpA to determine if immunoaccessible domains are on the bacterial surface. Positive-control antisera targeted confirmed surface proteins Asp55 and Msp5 (16, 39). Negative-control antiserum was specific for APH_0032, which is an A. phagocytophilum effector that localizes to the bacterium's vacuolar membrane and is not a surface protein (29). Anti-Asp55 is specific for a peptide epitope of a surface-exposed loop of the target protein (16). Considerably less detection of OmpA, Asp55, and Msp5 but not APH_0032 was observed for surface-trypsinized bacteria than for vehicle control-treated bacteria (Fig. 4A). As a complementary approach, transgenic A. phagocytophilum DC organisms expressing GFP (12, 56) were recovered from HL-60 cells and screened with anti-OmpA or positive-control antiserum obtained from an A. phagocytophilum-infected mouse by flow cytometry. The serum from the A. phagocytophilum-infected mouse (72), which recognizes Msp2 (P44) in a Western blot and intact A. phagocytophilum bacteria in indirect immunofluorescence microscopy (data not shown), recognized 1.7 ± 0.2-fold more DC organisms than preimmune mouse serum (Fig. 4B). Anti-OmpA recognized 6.4 ± 1.1-fold more DC bacteria expressing GFP than preimmune mouse serum. Similar results were obtained for RC organisms (data not shown).
Fig 4.
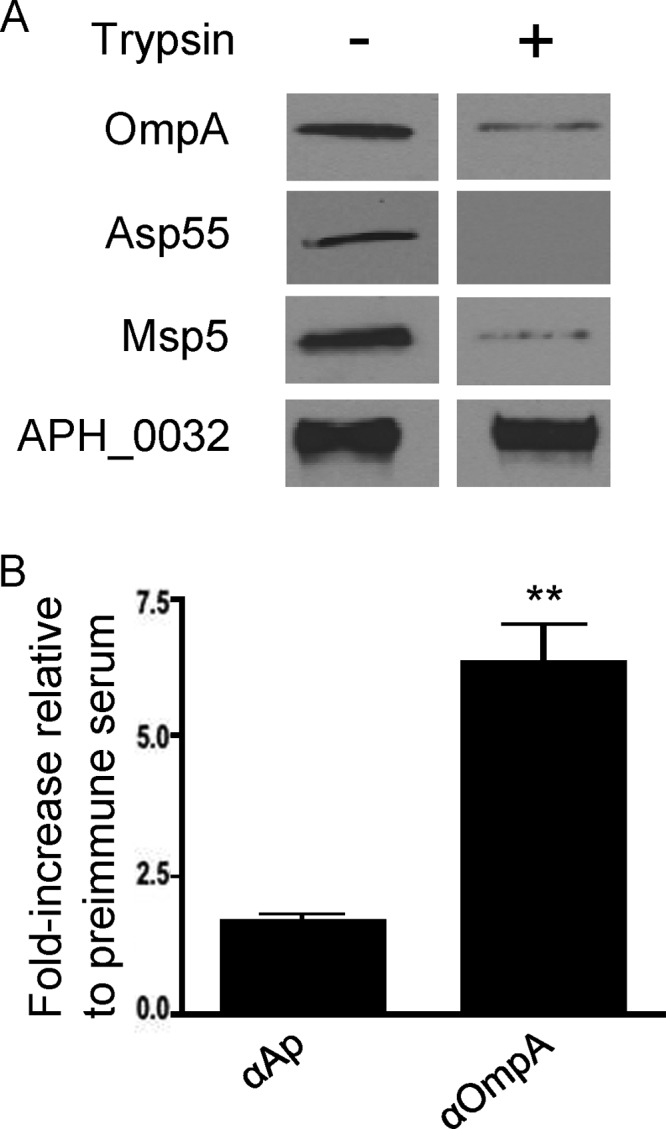
OmpA is on the A. phagocytophilum surface. (A) Intact DC bacteria were incubated with trypsin or vehicle control, fractionated by SDS-PAGE, and analyzed by Western blotting. Blots were screened with antisera targeting OmpA, Asp55, Msp5, or APH_0032. Data are representative of two experiments with similar results. (B) Live transgenic A. phagocytophilum DC organisms expressing GFP were incubated with preimmune mouse serum, mouse anti-OmpA, or serum recovered from an A. phagocytophilum-infected mouse. Primary antibodies were detected with anti-mouse IgG conjugated to Alexa Fluor 647. Flow cytometry was used to determine the percentage of Alexa Fluor 647- and GFP-positive DC organisms per sample. The fold increases in the percentages of Alexa Fluor 647 and GFP dual-positive DC organisms for each sample relative to preimmune serum are provided. Results presented are the means ± standard deviations of three experiments. Statistically significant (**, P < 0.005) values are indicated.
Pretreatment of A. phagocytophilum with anti-OmpA reduces infection of HL-60 cells.
Because OmpA is exposed on the A. phagocytophilum surface, we examined whether treating DC organisms with heat-inactivated anti-OmpA serum prior to incubation with HL-60 cells alters bacterial adhesion to, or infection of, host cells. Anti-OmpA had no effect on bacterial adhesion but significantly reduced infection (Fig. 5A through D). Pretreatment of bacteria with mouse polyclonal anti-GST serum had no effect on binding or infection.
Fig 5.

Pretreatment of A. phagocytophilum with anti-OmpA reduces infection of HL-60 cells. Host cell-free A. phagocytophilum (Ap) DC organisms were incubated with mouse polyclonal antiserum raised against GST-OmpA or GST alone. The treated bacteria were incubated with HL-60 cells for 60 min. After removal of unbound bacteria, the infection of HL-60 cells was allowed to proceed for 48 h, during which A. phagocytophilum binding and infection were assessed using antibody targeting Msp2 (P44) and confocal microscopy. (A) Percentages of HL-60 cells with bound A. phagocytophilum organisms. (B) Means ± standard deviations (SD) of bound A. phagocytophilum organisms per cell. (C) Percentages of infected HL-60 cells following incubation with A. phagocytophilum organisms in the presence of anti-OmpA or anti-GST. (D) Means ± SD of morulae per cell. Results in each panel are the means ± SD of three independent experiments. Statistically significant (**, P < 0.005) values are indicated.
In silico analyses of A. phagocytophilum OmpA and comparisons with homologs from other Rickettsiales pathogens.
Since anti-OmpA inhibits A. phagocytophilum infection, we hypothesized that OmpA may contribute to infection of host cells. We performed in silico analyses to identify the predicted extracellular region of OmpA, which would putatively contain any receptor-binding domain, and to assess whether this and other regions of OmpA are conserved among its homologs from other Rickettsiales bacteria. The OmpA N-terminal region extending through amino acid 86 is predicted to comprise the only extracellular domain, and amino acids 87 to 102 are predicted to form a transmembrane helix (Fig. 6A). A multiple-sequence alignment revealed that the A. phagocytophilum OmpA sequence has several stretches that exhibit identity or similarity with its homologs from other Anaplasma spp., Ehrlichia spp., and O. tsutsugamushi, an obligate intracellular bacterial pathogen that is in the order Rickettsiales (Fig. 6A and B).
Fig 6.

In silico analyses of OmpA sequences. (A and B) Alignments of A. phagocytophilum OmpA with its homologs from A. marginale St. Maries strain (AM854), A. marginale subsp. centrale Israel strain (ACIS_00486), E. chaffeensis Arkansas strain (ECH_0462), Ehrlichia canis Jake strain (Ecaj_0563), and Ehrlichia ruminantium Welgevonden strain (Erum_5620) (A) and with O. tsutsugamushi Ikeda strain OmpA (OTT_1320) (B). The signal sequences of all proteins have been removed. The A. phagocytophilum OmpA amino acid numbers are listed above each set of aligned sequences. A. phagocytophilum OmpA amino acids that contain the cellular adhesion domain, determined as shown in Fig. 9, are denoted in boldface. Amino acids that are highly similar or weakly similar are underscored by two dots or one dot, respectively. (A) A. phagocytophilum OmpA amino acids that are predicted to be extracellular and to form a transmembrane domain are denoted by an asterisk and a plus sign, respectively, above them. Amino acids that are identical among all six, five of six, four of six, and three of six sequences are highlighted in red, yellow, green, and blue, respectively. If a given amino acid is identical among the three Anaplasma species sequences and the amino acid at the same position is identical among the three Ehrlichia species sequences but is different from the one in the Anaplasma species sequences, then the amino acid for only the Anaplasma species sequences is highlighted in blue. (B) Amino acids that are identical in the A. phagocytophilum and O. tsutsugamushi OmpA sequences are highlighted in red. (C) Predicted tertiary structure for A. phagocytophilum OmpA. The OmpA mature protein sequence was analyzed using the Phyre2 algorithm. The highest-scoring model (confidence value of 99.97%) that exhibited the greatest amino acid identity was for A. phagocytophilum OmpA amino acids 23 to 158 threaded on the crystal structure for amino acids 1 to 133 of Bacillus subtilis chorismate mutase, which is in the OmpA superfamily. The orange portion corresponds to amino acids 44 to 56, which is predicted to form a surface-exposed helix and loop. The red portions correspond to Lys 49, Tyr 51, Phe 52, Asp 53, and Lys 56 of the K[IV]YFDaXK peptide (where “a” is a nonpolar amino acid and “X” is any amino acid), which corresponds to A. phagocytophilum OmpA residues 49 to 56 and is conserved among Anaplasma spp., Ehrlichia spp., and O. tsutsugamushi OmpA proteins.
The PHYRE2 server (www.sbg.bio.ic.ac.uk/phyre2) predicts tertiary structures for protein sequences and threads the predicted structures on known crystal structures (33). The highest-scoring model for A. phagocytophilum OmpA that exhibits the greatest amino acid sequence identity with the crystal structure on which it was threaded, Bacillus subtilis chorismate mutase, which is in the OmpA superfamily, is presented in Fig. 6C. Amino acids 44 to 56 are predicted to form a surface-exposed helix and loop. The peptide K[IV]YFDaXK (where “a” and “X” represent a nonpolar and any amino acid, respectively), which corresponds to A. phagocytophilum OmpA residues 49 to 56, is conserved among Anaplasma spp., Ehrlichia spp., and O. tsutsugamushi OmpA proteins (Fig. 6).
Interactions of GST-OmpA with endothelial cells.
We next tested if we could detect GST-OmpA binding to RF/6A cells. Since OmpA proteins of A. phagocytophilum and O. tsutsugamushi exhibit regions of identity (Fig. 6B), O. tsutsugamushi infects endothelial cells (17), and it is unknown whether O. tsutsugamushi OmpA interacts with endothelial cells, we also assessed whether GST-tagged O. tsutsugamushi OmpA (GST-OtOmpA) bound to RF/6A cells. Negative controls for cellular adhesion were GST alone and GST-tagged APH_1387 amino acids 112 to 579 (GST-APH_1387112-579). APH_1387 is an A. phagocytophilum effector that associates with the bacterium's vacuolar membrane (30). APH_1387 amino acids 112 to 579 lack the transmembrane domain that is required for interacting with eukaryotic cell membranes (unpublished observation). GST-OmpA but not GST bound to RF/6A cells (Fig. 7A and B). Neither GST-APH_1387112-579 nor GST-OtOmpA bound the host cells. GST-tagged A. phagocytophilum OmpA binding to RF/6A cells is therefore specific because the recombinant form of neither an irrelevant A. phagocytophilum protein nor OmpA derived from another Rickettsiales bacterium binds to RF/6A cells. GST-OmpA binding to RF/6A cells does not involve PSGL-1 or sLex since antibodies targeting either receptor fail to bind RF/6A cells (data not shown) and a previous report demonstrated that endothelial cells do not express PSGL-1 (34). We examined if preincubating RF/6A cells with GST-OmpA competitively inhibits A. phagocytophilum binding or infection. GST-OmpA but not GST significantly inhibited infection (Fig. 7C). Neither recombinant protein inhibited A. phagocytophilum adhesion (data not shown).
Fig 7.

GST-OmpA interactions with RF/6A endothelial cells. RF/6A cells were incubated with GST-OmpA, GST-OmpA19-74, GST-OmpA75-205, GST-O. tsutsugamushi OmpA (GST-OtOmpA), GST-APH_1387112-579 (negative control; does not associate with eukaryotic membranes), or GST alone for 60 min followed by extensive washing to remove unbound protein. (A) Indirect immunofluorescence microscopy analysis of GST-fusion proteins bound to RF/6A cells. The host cells were fixed and successively incubated with anti-GST antibody and anti-mouse IgG conjugated to Alexa Fluor 594. Gel mounting medium containing DAPI was added. Representative merged fluorescent confocal microscopic images are shown. Results are representative of two to four independent experiments. (B) Flow cytometric analysis of GST-fusion protein binding to RF/6A cells. The host cells were successively incubated with GST antibody and Alexa Fluor 488-conjugated anti-mouse IgG and analyzed by flow cytometry. (C) Preincubation of RF/6A cells with GST-OmpA competitively inhibits A. phagocytophilum infection. RF/6A cells were incubated with A. phagocytophilum DC organisms in the presence of GST or GST-OmpA for 1 h, after which confocal microscopy was used to assess bacterial infection at 48 h. Results shown are the percent infection of GST-treated host cells and are the means ± SD of 3 experiments. Statistically significant (***, P < 0.001) values are indicated.
Sialidase and trypsin treatments markedly reduce GST-OmpA binding to host cells.
Enzymatic removal of sialic acid residues from myeloid cell surfaces pronouncedly inhibits A. phagocytophilum binding and infection (5, 20). Sialic acid residues are also important for A. phagocytophilum infection of RF/6A cells, as pretreatment of RF/6A cells with sialidases reduced A. phagocytophilum infection by 52.8% ± 1.4% (Fig. 8A). The MAL-II lectin recognizes sialic acids that are attached to galactose units via α2,3 linkages (73). The SNA lectin preferentially binds to sialic acid attached to galactose in an α2,6 linkage (60). Sialidase treatment abolished MAL-II binding and markedly reduced SNA binding (Fig. 8B), indicating that the sialidase cocktail completely removed α2,3-linked sialic acids and partially removed α2,6-linked sialic acids. GST-OmpA did not bind as well to RF/6A cells that had been incubated in the vehicle control buffer as it did with cells incubated in other buffers (Fig. 8C and D). Nonetheless, GST-OmpA binding to sialidase-treated cells was reduced. These results suggest that OmpA recognizes α2,3-linked sialic acids but is also capable of interacting with α2,6-linked sialic acids. Pretreatment of RF/6A cells with trypsin, which would effectively digest protein and glycoprotein receptors, including terminally sialylated glycoproteins, nearly eliminated GST-OmpA binding (Fig. 8D).
Fig 8.

GST-OmpA binding to host cells is sialidase and trypsin sensitive. (A through C) RF/6A cells were treated with a sialidase cocktail or vehicle control (mock). (A) Sialidase- and mock-treated cells were incubated with A. phagocytophilum DC bacteria, after which the numbers of morulae per cell were assessed by indirect immunofluorescence microscopy at 48 h postinfection. The results of two separate experiments are presented. (B and C) Sialidase- and mock-treated cells were incubated with the sialic acid-specific lectin MAL II (preferentially recognizes α2,3-linked sialic acid) or SNA (preferentially recognizes α2,6-linked sialic acid) (B) or GST-OmpA (C). Binding of MAL II, SNA, and GST-OmpA was assessed by flow cytometry. (D) Binding of GST-OmpA to trypsin- and mock-treated RF/6A cells was assessed by flow cytometry. Control, RF/6A cells alone. Data presented in panels B through D are representative of at least two experiments with similar results.
GST-OmpA competitively inhibits A. phagocytophilum infection of HL-60 cells.
To define the relevance of OmpA to A. phagocytophilum infection of human myeloid cells and to delineate the OmpA region that is critical for cellular invasion, we examined if preincubating HL-60 cells with GST-OmpA or fragments thereof inhibits infection by A. phagocytophilum DC organisms. GST-tagged full-length OmpA and OmpA19-74, which comprises the majority of the predicted extracellular domain, but not GST-OmpA75-205 or GST alone significantly inhibited infection (Fig. 9) but had no effect on adhesion (data not shown).
Fig 9.

Preincubation of HL-60 cells with GST-OmpA or GST-OmpA19-74 competitively inhibits A. phagocytophilum infection. HL-60 cells were incubated with A. phagocytophilum (Ap) DC organisms in the presence of GST alone, GST-OmpA, GST-OmpA19-74, or GST-OmpA75-205 for 48 h, after which confocal microscopy was used to assess the percentage of infected cells (A) and the mean number ± SD of morulae per cell (B). Results shown are the percent infection of GST-treated host cells and are the means ± SD of 3 experiments. Statistically significant (**, P < 0.005; ***, P < 0.001) values are indicated.
GST-OmpA inhibits A. phagocytophilum binding to sLex-capped PSGL-1.
A. phagocytophilum binding to the α2,3-linked sialic acid determinant of sLex is necessary for the bacterium to optimally engage sLex-capped PSGL-1 and leads to infection of myeloid cells (20, 78). Since GST-OmpA recognizes α2,3-sialic acid and competitively inhibits A. phagocytophilum infection of HL-60 cells, we rationalized that GST-OmpA binds to α2,3-sialic acid of sLex. To test this, we incubated PSGL-1 CHO cells with GST-OmpA in an attempt to block A. phagocytophilum access to the α2,3-sialic acid determinant of sLex-capped PSGL-1 and thereby inhibit bacterial adherence to these cells. As a positive control for preventing bacterial access to the α2,3-linked sialic acid determinant of sLex, PSGL-1 CHO cells were incubated with CSLEX1 (13). PSGL-1 CHO cells treated with GST or mouse IgM served as negative blocking controls. GST-OmpA reduced A. phagocytophilum binding to sLex-modified PSGL-1 by approximately 60% relative to GST alone, and this degree of inhibition was comparable to the blocking afforded by CSLEX1 (Fig. 10).
Fig 10.
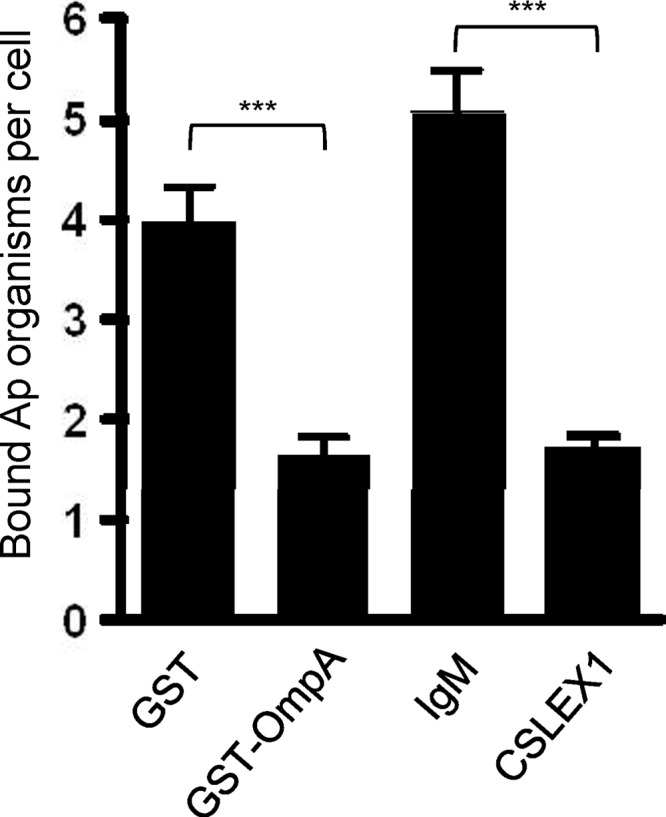
GST-OmpA inhibits A. phagocytophilum binding to sLex-capped PSGL-1. A. phagocytophilum DC organisms were added to PSGL-1 CHO cells that had been incubated with GST-OmpA, GST alone, CSLEX1, or mouse IgM, and incubation continued in the presence of recombinant protein or antibody for 1 h. After removal of unbound bacteria, the numbers of A. phagocytophilum DC organisms bound to PSGL-1 CHO cells were determined using indirect immunofluorescence microscopy. Results shown are representative of two independent experiments with similar results. Statistically significant (***, P < 0.001) values are indicated.
DISCUSSION
Sialic acid has long been known to be a determinant that is important for A. phagocytophilum infection (20). This study demonstrates that OmpA targets sialylated glycoproteins to promote A. phagocytophilum infection. Our results fit the model that A. phagocytophilum employs multiple surface proteins to bind three determinants of sLex-capped PSGL-1 to infect myeloid cells (Fig. 11A) (5, 78). When these data are examined in the context of results obtained from our studies (5, 55, 56, 58, 78) and those of others (20, 25), the respective contributions of sialic acid, α1,3-fucose, and PSGL-1 N-terminal peptide to A. phagocytophilum binding and entry become clearer. Treating myeloid cells with CSLEX1 to block A. phagocytophilum binding to the sialic acid determinant of sLex markedly reduces infection (Fig. 11C) (20), a phenomenon that is analogous to the inhibitory action of GST-OmpA. Moreover, the inhibitory effects of CSLEX1 and GST-OmpA on A. phagocytophilum binding to PSGL-1 CHO cells are nearly identical. Therefore, while OmpA is capable of binding sialic acid determinants of varied sialylated glycans, its specific interaction with the sialic acid residue of sLex is important for bacterial entry. GST-OmpA and GST-OmpA19-74 binding to host cells reduces A. phagocytophilum infection of HL-60 cells by approximately 52 and 57%, respectively, but has no inhibitory effect on bacterial adhesion. Thus, bacterial recognition of the PSGL-1 N terminus, α1,3-fucose of sLex, and perhaps sLex-/PSGL-1-independent interactions that still occur when the OmpA-sialic acid interaction is disrupted facilitates bacterial binding but leads to suboptimal infection (Fig. 11B) (5, 25, 55, 56, 58, 78). Antibodies that block access to the PSGL-1 N-terminal peptide determinant prevent bacterial binding and infection (25, 55, 78). Therefore, the collective avidity mediated by OmpA interaction with sialic acid together with A. phagocytophilum recognition of α1,3-fucose is insufficient to promote bacterial adhesion and, consequently, entry in the absence of PSGL-1 recognition (Fig. 11D). α1,3-Fucose is also important for bacterial binding and internalization (5, 25) and is the only known determinant that A. phagocytophilum requires to colonize I. scapularis ticks (51). Multiple surface proteins that play distinct roles in binding and invasion have also been demonstrated for spotted fever Rickettsia species (8).
Fig 11.

Models of how A. phagocytophilum multivalent binding to sLex-capped PSGL-1 promotes invasion and how GST-OmpA or antibodies targeting the α2,3-linked sialic acid determinant of sLex or the PSGL-1 N terminus inhibit infection. (A) A. phagocytophilum (Ap) surface proteins cooperatively bind three determinants of sLex-capped PSGL-1 to promote bacterial adhesion and entry. OmpA interacts with α2,3-sialic acid of sLex, while unidentified A. phagocytophilum surface proteins recognize α1,3-fucose of sLex and PSGL-1 N-terminal peptide. (B) GST-OmpA binds to α2,3-sialic acid of sLex, thereby competitively inhibiting access of OmpA on the A. phagocytophilum surface to the determinant. Since A. phagocytophilum binding to sialic acid is critical for internalization, this results in a marked decrease in A. phagocytophilum infection. (C) MAb CSLEX1 binds to the sialic acid determinant of sLex (20). Since CSLEX1 and GST-OmpA target the same determinant, they inhibit A. phagocytophilum interaction with sLex-capped PSGL-1 and retard sialic acid-dependent infection in analogous manners. In scenarios depicted in panels B and C, the A. phagocytophilum interactions with α1,3-fucose of sLex and PSGL-1 N-terminal peptide that occur when recognition of sialic acid is blocked are sufficient to enable at least some degree of bacterial adhesion. (D) MAbs that are specific for the PSGL-1 N terminus prevent A. phagocytophilum binding to the PSGL-1 N terminus (25, 55, and 78). The bacterial interactions with α2,3-sialic acid and α1,3-fucose that occur in the absence of concomitant binding to the PSGL-1 N terminus are insufficient to enable A. phagocytophilum adhesion to sLex-capped PSGL-1 and, consequently, cellular invasion.
GST-OmpA or GST-OmpA19-74 binding to sialic acids presented on HL-60 cell surfaces was sufficient to competitively block access of native OmpA on the bacterial surface to sialic acids and thereby retard infection. The inhibition was specific, as GST-OmpA75-205 and GST alone each failed to reduce infection. Yet we were unable to detect GST-OmpA binding to HL-60 cells or PSGL-1 CHO cells by indirect immunofluorescence or flow cytometry (data not shown). These phenomena are consistent with the facts that A. phagocytophilum binding to sialic acid on myeloid cell surfaces and to sLex-capped PSGL-1-modeled glycopeptides in the absence of concomitant binding to the PSGL-1 backbone cannot be detected (5, 25, 78). Thus, when it does not occur in the context of a multivalent interaction, GST-OmpA binding to the sialic acid determinant of PSGL-1 on HL-60 cell surfaces is insufficient to withstand the shear forces encountered during the multiple centrifugation steps of sample processing.
The ability of OmpA to recognize sialylated glycans other than sLex presumably contributes to A. phagocytophilum infection of sLex-deficient RF/6A endothelial cells. OmpA likely cooperates with additional bacterial surface proteins that interact with sialic acid-independent determinants to facilitate infection of endothelial cells. These premises are supported by our observations that while GST-OmpA binding to RF/6A cells was sialidase sensitive and GST-OmpA competitively inhibited A. phagocytophilum infection of RF/6A cells, neither treatment abolished infection. Completely removing α2,3-sialic acid residues and partially removing α2,6-sialic residues from RF/6A cell surfaces inhibited but did not eliminate GST-OmpA binding, an observation that is consistent with the reduced binding of A. phagocytophilum to host cells treated with the same sialidase cocktail as that used in this study (5, 20). OmpA may bind to sialic acid residues in α2,6 or α2,9 linkages or to another sugar that is insensitive to sialidase treatment. Trypsin-mediated cleavage of RF/6A surface proteins nearly abolishes GST-OmpA binding, which supports the involvement of a protein receptor and suggests that the sialylated glycans with which OmpA interacts most likely decorate glycoproteins as opposed to glycolipids.
The freshwater Gram-negative bacterium Caulobacter crescentus responds to initial contact to surfaces by triggering just-in-time adhesin production that promotes maximal binding (36). In a similar manner, A. phagocytophilum adherence to HL-60 cells upregulates ompA, which conceivably enhances binding and thereby promotes invasion. A. phagocytophilum engaging sLex-capped PSGL-1 itself does not increase ompA expression, as evidenced by the lack of change in ompA transcript levels following bacterial binding to PSGL-1 CHO cells. Therefore, A. phagocytophilum interaction with one or more sLex- and PSGL-1-independent receptors on HL-60 cells is coincident with an increase in ompA expression. A. phagocytophilum entry via sLex- and PSGL-1-independent as well as caveola-dependent routes has been demonstrated (37, 55, 56, 58). Whether these two internalization routes are one and the same and whether caveola-dependent entry upregulates ompA expression remain to be discerned. A. phagocytophilum DC binding to RF/6A cells, which lack PSGL-1 and sLex expression on their surfaces, does not upregulate ompA. This hints that the bacterium's preexisting level of OmpA is sufficient to infect endothelial cells.
OmpA is transcriptionally induced during tick transmission feeding, which implies that bacterial sensing of an undefined stimulus associated with the blood meal leads to ompA upregulation. E. chaffeensis ompA is part of a regulon that is under the transcriptional control of CtrA, a response regulator of the bacterium's two-component regulatory system (10). CtrA is conserved among all sequenced Anaplasmataceae members (9). It is unknown whether CtrA transcriptionally regulates ompA in A. phagocytophilum. Gene expression of the Lyme disease Borrelia sp. virulence factor, outer surface protein C, is induced during ixodid tick transmission feeding and is essential for establishing infection in mammals (11, 18, 21, 48, 67). OmpA may likewise be important for establishing infection in mammals. Further support for this idea comes from our observations that OmpA is expressed during A. phagocytophilum infection in humans and mice. OmpA is expressed throughout the A. phagocytophilum developmental cycle in HL-60 cells but is barely expressed in ISE6 cells. This observation is consistent with an A. phagocytophilum whole-genome transcriptional profiling study, which showed that ompA expression is over 3-fold higher during infection of HL-60 cells than during infection of ISE6 cells (47). Thus, OmpA appears to be critical for infection of and/or survival in myeloid cells but not in tick cells.
The most comprehensively studied OmpA proteins are those of invasive Escherichia coli strains that cause neonatal meningitis. E. coli OmpA binds to GlcNAcβ1,4-GlcNAc epitopes of the gp96 homolog, Ecgp96 (53, 61). Thus, both A. phagocytophilum and E. coli OmpA proteins engage glycoproteins. Both are also critically important for facilitating invasion of host cells once bacterial adhesion has been achieved, but neither is essential for adherence (53, 54). E. coli OmpA interaction with gp96 on neutrophils leads to decreases in transcript and protein levels of Rac1, Rac2, and gp91phox, which are essential for reactive oxygen species production (43). A. phagocytophilum was the first bacterium demonstrated to abrogate the respiratory burst by inhibiting gp91phox and rac2 transcription (1, 6, 44). While molecular means by which A. phagocytophilum downregulates gp91phox and rac2 transcription have been reported (15, 65), it will be important to determine if OmpA contributes to these phenomena.
OmpA and its Anaplasma marginale and E. chaffeensis homologs are emerging as important virulence factors and potential targets for protecting against infection. Serum against E. chaffeensis OmpA partially neutralizes ehrlichial infection of THP-1 cells (10). A. marginale AM854 exhibits 44% amino acid identity to A. phagocytophilum OmpA. A recent study by Palmer and colleagues demonstrated that vaccinating cattle against an A. marginale OMP mixture provided protection against A. marginale challenge. Meta-analysis suggested that AM854 and two additional OMPs may be responsible for immunity afforded by the complex vaccinogen (49). It is unknown if E. chaffeensis or A. marginale OmpA interacts with sialic acid. However, the ability of A. marginale to agglutinate erythrocytes is partially inhibited by sialidase treatment of the host cells (42). There is considerable conservation among OmpA proteins from Anaplasma and Ehrlichia species and O. tsutsugamushi. Yet GST-OtOmpA is unable to bind RF/6A cells, which suggests that the regions of A. phagocytophilum OmpA that share identity with corresponding regions of O. tsutsugamushi OmpA may not be directly involved in mediating infection. Alternatively, O. tsutsugamushi OmpA may not be involved in facilitating infection of endothelial cells. Since A. phagocytophilum OmpA residues 19 to 74 contain the protein's cellular invasion domain, it will be crucial to pinpoint the invasion domain within this region. Of particular interest are residues 49 to 56, which are predicted to form part of a surface-exposed loop and are conserved among Anaplasma species, Ehrlichia species, and O. tsutsugamushi OmpA proteins. It will also be important to verify if antisera against the specific invasion domain or at least residues 19 to 74 can improve upon the modest inhibition afforded by antisera raised against the entire protein.
Overall, this work advances knowledge of A. phagocytophilum pathogenesis by demonstrating that OmpA is an invasin, by delineating the OmpA region that mediates interactions with host cells, and by determining that OmpA interacts with sialylated glycoproteins. Given the conservation of the OmpA invasion domain-containing region between Anaplasma and Ehrlichia species, it is imperative to further explore the efficacy of targeting OmpA as a means for intervening against infection by the many pathogenic species of these genera.
ACKNOWLEDGMENTS
We thank Ulrike Munderloh of the University of Minnesota for providing transgenic A. phagocytophilum expressing GFP, J. Stephen Dumler of The Johns Hopkins University for providing MAb 20B4, Durland Fish of Yale University for providing A. phagocytophilum-infected ticks, Yasuko Rikihisa of The Ohio State University for donating antisera against Asp55 and Asp62, Rodger McEver of The Oklahoma Medical Research Foundation for providing CHO and PSGL-1 CHO cells, and David Seidman for critical review of the manuscript.
This study was supported by NIH grants R01 AI072683, R01 AI072683-04S1, and R21 AI090170 (to J.A.C.) and R01 AI141440 (to E. F.). The VCU Flow Cytometry and Imaging Shared Resource Facility is supported, in part, by funding from NIH-NCI Cancer Center Support Grant 5 P30 CA016059.
Footnotes
Published ahead of print 20 August 2012
REFERENCES
- 1. Banerjee R, Anguita J, Roos D, Fikrig E. 2000. Cutting edge: infection by the agent of human granulocytic ehrlichiosis prevents the respiratory burst by down-regulating gp91phox. J. Immunol. 164:3946–3949 [DOI] [PubMed] [Google Scholar]
- 2. Borjesson DL, et al. 2005. Insights into pathogen immune evasion mechanisms: Anaplasma phagocytophilum fails to induce an apoptosis differentiation program in human neutrophils. J. Immunol. 174:6364–6372 [DOI] [PubMed] [Google Scholar]
- 3. Carlyon JA. 2005. Laboratory maintenance of Anaplasma phagocytophilum, p 3A.2.1–3A.2.30 In Coico R, Kowalik TF, Quarles JM, Stevenson B, Taylor R. (ed), Current protocols in microbiology. J. Wiley and Sons, Hoboken, NJ: [DOI] [PubMed] [Google Scholar]
- 4. Carlyon JA, Abdel-Latif D, Pypaert M, Lacy P, Fikrig E. 2004. Anaplasma phagocytophilum utilizes multiple host evasion mechanisms to thwart NADPH oxidase-mediated killing during neutrophil infection. Infect. Immun. 72:4772–4783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Carlyon JA, et al. 2003. Murine neutrophils require alpha1,3-fucosylation but not PSGL-1 for productive infection with Anaplasma phagocytophilum. Blood 102:3387–3395 [DOI] [PubMed] [Google Scholar]
- 6. Carlyon JA, Chan WT, Galan J, Roos D, Fikrig E. 2002. Repression of rac2 mRNA expression by Anaplasma phagocytophila is essential to the inhibition of superoxide production and bacterial proliferation. J. Immunol. 169:7009–7018 [DOI] [PubMed] [Google Scholar]
- 7. Cascales E, Bernadac A, Gavioli M, Lazzaroni JC, Lloubes R. 2002. Pal lipoprotein of Escherichia coli plays a major role in outer membrane integrity. J. Bacteriol. 184:754–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chan YG, Riley SP, Martinez JJ. 2010. Adherence to and invasion of host cells by spotted fever group rickettsia species. Front. Microbiol. 1:139 doi:10.3389/fmicb.2010.00139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cheng Z, Kumagai Y, Lin M, Zhang C, Rikihisa Y. 2006. Intra-leukocyte expression of two-component systems in Ehrlichia chaffeensis and Anaplasma phagocytophilum and effects of the histidine kinase inhibitor closantel. Cell. Microbiol. 8:1241–1252 [DOI] [PubMed] [Google Scholar]
- 10. Cheng Z, Miura K, Popov VL, Kumagai Y, Rikihisa Y. 2011. Insights into the CtrA regulon in development of stress resistance in obligatory intracellular pathogen Ehrlichia chaffeensis. Mol. Microbiol. 82:1217–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Earnhart CG, et al. 2010. Identification of residues within ligand-binding domain 1 (LBD1) of the Borrelia burgdorferi OspC protein required for function in the mammalian environment. Mol. Microbiol. 76:393–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Felsheim RF, et al. 2006. Transformation of Anaplasma phagocytophilum. BMC Biotechnol. 6:42 doi:10.1186/1472-6750-6-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fukushima K, et al. 1984. Characterization of sialosylated Lewisx as a new tumor-associated antigen. Cancer Res. 44:5279–5285 [PubMed] [Google Scholar]
- 14. Galindo RC, et al. 2008. Differential expression of inflammatory and immune response genes in sheep infected with Anaplasma phagocytophilum. Vet. Immunol. Immunopathol. 126:27–34 [DOI] [PubMed] [Google Scholar]
- 15. Garcia-Garcia JC, Rennoll-Bankert KE, Pelly S, Milstone AM, Dumler JS. 2009. Silencing of host cell CYBB gene expression by the nuclear effector AnkA of the intracellular pathogen Anaplasma phagocytophilum. Infect. Immun. 77:2385–2391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ge Y, Rikihisa Y. 2007. Identification of novel surface proteins of Anaplasma phagocytophilum by affinity purification and proteomics. J. Bacteriol. 189:7819–7828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ge Y, Rikihisa Y. 2011. Subversion of host cell signaling by Orientia tsutsugamushi. Microbes Infect. 13:638–648 [DOI] [PubMed] [Google Scholar]
- 18. Gilmore RD, Jr, Piesman J. 2000. Inhibition of Borrelia burgdorferi migration from the midgut to the salivary glands following feeding by ticks on OspC-immunized mice. Infect. Immun. 68:411–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Godlewska R, Wisniewska K, Pietras Z, Jagusztyn-Krynicka EK. 2009. Peptidoglycan-associated lipoprotein (Pal) of Gram-negative bacteria: function, structure, role in pathogenesis and potential application in immunoprophylaxis. FEMS Microbiol. Lett. 298:1–11 [DOI] [PubMed] [Google Scholar]
- 20. Goodman JL, Nelson CM, Klein MB, Hayes SF, Weston BW. 1999. Leukocyte infection by the granulocytic ehrlichiosis agent is linked to expression of a selectin ligand. J. Clin. Invest. 103:407–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Grimm D, et al. 2004. Outer-surface protein C of the Lyme disease spirochete: a protein induced in ticks for infection of mammals. Proc. Natl. Acad. Sci. U. S. A. 101:3142–3147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hall-Baker PA, et al. 2010. Summary of notifiable diseases: United States, 2008. MMWR Morb. Mortal. Wkly. Rep. 57:1–94 [PubMed] [Google Scholar]
- 23. Heimburg-Molinaro J, Song X, Smith DF, Cummings RD. 2011. Preparation and analysis of glycan microarrays, chapt 12, unit 1210 In Coligan JE, et al. (ed), Current protocols in protein science. John Wiley and Sons, Hoboken, NJ: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Herron MJ, Ericson ME, Kurtti TJ, Munderloh UG. 2005. The interactions of Anaplasma phagocytophilum, endothelial cells, and human neutrophils. Ann. N. Y. Acad. Sci. 1063:374–382 [DOI] [PubMed] [Google Scholar]
- 25. Herron MJ, et al. 2000. Intracellular parasitism by the human granulocytic ehrlichiosis bacterium through the P-selectin ligand, PSGL-1. Science 288:1653–1656 [DOI] [PubMed] [Google Scholar]
- 26. Hotopp JC, et al. 2006. Comparative genomics of emerging human ehrlichiosis agents. PLoS Genet. 2:e21 doi:10.1371/journal.pgen.0020021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huang B, et al. 2010. The Anaplasma phagocytophilum-occupied vacuole selectively recruits Rab-GTPases that are predominantly associated with recycling endosomes. Cell. Microbiol. 12:1292–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huang B, Ojogun N, Ragland SA, Carlyon JA. 2012. Monoubiquitinated proteins decorate the Anaplasma phagocytophilum-occupied vacuolar membrane. FEMS Immunol. Med. Microbiol. 64:32–41 [DOI] [PubMed] [Google Scholar]
- 29. Huang B, et al. 2010. Anaplasma phagocytophilum APH_0032 is expressed late during infection and localizes to the pathogen-occupied vacuolar membrane. Microb. Pathog. 49:273–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Huang B, et al. 2010. Anaplasma phagocytophilum APH_1387 is expressed throughout bacterial intracellular development and localizes to the pathogen-occupied vacuolar membrane. Infect. Immun. 78:1864–1873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. IJdo JW, Mueller AC. 2004. Neutrophil NADPH oxidase is reduced at the Anaplasma phagocytophilum phagosome. Infect. Immun. 72:5392–5401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ismail N, Bloch KC, McBride JW. 2010. Human ehrlichiosis and anaplasmosis. Clin. Lab. Med. 30:261–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kelley LA, Sternberg MJ. 2009. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4:363–371 [DOI] [PubMed] [Google Scholar]
- 34. Laszik Z, et al. 1996. P-selectin glycoprotein ligand-1 is broadly expressed in cells of myeloid, lymphoid, and dendritic lineage and in some nonhematopoietic cells. Blood 88:3010–3021 [PubMed] [Google Scholar]
- 35. Li F, et al. 1996. Post-translational modifications of recombinant P-selectin glycoprotein ligand-1 required for binding to P- and E-selectin. J. Biol. Chem. 271:3255–3264 [PubMed] [Google Scholar]
- 36. Li G, et al. 2012. Surface contact stimulates the just-in-time deployment of bacterial adhesins. Mol. Microbiol. 83:41–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lin M, Rikihisa Y. 2003. Obligatory intracellular parasitism by Ehrlichia chaffeensis and Anaplasma phagocytophilum involves caveolae and glycosylphosphatidylinositol-anchored proteins. Cell. Microbiol. 5:809–820 [DOI] [PubMed] [Google Scholar]
- 38. Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- 39. Lopez JE, et al. 2007. Immunogenicity of Anaplasma marginale type IV secretion system proteins in a protective outer membrane vaccine. Infect. Immun. 75:2333–2342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mastronunzio JE, Kurscheid S, Fikrig E. 2012. Post-genomic analyses reveal development of infectious Anaplasma phagocytophilum during transmission from ticks to mice. J. Bacteriol. 194:2238–2247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McEver RP, Cummings RD. 1997. Perspectives series: cell adhesion in vascular biology. Role of PSGL-1 binding to selectins in leukocyte recruitment. J. Clin. Invest. 100:485–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McGarey DJ, Allred DR. 1994. Characterization of hemagglutinating components on the Anaplasma marginale initial body surface and identification of possible adhesins. Infect. Immun. 62:4587–4593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mittal R, Prasadarao NV. 2011. gp96 expression in neutrophils is critical for the onset of Escherichia coli K1 (RS218) meningitis. Nat. Commun. 2:552 doi:10.1038/ncomms1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mott J, Rikihisa Y. 2000. Human granulocytic ehrlichiosis agent inhibits superoxide anion generation by human neutrophils. Infect. Immun. 68:6697–6703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Munderloh UG, et al. 1999. Invasion and intracellular development of the human granulocytic ehrlichiosis agent in tick cell culture. J. Clin. Microbiol. 37:2518–2524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Munderloh UG, et al. 2004. Infection of endothelial cells with Anaplasma marginale and A. phagocytophilum. Vet. Microbiol. 101:53–64 [DOI] [PubMed] [Google Scholar]
- 47. Nelson CM, et al. 2008. Whole genome transcription profiling of Anaplasma phagocytophilum in human and tick host cells by tiling array analysis. BMC Genomics 9:364 doi:10.1186/1471-2164-9-364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pal U, et al. 2004. OspC facilitates Borrelia burgdorferi invasion of Ixodes scapularis salivary glands. J. Clin. Invest. 113:220–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Palmer GH, Brown WC, Noh SM, Brayton KA. 2012. Genome-wide screening and identification of antigens for rickettsial vaccine development. FEMS Immunol. Med. Microbiol. 64:115–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Park J, Choi KS, Dumler JS. 2003. Major surface protein 2 of Anaplasma phagocytophilum facilitates adherence to granulocytes. Infect. Immun. 71:4018–4025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pedra JH, et al. 2010. Fucosylation enhances colonization of ticks by Anaplasma phagocytophilum. Cell. Microbiol. 12:1222–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Popov VL, et al. 1998. Ultrastructural differentiation of the genogroups in the genus Ehrlichia. J. Med. Microbiol. 47:235–251 [DOI] [PubMed] [Google Scholar]
- 53. Prasadarao NV. 2002. Identification of Escherichia coli outer membrane protein A receptor on human brain microvascular endothelial cells. Infect. Immun. 70:4556–4563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Prasadarao NV, et al. 1996. Outer membrane protein A of Escherichia coli contributes to invasion of brain microvascular endothelial cells. Infect. Immun. 64:146–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Reneer DV, et al. 2006. Characterization of a sialic acid- and P-selectin glycoprotein ligand-1-independent adhesin activity in the granulocytotropic bacterium Anaplasma phagocytophilum. Cell. Microbiol. 8:1972–1984 [DOI] [PubMed] [Google Scholar]
- 56. Reneer DV, Troese MJ, Huang B, Kearns SA, Carlyon JA. 2008. Anaplasma phagocytophilum PSGL-1-independent infection does not require Syk and leads to less-efficient AnkA delivery. Cell. Microbiol. 10:1827–1838 [DOI] [PubMed] [Google Scholar]
- 57. Rikihisa Y. 2011. Mechanisms of obligatory intracellular infection with Anaplasma phagocytophilum. Clin. Microbiol. Rev. 24:469–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sarkar M, Reneer DV, Carlyon JA. 2007. Sialyl-Lewis x-independent infection of human myeloid cells by Anaplasma phagocytophilum strains HZ and HGE1. Infect. Immun. 75:5720–5725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Scorpio DG, Caspersen K, Ogata H, Park J, Dumler JS. 2004. Restricted changes in major surface protein-2 (msp2) transcription after prolonged in vitro passage of Anaplasma phagocytophilum. BMC Microbiol. 4:1 doi:10.1186/1471-2180-4-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Shibuya N, et al. 1987. The elderberry (Sambucus nigra L.) bark lectin recognizes the Neu5Ac(alpha 2-6)Gal/GalNAc sequence. J. Biol. Chem. 262:1596–1601 [PubMed] [Google Scholar]
- 61. Shin S, Lu G, Cai M, Kim KS. 2005. Escherichia coli outer membrane protein A adheres to human brain microvascular endothelial cells. Biochem. Biophys. Res. Commun. 330:1199–1204 [DOI] [PubMed] [Google Scholar]
- 62. Sukumaran B, Carlyon JA, Cai JL, Berliner N, Fikrig E. 2005. Early transcriptional response of human neutrophils to Anaplasma phagocytophilum infection. Infect. Immun. 73:8089–8099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sukumaran B, et al. 2011. Anaplasma phagocytophilum AptA modulates Erk1/2 signalling. Cell. Microbiol. 13:47–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Thomas RJ, Dumler JS, Carlyon JA. 2009. Current management of human granulocytic anaplasmosis, human monocytic ehrlichiosis and Ehrlichia ewingii ehrlichiosis. Expert Rev. Anti Infect. Ther. 7:709–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Thomas V, Samanta S, Wu C, Berliner N, Fikrig E. 2005. Anaplasma phagocytophilum modulates gp91phox gene expression through altered interferon regulatory factor 1 and PU.1 levels and binding of CCAAT displacement protein. Infect. Immun. 73:208–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tilly K, et al. 2006. Borrelia burgdorferi OspC protein required exclusively in a crucial early stage of mammalian infection. Infect. Immun. 74:3554–3564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Troese MJ, Carlyon JA. 2009. Anaplasma phagocytophilum dense-cored organisms mediate cellular adherence through recognition of human P-selectin glycoprotein ligand 1. Infect. Immun. 77:4018–4027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Troese MJ, et al. 2011. Proteomic analysis of Anaplasma phagocytophilum during infection of human myeloid cells identifies a protein that is pronouncedly upregulated on the infectious dense-cored cell. Infect. Immun. 79:4696–4707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Troese MJ, et al. 2009. Differential expression and glycosylation of anaplasma phagocytophilum major surface protein 2 paralogs during cultivation in sialyl Lewis x-deficient host cells. Infect. Immun. 77:1746–1756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Varki NM, Varki A. 2007. Diversity in cell surface sialic acid presentations: implications for biology and disease. Lab. Invest. 87:851–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wang T, et al. 2002. Superoxide anion production during Anaplasma phagocytophila infection. J. Infect. Dis. 186:274–280 [DOI] [PubMed] [Google Scholar]
- 73. Wang WC, Cummings RD. 1988. The immobilized leukoagglutinin from the seeds of Maackia amurensis binds with high affinity to complex-type Asn-linked oligosaccharides containing terminal sialic acid-linked alpha-2,3 to penultimate galactose residues. J. Biol. Chem. 263:4576–4585 [PubMed] [Google Scholar]
- 74. Wang Y, et al. 2006. Identification of surface-exposed components of MOMP of Chlamydia trachomatis serovar F. Protein Sci. 15:122–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Woldehiwet Z, et al. 2002. Cultivation of an ovine strain of Ehrlichia phagocytophila in tick cell cultures. J. Comp. Pathol. 127:142–149 [DOI] [PubMed] [Google Scholar]
- 76. Xia L, et al. 2003. N-terminal residues in murine P-selectin glycoprotein ligand-1 required for binding to murine P-selectin. Blood 101:552–559 [DOI] [PubMed] [Google Scholar]
- 77. Xiong Q, Rikihisa Y. 2011. The prenylation inhibitor manumycin A reduces the viability of Anaplasma phagocytophilum. J. Med. Microbiol. 60:744–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Yago T, et al. 2003. Structurally distinct requirements for binding of P-selectin glycoprotein ligand-1 and sialyl Lewis x to Anaplasma phagocytophilum and P-selectin. J. Biol. Chem. 278:37987–37997 [DOI] [PubMed] [Google Scholar]