

Abstract
Staphylococcus aureus is a prevalent and significant human pathogen. Among the repertoire of virulence factors produced by this bacterium are the 14 staphylococcal superantigen-like (SSL) proteins. SSL protein 4 (SSL4) is one member of this family and contains a highly conserved carbohydrate binding site also found in SSL2, SSL3, SSL5, SSL6, and SSL11. Recombinant SSL4t, comprising amino acids 109 to 309 of Newman strain SSL4 (SSL4-Newman), has been shown to bind and be internalized by human granulocytes and macrophages in a sialic-acid (Sia)-dependent manner. SSL4t can compete with itself for cell binding, indicating that binding is target specific. A 2.5-Å-resolution crystal structure of SSL4t complexed with sialyl Lewis X (sLex) [sLex-Neu5Acα2-3Galβ1-4(Fucα1-3)GlcNAc] revealed a similar binding site to SSL5 and SSL11. These data, along with data on SSL4t binding to a glycan array and biosensor analysis of sLex and sialyllactosamine (sLacNac) binding are compared with those for SSL11. Although these proteins show great similarity in their carbohydrate binding sites, with a root mean square (RMS) difference between main chain atom positions of only 0.34 Å, these proteins differ in detail in their affinity for sLex and sLacNac, as well as their glycan preference. Together with cell binding data, this shows how S. aureus produces multiple related proteins that target myeloid cells through specific sialyllactosamine-containing glycoproteins.
INTRODUCTION
Staphylococcus aureus is the most common cause of hospital-acquired infection (18, 19, 29) and also causes serious acute community-acquired infections in otherwise healthy individuals. S. aureus infects almost any tissue in the body and produces an array of cell surface and secreted virulence factors that contribute to its capacity to cause disease (9, 17, 20, 22, 30, 37, 41). Among its repertoire of immune-suppressive virulence proteins are the staphylococcal superantigen (Sag)-like (SSL) proteins, which share structural similarity with the classical superantigens (2, 25).
A total of 11 unique ssl genes reside within an almost contiguous region of the staphylococcal genome in a genomic island known as vSaα (24, 28). vSaα is present in all S. aureus genomes sequenced thus far and has been hypothesized to play an essential role in the evolution of this microbe as a major human pathogen (3). Despite their structural similarity to superantigens, SSLs do not bind to T-cell receptors or major histocompatibility complex class II molecules and do not stimulate T cells (2). Rather they target components of innate immunity, such as complement, Fc receptors, and myeloid cells. SSL protein 7 (SSL7) is the best characterized of the SSLs and binds complement factor C5 and IgA with high affinity and inhibits the end stage of complement activation and IgA binding to FcαR (27, 36). SSL10 binds to the Fc region of human IgG1 and blocks FcγR-mediated phagocytosis (35).
Unlike SSL7 and SSL10, SSL5 and SSL11 target myeloid cells by binding to cell surface glycoproteins containing the minimal trisaccharide siallylactosamine (sLacNac) (sLacNac-NeuAcα2-3-Galβ1-4-NAGluc). The crystal structures of SSL5 and SSL11 complexed with the blood group antigen sialyl Lewis X (sLex) [sLex-Neu5Acα2-3Galβ1-4(Fucα1-3)GlcNAc] have revealed a conserved binding site located in the C-terminal β-grasp domain (4, 11). sLex is essential to the inflammatory response and is constitutively expressed on granulocytes, regulating selectin-mediated tethering to endothelial cells and extravasation of neutrophils into the tissue to fight infection (32). Both SSL5 and SSL11 bind to neutrophils and inhibit P-selectin-mediated neutrophil rolling at a nanomolar concentration (7, 11). Interestingly SSL5 and SSL11 are rapidly internalized by neutrophils in an energy-dependent fashion, but it remains to be determined which intracellular molecules are targeted once inside the cell (4, 11). Structural modeling of all of the SSLs suggests that carbohydrate binding is shared by a subgroup that includes SSL2, SSL3, SSL4, SSL5, SSL6, and SSL11 (4). Recently it was shown SSL3 binds and inhibits Toll-like receptor 2 (TLR2) activation at the surface of human and murine neutrophils and monocytes in a partially glycan-dependent manner (5).
Here we show that SSL4 also binds to myeloid cells in a glycan-dependent fashion, similar to SSL11, and is rapidly internalized by neutrophils and macrophages and, to a lesser extent, monocytes. The crystal structure of SSL4 complexed with sLex reveals the carbohydrate binding site in atomic detail and enables us to explain small but significant differences in binding affinity and specificity compared with SSL11.
MATERIALS AND METHODS
Sequence comparisons were made using ClustalW (10), protein structure figures were prepared using PyMOL (http://www.pymol.org), flow cytometry figures were prepared using FlowJo, and confocal microscopy figures were prepared using FV10-ASW (Olympus).
Production of rSSL4 and mutant SSL4.
The gene encoding SSL4 from the published Newman strain (NCBI GeneID no. 5332478) of S. aureus (3) was amplified by PCR using the primers SSL4-NWM-Fwd (GCGGATCCACAAAACAAGTACCAACAG) and SSL4-NWM-Rev (CCGGAAGCTTTTATTTTATATTCACTTCAATGTTATC). The resulting products were cut using the restriction enzymes BamHI (underlined in the forward primer) and HindIII (underlined in the reverse primer) and cloned into the pET32a.3C vector. This construct comprises amino acids 109 to 309 of SSL4-Newman and is designated SSL4t. The gene encoding SSL11 from the naturally occurring US6610 strain of S. aureus had been previously cloned into the pET32a.3c vector by Chung et al. (11). Recombinant SSL4t and SSL11 (rSSL4t and rSSL11, respectively) were expressed in Escherichia coli AD494 as thioredoxin fusion proteins, as described previously (2, 26). Thioredoxin was cleaved off with 3C protease; rSSL4t and rSSL11 were further purified using ion-exchange chromatography (MonoS and MonoQ columns; GE Healthcare). Site-directed mutagenesis of SSL4t by overlap PCR (4, 11) using the primers R182A-Fwd (CAAGAAAATGCCATGGCAGATGTCATAGATG) and R182A-Rev (CTGCCATGGCATTTTCTTGTAATTTTTTGTGC) was performed to introduce the Arg-182→Ala mutation. The Asn-181→His mutation was introduced using the primers N181H-Fwd (TTACAAGAACATCGCATGGCAGATGTCATAG) and N181H-Rev (CCATGCGATGTTCTTGTAATTTTTTGTGCAA). The amplified products were cloned into pET32a.3C vector, and the mutations were confirmed by sequencing. The rSSL4t-R182A and rSSL4t-N181H mutant proteins were expressed and purified as for SSL4t. SSL proteins were labeled with one of the amine-reactive dyes Alexa Fluor 488, Alexa Fluor 647 (Invitrogen), or fluorescein isothiocyanate (FITC) (Molecular Probes) as per the manufacturer's instructions.
Crystallization of SSL4 and its complex with sLex.
SSL4t crystallization trials were carried out with sitting drops (100 nl protein plus 100 nl precipitant) dispensed by a Cartesian Honeybee nanoliter-dispensing robot (Genomic solutions) using a 480-condition crystallization screen (31) at 18°C. Following optimization, the highest-quality SSL4t crystals grew in sitting drops of 1 μl SSL4t (14.5 mg/ml in 20 mM Tris [pH 7.4], 100 mM NaCl) and 1 μl precipitant solution (0.2 M bis-tris-propane [pH 6.5], 21% methyl polyethylene glycol 5000 [MPEG 5000]). Crystallization trials were carried out for SSL4t complexed with sLex (Dextra Laboratories, United Kingdom) as for SSL4t. Following optimization, the highest-quality crystals grew in sitting drops of 1 μl protein solution (13.5 mg/ml rSSL4t, 10 mM sLex in 10 mM Tris [pH 7.4], 150 mM NaCl) and 2 μl precipitant solution {0.18 M PIPES [piperazine-N,N′-bis(2-ethanesulfonic acid)] (pH 6.7), 28% MPEG 5000}. The crystals were mounted in cryoloops, transferred to a cryoprotectant solution that consisted of the precipitant solution plus 20% glycerol for the SSL4t crystal or 17% glycerol for the SSL4t sLex complex crystal and flash-cooled by being plunged into liquid nitrogen.
Data collection and processing.
X-ray diffraction data for both crystals were collected at 100 K using CuKα radiation (λ = 1.5418 Å) from a Rigaku Micromax-007 HF rotating anode generator equipped with Osmic mirrors, an Oxford cryostream, and a Mar345 imaging plate system. SSL4t crystal data were processed with MOSFLM and scaled with SCALA from the CCP4 suite (12). The data set was more than 99% complete to a 1.7-Å resolution with a space group of C2. SSL4t and sLex complex data were integrated using XDS (23) and scaled with SCALA. The data set was more than 99% complete to a 2.5-Å resolution with a space group of I222.
Structure determination and refinement.
The SSL4t structure was solved by molecular replacement using PHASER (39) with the structures of SSL5 (Protein Data Bank [PDB; http://www.rcsb.org] ID no. 2Z8L) (2) and SSL11 (PDB ID no. 2RDG) (11) as the search models. The molecular replacement phases were then used with PHENIX (1) to build a contiguous model. This structure was refined through cycles of manual model building using COOT (15) and refinement using REFMAC5 (33). Water molecules were included in the model if they had good spherical density, reasonable B-factors, and favorable hydrogen bonding contacts. Other solvent species were identified from their densities and environment. The structure was validated using MOLPROBITY (14), and the refinement statistics are shown in Table 1. The structure of SSL4t in complex with sLex was solved by molecular replacement using PHASER (39) with the SSL4t structure as a search model. This structure was refined as for the native SSL4t structure. Well-defined density was immediately apparent for the complete sLex molecule; however, this was not modeled until the protein structure was nearing completion. A restraint library for sLex was developed through the program SKETCHER of the CCP4 suite (12). Water molecules and other solvent species were added as described above, and the refinement statistics are shown in Table 1.
Table 1.
Data collection and refinement statistics parameters
| Parameter | Value(s) fora: |
|
|---|---|---|
| SSL4 | sLex complex | |
| Crystal data | ||
| Space group | C2 | I222 |
| Cell | ||
| Axial lengths (Å) | 96.1, 81.2, 57.3 | 61.9, 78.9, 97.7 |
| Angles (°) | 90.00, 105.45, 90.00 | 90.00, 90.00, 90.00 |
| Data collection | ||
| Resolution range (Å) | 27.62–1.70 (1.79–1.70) | 24.42–2.50 (2.64–2.5) |
| Data collection temp (K) | 113 | 113 |
| No. of unique reflections | 46,435 | 8,563 |
| Completeness (%) | 99.7 (99.3) | 99.9 (100.00) |
| Mosaicity (°) | 1.02 | 0.214 |
| Rmerge (%) | 0.058 (0.698) | 16.9 (74.7) |
| I/σa | 17.4 (2.4) | 12.0 (3.1) |
| Refinement | ||
| Resolution range (Å) | 27.62–1.70 | 24.42–2.50 |
| R/Rfree | 0.2086/0.2614 | 0.1847/0.2543 |
| Mean B-value (Å2) | 29.7 | 25.6 |
| RMSD from standard values | ||
| Bond lengths (Å) | 0.025 | 0.017 |
| Bond angles (°) | 2.1 | 1.8 |
| Ramachandran plot | ||
| % of residues in favored regions | 97.1 | 95.3 |
| Outliers | None | None |
Throughout, values in parentheses are for the outermost resolution shell.
SSL4 affinity for sLex by surface plasmon resonance (SPR).
For surface plasmon resonance (SPR), SLex-bovine serum albumin (BSA) and sLacNac-BSA (Dextra Laboratories, United Kingdom) were coupled by carbodiimide chemistry to a CM5 biosensor chip surface using a Biacore T200 (GE Healthcare Biacore, Australia) as per the manufacturer's instructions. Coupling was typically 100 to 200 response units (RU). rSSL4t, rSSL11, rSSL4t-N181H, and rSSL4t-R182A were freshly purified by gel filtration chromatography (Superdex; GE Healthcare); the trailing edges of the peaks were used as the analytes (8 μM to 6.125 nM) in HBS-EP+ (0.01 M HEPES [pH 7.4], 0.15 M NaCl, 3 mM EDTA, 0.05% surfactant P20; GE Healthcare) and were passed over the immobilized ligand at 30 μl/min. The response at equilibrium (Req) was measured as the binding response plateau at 6 min. Equilibrium binding data were fitted to a single-binding-site model using the Biacore T200 evaluation software: Req/Bmax = ([rSSL4t])/(KD + [rSSL4t]), where Req is the plateau binding response, Bmax is the maximal bound analyte at calculated saturation, Req/Bmax is the fraction of immobilized ligand bound, and KD is the affinity constant at equilibrium.
SSL4 glycan binding specificity by glycomics array.
Purified FITC-rSSL4t was submitted to the Centre for Functional Glycomics (http://www.functionalglycomics.org; Scripps Research Institute, San Diego, CA). Binding was analyzed at 100 μg/ml to 320 glycans arrayed on a glass slide. Relative binding was measured as relative fluorescent units (RFU). The data from the glycomics array are available in the supplemental material.
SSL4 binding cell populations by flow cytometry.
Human leukocytes were purified from blood collected in sodium heparin-containing Vacuette tubes (Griener Bio One). Five milliliters of concentrated erythrocyte lysis buffer (100 mM KHCO3, 9% [wt/vol] NH4Cl, 1 mM EDTA) was added to 40 ml H2O and warmed to 20°C. Five milliliters of fresh human blood was added, and the mixture was incubated with constant rotation until the erythrocytes were lysed, before centrifugation. The cells were washed and resuspended in phosphate-buffered saline (PBS)–0.5% bovine serum albumin (BSA) at 1 × 106 cells/ml. A 0.5 μM concentration of FITC-labeled rSSL4t and 5 μl/ml CD14-phycoerythrin (PE) conjugate (M5E2-PE; BD Pharmingen), CD3-PE, CD10-PE, or CD19-PE (SN5c-PE, MEM-57-PE, and LT19-PE, respectively; Serotec) were added to the cells for 30 min before they were washed, resuspended in PBS–0.5% BSA, and analyzed on a BD LSR II flow cytometer. Neutrophils were purified from blood collected in potassium EDTA-containing Vacutainer tubes (BD Biosciences). Five milliliters of blood was layered over 2.5 ml of Histopaque 1077 (Sigma), which was layered over 2.5 ml of Histopaque 1119 (Sigma) and then centrifuged. The interface was collected, contaminating erythrocytes were lysed by hypotonic saline lysis, and the neutrophils were resuspended in PBS–0.5% BSA at 1 × 106 cells/ml. A 0.5 μM concentration of FITC-labeled rSSL4t or rSSL4t-R182A was added to the neutrophils for 30 min before they were washed, resuspended in PBS–0.5% BSA, and analyzed on a BD LSR II flow cytometer. For neuraminidase treatment, neutrophils were washed and resuspended at 1 × 107 cells/ml in 150 mM NaCl–5 mM CaCl2 (pH 6.0) and then incubated with 25 U/ml neuraminidase (New England BioLabs) for 1 h at 37°C in 5% CO2. For complementary binding studies, 0.01 μM Alexa Fluor 488-labeled rSSL4t and rSSL11 were added to neutrophils with 0.1 μM rSSL4t, rSSL11, or rSSL4t-N181H for 30 min before being washed, resuspended in PBS–0.5% BSA, and analyzed on a BD LSR II flow cytometer.
Cell staining of SSL4 by confocal microscopy.
Freshly isolated neutrophils were suspended at 1 × 106 cells/ml and allowed to adhere to a poly-d-lysine-coated 2.3-cm glass plate (World Precision Instruments) in Dulbecco's PBS without Ca2+ and Mg2+ (Sigma) for 30 min. Monocyte-derived macrophages were prepared as described previously (13). In short, human peripheral blood mononuclear cells (PBMCs) were collected by Histopaque density centrifugation as for neutrophils. Blood for human serum was collected in a serum nongel with a silica Vacutainer tube (BD Biosciences), kept at 20°C for 30 min to clot, and centrifuged. PBMCs at 1 × 106 cells/ml were plated onto a poly-d-lysine-coated 2.3-cm glass plate in RPMI 1640 medium with l-glutamine and sodium bicarbonate (Gibco) containing 10% human serum and incubated at 37°C and 5% CO2. After 24 h, nonadherent cells were removed and adherent cells were cultured in RPMI with 10% heat-treated fetal bovine serum (FBS) (Invitrogen) at 37°C and 5% CO2 for 14 days. To prepare primary monocytes, PBMCs at 2 × 106 cells/ml were plated onto a plastic 2.3-cm plate in RPMI 1640 medium with l-glutamine and sodium bicarbonate (Gibco) containing 10% heat-treated FBS (Invitrogen) and incubated at 37°C and 5% CO2 for 1 h. Selection was by plastic adhesion, and nonadherent cells were removed through PBS washing. Cell-coated plates were washed and incubated with 0.2 μM Alexa Fluor 647 (Invitrogen)-labeled rSSL4t and 0.2 μM Alexa Fluor 488 (Invitrogen)-labeled rSSL11 in Dulbecco's PBS with Ca2+ and Mg2+ (Sigma) for 30 min at 37°C or 4°C. The cells were washed with Dulbecco's PBS without Ca2+ and Mg2+ and visualized using an Olympus FV1000 confocal laser scanning microscope.
Protein structure accession numbers.
The atomic coordinates and structure factors for both SSL4t and SSL4t complexed with sLex have been deposited in the PDB under code no. 4dXF and 4DXG, respectively.
RESULTS
SSL4 was cloned and expressed as a construct that extended from the beginning of the conserved oligosaccharide/oligonucleotide binding (OB)-fold domain and β-grasp domain shared with all SSLs. This removed a variable N-terminal region predicted to have low complexity. When expressed as a full-length peptide, it was observed that this protein degraded to a stable form of identical molecular weight to SSL4t. Additionally a crystal structure of this full-length protein showed there was no electron density for the additional amino acids at the N terminus, which were therefore assumed to be disordered (data not shown). This study has focused on the carbohydrate specificity of the SSLs, which is associated with the conserved glycan binding site in the β-grasp domain of the protein. Therefore, it was decided to clone and express SSL4 without this N-terminal sequence. This does not exclude this region from being of functional significance, where it may augment the cellular effect of this protein.
Comparison of 12 published SSL4t sequences identified 4 alleles. A sequence alignment of the Newman (identical to MW2, USA300 TCH1516, USA300 FPR3757, NCT88325, and MSSA476), N315 (identical to Mu3, JH1, and JH9), COL, and MRSA252 SSL4t alleles revealed sequence variation of up to 25% (Fig. 1a). This is primarily attributable to the MRSA252 allele, which displays 23 to 25% sequence variation with the other identified alleles. Sequence variation between the remaining alleles is no more than 8%. Previous amino acid sequence alignments of SSL2, SSL3, SSL4, SSL5, SSL6, and SSL11 from the MW2 S. aureus strain revealed remarkable conservation in the sLex binding region (4), with 12 of the 17 residues between positions 169 and 185 (SSL4 numbering) being identical in all six SSLs. This is in contrast to the modest similarity seen in the rest of the sequences. An alignment of SSL4t, SSL5, and SSL11 highlights this (Fig. 1b), with only 34% overall sequence identity shared between SSL4t and SSL5 and 36% identity shared between SSL4t and SSL11.
Fig 1.

Sequence alignments of SSL4t alleles and SSL4t, SSL5, and SSL11. (a) Amino acid alignment of known SSL4t allele variants. Identity (*), strong similarity (:), and weak similarity (.) are indicated. (b) Amino acid alignment of SSL4t from S. aureus strain Newman, SSL5 from strain GL10, and SSL11 from strain US6610. The 17-amino-acid binding site is underlined, and sequence similarity is labeled as described above.
The crystal structure of SSL4t was solved by molecular replacement using the structures of SSL5 and SSL11 as search models. The structure was refined to a resolution of 1.7 Å, with excellent geometry and an R value of 20.86% (Rfree, 26.14%) (Table 1). The final structure contains two SSL4t chains, residues 7 to 200 in molecule A, residues 11 to 200 in molecule B, 316 water molecules, 1 glycerol molecule, and 4 chloride ions. SSL4t shows the same characteristic two-domain fold as other superantigens (Sags) and SSLs. The N-terminal domain (residues 1 to 100) forms an OB-fold domain, a five-stranded β-barrel structure comprising β-strands β1 to β5. The C-terminal domain (residues 101 to 200) forms a β-grasp fold in which a central helix (α4) rests against a mixed β-sheet formed from β-strands: β7-β6-β12-β9-β10.
The crystal structure of SSL4t in complex with sLex was then solved by molecular replacement using the high-resolution SSL4t structure as a model. The structure was refined to a resolution of 2.5 Å, with excellent geometry and an R value of 18.47% (Rfree, 25.43%) (Table 1). The final structure contained one chain of SSL4 from residues 7 to 200, 1 sLex molecule, 1 PIPES molecule, 2 chloride ions, and 62 water molecules (Fig. 2a).
Fig 2.
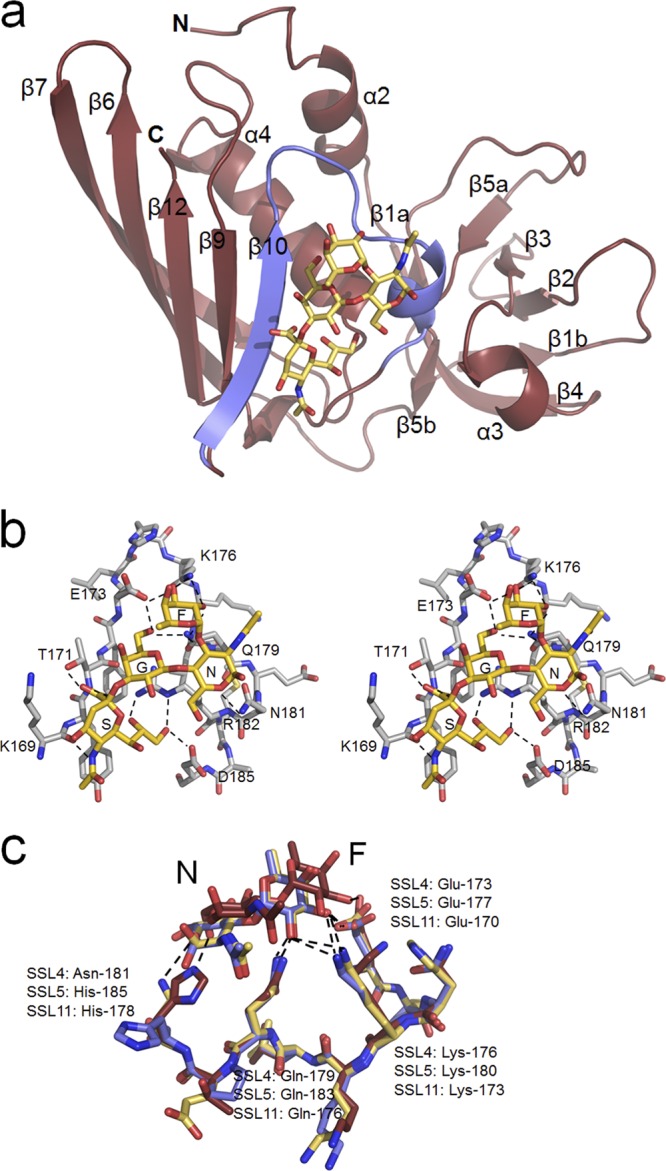
Structure of the SSL4t-sLex complex. (a) Ribbon diagram showing the SSL4t-sLex complex with secondary structure elements labeled according to standard Sag nomenclature. The N-terminal OB-fold domain is on the right, with the C-terminal β-grasp domain on the left. The bound sLex is shown as sticks and sits in a surface depression made by a V-shaped loop between strand β10 and a deformed 310 helix (blue). (b) Stereo view of the sLex binding site of SSL4t. sLex atoms are colored gold and SSL4t atoms silver, with element coloring. Hydrogen bonds between sLex and protein atoms are shown by dashed lines. sLex residues are labeled S (Sia), G (Gal), N (NAG), and F (Fuc), and SSL4t residues that form direct hydrogen bonds to sLex are labeled. (c) Overlay of the interaction between NAG (N) and Fuc (F) of sLex and Glu-173, Lys-176, Gln-179, and Asn-181 of SSL4t (yellow), Glu-170, Lys-173, Gln-176, and His-178 of SSL11 (red), and Glu-177, Lys-180, Gln-183, and His-185 of SSL5 (blue).
Dimers of SSL4t were formed in both crystal structures as a result of crystal packing. The β7 strands from adjacent symmetry-related molecules aligned in antiparallel fashion, creating a 10-stranded β-sheet between the C-terminal domains of both molecules. The same interaction is seen in all crystal forms of SSL5 and SSL11, indicating that this form of association is of possible functional significance—for example, at the target cell surface where the proteins could be concentrated. Isothermal titration calorimetry was used to test dimer formation of SSL4t in solution. SSL4t was shown not to form dimers at high concentrations in solution (data not shown).
In its complex with SSL4t, sLex occupies the same V-shaped loop between strand β10 and a distorted 310 helix, as seen in both SSL5 and SSL11 (Fig. 2b). Comparisons of the RMSDs between main chain atom positions in the 17-residue binding sites and the entire protein chains when aligned reveal the remarkable conservation of the binding loop between these proteins. These are 0.34 Å and 6.31 Å, respectively, between SSL4t and SSL11 and 0.35 Å and 6.10 Å, respectively, between SSL4t and SSL5. Key residues of SSL4t that interact with the sLex molecule are Lys-169, Thr-171, and Glu-173 from the β10 strand, Lys-176 from the top of the V-shaped loop, and Gln-179, Asn-181, Arg-182, and Asp-185 from the descending α-helix. SSL4t makes 14 direct hydrogen bonds with the sLex molecule; the majority of these interactions occur with the Sia and galactose (Gal) units of the tetrasaccharide and are identical to those made by SSL5 and SSL11. Differences between the binding sites of SSL4t, SSL5, and SSL11 are seen in the N-acetylglucosamine (NAG) and fucose (Fuc) interactions (Fig. 2c). Asn-181 of SSL4t makes a direct hydrogen bond with O5 of NAG, which allows the conserved Glu-173, Lys-176, and Gln-179 residues of SSL4t to make four additional hydrogen bonds with the Fuc residue. Asn-181 is replaced in SSL11 by His-178 which, due the longer side chain, forms a direct hydrogen bond with the O6 of NAG and also causes the NAG and Fuc residues to be held 2 to 3 Å further out of the binding pocket in SSL11. This prevents any hydrogen bonding between Lys-173 and Gln-176 of SSL11 and the Fuc of sLex. The two available crystal structures of SSL5 and sLex show that His-185, equivalent to Asn-181 in SSL4t and His-178 in SSL11, can have two positions. In position 1, it forms a hydrogen bond with the O6 of NAG and pushes the NAG and Fuc residues out of the binding pocket as for SSL11. In position 2, however, the His flips away, allowing the NAG and Fuc residues to move in closer, just as in SSL4t (Fig. 2c).
SPR was used to calculate and compare the affinities of SSL4 and SSL11 for the blood group antigen sLex and the carbohydrate sLacNac, a subcomponent of sLex lacking the terminal fucose. The sLex and sLacNac carbohydrates were immobilized onto a chip surface, and highly purified recombinant SSL4t and SSL11 were then used as soluble analyte. An initial SSL4t concentration series revealed more than one binding species affecting the binding and dissociation kinetics (data not shown), suggesting the presence of protein aggregates as seen by Chung et al. for SSL11 (11). SSL4t and SSL11 were further purified by size exclusion chromatography on a Sephadex 10/200 GL fast protein liquid chromatography (FPLC) (GE) column, and fractions from the trailing edge of the protein peaks, presumed to be dominantly monomeric protein in solution, were then analyzed immediately and separately for sLex and sLacNac binding. Equilibrium binding curves were obtained from the trailing edge of both SSL4t and SSL11 binding sLex and sLacNac (Fig. 3a, b, e, and f). The equilibrium dissociation constants (KD) of SSL4t binding sLex and sLacNac were calculated to be 87.0 ± 4.5 nM and 471.7 ± 18.7 nM, respectively, compared with the values 2.32 ± 0.20 μM and 2.43 ± 0.02 μM, respectively, for the same carbohydrates bound by SSL11.
Fig 3.

Quantitative measure of SSL4t, SSL4t-N181H, and SSL11 binding to sLex and sLacNac. Binding responses (Resp.) at equilibrium (Req) are shown against the concentration and fitted to a steady-state affinity binding model to calculate an equilibrium affinity constant (KD). (a) sLex sensor chip binding and equilibrium binding analysis of 6.125 to 500 nM SSL4t in duplicate. The KD was calculated to be 87.0 ± 4.5 nM. (b) sLacNac sensor chip binding and equilibrium binding analysis of 25 to 850 nM SSL4t in duplicate. The KD was calculated to be 471.7 ± 18.7 nM. (c) sLex sensor chip binding and equilibrium binding analysis of 6.125 to 500 nM SSL4t-N181H in duplicate. The KD was calculated to be 90.8 ± 6.4 nM. (d) sLacNac sensor chip binding and equilibrium binding analysis of 25 to 500 nM SSL4t-N181H in duplicate. The KD was calculated to be 119.1 ± 10.3 nM. (e) sLex sensor chip binding and equilibrium binding analysis of 0.5 to 8 μM SSL11 in duplicate. The KD was calculated to be 2.32 ± 0.20 μM. (f) sLacNac sensor chip binding and equilibrium binding analysis of 0.125 to 8 μM SSL11 in duplicate. The KD was calculated to be 2.43 ± 0.02 μM. The plots shown are representative of two independent experiments, Rmax and χ2 values can be found in Table S1 in the supplemental material.
It was hypothesized from the crystal structures of these proteins complexed with sLex that the Asn-181 residue of SSL4t may be responsible for this higher affinity for sLex over SSL11. This was tested by mutagenesis; the Asn-181 residue of SSL4t was mutated to His, the equivalent residue in SSL11. The affinity of the SSL4t-N181H mutant for sLex was, however, similar to that of SSL4t, with a calculated KD of 90.8 ± 6.4 nM. Unlike SSL4t, but identical to SSL11, the dissociation constant of this mutant for sLacNac was very similar to that of sLex, with a calculated KD of 119.1 ± 10.3 nM. The SSL4t structure also showed that Arg-182 formed the floor of the carbohydrate binding pocket, analogous to Arg-186 in SSL5 and Arg-179 in SSL11. This was confirmed by mutagenesis, with SSL4t-R182A displaying no detectable binding to sLex (see the supplemental material). Modeling indicated that the Asn-181→His and Arg-182→Ala mutations should have no effect on the folding or stability of the SSL4t molecule. Recombinant SSL4t-N181H and SSL4t-R182A were expressed and purified at similar levels in E. coli to the parental protein.
SSL4 was tested for binding against 320 different glycans, using a microarray format provided by the Consortium for Functional Glycomics. Glycans were attached through spacer groups SP0 and SP8 to SP21. SSL4t was tested at 100 μg/ml, and the results for the 10 strongest glycans are presented in Table 2. The core ligand for SSL4t, present in almost all of the top-scoring glycans, was 3′-sLacNac, which was itself represented at position 9 on Table 2. As mentioned earlier, sLacNac is a subcomponent of sLex, which is represented in row 3 in Table 2. Like SSL11 (11), SSL4t does not bind sLacNac, with a β1-3 linkage between the Gal and NAG residues highlighting its preference for this ligand. However, whereas SSL11 does not bind to very similar glycans such as sGal (Neu5Acα2-3Galβ) and sialyllactose (Neu5Acα2-3Galβ1-4Glcβ), SSL4t displayed moderate binding (Table 2).
Table 2.
SSL4 glycomics array resultsa
| Glycan name | Avg no. of RFU | SD |
|---|---|---|
| Neu5Acα2-3Galβ1-4(Fucα1-3)GlcNAcβ1-3Galβ1-4GlcNAcβ-Sp8 | 10,246 | 1,636 |
| Neu5Acα2-3Galβ1-4(Fucα1-3)GlcNAcβ1-3Galβ1-4(Fucα1-3)GlcNAcβ1-3Galβ1-4(Fucα1-3)GlcNAcβ–Sp0 | 7,555 | 449 |
| Neu5Acα2-3Galβ1-4(Fucα1-3)GlcNAcβ–Sp8 (sLex) | 6,916 | 2,675 |
| Neu5Acα2-3Galβ1-4GlcNAcβ1-3Galβ1-4GlcNAcβ1-3Galβ1-4GlcNAcβ–Sp0 | 4,871 | 967 |
| Neu5Acα2-3Galβ1-4[6OSO3]GlcNAcβ-Sp8 | 4,717 | 305 |
| Neu5Acα2-3Galβ1-4(Fucα1-3)GlcNAcβ1-3Galβ-Sp8 | 4,496 | 405 |
| Neu5Gca2-3Galβ1-4(Fucα1-3)GlcNAcβ-Sp0 | 4,444 | 900 |
| Neu5Acα2-3Galβ1-4GlcNAcβ1-2Manα1-3(Neu5Acα2-3Galβ1-4GlcNAcβ1-2Manα1-6)Manβ1-4GlcNAcβ1-4GlcNAcβ-Sp12 | 4,253 | 297 |
| Neu5Acα2-3Galβ1-4GlcNAcβ–Sp8 (sLacNac) | 3,774 | 215 |
| Galβ1-3(Neu5Acα2-3Galβ1-4(Fucα1-3)GlcNAcβ1-6)GalNAc–Sp14 | 3,525 | 495 |
| Neu5Acα2-3Galβ1-3GlcNAcβ–Sp8 | 307 | 87 |
| Neu5Acα2-3Galβ-Sp8 (sGal) | 2,327 | 172 |
| Neu5Acα2-3Galβ1-4Glcβ–Sp0 (sLac) | 517 | 97 |
| Neu5Acα2-3Galβ1-4Glcβ–Sp8 (sLac) | 1,627 | 167 |
The 10 strongest glycans are presented in descending order of fluorescence in the top portion of the table.
FITC-labeled SSL4t bound strongly to human monocytes (CD14) and neutrophils (CD10) and weakly to T lymphocytes. No binding was observed to B lymphocytes (Fig. 4a). This binding specificity was consistent with multiple individual blood samples tested and was similar in profile to the cell specificity of SSL11 (11). SSL4t binding was substantially reduced on granulocytes pretreated with 25 U/ml neuraminidase (Fig. 4b), and the SSL4t-R182A mutant displayed no detectable binding to human granulocytes (Fig. 4c). Together, these data indicate that this interaction is carbohydrate dependent. To determine whether SSL4 and SSL11 compete for the same glycosylated targets, complementary binding studies were performed with a low concentration of either fluorescent SSL4t or SSL11 (0.01 μM) mixed with a 10-fold excess (0.1 μM) of the unlabeled protein. SSL4t competed effectively and was saturable with itself, suggesting that the majority of binding was to specific surface receptor(s) (Fig. 4d). Excess unlabeled SSL11 had no effect on SSL4t-488 binding (Fig. 4d), indicating that despite a shared affinity for sialylated glycoproteins, the target molecules bound by SSL4 and SSL11 are likely not to be identical. This is consistent with the binding preference differences observed in the glycomics array data. The unlabeled SSL4t-N181H mutant competed effectively with SSL4t-488 to a similar degree as unlabeled SSL4t. Competition using unlabeled SSL11, SSL4t, and SSL4t-N181H with SSL11-488 showed no change in SSL11-488 binding (Fig. 4e), indicating a wider binding specificity from this SSL.
Fig 4.

Sialic-acid-dependent binding of SSL4t to human myeloid cells. (a) Human leukocytes double stained with rSSL4t-FITC (x axis) and PE-conjugated antibodies specific to monocytes (CD14), neutrophils (CD10), B lymphocytes (CD19), and T lymphocytes (CD3). (b) Binding of 0.1 μM SSL4t-FITC to human granulocytes untreated (peak 1) or pretreated (peak 2) with neuraminidase. (c) Human granulocytes stained with 0.1 μM SSL4t-FITC (peak 1) or 0.4 μM SSL4t-R182A-FITC (peak 2). (d) Human granulocytes stained with 0.01 μM SSL4t-488 (peak 1) plus 10-fold (0.1 μM) excess of either unlabeled SSL11 (peak 2), unlabeled SSL4t (peak 3), or unlabeled SSL4t-N181H (peak 4). (e) Human granulocytes stained with 0.01 μM SSL11-488 (peak 1) plus 10-fold (0.1 μM) excess of unlabeled SSL11 (peak 2), unlabeled SSL4t (peak 3), or unlabeled SSL4t-N181H (peak 4).
SSL11 is rapidly internalized by human granulocytes in an energy-dependent manner (11). To determine the fate of SSL4, SSL4t was labeled with Alexa Fluor 647, incubated with human granulocytes at 37°C or 4°C, and monitored by live-cell confocal microscopy. SSL4t was rapidly internalized by human neutrophils into sharply distinct vesicles at 37°C, whereas at 4°C, it remained surface bound (Fig. 5). We observed that at 4°C, neutrophils appear spherical, while at 37°C, they appear to flatten and become amorphous in shape. This is independent of SSL4 binding as we observed these differences in neutrophil morphology in the absence of SSL4 (data not shown). SSL4t also binds human monocytes, and staining of these cells revealed moderate internalization into vesicles at 37°C but not at 4°C (Fig. 5). Unlike neutrophils, a significant amount of cell-bound SSL4t appeared to remain at the cell surface; internalization is not as complete in these cells. SSL4t was also rapidly internalized into activated human macrophages at 37°C but not at 4°C (Fig. 5). These data reveal SSL4t binding and complete internalization into neutrophils and macrophages, but only binding and moderate internalization by monocytes. The fluorescent staining pattern of neutrophils and monocytes showed strong intracellular punctate staining, suggesting concentration into vesicles. Macrophages displayed a more diffuse intracellular staining pattern similar to cytoskeletal staining. To confirm that SSL4 and SSL11 were internalized to the same intracellular compartment, neutrophils, monocytes, and macrophages were stained with both SSL4t-647 (red) and SSL11-488 (green) at 37°C. Two-color overlay (yellow) confirmed that they both trafficked to the same intracellular location (Fig. 5).
Fig 5.

Binding and internalization of SSL4t-647 into neutrophils, monocytes, and macrophages. Cells were incubated with 0.2 μM SSL4t-647 and examined at either 4°C or after cells had been warmed to 37°C. In the lower panel, human neutrophils, monocytes, and macrophages were incubated with 0.2 μM both SSL11-488 and SSL4t-647, and the cells were warmed to 37°C for 30 min. Two color channels were overlaid to identify coincident fluorescence (yellow).
DISCUSSION
The staphylococcal superantigens (Sags) form a family of structurally and functionally related proteins, of which 23 serologically distinct molecules have been described (25, 42). Despite their common structure, Sags utilize a remarkable range of binding properties to carry out their role in stimulating T cells through cross-linking major histocompatibility complex II with the T-cell receptor (34). The SSLs have emerged as a family of structurally related molecules that carry out an entirely different function; these molecules target multiple components of the innate immune response through a variety of structural mechanisms (4, 11, 27). This highlights how S. aureus has come to use the simple two-domain fold of the Sags as an adaptable framework in which to generate a plethora of virulence factors that target both the adaptive and innate immune responses.
Both SSL5 and SSL11 have been shown to bind sialated glycans through a conserved binding site in the C-terminal β-grasp domain that is present in several SSLs (4, 11). Here we show that SSL4, a member of this subfamily, binds to sLex in an almost identical fashion. The key amino acid residues that interact directly with sLex are almost entirely conserved between SSL5, SSL11, and SSL4. Despite this, SSL4t displays a much higher affinity than SSL11 for sLex (KD = 87.0 nM compared with 2.32 μM), in quantitative binding studies. This is largely due to the substitution for Asn-181 in SSL4t by His-178 in SSL11, which holds the NAG and Fuc residues 2 to 3 Å further out of the binding pocket compared to SSL4, resulting in three fewer direct hydrogen bonds made between the protein and ligand. Mutation of the equivalent residue of SSL4t, Asn-181, to histidine had little effect on the affinity for sLex (KD = 90.8 nM). We hypothesize that this is because His-181 of SSL4t-N181H can flip out, as seen in SSL5, allowing NAG and Fuc to bind closer to the binding site; although one hydrogen bond to NAG is lost by this, the three additional hydrogen bonds made to Fuc remain. In SSL11, the His-178 cannot flip away as it is held in place by a water-mediated hydrogen bond with the preceding Thr-177. This hydrogen bond could not occur in SSL4 and SSL5, in which the residues equivalent to Thr-177 are glutamic acid and proline, respectively. We further predict SSLs with an asparagine or aspartic acid at the equivalent site of Asn-181 of SSL4t, such as SSL5 and SSL2 of strain MW2, bind sLex in a position to form multiple hydrogen bonds to Fuc and have increased affinity for sLex. Those with histidine at this position and threonine at the preceding position bind sLex, as for SSL11, with reduced recognition of Fuc and a lower affinity.
SSL11 shows a similar affinity for the carbohydrate sLacNac (KD = 2.43 μM), a truncated form of sLex lacking the terminal fucose, as for sLex (KD = 2.32 μM). This is unlike SSL4t, which shows a reduced affinity for sLacNac (KD = 471.7 nM) compared with sLex (KD = 87.0 nM). Like SSL11, the SSL4t-N181H mutant shows similar affinities for sLacNac (KD = 119.1 nM) and sLex (KD = 90.8 nM). This indicates that a histidine at this position of the carbohydrate binding site forms a stronger bond to the O6 of NAG, independent of the presence of a Fuc residue, whereas an asparagine at this position forms a weaker bond to the O5 of NAG and the overall carbohydrate specificity is more dependent on Fuc. Together, this indicates a preference of SSL4 for glycans terminating in sLex, which includes Fuc.
Glycomics array data reflect variations in the fine specificity of SSL4 for other glycans. As for SSL11 (11), sLacNac is the core ligand for SSL4t, but the affinity is increased in the presence of an attached Fuc residue, as in sLex. Whereas SSL11 displayed weak or no binding to similar glycans, such as sGal or sLac, trace binding was observed for SSL4t. These observations support structural and quantitative affinity data highlighting differences between the binding sites and affinity to core glycans between these proteins. Reflected in this differential glycan preference is the finding that SSL4t and SSL11 do not compete for binding to cells. SSL4t is able to compete with itself, indicating that the majority of binding is target specific. It is apparent that the target molecules of these proteins are likely not identical. It has been suggested that variant carbohydrate sequences related to the sLex sequence, among others, are preferentially bound by one or the other of the selectins, resulting in subtle differences in their binding specificities (16). It appears that the same could be true for the carbohydrate-binding SSLs and might explain why S. aureus produces an array of homologous proteins to target sialyated glycoproteins with variations in their ligand preferences.
SSL4t has a similar leukocyte binding specificity to SSL11, binding preferentially to myeloid cells while not interacting to any significant degree with lymphocytes. Sialic acids are found on the terminating residues of many human-secreted and cell surface glycoproteins (40). While many glycoproteins express sLacNac, the core ligand for SSL4t, it is known that sLex is expressed constitutively on granulocytes, monocytes, and a small percentage of T cells (32). This matches the leukocyte binding profile of SSL4t and supports quantitative affinity data, which indicate the target receptor(s) of SSL4 is more specific than just the core sLacNac ligand and likely includes sLex. In additional support of this, the SSL4t-N181H mutant, which has an affinity for sLex similar to that of SSL4t but enhanced affinity for sLacNac, competes for cell binding with SSL4t to a level identical to that of SSL4t.
We believe that SSL4 is targeting sialated glycoproteins at the surface of myeloid cells to gain entry and affect cellular function through an as yet unknown mechanism. TLR2 has been shown to be the target of SSL3, which also contains the conserved carbohydrate binding site. This binding inhibited the activation of TLR2 at the surface of human and murine neutrophils and monocytes in a partially glycan-dependent manner (5). The activation of TLR2 was also shown to be inhibited by SSL4, a function shared with no other SSL studied (5). This activity, however, was several-hundred-fold less potent than that observed for SSL3 and likely reflects the fact that SSL4 is the most homologous SSL to SSL3. MRSA252 SSL4 displayed enhanced TLR2 inhibition activity, although this allele shares greater homology with SSL3 from the same strain than it does any SSL4 allele identified in this study. At the time it was sequenced, MRSA252 was the most genetically diverse S. aureus strain identified (21). This suggests the SSL4 and SSL3 of MRSA252 have undergone less divergence following the duplication event from where these proteins arose than in other strains. This piece of data supports our observations that the target molecules of the glycan-binding SSLs are likely not to be identical. We believe SSL4 is targeting additional receptors on the surface of these cells.
SSL4t is actively internalized into neutrophils, macrophages, and, to a lesser extent, monocytes, in a totally glycan-dependent manner. SSL4t-R182A, which does not bind sLex in quantitative binding analysis, also shows a complete loss of cell binding. Identification of the target receptor(s) for SSL4 at the surface of these cells would lead to further insights into the function of this virulence factor. Interestingly, SSL4t displays different staining patterns for these cell types. While being actively internalized into discrete intracellular vesicles by neutrophils and monocytes, SSL4t staining of macrophages shows a distinct cytoskeleton-like diffuse staining. If SSL4 is targeting the cytoskeleton of macrophages, it will be interesting to determine the effects this would have on cellular mobility and intracellular functions.
Sialic acids are a convenient target for microbes, which utilize them as a scaffold for host recognition, attachment, and invasion (8). A wide variety of microbial and viral adhesins, pili, fimbriae, and hemagglutinins all target sialated glycoproteins. S. aureus extends this by utilizing the adaptable Sag structure to produce six homologous proteins that contain a highly conserved sialated glycan binding site. Recent studies have shown the ssl genes to be negatively regulated by the quorum-sensing accessory gene regulator (Agr) and to be expressed in large quantities following dysfunction in this gene regulator (6). Natural agr-defective strains of S. aureus frequently occur during infection, and these strains have been shown to have residual virulence due to overproduction of these immunomodulatory proteins (6, 38). It is interesting to speculate as to what potent effect these SSL proteins may have when expressed together to promote staphylococcal survival and persistence during colonization and infection.
Supplementary Material
ACKNOWLEDGMENTS
We thank F. Clow of the Biacore Bionsensing Facility, J. Ross of the Biomedical Imaging Research Unit, and S. Edgar of the Flow Cytometry Facility of the University of Auckland for technical support.
We acknowledge the support of the Functional Glycomics Consortium, San Diego, funded by NIH grant GM62116. This work was supported by the Maurice Wilkins Centre for Molecular Biodiscovery and grants from the Health Research Council of New Zealand.
Footnotes
Published ahead of print 4 September 2012
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1. Adams PD, et al. 2002. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58:1948–1954 [DOI] [PubMed] [Google Scholar]
- 2. Arcus VL, Langley R, Proft T, Fraser JD, Baker EN. 2002. The three-dimensional structure of a superantigen-like protein, SET3, from a pathogenicity island of the Staphylococcus aureus genome. J. Biol. Chem. 277:32274–32281 [DOI] [PubMed] [Google Scholar]
- 3. Baba T, Bae T, Schneewind O, Takeuchi F, Hiramatsu K. 2008. Genome sequence of Staphylococcus aureus strain Newman and comparative analysis of staphylococcal genomes: polymorphism and evolution of two major pathogenicity islands. J. Bacteriol. 190:300–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Baker HM, et al. 2007. Crystal structures of the staphylococcal toxin SSL5 in complex with sialyl Lewis X reveal a conserved binding site that shares common features with viral and bacterial sialic acid binding proteins. J. Mol. Biol. 374:1298–1308 [DOI] [PubMed] [Google Scholar]
- 5. Bardoel BW, et al. 20 June 2012. Evasion of Toll-like receptor 2 activation by staphylococcal superantigen-like protein 3. J. Mol. Med. [Epub ahead of print.] doi:10.1007/s00109-012-0926-8 [DOI] [PubMed] [Google Scholar]
- 6. Benson MA, et al. 2011. Staphylococcus aureus regulates the expression and production of the staphylococcal superantigen-like secreted proteins in a Rot-dependent manner. Mol. Microbiol. 81:659–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bestebroer J, et al. 2007. Staphylococcal superantigen-like 5 binds PSGL-1 and inhibits P-selectin-mediated neutrophil rolling. Blood 109:2936–2943 [DOI] [PubMed] [Google Scholar]
- 8. Bishop JR, Gagneux P. 2007. Evolution of carbohydrate antigens—microbial forces shaping host glycomes? Glycobiology 17:23R–34R [DOI] [PubMed] [Google Scholar]
- 9. Bronner S, Monteil H, Prevost G. 2004. Regulation of virulence determinants in Staphylococcus aureus: complexity and applications. FEMS Microbiol. Rev. 28:183–200 [DOI] [PubMed] [Google Scholar]
- 10. Chenna R, et al. 2003. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 31:3497–3500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chung MC, et al. 2007. The crystal structure of staphylococcal superantigen-like protein 11 in complex with sialyl Lewis X reveals the mechanism for cell binding and immune inhibition. Mol. Microbiol. 66:1342–1355 [DOI] [PubMed] [Google Scholar]
- 12. Collaborative Computational Project No. 4 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50:760–763 [DOI] [PubMed] [Google Scholar]
- 13. Daigneault M, Preston JA, Marriott HM, Whyte MKB, Dockrell DH. 2010. The identification of markers of macrophage differentiation in PMA-stimulated THP-1 cells and monocyte-derived macrophages. PLoS One 5:e8668 doi:10.1371/journal.pone.0008668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Davis IW, Murray LW, Richardson JS, Richardson DC. 2004. MolProbity: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 32:W615–W619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126–2132 [DOI] [PubMed] [Google Scholar]
- 16. Feizi T. 2000. Carbohydrate-mediated recognition systems in innate immunity. Immunol. Rev. 173:79–88 [DOI] [PubMed] [Google Scholar]
- 17. Foster TJ. 2005. Immune evasion by staphylococci. Nat. Rev. Microbiol. 3:948–958 [DOI] [PubMed] [Google Scholar]
- 18. Foster TJ. 2004. The Staphylococcus aureus “superbug.” J. Clin. Invest. 114:1693–1696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fridkin SK, et al. 2005. Methicillin-resistant Staphylococcus aureus disease in three communities. N. Engl. J. Med. 352:1436–1444 [DOI] [PubMed] [Google Scholar]
- 20. Herbert S, et al. 2007. Molecular basis of resistance to muramidase and cationic antimicrobial peptide activity of lysozyme in staphylococci. PLoS Pathog. 3:e102 doi:10.1371/journal.ppat.0030102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Holden MTG, et al. 2004. Complete genomes of two clinical Staphylococcus aureus strains: evidence for the evolution of virulence and drug resistance. Proc. Natl. Acad. Sci. U. S. A. 101:9786–9791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jongerius I, et al. 2007. Staphylococcal complement evasion by various convertase-blocking molecules. J. Exp. Med. 204:2461–2471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kabsch W. 1993. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry land cell constants. J. Appl. Crystallogr. 26:795–800 [Google Scholar]
- 24. Kuroda M, et al. 2001. Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet 357:1225–1240 [DOI] [PubMed] [Google Scholar]
- 25. Langley R, Patel D, Jackson N, Clow F, Fraser JD. 2010. Staphylococcal superantigen super-domains in immune evasion. Crit. Rev. Immunol. 30:149–165 [DOI] [PubMed] [Google Scholar]
- 26. Langley R, et al. 2005. The staphylococcal superantigen-like protein 7 binds IgA and complement C5 and inhibits IgA-FcαRI binding and serum killing of bacteria. J. Immunol. 174:2926–2933 [DOI] [PubMed] [Google Scholar]
- 27. Laursen NS, et al. 2010. Structural basis for inhibition of complement C5 by the SSL7 protein from Staphylococcus aureus. Proc. Natl. Acad. Sci. U. S. A. 107:3681–3686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lindsay JA, Holden MTG. 2006. Understanding the rise of the superbug: investigation of the evolution and genomic variation of Staphylococcus aureus. Funct. Integr. Genomics 6:186–201 [DOI] [PubMed] [Google Scholar]
- 29. Lowy FD. 1998. Medical progress: Staphylococcus aureus infections. N. Engl. J. Med. 339:520–532 [DOI] [PubMed] [Google Scholar]
- 30. Luong TT, Lee CY. 2002. Overproduction of type 8 capsular polysaccharide augments Staphylococcus aureus virulence. Infect. Immun. 70:3389–3395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Moreland N, et al. 2005. A flexible and economical medium-throughput strategy for protein production and crystallization. Acta Crystallogr. D Biol. Crystallogr. 61:1378–1385 [DOI] [PubMed] [Google Scholar]
- 32. Munro JM, et al. 1992. Expression of sialyl-Lewis X, an E-selectin ligand, in inflammation, immune processes, and lymphoid tissues. Am. J. Pathol. 141:1397–1408 [PMC free article] [PubMed] [Google Scholar]
- 33. Murshudov GN, Vagin AA, Dodson EJ. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53:240–255 [DOI] [PubMed] [Google Scholar]
- 34. Papageorgiou AC, Acharya KR. 2000. Microbial superantigens: from structure to function. Trends Microbiol. 8:369–375 [DOI] [PubMed] [Google Scholar]
- 35. Patel D, Wines BD, Langley RJ, Fraser JD. 2010. Specificity of staphylococcal superantigen-like protein 10 toward the human IgG1 Fc domain. J. Immunol. 184:6283–6292 [DOI] [PubMed] [Google Scholar]
- 36. Ramsland PA, et al. 2007. Structural basis for evasion of IgA immunity by Staphylococcus aureus revealed in the complex of SSL7 with Fc of human IgA1. Proc. Natl. Acad. Sci. U. S. A. 104:15051–15056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rooijakkers SHM, Van Kessel KPM, Van Strijp JAG. 2005. Staphylococcal innate immune evasion. Trends Microbiol. 13:596–601 [DOI] [PubMed] [Google Scholar]
- 38. Shopsin B, et al. 2008. Prevalence of agr dysfunction among colonizing Staphylococcus aureus strains. J. Infect. Dis. 198:1171–1174 [DOI] [PubMed] [Google Scholar]
- 39. Storoni LC, McCoy AJ, Read RJ. 2004. Likelihood-enhanced fast rotation functions. Acta Crystallogr. D Biol. Crystallogr. 60:432–438 [DOI] [PubMed] [Google Scholar]
- 40. Varki A. 2007. Glycan-based interactions involving vertebrate sialic-acid-recognizing proteins. Nature 446:1023–1029 [DOI] [PubMed] [Google Scholar]
- 41. Voyich JM, et al. 2005. Insights into mechanisms used by Staphylococcus aureus to avoid destruction by human neutrophils. J. Immunol. 175:3907–3919 [DOI] [PubMed] [Google Scholar]
- 42. Wilson GJ, et al. 2011. A novel core genome-encoded superantigen contributes to lethality of community-associated MRSA necrotizing pneumonia. PLoS Pathog. 7:e1002271 doi:10.1371/journal.ppat.1002271 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.