

Abstract
Here we report the isolation of 6 temperate bacteriophages (phages) that are prevented from replicating within the laboratory strain Pseudomonas aeruginosa PA14 by the endogenous CRISPR/Cas system of this microbe. These phages are only the second identified group of naturally occurring phages demonstrated to be blocked for replication by a nonengineered CRISPR/Cas system, and our results provide the first evidence that the P. aeruginosa type I-F CRISPR/Cas system can function in phage resistance. Previous studies have highlighted the importance of the protospacer adjacent motif (PAM) and a proximal 8-nucleotide seed sequence in mediating CRISPR/Cas-based immunity. Through engineering of a protospacer region of phage DMS3 to make it a target of resistance by the CRISPR/Cas system and screening for mutants that escape CRISPR/Cas-mediated resistance, we show that nucleotides within the PAM and seed sequence and across the non-seed-sequence regions are critical for the functioning of this CRISPR/Cas system. We also demonstrate that P. aeruginosa can acquire spacer content in response to lytic phage challenge, illustrating the adaptive nature of this CRISPR/Cas system. Finally, we demonstrate that the P. aeruginosa CRISPR/Cas system mediates a gradient of resistance to a phage based on the level of complementarity between CRISPR spacer RNA and phage protospacer target. This work introduces a new in vivo system to study CRISPR/Cas-mediated resistance and an additional set of tools for the elucidation of CRISPR/Cas function.
INTRODUCTION
Clustered regularly interspaced short palindromic repeats (CRISPR) are found in roughly 50% of sequenced bacterial genomes and 90% of archaeal genomes (2, 24). CRISPR regions are composed of multiple repeated sequences ranging from 21 to 48 bp in length separated by 26- to 72-bp spacers (1, 2). The sequences of spacer regions are variable but are often identical to sequences found within phages, plasmids, or other foreign DNA (24). CRISPR loci are transcribed as one large transcript that is processed within the identical repeat sequence into mature, small CRISPR RNAs (crRNAs) by either CRISPR-associated (Cas) proteins alone or RNase III associated with Cas proteins (5, 11, 15). These mature crRNAs are then complexed with subtype-specific Cas proteins, and this complex specifically interacts with nucleotide target sequences complementary to spacer sequences (5, 14, 16, 26). Complementarity between mature crRNAs and sequences found within phages or plasmids leads to inhibition of their replication through target nucleotide cleavage (12, 14, 25). The ability of CRISPR/Cas systems to also incorporate DNA sequences from newly encountered foreign DNA and subsequently resist phages or plasmids containing these sequences has led these systems to be referred to as bacterial adaptive immune systems (24).
Pseudomonas aeruginosa is an opportunistic pathogen of humans and animals that is capable of becoming highly antibiotic resistant (18); thus, it has become a model for new or previously overlooked antibacterial treatments, such as phage therapy (22). To investigate the interplay between the P. aeruginosa CRISPR/Cas systems and mobile genetic elements, such as phage, we previously analyzed the location, prevalence, and CRISPR content of 122 human clinical P. aeruginosa isolates (8). We found that 36% of tested P. aeruginosa clinical isolates harbored CRISPR/Cas systems, 6% of the type I-E (Escherichia) subtype and 33% of the type I-F (Yersinia) subtype (8). Furthermore, many CRISPR spacers found within these clinical strains were 100% identical to chromosomally integrating P. aeruginosa mobile genetic elements, such as temperate phages, prophages, pathogenicity islands, and transposons, suggesting that the P. aeruginosa CRISPR/Cas systems interact with these elements in a manner similar to that of other characterized CRISPR/Cas systems (8). Consistent with this idea, all of the P. aeruginosa isolates tested produced mature (processed) crRNAs, indicating the expression of both large CRISPR transcripts and functional Cas proteins (8). However, using the laboratory strain P. aeruginosa UCBPP-PA14 (PA14) and 6 crRNA-expressing clinical isolates harboring spacers 100% identical to those of P. aeruginosa bacteriophages DMS3, MP22, F116, and D3, we were unable to demonstrate CRISPR/Cas-mediated phage resistance (8). These data were particularly perplexing in light of our previous observation that the P. aeruginosa CRISPR/Cas system does interact with a specific protospacer sequence in bacteriophage DMS3 to inhibit biofilm formation (7, 29).
Motivated by the paradoxical behavior of the P. aeruginosa CRISPR/Cas system, in this work, we performed a large-scale screen to identify phages that are inhibited by this system. To this end, we generated a large collection of temperate phages by inducing and isolating them from diverse P. aeruginosa isolates. We then tested their ability to infect wild-type (WT) P. aeruginosa PA14 and an isogenic strain with its CRISPR-encoding region deleted (the ΔCR/cas mutant, lacking both CRISPR loci and all cas genes). Using this alternative approach, we were able to obtain previously uncharacterized phages whose replication is blocked by the endogenous CRISPR/Cas system. Further analysis of these new phages and engineered derivatives of phage DMS3 have allowed us, for the first time, to investigate phage resistance mediated by the P. aeruginosa type I-F CRISPR/Cas system.
MATERIALS AND METHODS
Strains and media.
The strains, plasmids, and primers used in this study are listed in Table 1. P. aeruginosa strain PA14 was used in this study. P. aeruginosa and Escherichia coli strains were routinely cultured in lysogeny broth (LB) at 37°C. Growth media were supplemented with the following antibiotics at the indicated concentrations: ampicillin (Ap), 150 μg ml−1 (E. coli); gentamicin (Gm), 10 μg ml−1 (E. coli) and 50 μg ml−1 (P. aeruginosa); and carbenicillin, 50 μg ml−1 (E. coli) and 250 μg ml−1 (P. aeruginosa).
Table 1.
Bacterial strains, bacteriophage, and plasmids utilized in this study
| Strain, bacteriophage, or plasmid | Relevant characteristic (sequence) | Reference or source |
|---|---|---|
| Cloning strains | ||
| S. cerevisiae INVSc1 | MATa his3D1 leu2 trp1-289 ura3-52 MAT his3D1 leu2 trp1-289 ura3-52 | 21 |
| E. coli S17-1(λ pir) | thi pro hsdR negativehsdM positive ΔrecA RP4-2::Tc Mu-Km::Tn7 | 21 |
| Bacteriophage | ||
| SMC3884 | Wild-type DMS3 lysogen | 29 |
| SMC5486 | ΔCR mutant JBD18 lysogen | This study |
| SMC5487 | ΔCR mutant JBD25 lysogen | This study |
| SMC5488 | ΔCR mutant JBD67 lysogen | This study |
| SMC5489 | DMS3vir mutant (no bacteria) | This study |
| SMC5361 | Δcsy3 lysogen of DMS3 with DMS3-42 C253G allele | This study |
| SMC5362 | ΔCRISPR2 lysogen of DMS3 with DMS3-42 C253G allele | This study |
| SMC5363 | Δcsy3 lysogen of DMS3 with DMS3-42 T255C allele | This study |
| SMC5364 | ΔCRISPR2 lysogen of DMS3 with DMS3-42 T255C allele | This study |
| DMS3-42 T255C evasion mutants | ||
| SMC5429 | Evasion mutant 1 T255C/A245G | This study |
| SMC5430 | Evasion mutant 2 T255C/T244C | This study |
| SMC5431 | Evasion mutant 3 T255C/A245G | This study |
| SMC5432 | Evasion mutant 4 T255C/T239G | This study |
| SMC5433 | Evasion mutant 5 T255C/G264A | This study |
| SMC5434 | Evasion mutant 6 T255C/T239A | This study |
| SMC5391 | Evasion mutant 7 T255C/A251G | This study |
| SMC5392 | Evasion mutant 8 T255C/A251G | This study |
| SMC5393 | Evasion mutant 9 T255C/A251G | This study |
| SMC5424 | Evasion mutant 10 T255C/A248G | This study |
| SMC5394 | Evasion mutant 11 T255C/A251G | This study |
| SMC5395 | Evasion mutant 12 T255C/A245G | This study |
| SMC5396 | Evasion mutant 13 T255C/G257T | This study |
| SMC5425 | Evasion mutant 14 T255C/C255T | This study |
| SMC5397 | Evasion mutant 15 T255C/A251G | This study |
| SMC5398 | Evasion mutant 16 T255C/A251G | This study |
| SMC5399 | Evasion mutant 17 T255C/A262G | This study |
| SMC5400 | Evasion mutant 18 T255C/A245G | This study |
| SMC5426 | Evasion mutant 19 T255C/A248T | This study |
| SMC5401 | Evasion mutant 20 T255C/A248G | This study |
| SMC5402 | Evasion mutant 21 T255C/A251G | This study |
| SMC5403 | Evasion mutant 22 T255C/A251G | This study |
| SMC5404 | Evasion mutant 23 T255C/A245G | This study |
| SMC5405 | Evasion mutant 24 T255C/A251G | This study |
| SMC5406 | Evasion mutant 25 T255C/A245G | This study |
| SMC5407 | Evasion mutant 26 T255C/A251G | This study |
| SMC5408 | Evasion mutant 27 T255C/A251G | This study |
| SMC5409 | Evasion mutant 28 T255C/A251G | This study |
| SMC5410 | Evasion mutant 29 T255C/G257T | This study |
| SMC5428 | Evasion mutant 30 T255C/C236A | This study |
| SMC5411 | Evasion mutant 31 T255C/T244C | This study |
| SMC5427 | Evasion mutant 32 T255C/A248G | This study |
| SMC5412 | Evasion mutant 33 T255C/G264A | This study |
| SMC5413 | Evasion mutant 34 T255C/A248G | This study |
| SMC5414 | Evasion mutant 35 T255C/G264A | This study |
| SMC5415 | Evasion mutant 36 T255C/A245G | This study |
| SMC5416 | Evasion mutant 37 T255C/A251G | This study |
| SMC5417 | Evasion mutant 38 T255C/A248G | This study |
| SMC5418 | Evasion mutant 39 T255C/A251G | This study |
| SMC5419 | Evasion mutant 40 T255C/A262G | This study |
| SMC5420 | Evasion mutant 41 T255C (no secondary mutation found) | This study |
| SMC5421 | Evasion mutant 42 T255C/A245G | This study |
| SMC5422 | Evasion mutant 43 T255C/G261A | This study |
| CRISPR deletions | ||
| SMC3893 | ΔCRISPR1 | 29 |
| SMC3895 | ΔCRISPR2 | 29 |
| SMC5454 | ΔCRISPR1 ΔCRISPR2 double mutant (cas genes intact) | This study |
| SMC4279 | ΔCRISPR region (no remaining cas genes) | 8 |
| Plasmids | ||
| SMC3505 | E. coli/pMQ70-derived vector (Carbr/Ampr) | This study |
| SMC5490 | E. coli/pMQ70 DMS3-42 target allele (Carbr/Ampr) | This study |
| SMC5491 | E. coli/pMQ70 DMS3-42 C253G target allele (Carbr/Ampr) | This study |
| SMC5492 | E. coli/pMQ70 DMS3-42 T255C target allele (Carbr/Ampr) | This study |
| Spacer integration screen | ||
| SMC5467 | CRISPR1 spacer α (DMS3-42) (AACGGCCGACGCTTCTGGGTCGTCGTGAAAGT) | This study |
| SMC5457 | CRISPR2 spacer α (DMS3-42) (GACGTCTGACCAGCGAGTTGCAACGTCACCAC) | This study |
| SMC5460 | CRISPR2 spacer β (DMS3-41) (CTGGCCACCCAGGCGAAACTGGCGGCAGTGCT) | This study |
| SMC5465 | CRISPR2 spacer χ (DMS3-41) (AGTCGTTGTCCAGCGGCATCATGGGGCTGTTT) | This study |
| SMC5464 | CRISPR2 spacer δ (DMS3-42) (ACTTTCACGACGACCCAGAAGCGTCGGCCGTT) | This study |
| SMC5466 | CRISPR2 spacer ε (DMS3-45) (TCGGACACGCACGTTATGCGACTGCTGTCCAC) | This study |
| SMC5463 | CRISPR2 spacer ϕ (DMS3-42) (ACTTTCACGACGACCCAGAAGCGTCGGCCGTT) | This study |
| SMC5461 | CRISPR2 spacer γ (DMS3-41) (GGAACAGGCACAAGCGACAGTATCCCGATTCT) | This study |
| SMC5459 | CRISPR2 spacer η (DMS3-42) (GACGTCTGACCAGCGAGTTGCAACGTCACCAC) | This study |
Plaque assay.
Phage production was determined using a plaque assay, essentially as described by Budzik et al. (6). Briefly, 200 μl of WT P. aeruginosa PA14 or mutant was added to 4 ml molten top agar (0.8%) and poured over a prewarmed LB agar plate. Strains to be tested for phage production were grown overnight at 37°C in LB and filter sterilized using a 0.22-μm-pore-size filter. After solidification of top agar lawns, 3 μl of serially diluted, filter-sterilized control and test lysates was spotted onto the top agar lawn and incubated at 37°C overnight. Plaques were counted and expressed as the number of PFU/ml.
Strain construction.
Most strains and constructs were created using a Saccharomyces cerevisiae recombineering technique described previously (7, 21). During attempts to mutate the protospacer in phage DMS3 gene 42 (DMS3-42), we could create only three of the five mutations in a CRISPR/Cas-intact background (see Fig. 2 and associated text for details). The other two mutations (C253G and T255C) were constructed in the ΔCRISPR2 (ΔC2) or Δcsy3 mutant, which are both CRISPR/Cas-deficient backgrounds.
Fig 2.
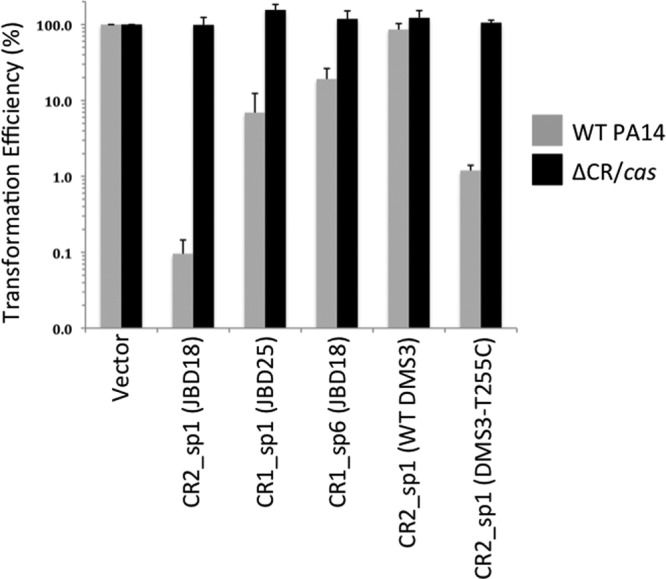
CRISPR/Cas-dependent replication of plasmids harboring protospacer sequences. The transformation efficiency of plasmids harboring no protospacer (vector) or the protospacer is indicated on the axis of the graph in the presence or absence of the CRISPR/Cas system. The transformation efficiency of each spacer-bearing plasmid was quantified relative to the transformation efficiency of the empty vector introduced into the same strain. Transformations were performed by electroporation as described in Materials and Methods. The CR2_sp1 (JBD18) protospacer sequence is identical to that of DMS3100%.
CRISPR/Cas evasion screen.
Phage DMS3 harboring a chromosomal DMS3-42 T255C mutation was purified from an overnight-grown ΔCRISPR2 mutant P. aeruginosa lysogen (SMC5364) using a 0.22-μm-pore-size filter. Purified T255C mutant phage (200 μl lysate) and WT P. aeruginosa PA14 (200 μl) were then mixed 1:1 in 4 ml molten top agar (0.8%) and poured over a prewarmed LB agar plate. After solidification, LB top agar plates were incubated at 37°C overnight. A few single plaques could be observed on each plate. Individual CRISPR/Cas evader plaques were picked and cultured at 37°C overnight in 5 ml of liquid LB medium. Following overnight growth, cultures were passed through a 0.22-μm-pore-size filter, serially diluted, and re-plaque purified on WT P. aeruginosa. This process was repeated a final time, before WT P. aeruginosa PA14 lysogenized by the escaper mutant phage was streaked out to single colonies and stored at −80°C. The DMS3-42 gene was PCR amplified from each strain and sequenced, revealing each crRNA target site mutation. Evasion mutant phages were purified twice on lawns of WT P. aeruginosa PA14 before having their DMS3-42 gene sequenced. In total, 43 such evasion mutants were sequenced.
crRNA target plasmid transformation assay.
A shuttle vector called pHERD30T that replicates in both E. coli and P. aeruginosa was used to clone predicted protospacers for the purposes of plasmid transformation assays. Oligonucleotides (42 nucleotides [nt]) corresponding to a protospacer of interest (32 nt), along with 5 nt up- and downstream, were synthesized. Extra bases corresponding to digested NcoI and HindIII restriction sites were added onto the synthesized oligonucleotides, such that sticky ends were created when the oligonucleotides were annealed. After annealing, these protospacers were ligated to pHERD30T digested with NcoI and HindIII and transformed into strain DH5α, and positive clones were confirmed by sequencing. Plasmids containing protospacers matching the sequence of spacer 1 from CRISPR2 (abbreviated CR2_sp1) from WT DMS3 (5 mismatches), T255C (4 mismatches), and JBD18/DMS3100% (DMS3100% is a phage mutant bearing a protospacer that is 100% complementary to the spacer portion of crRNACR2_sp1; 0 mismatches) as well as protospacers matching CR1_sp1 (from JBD25) or CR1_sp6 (from JBD18) were isolated from E. coli using a BioBasic miniprep kit. P. aeruginosa cells with CRISPR/Cas intact (WT cells) or deficient in CRISPR/Cas (ΔCR/cas cells) were grown in 5 ml of LB at 37°C in a shaker for 18 h. Following growth, 1.0 ml of the culture was washed twice in 1.0 ml 300 mM sucrose, resuspended in 100 μl 300 mM sucrose, and mixed with 300 to 400 ng of the indicated plasmid DNA. Each sample was electroporated and recovered in 1.0 ml of LB medium for 1 h at 37°C. Following incubation, each sample was diluted 10- and 100-fold and 100 μl was plated onto a prewarmed LB plate containing 30 μg/ml gentamicin. Each plate was incubated at 37°C for 18 h, and the resulting colonies on each plate were counted and used to determine the transformation efficiency as a function of the number of CFU per nanogram of plasmid added to each plate. The transformation efficiency of plasmids containing protospacers was compared to that of the empty vector for each individual recipient strain and shown as a percentage. Three separate transformations were performed for each construct tested.
JBD phage isolation.
A panel of 88 diverse Pseudomonas aeruginosa isolates was screened for the presence of prophages, with 20 of these strains also being used as indicator lawns. Indicators were chosen on the basis of an initial screen with 8 previously characterized phages used to infect all 88 strains. This group of 20 was chosen on the basis of the diverse plaquing patterns (not shown). The prophage screening was performed by individually treating each of the 88 strains with mitomycin C (3 μg/ml) during log-phase growth (optical density at 600 nm [OD600] = 0.5). After 3 to 4 h of treatment, lysis of the culture was apparent and chloroform was added for 15 min to complete lysis. Debris was pelleted by centrifugation, and the supernatant was stored at 4°C over chloroform. Lysates were then screened against the panel of 20 P. aeruginosa indicator strains using traditional plaque assays. Plaques were picked and purified three times on the same host.
JBD phage purification.
High-titer phage lysates (1011 to 1012 PFU/ml) were produced by soaking plates with near confluent lysis in SM buffer (100 mM NaCl, 8 mM MgSO4, 50 mM Tris-HCl, pH 7.5, 0.01% gelatin). Lysates from 10 plates were pooled, subjected to chloroform treatment, and centrifuged to clear bacterial debris. The supernatant was then treated with DNase (5 μg/ml) and RNase (5 μg/ml) for 1 h at room temperature. Sodium chloride was then added to a final concentration of 1 M, and the mixture was placed on ice for 1 h. Finally, the lysate was passed through a 0.45-μm-pore-size filter, polyethylene glycol (PEG) 8000 (10%, wt/vol) was added, and the mixture was dissolved before being placed on ice at 4°C for 16 h. The resulting suspension was centrifuged, and the PEG pellet was resuspended with 500 μl of SM buffer and chloroform extracted in an equal volume with centrifugation at 5,000 × g for 15 min at 4°C. The phage preparations were purified by equilibrium density gradient centrifugation in SM buffer-CsCl (0.74 mg/ml), performed two times in succession. Ultracentrifugation was conducted for 24 h at 50,000 rpm at 4°C in a 75Ti rotor in a Beckman Coulter Optima L-90K ultracentrifuge. Bands were extracted with a 16.5-gauge needle and dialyzed twice in a 1,000-times volume of SM buffer.
Transmission electron microscopy.
Cesium chloride-purified phages were negatively stained with 2% (wt/vol) uranyl acetate on 400-mesh copper grids coated with carbon. Microscopy was conducted with an Hitachi 7000 transmission electron microscope at magnifications ranging from ×150,000 to ×300,000.
K+ efflux assays.
Bacteria were grown in LB with 10 mM MgSO4 to mid-log phase (OD600 = 0.5), and 5 ml of the culture was washed twice in SM buffer without gelatin. Cells were placed on ice until use. To measure efflux, cells were brought to 37°C in a water bath and a potassium selective electrode was inserted, with measurements acquired every 5 s. CsCl-purified phage was added to the cells at a multiplicity of infection (MOI) of 10. Mock-treated cells received SM buffer treatment and were also subjected to the same protocol in parallel to control for spontaneous leakage of ions from cells. All assays were performed in triplicate. Results of single representative experiments are shown in the figures.
Spacer integration screen.
Three hundred microliters of WT P. aeruginosa PA14 grown overnight in LB medium was coinoculated with ∼1010 PFU of phage DMS3 vir (MOI, ∼10 to 100) in 5 ml of fresh LB medium overnight at 37°C. Overnight cocultures were serially diluted in LB medium and plated onto predried LB plates before incubation at 37°C overnight. Plates that yielded single colonies were retained and analyzed visually for twitching motility-positive colonies (rough edges). It was noted by eye, with a dissecting scope, and in twitching motility assays (4) that the vast majority of colonies resistant to DMS3 vir infection had lost type IV pilus (T4P) function, an expected finding, as we previously demonstrated that T4P is the receptor of DMS3 (6). However, a few (∼1 to 3) rough-edged, twitch-positive colonies were observed per plate. These colonies were repatched onto new LB plates and tested for T4P activity by twitch assay. Following confirmation of T4P activity, both CRISPR loci from each screen candidate were PCR amplified and sequenced.
Nucleotide sequence accession numbers.
The full genome sequences of JBD18, JBD25, and JBD67 are available in the NCBI database under accession numbers JX495041, JX495042, and JX495043, respectively.
RESULTS
The replication of six different temperate phages is inhibited by the P. aeruginosa type I-F CRISPR/Cas system.
A panel of 88 clinical and environmental isolates of P. aeruginosa was treated with mitomycin C to induce prophages, the resulting cell lysates were spotted on lawns of 20 diverse P. aeruginosa indicator strains, and any observed plaques were subsequently purified. Approximately 60 of the isolated temperate phages were able to form plaques on strain P. aeruginosa PA14. However, 30 phage isolates could generate plaques on other P. aeruginosa isolates in our collection but were unable to develop plaques on P. aeruginosa PA14.
To assess the possible role of CRISPR/Cas-mediated immunity in preventing these phages from forming plaques on P. aeruginosa PA14, we assessed the ability of this collection of phages to form plaques on a P. aeruginosa PA14 strain lacking a functional CRISPR/Cas system (the ΔCR/cas mutant) (8). Six of these 30 phages (designated JBD18, JBD25, JBD37, JBD54, JBD55, and JBD67; see Fig. 1A for electron microscopy images of selected phages) were able to form plaques on the P. aeruginosa PA14 ΔCR/cas strain but not on WT P. aeruginosa PA14 (Fig. 1B; compare the leftmost and rightmost panels). Upon generation of high-titer lysates of these six phages by growth on the ΔCR/cas mutant, we observed 107- to 109-fold decreases in the efficiency of plaquing (EOP) on WT compared to that on the ΔCR/cas mutant. These data strongly imply that the CRISPR/Cas system in the WT P. aeruginosa PA14 strain drastically inhibits the growth of these phages.
Fig 1.

Isolation of bacteriophage blocked for replication by the native CRISPR/Cas system of P. aeruginosa. (A) Representative negative-stained images of bacteriophages JBD18, JBD25, and JBD67 obtained using transmission electron microscopy. All three bacteriophages display the head and noncontractile tail characteristic of the double-stranded DNA viral family Siphoviridae. (B) Ability of bacteriophages DMS3, JBD18, JBD25, and JBD67 to replicate on WT P. aeruginosa PA14 and strains lacking the crRNA encoding the CRISPR1 (the ΔCRISPR1 strain) or CRISPR2 (the ΔCRISPR2 strain) region or both CRISPR loci (the ΔCRISPR1/2 strain). Serial 10-fold dilutions of bacteriophage lysates are shown by spot titration onto top agar lawns of the indicated P. aeruginosa strains. The presence of plaques indicates that the denoted strain can no longer mediate resistance to that bacteriophage. Bacteriophages JBD18 and JBD67 are unable to plaque on strains harboring functional CRISPR1 and CRISPR2, while JBD25 is blocked for replication only by strains with CRISPR1. (C) A K+ efflux assay was performed using phage JBD18 to infect WT, ΔCR/cas, and ΔpilA P. aeruginosa strains to demonstrate that phage DNA is still injected in WT P. aeruginosa PA14. (D) Diagram of the CRISPR and cas genes found in P. aeruginosa PA14. CRISPR spacer content that is 100% identical over all 32 nucleotides to a region of bacteriophage JBD18 is indicated with spotted boxes, while those identical to regions of JBD25 and JBD67 are depicted in hashes and dark gray, respectively. CRISPR1 and CRISPR2 are thought to be encoded on opposing DNA strands and are numbered according to convention. (E) Candidate CRISPR spacers that likely interact with bacteriophages JBD18, JBD25, and JBD67 shown pictorially in panel D.
If resistance to the JBD phages was mediated by a CRISPR/Cas system, we would predict that these phages, while unable to replicate, should still be capable of injecting their DNA into the host. To test this idea, K+ efflux assays were conducted to quantify the phage genome entry kinetics of one of these phages, JBD18. This efflux assay directly measures the increase in extracellular K+ ions that occurs as a result of cytoplasmic ion leakage as the phage genome passes through the inner bacterial membrane (3). JBD18 infection resulted in the same kinetics and magnitude of K+ ion efflux from the WT and ΔCR/cas strains (Fig. 1C), demonstrating that the JBD18 genome is able to enter WT cells at a normal rate. In contrast to the WT strain, infection of a ΔpilA mutant, which is resistant to JBD18 because it lacks the type IV pilus required for cell surface adsorption of this phage (6), resulted in little K+ efflux. This result confirms that the observed K+ efflux requires attachment of phage to cells and that mutating the CRISPR/Cas system does not alter injection kinetics.
To confirm the requirement of cas genes for the apparent CRISPR/Cas system-mediated inhibition of the phages under investigation here, strains containing single gene deletions of each of the P. aeruginosa PA14 cas genes were assayed. As shown in Table 2 (first 3 rows), deletion of any of the cas genes, with the exception of cas1, resulted in sensitivity to infection by JBD18, JBD25, and JBD67. The absence of an effect for the Δcas1 strain was expected, as the Cas1 protein is believed to play a role in the acquisition of new spacers but not in CRISPR-mediated interference (1, 27, 29). Deletions of cas3, csy1, csy2, csy3, and csy4 in P. aeruginosa PA14 have been shown to reduce or eliminate crRNA accumulation, while Δcas1 strains display normal levels of processed crRNA (7).
Table 2.
Summary of results of plaque assays with P. aeruginosa PA14 CRISPR/cas mutantsa
| Phage | Genotypeb |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| WT | ΔCR/cas | ΔCR1 | ΔCR2 | Δcas1 | Δcas3 | Δcsy1 | Δcsy2 | Δcsy3 | Δcsy4 | |
| JBD18 | − | + | − | − | − | + | + | + | + | + |
| JBD25 | − | + | + | − | − | + | + | + | + | + |
| JBD67 | − | + | − | − | − | + | + | + | + | + |
| WT DMS3 | + | + | + | + | + | + | + | + | + | + |
| DMS3 C253G | - | + | - | + | - | + | + | + | + | + |
| DMS3 T255C | - | + | - | + | - | + | + | + | + | + |
| DMS3100% | − | + | − | + | − | + | + | + | + | + |
Plaque assays conducted with the indicated phages used to infect lawns of WT or various mutant strains.
+, formation of plaques (EOP, 1 relative to ΔCR/cas); -, reduced plaquing efficiency with a >104-fold reduction; −, reduced plaquing efficiency with a >106-fold reduction.
Sequencing the genomes of JBD18, JBD25, and JBD67 reveals functional protospacer sequences.
To further elucidate the mechanism by which the phages identified here were targeted by the CRISPR/Cas system, we sequenced the genomes of three of these phages (JBD18, JBD25, and JBD67). As shown in Fig. 1D, P. aeruginosa PA14 possesses two different CRISPR loci, designated CRISPR1 and CRISPR2, with the cas genes located between the CRISPR loci (29). Phage JBD18 contains regions with 100% matches to spacer 6 in the CRISPR1 locus (CR1_sp6) and spacers 1 and 2 in the CRISPR2 locus (CR2_sp1, and CR2_sp2, respectively) (Fig. 1D and E). Importantly, these putative protospacers also display the required protospacer adjacent motif (PAM), which is GG in the type I-F CRISPR/Cas system found in P. aeruginosa PA14 (19). JBD67 displays the same putative protospacers as JBD18, with the exception of a single mismatch with CR2_sp2, where the A highlighted in bold in Fig. 1E is a G. JBD25 possesses only one predicted spacer match to CR1_sp1, and the PAM is also present. Plaque assays showed that deletion of either CRISPR1 (ΔCRISPR1 strain) or CRISPR2 (ΔCRISPR2 strain) alone did not alleviate the inhibition of JBD18 and JBD67, whereas JBD25 was not inhibited in the ΔCRISPR1 strain (Fig. 1B). These results are consistent with the protospacer matches found in the genomes of these phages, as outlined above.
To demonstrate that the protospacers found in JBD18, JBD25, and JBD67 were authentic targets of the PA14 CRISPR/Cas system, the putative protospacer sequences and PAMs from each phage genome were synthesized and cloned into a high-copy-number shuttle vector capable of replication in E. coli and P. aeruginosa. The protospacer-containing plasmids or the vector with no insert was electroporated into WT P. aeruginosa PA14 or the ΔCR/cas strain, and transformation efficiencies were calculated. The plasmid containing the CR2_sp1 protospacer transformed WT P. aeruginosa PA14 with less than 0.1% of the efficiency of the empty vector, but no difference in transformation efficiency was observed in the ΔCR/cas strain (Fig. 2). Similarly, transformation of WT P. aeruginosa PA14 by plasmids containing either the CR1_sp1 or CR1_sp6 protospacer was reduced by ≥80% compared to that achieved by the empty vector (Fig. 2). In contrast, no reduction of transformation efficiency was observed for these plasmids when they were introduced into the ΔCR/cas strain. Although the magnitude of the reduction in transformation efficiency caused by the action of the CRISPR/Cas system varied depending on the particular protospacer being tested, these results clearly demonstrate that the P. aeruginosa PA14 CRISPR/Cas system is active against protospacers present in phages that are inhibited by the system. Thus, the presence of these protospacers is likely the source of the vulnerability of these phages to the CRISPR/Cas system.
In summary, the results described above demonstrate for the first time that the type I-F CRISPR/Cas system of P. aeruginosa functions in a manner similar to that of other CRISPR/Cas systems to inhibit the replication of phages bearing matches to spacers contained within the CRISPR loci.
Engineering of DMS3 variants blocked for infection by CRISPR/Cas system.
We previously demonstrated that P. aeruginosa crRNACR2_sp1 (crRNA encoded by CR2_sp1) interacts with a protospacer in phage DMS3 gene 42 (DMS3-42) to inhibit biofilm formation in DMS3 lysogens (7, 29). However, we observed no resistance mediated by this crRNA interaction with phage DMS3 (8, 29). Since the transformation efficiency of a plasmid bearing the CR2_sp1 protospacer from JBD18, which is 100% identical to crRNACR2_sp1, was strongly inhibited by the P. aeruginosa PA14 CRISPR/Cas system (Fig. 2), we hypothesized that the lack of inhibition of DMS3 by the CRISPR/Cas system might be due to the lack of complementarity between crRNACR2_sp1 and the DMS3-42 protospacer (Fig. 3A).
Fig 3.

Engineering of DMS3 variants blocked for infection by the CRISPR/Cas system. (A) Model of Csy-crRNACR2_sp1 riboprotein complex (comp.) interacting with the DMS3-42 target sequence. The model is based on previous work performed by the Doudna group (15, 26). Csy proteins (various shades of gray) are shown coating crRNACR2_sp1, while lines denote Watson-Crick base pairing between crRNACR2_sp1 and its bacteriophage target sequence in gene 42 of phage DMS3 (DMS3-42). The crRNA seed sequence and bacteriophage PAM, which is thought to be critical for crRNA-target interaction, are shown within shadowed boxes. Thick black arrows show the location of mutant alleles C253G and T255C; note their proximity but exclusion from seed and PAM sequences. (B) Ability of bacteriophage DMS3 harboring WT, C253G, and T255C DMS3-42 alleles to infect WT (CRISPR/Cas-intact) and ΔCR (CRISPR/Cas-deficient) bacteria. WT DMS3 can readily replicate in the presence and absence of the CRISPR/Cas system (compare the tops row in both panels). Bacteriophages harboring either the DMS3-42 C253G or the DMS3-42 T255C allele purified from two different genetic backgrounds are not able to replicate in CRISPR/Cas-intact bacteria (compare the bottom four rows of each panel). (C and D) Ability of bacteriophages harboring C253G (C) and T255C (D) alleles to inject their genomic DNA into WT (CRISPR/Cas intact) or the ΔCR/cas (CRISPR/Cas-deficient) or ΔpilA (lacking bacteriophage receptor) mutant determined by the K+ efflux assay.
To test the effect of creating complementarity between crRNACR2_sp1 and the DMS3 protospacer, single-base-pair mutant alleles of DMS3 were created (denoted by thick black arrows in Fig. 3A). Strikingly, the single nucleotide changes in the DMS3 genome, C253G or T255C, led to strong inhibition of DMS3 replication by the CRISPR/Cas system, even though 4 mismatches with crRNACR2_sp1 remained (Fig. 3B). Conversely, other changes in the protospacer (C237G, G240C, and C258G) had no effect on the ability of DMS3 to replicate in WT P. aeruginosa PA14 (not shown). These data indicate that for crRNACR2_sp1 to mediate resistance to phage DMS3, complementarity between the crRNA and the protospacer at nucleotide position 253 or 255 but not at positions 237, 240, and 258 is required. Like phage JBD18, K+ efflux assays demonstrated that this resistance was manifested after genome injection (Fig. 3C and D), as would be expected for resistance mediated by the CRISPR/Cas system.
To further investigate the effects of the point mutations described above, the transformation efficiencies of plasmids harboring the WT DMS3 CR2_sp1 protospacer (5 mismatches) and the T255C mutant protospacer (4 mismatches) were compared. As shown in Fig. 2 (rightmost two sets of bars), the presence of the T255C mutation in the DMS3 protospacer sequence caused an 80-fold decrease in the transformation efficiency of P. aeruginosa PA14 compared to that achieved with the plasmid bearing the WT DMS3 protospacer sequence. This difference was not observed when the same plasmids were used to transform the PA14 ΔCR/cas mutant strain. These data show that the same point mutation that led to a dramatic CRISPR/Cas-mediated inhibition of phage DMS3 replication (Fig. 3) also imparted a reduction in transformation efficiency similar in magnitude to the reductions described for the JBD phage-derived sequences.
To further enhance our understanding of the specificity requirements of this CRISPR/Cas system for the protospacer target sequence, we undertook a screen to identify phage mutants that evade targeting. We plated high titers of phage DMS3 harboring the DMS3-42 T255C allele (MOI, ∼100) onto WT (CRISPR/Cas-intact) P. aeruginosa PA14 lawns. As was previously observed in E. coli and Streptococcus thermophilus (1, 5), this procedure allowed us to isolate mutant phages with the ability to escape the CRISPR/Cas system. Consistent with the previous studies, such escape mutants (42 out 43 sequenced) were found to possess single base mutations within the CR2_sp1 protospacer or PAM in DMS3-42 (Fig. 4). We could not identify the 43rd mutation, which presumably maps outside the DMS3-42 gene.
Fig 4.
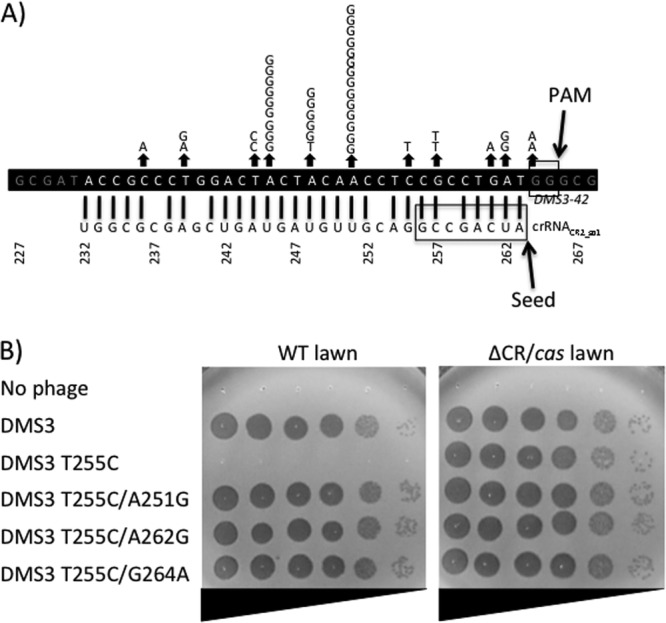
Isolation and characterization of bacteriophages that have evaded CRISPR/Cas-mediated resistance. (A) Diagram showing crRNACR2_sp1 (bottom) interacting with the bacteriophage DMS3-42 T255 target allele (top). The crRNA seed (26) and bacteriophage PAM (19) sequences are boxed to simplify the orientation and interpretation of results. Each letter depicted above an arrow indicates an individual CRISPR/Cas evasion mutation harboring the given single-base-pair mutation. (B) Susceptibility of bacterial strains with intact CRISPR/Cas (WT) and deficient in CRISPR/Cas (ΔCR/cas) to bacteriophage DMS3 harboring the indicated DMS3-42 alleles.
Mismatches between crRNA and protospacer can result in intermediate resistance.
In analyzing the effects of point mutations in the DMS3 CR2_sp1 protospacer, we created a phage mutant, which we refer to as DMS3100%, bearing a protospacer that is 100% complementary to the spacer portion of crRNACR2_sp1. Interestingly, while the plaquing efficiency of the DMS3 mutant bearing the T255C mutation was inhibited by ∼104-fold when plated on P. aeruginosa PA14, that of the DMS3100% phage was inhibited by greater than 106-fold (Table 2).
This discrepancy was even more apparent when experiments were performed at 30°C, where the plaquing efficiency of DMS3 T255C was reduced by only ∼100-fold compared to that for WT DMS3, while the plaquing efficiency of DMS3100% was still reduced by >106-fold (Fig. 5A). The DMS3 C253G mutant also displayed at least a 1,000-fold higher plaquing efficiency on P. aeruginosa PA14 at 30°C than DMS3100% (Fig. 5C and inset). These data imply that although this CRISPR/Cas system still functions in the face of crRNA mismatches, the efficiency of the system may be reduced. This finding was mirrored by plasmid transformation assays with P. aeruginosa PA14, where the transformation efficiency of a plasmid bearing a protospacer with 100% identity to CR2_sp1 was 10-fold lower than that for the one bearing the T255C mutant protospacer, which still had four mismatches with the crRNA (Fig. 2).
Fig 5.

Effect of temperature on CRISPR/Cas-mediated inhibition of bacteriophage replication. Plaque assays showing the replication of WT DMS3, DMS3 C253G, DMS3 T255C, DMS3100%, and JBD18 in the presence (A, C) or absence (B) of the CRISPR/Cas system at 30°C. Arrows, locations of single plaques. Phages were spotted in 10-fold serial dilutions.
It should be noted that we do not have an explanation for the weaker CRISPR/Cas system effect in plaquing assays performed at 30°C. The results in plasmid transformation efficiency assays were not affected by temperature (data not shown), suggesting that the CRISPR/Cas functions with similar efficiency at the two temperatures. We speculate that phage replication may be more efficient at a lower temperature, and thus, evasion of the CRISPR/Cas system occurs more readily.
The P. aeruginosa type I-F CRISPR/Cas system is adaptive to lytic phage attack.
In the context of a bacterial immune system, it is critical that bacteria acquire new spacer content in response to phage challenge (2, 24). To date, only the S. thermophilus CRISPR/Cas system has been shown to undergo spacer acquisition in response to phage attack using a native CRISPR/Cas system (1), although other systems have demonstrated spacer acquisition under engineered conditions, such as plasmid overexpression or misregulation of cas genes (10, 23, 28).
To analyze CRISPR adaptation in P. aeruginosa, we created a nonreverting lytic mutant (vir mutant) of phage DMS3 (DMS3 vir). This lytic variant was made by deleting the nucleotides that encode the first 76 amino acids of the 185-amino-acid c repressor protein, which is required for lysogeny (13). A lytic phage similar to DMS3 that completely lacked its c repressor was previously isolated (17). Figure 6A illustrates that addition of increasing levels of DMS3 vir to WT P. aeruginosa cultures results in a concurrent rise in bacterial lysis, clearly demonstrating the lytic activity of DMS3 vir.
Fig 6.

Screen to identify integration of new bacteriophage-derived spacer content into CRISPR. (A) Image showing greater lysis of P. aeruginosa cells as increasing levels of lytic bacteriophage DMS3 vir are added. (B) Dissecting microscope image of twitching-negative (left) and twitching-positive (right) colony edges. This phenotype is also visible by eye for well-grown P. aeruginosa colonies (data not shown). The majority of DMS3 vir-resistant colonies that retained T4P function (irregularly edged colonies that were twitching motility positive) had obtained a new CRISPR spacer that provided resistance to bacteriophage DMS3 vir. (C to F) Shown are the newly inserted spacers carried in each strain, with the WT and the ΔCRISPR/cas mutant included as controls (C). The sequence of the inserted spacers (D), their putative target gene (E), and the target strand (F) are also indicated. All newly integrated spacers target phage regions with an intact GG PAM motif. (G) Susceptibility of each strain to bacteriophage DMS3 vir. Each spot marks a 10-fold dilution of the DMS3 vir lysate from left to right. Taken together, these data demonstrate that the P. aeruginosa CRISPR/Cas system can integrate new spacer content conferring resistance in response to bacteriophage challenge.
Although it was possible to isolate small numbers of DMS3 vir-resistant colonies from these lysed cultures by plating the lysate on LB agar, most resistant strains carried mutations that resulted in loss of type IV pilus (T4P) function. T4P is the known receptor for DMS3 and many other P. aeruginosa phages (9). The T4P of P. aeruginosa can be extended and retracted to move the bacteria across a hard agar surface in a process known as twitching motility (4). P. aeruginosa twitching-deficient colonies have a smooth edge due to their inability to undergo twitching motility at the agar-colony edge (Fig. 6B) and are unable to produce a motility zone in the standard plate-based twitching motility assay (4). This loss of T4P production was assessed for the DMS3 vir-resistant strains first by colony morphology and then via the standard plate-based twitching motility assay.
A small number of strains resistant to DMS3 vir infection (<1%) retained T4P function, as judged by the colony assay. The CRISPR loci of nine confirmed twitch-positive, DMS3 vir-resistant isolates (Fig. 6G) from this selection were sequenced (Table 1), revealing the insertion of new spacer sequences into CRISPR1 or CRISPR2, and these spacers were 100% complementary to regions of phage DMS3 (Fig. 6C to E). Importantly, each of the newly inserted spacer sequences is complementary to a DMS3 protospacer with a conserved GG dinucleotide PAM sequence. The newly integrated CRISPR spacers provided resistance to phage DMS3 vir and are predicted to encode small crRNAs complementary to either the coding or noncoding strand of phage DMS3 (Fig. 6F).
DISCUSSION
Here we demonstrate that the type I-F CRISPR/Cas system, like the type I-E (E. coli) (5) and type II-B (S. thermophilus) (1) CRISPR/Cas systems, can provide sequence-specific and adaptive resistance to phage challenge. Using a diverse temperate phage library isolated from environmental and clinical strains of P. aeruginosa, we isolated six phages that are natively targeted and blocked for infection by the endogenous P. aeruginosa CRISPR/Cas system. In addition, we used single nucleotide point mutations to engineer a phage which was not susceptible to this CRISPR/Cas system to become resisted. Lastly, we devised a screening technique to observe CRISPR spacer acquisition in response to phage challenge. This work provides the first evidence for an endogenously functioning CRISPR/Cas adaptive immune system in P. aeruginosa and has opened up a new in vivo system for future study. This is an important advance because many key structural and in vitro functional studies on Cas proteins have been performed on the P. aeruginosa PA14 type I-F system (15, 26, 27).
Our work has provided insight into the protospacer sequence requirements of this type I-F system. In engineering phage DMS3 to be targeted by the CRISPR/Cas system (Fig. 3) and in selecting for escape mutants of DMS3 (Fig. 4), we found that mismatches in nucleotide positions across the whole protospacer region and PAM led to abrogation of targeting (Fig. 4B). These results contrast to those of other studies of the type I-E CRISPR/Cas system, where only mismatches within the PAM and an 8-nucleotide region adjacent to the PAM, designated the seed, affected targeting by the CRISPR/Cas system (20). Our identification of escape phages with mutations in the PAM provides the first experimental evidence that the conserved GG dinucleotide PAM of the type I-F CRISPR/Cas system is required for phage targeting in vivo, as has been observed in other CRISPR/Cas systems (19, 20). Similar to previous work (20), we found that a 1-nucleotide gap at position 6 of the seed sequence could be tolerated. Overall, these data support the importance of the crRNA seed sequence and phage PAM but highlight the critical importance of regions external to the seed sequence and PAMs for crRNA-mediated resistance.
Using the plasmid transformation efficiency assay, we demonstrated that protospacers with 100% identity to the CR1_sp1, CR1_sp6, and CR2_sp1 spacers are targeted by the system. Plasmids containing these sequences displayed reduced transformation efficiencies. Surprisingly, the degree of inhibition of transformation varied widely depending on the protospacer tested. For example, the CR1_sp6 protospacer from JBD18 and JBD67, when placed on a plasmid, resulted in only a 5-fold inhibition of transformation, while the CR2_sp1 protospacer from JBD18 and JBD67 (or DMS3100%) caused a 1,000-fold inhibition. Surprisingly, the replication of phages JBD18 and JBD67, which contained the CR1_sp6 protospacer, was still inhibited 107-fold when plated on a strain with the CRISPR2 locus deleted. In this background, the CR1_sp6 protospacer is the only sequence in these phages that displays any significant similarity to a CRISPR spacer; thus, its presence in the JBD18 and JBD67 genomes is almost certainly the cause of the poor plating efficiency of these phages, despite the weak effect of this protospacer in the transformation assay. The various effects on transformation efficiency of different protospacers are difficult to explain, but these differences were observed consistently and likely reflect subtleties in the functioning of this system that are yet to be elucidated.
Through engineering of phage DMS3, we found that the protospacer within the DMS3-42 gene could be efficiently targeted by the CRISPR/Cas system, even when it possessed 4 mismatches with crRNACR2_sp1. However, the DMS3100% phage, which matches crRNACR2_sp1 with 100% identity, was clearly targeted more efficiently than phage containing mismatches, as was observed most noticeably in the plaquing assays performed at 30°C (Fig. 5). This decreased targeting of the mismatched protospacer was also reflected in the plasmid transformation assay, where the protospacer with no mismatches caused a 10-fold greater decrease in transformation efficiency than a protospacer with mismatches (Fig. 2). These data show that the CRISPR/Cas system is not all or nothing and that the degree of complementarity between the crRNA and protospacer sequences can affect the efficiency with which the system operates.
Consistent with a gradient of effectiveness for the P. aeruginosa CRISPR/Cas system, the WT DMS3-42 protospacer, which has 5 mismatches with crRNACR2_sp1, does not cause resistance to phage DMS3. However, we have previously shown that this protospacer does interact with the CRISPR/Cas system to inhibit biofilm formation in P. aeruginosa PA14 when it contains a DMS3 prophage. These data suggest that the effect on biofilm formation (in the absence of detectable resistance) may reflect a weak or altered interaction between the CRISPR/Cas system and phage DMS3.
Previously, we identified, sequenced, and assayed the function of CRISPR/Cas systems found in a diverse array of clinical P. aeruginosa strains (8). In that study, we were unable to detect CRISPR/Cas-mediated resistance to phages DMS3, MP22, F116, and D3, even though the strains on which they were tested (P. aeruginosa PA14 and 6 other clinical isolates) were shown to express fully processed crRNA and harbor spacers 100% identical to the tested phages (8). In the work described here, by screening a large collection of temperate phages isolated from diverse P. aeruginosa strains, we identified a small group of phages that are inhibited by the CRISPR/Cas system of P. aeruginosa PA14. At this point, it is unclear why some phages are inhibited by the CRISPR/Cas system, while others possessing protospacers and intact PAM sequences, which would thus be predicted to be targeted, are not inhibited. It is possible that crRNA molecules complementary to certain spacers do not accumulate to levels high enough to be equally effective or lack activity for other reasons. Additionally, some phages may possess mechanisms for overcoming the CRISPR/Cas system, though no such mechanisms have yet been described.
To date, only the S. thermophilus CRISPR/Cas system has been shown to undergo spacer acquisition in response to phage attack using a native CRISPR/Cas system (1), although other systems have been shown to acquire spacer content under engineered conditions, such as plasmid overexpression or misregulation of cas genes (23, 28). Using a nonreverting lytic mutant of phage DMS3, we demonstrate that in P. aeruginosa new CRISPR spacers can be obtained through a plate-based screen. This method should drastically improve the ability to perform future CRISPR adaptation studies. Further, this technique should be applicable to many of the pilus-attaching phages known to infect P. aeruginosa, including those with single-stranded RNA genomes, such as the levivirus phages PP7 and PRR1 (9).
In conclusion, this work presents an important advance for the investigation of CRISPR/Cas in vivo function. Our results provide the first evidence for phage and plasmid resistance by a type I-F CRISPR/Cas system, as well as a collection of phage-crRNA interactions for future analysis. Furthermore, our demonstration of variable effects of different protospacers in the plasmid transformation assay and the potential modulation of sensitivity to the CRISPR/Cas system through reduction of complementarity between protospacer and crRNA highlights a potential for subtlety in this system that has not previously been recognized.
ACKNOWLEDGMENTS
This work was supported by the John H. Copenhaver, Jr., and William H. Thomas fellowship and T32 AI007519 to K.C.C., NIH grant R01A1003256 and NSF grant MCB-9984521 to G.A.O., CIHR Canada Graduate Scholarship Doctoral Award to J.B.-D., and CIHR Emerging Team grant XNE86943 to A.R.D.
Footnotes
Published ahead of print 10 August 2012
REFERENCES
- 1. Barrangou R, et al. 2007. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315:1709–1712 [DOI] [PubMed] [Google Scholar]
- 2. Bhaya D, Barrangou R. 2011. CRISPR/Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annu. Rev. Genet. 45:273–297 [DOI] [PubMed] [Google Scholar]
- 3. Boulanger P, Letellier L. 1992. Ion channels are likely to be involved in the two steps of phage T5 DNA penetration into Escherichia coli cells. J. Biol. Chem. 267:3168–3172 [PubMed] [Google Scholar]
- 4. Bradley DE. 1980. A function of Pseudomonas aeruginosa PAO polar pili: twitching motility. Can. J. Microbiol. 26:146–154 [DOI] [PubMed] [Google Scholar]
- 5. Brouns SJJ, et al. 2008. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321:960–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Budzik JM, Rosche WA, Rietsch A, O'Toole GA. 2004. Isolation and characterization of a generalized transducing phage for Pseudomonas aeruginosa strains PAO1 and PA14. J. Bacteriol. 186:3270–3273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cady KC, O'Toole GA. 2011. Non-identity-mediated CRISPR-bacteriophage interaction mediated via the Csy and Cas3 proteins. J. Bacteriol. 193:3433–3445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cady KC, et al. 2011. Prevalence, conservation and functional analysis of Yersinia and Escherichia CRISPR regions in clinical Pseudomonas aeruginosa isolates. Microbiology 157:430–437 [DOI] [PubMed] [Google Scholar]
- 9. Ceyssens PJ, Lavigne R. 2010. Bacteriophages of Pseudomonas. Future Microbiol. 5:1041–1055 [DOI] [PubMed] [Google Scholar]
- 10. Datsenko KA, et al. 2012. Molecular memory of prior infections activates the CRISPR/Cas adaptive bacterial immunity system. Nat. Commun. 3:945. [DOI] [PubMed] [Google Scholar]
- 11. Deltcheva E, et al. 2011. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471:602–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Garneau JE, et al. 2010. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468:67–71 [DOI] [PubMed] [Google Scholar]
- 13. Geuskens V, et al. 1991. Frameshift mutations in the bacteriophage Mu repressor gene can confer a trans-dominant virulent phenotype to the phage. J. Bacteriol. 173:6578–6585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hale CR, et al. 2009. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell 139:945–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Haurwitz RE, Jinek M, Wiedenheft B, Zhou K, Doudna JA. 2010. Sequence-and structure-specific RNA processing by a CRISPR endonuclease. Science 329:1355–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jinek M, et al. 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim S, Rahman M, Kim J. 2012. Complete genome sequence of Pseudomonas aeruginosa lytic bacteriophage PA10 which resembles temperate bacteriophage D3112. J. Virol. 86:3400–3401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mah TF, et al. 2003. A genetic basis for Pseudomonas aeruginosa biofilm antibiotic resistance. Nature 426:306–310 [DOI] [PubMed] [Google Scholar]
- 19. Mojica F, Diez-Villasenor C, Garcia-Martinez J, Almendros C. 2009. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 155:733–740 [DOI] [PubMed] [Google Scholar]
- 20. Semenova E, et al. 2011. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc. Natl. Acad. Sci. U. S. A. 108:10098–10103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shanks RMQ, Caiazza NC, Hinsa SM, Toutain CM, O'Toole GA. 2006. Saccharomyces cerevisiae-based molecular tool kit for manipulation of genes from gram-negative bacteria. Appl. Environ. Microbiol. 72:5027–5036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sulakvelidze A, Alavidze Z, Morris JG., Jr 2001. Bacteriophage therapy. Antimicrob. Agents Chemother. 45:649–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Swarts DC, Mosterd C, van Passel MWJ, Brouns SJJ. 2012. CRISPR interference directs strand specific spacer acquisition. PLoS One 7:e35888 doi:10.1371/journal.pone.0035888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Van der Oost J, Jore MM, Westra ER, Lundgren M, Brouns SJJ. 2009. CRISPR-based adaptive and heritable immunity in prokaryotes. Trends Biochem. Sci. 34:401–407 [DOI] [PubMed] [Google Scholar]
- 25. Westra ER, et al. 2012. CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by Cascade and Cas3. Mol. Cell 5:595–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wiedenheft B, et al. 2011. RNA-guided complex from a bacterial immune system enhances target recognition through seed sequence interactions. Proc. Natl. Acad. Sci. U. S. A. 108:10092–10097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wiedenheft B, et al. 2009. Structural basis for DNase activity of a conserved protein implicated in CRISPR-mediated genome defense. Structure 17:904–912 [DOI] [PubMed] [Google Scholar]
- 28. Yosef I, Goren MG, Qimron U. 2012. Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res. 40:5569–5576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zegans ME, et al. 2009. Interaction between bacteriophage DMS3 and host CRISPR region inhibits group behaviors of Pseudomonas aeruginosa. J. Bacteriol. 191:210–219 [DOI] [PMC free article] [PubMed] [Google Scholar]