

Abstract
Despite increasing interest in coagulase-negative staphylococci (CoNS), little information is available about their bacteriophages. We isolated and sequenced three novel temperate Siphoviridae phages (StB12, StB27, and StB20) from the CoNS Staphylococcus hominis and S. capitis species. The genome sizes are around 40 kb, and open reading frames (ORFs) are arranged in functional modules encoding lysogeny, DNA metabolism, morphology, and cell lysis. Bioinformatics analysis allowed us to assign a potential function to half of the predicted proteins. Structural elements were further identified by proteomic analysis of phage particles, and DNA-packaging mechanisms were determined. Interestingly, the three phages show identical integration sites within their host genomes. In addition to this experimental characterization, we propose a novel classification based on the analysis of 85 phage and prophage genomes, including 15 originating from CoNS. Our analysis established 9 distinct clusters and revealed close relationships between S. aureus and CoNS phages. Genes involved in DNA metabolism and lysis and potentially in phage-host interaction appear to be widespread, while structural genes tend to be cluster specific. Our findings support the notion of a possible reciprocal exchange of genes between phages originating from S. aureus and CoNS, which may be of crucial importance for pathogenesis in staphylococci.
INTRODUCTION
Staphylococcus species are common colonizers of the skin and mucous membranes in humans (9, 19, 48, 63). The most prominent pathogen of the genus is the coagulase-positive Staphylococcus aureus, which is the causative agent of a broad range of diseases and is responsible for numerous hospital- and community-acquired infections (9, 19). While primarily considered innocuous commensal microorganisms, coagulase-negative staphylococcus (CoNS) species represent an increasingly severe threat to public health. S. epidermidis is a major cause of nosocomial infections due to its ability to develop biofilms on medical devices and implants (48). In addition, other species, such as S. haemolyticus, S. lugdunensis, and S. hominis, are associated with various infections with possible fatal outcomes in newborns or immunocompromised patients (48, 63).
Given the public health relevance of staphylococcal infections, the genomes of numerous S. aureus strains have been sequenced (19). As with other human pathogens, phages play pivotal roles in S. aureus pathogenesis (9, 19). They encode major virulence factors, such as staphylokinases, superantigens, Panton-Valentine leukocidin (PVL), and other innate immune modulators, but are also responsible for the mobilization of pathogenicity islands (SaPIs) (9, 19). Hence, phages are considered to be major vehicles for horizontal transfer of virulence genes among S. aureus strains. In addition, their movement within bacterial genomes contributes to genome plasticity and allows rapid adaptation of pathogens to various host conditions, such as escaping host immunity (19, 25–27).
Numerous S. aureus phages have been isolated and characterized at the molecular level (2, 6, 22, 32, 33, 36, 37, 39, 42, 44, 46, 47). They all belong to the order Caudovirales, with the most abundant being Siphoviridae phages, typically composed of a long noncontractile tail and an icosahedral head filled with double-stranded DNA. Genome studies revealed extensive mosaicism among phages, with genes organized into functional modules that are frequently exchanged between elements (10, 37). Virulence genes in general are located far from the integrase gene, between the lysis module and the right attachment site (9, 10, 37).
The mosaic organization renders the analysis of phage evolution challenging. Accordingly, several classification systems were proposed for staphylococcal phages based on morphology and serology (10, 49) or on comparative genomics (14, 37) or using specific marker genes representative of functional modules (e.g., integrase genes) (26, 34). A recent classification proposed by Kwan et al. (37) groups 27 S. aureus phages into three classes depending on their genome sizes (class I, Podoviridae; class II, Siphoviridae; and class III, Myoviridae). Using comparative genomics, class II was further divided into three clades. A new clade was added later in class II by Daniel et al. after including additional Siphoviridae phage sequences, including S. epidermidis phages (14).
So far, only six phages from CoNS have been sequenced and characterized, and little information regarding the evolution of coagulase-negative Staphylococcus phages is available in the literature (14, 28, 43, 56a). In the present work, we sequenced and characterized two phages from S. hominis and one from S. capitis. Evolutionary relationships within Staphylococcus phages were reassessed by bioinformatics analysis, including 56 additional S. aureus Siphoviridae phage genomes and 15 sequences of phages and prophages from coagulase-negative species. We found that within class II, staphylococcal phages differentiate into distinct clusters on the basis of their structural genes, while other functional modules are more conserved in the different clusters. Therefore, structural features may be considered cluster-specific characteristics of staphylococcal Siphoviridae phages, even though they share similar morphologies. In addition, our analysis refines the subgroups in class II, notably by generating a novel cluster grouping StB20 with other non-aureus phages and by grouping StB12, StB27, and other CoNS prophages with S. aureus phages in the large cluster 1. This suggests that gene exchange might have occurred or might still occur between CoNS strains and pathogenic S. aureus strains.
MATERIALS AND METHODS
Bacterial strains, phages, and growth conditions.
StB12, StB27, and StB20 phages were isolated from S. hominis T0C2HA1, S. hominis T0C6HA2, and S. capitis R104, respectively. The strains were identified by the Vitek 2 (Bio Mérieux SA) and the Api Id 32 Staph (Bio Mérieux SA) methods. Strains were grown in LB or BHI medium at 37°C with agitation. For the induction of StB12, StB27, and StB20 prophages, mitomycin C (0.5 μg/ml) was added to exponentially growing cultures (400 ml; optical density at 600 nm [OD600], ∼0.2) of S. hominis T0C2HA1, S. hominis T0C6HA2, and S. capitis R104, respectively. After 4 h of incubation at 37°C with agitation, culture lysates were centrifuged at 3,250 × g for 15 min at 4°C. The phages were then purified from supernatants by CsCl gradient centrifugation as described elsewhere (57).
Electron microscopy (EM).
For transmission electron microscopy (TEM) analysis, phage particles purified by CsCl gradient centrifugation were dialyzed against SM buffer (57) and negatively stained with 2% uranyl acetate on carbon-coated copper grids by standard procedures. Observations were done on a Tecnai 10 (FEI) microscope coupled to a Veleta charge-coupled device (CCD) camera (Olympus iTEM), and images were captured and analyzed using SIS Olympus iTEM software. Phage dimensions are the means of at least four measurements.
Genome sequencing and annotation and genome comparison at the nucleotide level.
StB12, StB27, and StB20 genomes were sequenced and assembled by GATC Biotech AG (Germany). The annotation was performed using ORF Finder (56), Genemark (7), BLAST (3, 4), Pfam (51), and ACLAME searches (40). Mauve software (14a) and the “align sequences nucleotide” BLAST tool at the NCBI (http://blast.ncbi.nlm.nih.gov/Blast) were used for sequence comparison at the nucleotide level.
Proteomic analysis.
Purified phage particles were either resuspended in SDS loading buffer and boiled for 20 min or treated with 0.1% trichloroacetic acid (TCA). The extracted phage proteins were then separated on 10% SDS-polyacrylamide gel electrophoresis. The Coomassie-stained protein bands were reduced, alkylated, and trypsin digested according to the method of Shevchenko et al. (58). The tryptic peptides were separated on a nano-HPLC Easy-nLCII (Thermo Fisher Scientific, Odense, Denmark) coupled to a quadrupole time-of-flight (Q-TOF) mass spectrometer (Waters, Milford, MA). The peptides were loaded on a 10-cm column with a 75-μm inner diameter packed with 3-μm C18 particles. Reverse chromatography was performed with a binary buffer system consisting of 0.1% formic acid (FA) (buffer A) and 95% acetonitrile in 0.1% FA (buffer B) for a 1-hour gradient run with a flow rate of 300 nl/min. The QTOF was operated in the data-dependent mode, and the three most abundant isotope patterns with +2 and +3 charges were selected. The raw files were processed using the Mascot daemon platform. The fragmentation spectra were searched against annotated open reading frame (ORF) sequences of the appropriate phages with the parent and fragment ions mass tolerances set to 100 ppm and 0.8 Da, respectively, and with 1 miscleavage. Carbamidomethylation of cysteine was set as a fixed modification and methionine oxidation and pyroglutamic acid as variable modifications for database searching.
Restriction profile analysis.
Phage DNA was extracted from purified phage particles of StB12, StB27, and StB20, as previously described (59). Genomic DNA (1 μg) was used for digestion with the EcoRI endonuclease (Roche), and the digestion products were visualized on electrophoresis gels stained with ethidium bromide. For the heating/cooling experiment performed on StB20 DNA, EcoRI-digested DNA was heated at 75°C for 10 min; one half of the digested DNA was then cooled on ice (“fast cooling”), and one half was cooled at 37°C for 10 min (“slow cooling”). Heating/cooling had no effect on restriction profiles obtained for StB12 and StB27 (data not shown).
Integration site determination.
Determination of the integration sites of StB12, StB27, and StB20 phages in their host genomes were determined by direct sequencing of bacterial genomic DNA, which was performed by Base Clear, Netherlands. Primers were designed to sequence genomic DNA from both ends of the prophages (see Results and Discussion). Primer sequences are given in Table S1 in the supplemental material.
Integrase phylogeny.
The integrases of the serine recombinase family were detected by HMM search using HMMER 3.0 (16) among all the staphylococcal phages and the 110 Staphylococcus genomes. A protein was considered a serine recombinase when both the N-terminal γδ resolvase domain (Pfam ID PF00239) and the catalytic recombinase domain (Pfam ID PF07508) were detected (reviewed in reference 60). Serine recombinases detected in the Staphylococcus genomes were considered putatively of phage origin when they displayed at least 40% similarity in amino acid sequence and less than 20% difference in protein length with a serine recombinase detected in a Staphylococcus phage. The positions of these hits in the genome were then checked for phage-related proteins nearby. Thus, 19 serine recombinases of phage origin were detected within the Staphylococcus genomes, and 13 were found among the staphylococcal phages.
The molecular phylogeny of all integrase proteins homologous to the integrases of StB12, StB20, and StB27 has been explored by the construction of multiple-sequence alignments with MUSCLE v3.6 (17). After alignment, ambiguous regions (i.e., those containing gaps and/or poorly aligned) were removed with Gblocks (v0.91b) (12). The phylogenetic tree was reconstructed using the maximum-likelihood method implemented in the PhyML program (v3.0 aLRT) with the WAG matrix and a gamma correction for variable evolutionary rates (5). Statistical support for internal branches was assessed using the aLRT test (5). The tree was rooted using the midpoint rooting method.
Prophage detection in Staphylococcus genomes.
BLASTN was used for similarity searches between the ORFs of the three phages (StB12, StB20, and StB27) and all the Staphylococcus genomes available on PATRIC (24). Only matches showing an E value of <1 × 10−3 were retained. In order to detect prophages closely related to StB12, StB20, and StB27 phages, only the regions containing at least 15 consecutive colocalized matches were retained; these phages shared on average 15 homologous ORFs with all the other phages of the data set. Two matches were considered colocalized when they were separated by less than 10 kb on the chromosome. To precisely determine the ends of the prophage and to correlate predictions and annotations of ORFs, these regions were extended to about 50 kb and were submitted to PHAST (64). Thus, 23 prophages were detected (13 in S. aureus and 10 in other Staphylococcus genomes) among 22 Staphylococcus genomes. These 23 prophages have sizes ranging from 38 to 63 kb, suggesting that they have not endured extensive pseudogenization and that they are still functional.
Phage and prophage relationships.
We analyzed 62 complete genomes of Staphylococcus phages and prophages downloaded from GenBank (http://www.ncbi.nlm.nih.gov/GenBank/) and EMBL (http://www.ebi.ac.uk/) and 23 prophages detected in the 110 Staphylococcus complete or draft genomes available on the PATRIC server (http://www.patricbrc.org) (24). Among the 85 genomes of the data set, 69 sequences originate from phages and prophages of S. aureus, 7 from phages and prophages of S. epidermidis, and 1 each from phages and prophages of S. capitis, S. caprae, S. haemolyticus, S. lugdunensis, and S. pseudintermedius. A detailed list of these phages and prophages and their main characteristics is given in Data Set S1 in the supplemental material.
To compare the protein contents of all phage and prophage genomes, the protein repertoire relatedness was measured for all possible pairwise combinations of genomes as the fraction of shared homologous proteins in the smallest genomes (61). When considering two genomes, A and B, the value of relatedness between A and B corresponds to the division of the number of homologous proteins common to genomes A and B by the minimum number of proteins in A or B. The homologous proteins were defined by identifying unique pairwise reciprocal best hits, which exhibited at least 40% similarity in amino acid sequence and less than 20% difference in protein length.
Protein repertoire relatedness was used to compute a distance matrix of all pairwise combinations of genomes. This matrix was then used to calculate a phylogenetic tree using the BIONJ algorithm (23). The Markov clustering algorithm (18) was used to cluster and organize the genomes into distinct groups from this distance matrix. Using default parameters (i.e., inflation parameter = 3), we defined nine phage groups (called clusters), all of which contain at least four phages. A network representation of protein repertoire relatedness was built using BioLayout Express 3D (21). For clarity, the links between two phages having a protein repertoire relatedness below 0.3 were not represented.
Modular evolution.
To investigate the modularity of the three sequenced phages, we built the families of homologous proteins from the 85 phages of Staphylococcus. Protein sequences were defined as homologous if they showed at least 40% similarity in amino acid sequence and less than 20% difference in protein length. Homologous proteins were then expanded to homologous protein families by transitivity. Each family of homologous proteins was described by its relative representation among the different groups of phages (i.e., clusters previously defined).
Nucleotide sequence accession numbers.
The StB12, StB27, and StB20 sequences are available in GenBank under accession numbers JN700520, JN700519, and JN700521, respectively.
RESULTS AND DISCUSSION
Three novel Siphoviridae phages isolated from coagulase-negative staphylococci.
To date, only four phages and two prophages of CoNS have been sequenced and characterized (14, 28, 43, 56a). To isolate additional temperate phages from coagulase-negative staphylococci, such strains were tested for the presence of prophages by mitomycin C treatment (data not shown; see Materials and Methods). This allowed the isolation of two phages from S. hominis strains (StB12 and StB27) and one phage from an S. capitis strain (StB20). Electron microscopy analysis of StB12, StB27, and StB20 showed similar icosahedral heads (60 nm on average) and long, flexible, noncontractile tails (206 nm for StB12, 213 nm for StB27, and 250 nm for StB20) (Fig. 1). The tails end in multiple-disc baseplate structures reminiscent of those observed for the PH15 and CNPH82 phages previously isolated from the coagulase-negative S. epidermidis species (14). Hence, the three phages belong to the family Siphoviridae within the order Caudovirales (1).
Fig 1.

Morphology of StB20, StB12, and StB27 phages. Transmission electron micrographs show StB12 (A), StB27 (B), and StB20 (C) phage particles. The three phages are characterized by an icosahedral head and a long noncontractile tail. The arrowheads indicate baseplate structures found at the ends of the tails. (A) The arrow indicates the collar structure visible on the virion head of StB12 detached from its tail. Bars, 100 nm.
Complete genome sequences and their comparison.
The genome sequences of StB12, StB20, and StB27 were determined and annotated (Table 1; see Tables S2, S3, and S4 in the supplemental material). Among the 193 proteins predicted for the three genomes, we could assign a function to 99 proteins (51%). A total of 81 (42%) are conserved proteins with unknown functions, and 13 proteins (7%) did not match any sequences in the current databases (Table 1; see Tables S2, S3, and S4 in the supplemental material). As observed in general, the ORFs of the StB12, StB27, and StB20 genomes are tightly packed and organized in functional modules. They are arranged in the following order: lysogeny; DNA metabolism; DNA packaging; and head, tail, and host cell lysis (Fig. 2).
Table 1.
General features of StB12, StB27, and StB20 phage genomes
| Feature | Value for phage: |
||
|---|---|---|---|
| StB12 | StB27 | StB20 | |
| Bacterial host | S. hominis | S. hominis | S. capitis |
| Genome size (bp) | 44,714 | 40,071 | 40,917 |
| GC content (%) | 34.17 | 33.22 | 34.07 |
| Predicted no. of genes | 74 | 53 | 66 |
| Gene density (no. of genes/kb) | 1.65 | 1.32 | 1.61 |
| % Coding | 93.8 | 93.7 | 94.5 |
| Putative functions [no. (%)] | 33 (44.5) | 34 (64) | 32 (48.5) |
| Unknown functions [no. (%)] | 33 (44.5) | 16 (30) | 32 (48.5) |
| No database match [no. (%)] | 8 (11) | 3 (6) | 2 (3) |
Fig 2.

StB12, StB27, and StB20 genome comparison at the nucleotide level. Functional modules, as defined following genome annotation, are represented in different colors: red, lysogeny; yellow, DNA metabolism; green, DNA packaging and head; blue, tail; dark pink, tail-associated holin and peptidoglycan hydrolases belonging to tail modules; light pink, lysis. The three genomes were compared by using Mauve software, and similarity profiles are shown. The levels of similarity calculated by BLAST alignments are also indicated for specific regions. The four regions conserved within the three phages are indicated by the circled numbers. Int, integrase; TerS, terminase small subunit; TerL, terminase large subunit; Port, portal protein; Prohead prot, prohead protease; MHP, major head protein; MTP, major tail protein; TMP, tail measure protein; H, holin; PG hydrolase, peptidoglycan hydrolase.
Comparison of the three sequences at the DNA level using Mauve showed that the tail morphogenesis module constitutes the most conserved region between the StB12 and StB27 genomes, while the StB12 DNA replication and metabolism module is more closely related to StB20 than to StB27. This was further confirmed by DNA sequence alignments (using the BLAST alignment tool at NCBI [http://blast.ncbi.nlm.nih.gov/Blast]): identity reached 91 to 93% over ∼9 kb between StB12 and StB27 in the tail region and 70 to 74% over an ∼2.4-kb region between StB12 and StB20 in the DNA metabolism module (Fig. 2). Interestingly, four small regions are conserved within the three genomes. These regions encode the integrase, the transcriptional regulator RinB, and two small regions of the tail morphogenesis module (a Zn carboxypeptidase described below and a putative tail fiber) (Fig. 2, numbers 1 to 4). Thus, conservation at the nucleotide level between the three genomes is confined to regions encoding important determinants of the phage life cycle (lysogeny and host specificity).
Identification of structural proteins.
In addition to bioinformatics analysis and predictions of functional assignments, structural proteins were validated by SDS-PAGE and proteins were identified by liquid chromatography-mass spectrometry (LC-MS) analysis (Fig. 3). In most cases, the isolated bands were composed of several proteins, including degradation products (not shown in Fig. 3). These protein fragments may reflect specific proteolytic cleavages, as observed for the StB20 major head protein (see below). In particular, carboxy-terminal degradation was observed for StB27 and StB20 tail tape measure proteins (TMP) (Fig. 3B, band 1, and C, bands 1 and 2), as previously reported in staphylococcal phages (62). A structural function could be assigned to 32 proteins, including 11 proteins listed as hypothetical proteins in GenBank (see Tables S2, S3, and S4 in the supplemental material). In addition, three recombination proteins (encoded by StB12 ORF13, StB27 ORF13, and StB27 ORF14) and a putative protein of unknown function belonging to the lysogeny module (encoded by StB27 ORF3) were also detected (Fig. 3A and B). The presence of nonstructural proteins in phage particles was previously reported for the S. aureus phage 80α (50), suggesting that some proteins involved in DNA metabolism might be associated with structural components.
Fig 3.

Structural proteins of STB12, STB27, and STB20 phages. On the left are shown SDS-PAGE gels of proteins extracted from StB12 (A), StB27 (B), and StB20 (C) purified phage particles. The numbers on the left of the gels indicate the sizes of the broad-range mass standard (M). (Right) Identification by LC-MS analysis of proteins extracted from the corresponding SDS-PAGE. For each band, only proteins identified with a Mascot score of ≥120 are considered. Discrepancies between observed and calculated molecular weights (Mw) (in thousands) may be due to specific proteolytic cleavage (see the text) or protein degradation or may be gel migration artifacts.
Head modules and DNA-packaging strategies.
Besides proteins required for head morphogenesis, the DNA-packaging and head module encodes a specific molecular machine involved in DNA head filling (10, 11, 52) (Fig. 2; see Tables S2, S3, and S4 in the supplemental material). This machine is composed of a portal protein forming a channel for DNA entry and a terminase constituted from the TerS and TerL subunits. The terminase is required for concatemeric DNA resolution into genome lengths and for subsequent translocation of DNA into phage heads (11, 52).
In the cases of StB12 and StB27, the composition and organization of the DNA-packaging and head module indicate that these phages use the headful packaging mechanism, which relies on the recognition of a pac site by the terminase protein (Fig. 2; see Tables S2 and S3 in the supplemental material) (10). In particular, the StB12 ORF44 and StB27 ORF31 proteins that have been identified by the proteomics approach (Fig. 3A, band 3, and B, band 4, respectively) belong to the portal protein family of SPPI, a phage using the headful packaging strategy (52). Enzymatic digestion of the StB12 and StB27 genomes led to the production of a single submolar restriction fragment corresponding to a pac fragment (Fig. 4A and B, arrowheads), confirming the use of this packaging mechanism. In contrast, the StB20 ORF38-encoded portal protein (Fig. 3C, band 6) is related to the HK97 portal family, typical of λ-like phages with cohesive ends. Enzymatic digestion of the StB20 genome produced two submolar fragments typical of the cos-type strategy (Fig. 4C, line 1, asterisks). This was further confirmed by heating/cooling DNA after enzymatic digestion. Slow cooling promoted end-to-end hybridization of the terminal fragments, while fast cooling prevented annealing (Fig. 4C, lines 2 and 3). In addition, the actual molecular mass of the major head protein (StB20 ORF40, migrating at the 37-kDa level) (Fig. 3C, band 8) was lower than the 50.4 kDa predicted on the basis of the sequence. This might indicate that the major head protein is cleaved by a specific protease, as is usually observed in cos-type phages (10). In agreement with this hypothesis, StB20 ORF39 encodes a putative prohead protease.
Fig 4.
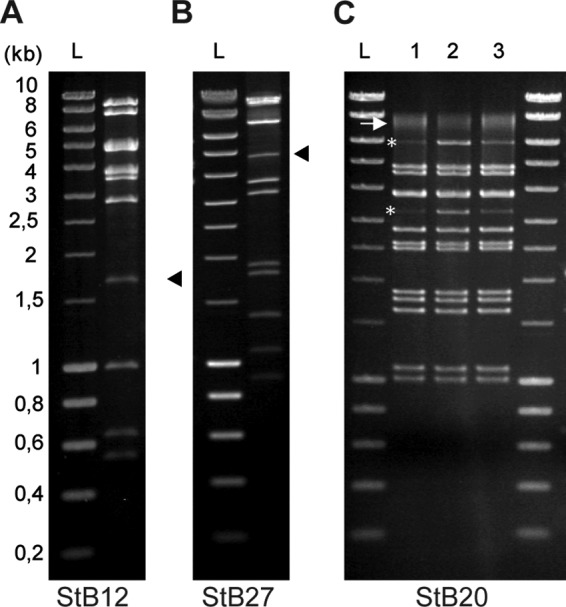
Determination of the DNA-packaging strategy used by StB12, StB20, and StB27 phages. Phage DNA of StB12 (A), StB27 (B), and StB20 (C) was digested by EcoRI, and DNA fragments were visualized on electrophoresis gels stained with ethidium bromide. (A and B) Submolar fragments corresponding to pac fragments observed in the StB12 and StB27 restriction profiles are indicated by arrowheads. (C) (Lane 1) Asterisks indicate the two terminal fragments observed in the StB20 restriction profile. The arrow indicates the two terminal fragments annealed together. (Lanes 2 and 3) Terminal fragments are more visible on the restriction profile obtained after heating and fast cooling of digested DNA (lane 2) than after slow cooling (lane 3), improving the annealing of terminal fragments. Lanes L, size ladders.
Tail modules: identification of three putative tail proteins with host cell wall lytic activity.
The proteomics approach also allowed the identification of tail proteins (7 in StB12, 8 in StB27, and 5 in StB20) (Fig. 3; see Tables S2, S3, and S4 in the supplemental material). As expected for long-tail phages, major tail proteins are among the most abundant (38). Interestingly, tail proteins predicted to be involved in cell wall degradation were also identified (Fig. 3; see Tables S2, S3, and S4 in the supplemental material). The minor tail proteins encoded by StB12 ORF61, StB27 ORF45, and StB20 ORF55 are characterized by the presence of a Zn-carboxypeptidase domain (PF00246). This domain is characteristic of a metallopeptidase family commonly found in eukaryotes, where it is known to catalyze peptide bond cleavage in a large number of cellular functions (35). In addition, three other ORFs are also related to proteases, StB12 ORF60, StB27 ORF44, and StB20 ORF53, which contain an endopeptidase domain (PF06605). Although conserved among phage tail proteins, the function of this domain is still unclear. In addition to these functions, the StB27 and StB12 protein profiles revealed the presence of a peptidoglycan hydrolase corresponding to StB27 ORF48 and StB12 ORF64 to -66, respectively (Fig. 3A and B). These proteins are related to the well-characterized and highly conserved tail-associated GP61 protein of the staphylococcal phage phiMR11. GP61 is thought to be involved in cell host lysis during phage adsorption (13, 14, 53, 62). The GP61 homolog in StB12 is encoded by the three successive ORF64, ORF65, and ORF66 that are interrupted by two putative group I introns at positions 37063 to 37307 and 37542 to 37918. It is likely that these introns are spliced in vivo to generate a functional 70-kDa protein (Fig. 3A, band 1). Group I introns are frequently found in phages, including staphylococcal phages (14, 37). Interestingly, putative tail fiber proteins were also identified in the 3 protein profiles (corresponding to StB12 ORF62 and ORF67, StB27 ORF46 and ORF49, and StB20 ORF56) (Fig. 3; see Tables S2, S3, and S4 in the supplemental material), although fibers were not visible on EM images (Fig. 1).
Integrases and integration site specificity.
The insertion sites of StB12, StB20, and StB27 phages in their host genomes were mapped by direct sequencing on genomic DNA, using primers complementary to the prophage ends (i.e., the integrase and lysine genes for StB20 and StB27 and the integrase gene and ORF74 for StB12). Interestingly, the three phages are integrated within a conserved locus encoding a putative mercuric resistance system. The StB12, STB20, and StB27 integrases are related to each other and to the experimentally described serine recombinase of the S. aureus phage phiMR11 (54). Serine recombinases appear to be rarely detected in staphylococcal phages (26). To complement this analysis, we detected serine recombinase sequences of phage and prophage origin in staphylococci and inferred the corresponding phylogenetic tree (see Materials and Methods). Staphylococcus serine recombinases are classified into three clades (Fig. 5), two of them including only S. aureus phages and the other including mostly non-S. aureus phages. The analysis of the integration sites revealed that the three groups have specific and distinct integration loci. Clade 1 S. aureus phages integrate in the ribosomal protein L32p locus, clade 2 phages in a putative mercuric resistance system, and clade 3 S. aureus phages in the signal recognition particle locus.
Fig 5.

Phylogenetic tree of phage integrases of the serine recombinase family detected in Staphylococcus genomes and Staphylococcus phages. Three major clades are defined, which correspond to 3 different integration loci. Clade 1, ribosomal protein L32p locus; clade 2, mercuric ion reductase locus; clade 3, signal recognition particle locus, subunit Ffh SRP54. LRT values are indicated for each node. The tree was rooted following the midpoint root method.
Evolutionary relationships among Staphylococcus phages.
So far, studies on the diversity and evolution of Staphylococcus phages have mainly focused on S. aureus phages (37). At the time we started this work, 59 staphylococcal phage sequences were available in GenBank, with only 2 from coagulase-negative staphylococci (14). Recently, two additional S. epidermidis phages have been isolated and sequenced (28). However, little is known about phages from coagulase-negative species and their relationships with other staphylococcal phages. The increasing number of genome sequences of CoNS represent a useful reservoir for extracting prophage sequences. To gather non-S. aureus phage sequences, prophages related to StB12, StB27, and StB20 were searched in Staphylococcus genomes (see Materials and Methods). Twenty-six prophages were detected, 13 of which were found in non-S. aureus species, including the StB12, StB20, and StB27 sequences (see Data Set S1 in the supplemental material).
Classification based on the similarity of gene repertoires between phages can be done using tree-like approaches (55) or network-like approaches (41). Using both approaches, we classified the 85 phages and prophages into 9 distinct clusters (see Materials and Methods) (Fig. 6; see Fig. S1 in the supplemental material). Each cluster contains at least four phages/prophages. Seven of these clusters (1 to 6 and 9) are composed of Siphoviridae and constitute class II according to the Kwan et al. classification (37). Clusters 7 and 8 are unrelated to these, since they are composed of Myoviridae and Podoviridae, respectively, and constitute classes III and I.
Fig 6.

Network representation of phage relationships based on the protein content (see Materials and Methods). Each node represents a phage or a prophage. Links between two phages are represented when they share at least 30% of homologs. The circles delimit the different clusters defined with the Markov cluster algorithm (MCL). The different colors represent the host species (red, S. aureus; gray, S. pseudintermedius; yellow, S. hominis; blue, S. capitis; brown, S. lugdunensis; green, S. epidermidis; mauve,: S. caprae; pink, S. haemolyticus). The letter “S” tags phages and prophages containing an integrase of the serine recombinase family. The classification is in agreement with the classes and clades previously defined, while a more precise distinction was obtained among Siphoviridae phages (14, 37). Cluster 8 corresponds to the class I (Podoviridae; genome < 20 kb), clusters 1 to 6 and 9 to class II (Siphoviridae; genome ∼40 kb), and cluster 7 to class III (Myoviridae; genome > 125 kb). Cluster 1 corresponds to clade A, cluster 4 to clade B, and clades C and D were split into two subclades (clusters 2 and 3 and clusters 5 and 6, respectively). Cluster 9 constitutes a new clade. In addition, the previously unclassified phages 187 and 2638A are attached to clusters 1 and 6, respectively. In cluster 7, the phage K is able to infect both S. aureus and coagulase-negative staphylococci (47). A list of (pro)phages is provided in Data Set S1 in the supplemental material.
Compared to the Kwan classification, a more precise resolution was obtained in class II in the present analysis (14, 37). Within class II, our analysis generates seven related clusters instead of the four clades previously proposed. Most importantly, one cluster (cluster 9) is composed exclusively of non-S. aureus phages and constitutes an entirely new clade compared to the earlier classification. It includes StB20 and its closest relative, a prophage of S. caprae C87 (31 ORFs out of 66 [47%]). Four of the seven clusters are composed exclusively of S. aureus phages (clusters 2, 4, 5, and 6), and the last two clusters (clusters 1 and 3) are composed of S. aureus and non-S. aureus phages. Cluster 1 contains StB12 and StB27 with a majority of S. aureus phages (25/32), and cluster 3 contains a majority of non-S. aureus phages (6/8). Interestingly, the most closely related phages of StB12 and StB27 are the S. aureus A8117 (23 ORFs out of 74 [31%]) and phiMR25 (18 ORFs out of 53 [34%]) phages, respectively. Such close relationships with S. aureus phages were also observed for S. epidermidis phages within cluster 3 (14). Thus, Siphoviridae phages from S. aureus and CoNS are closely related. This raises the question of gene exchange between phages isolated from different Staphylococcus species. One possibility is that they have coexisted in a common host where gene exchange has occurred, either before the last common ancestor of these species or after. In the latter case, it raises the hypothesis that they could occasionally cross species barriers. We were not able to find any Staphylococcus strain in our laboratory collection, including S. aureus, sensitive to StB12, StB27, or StB20 phages under our experimental conditions (data not shown). However, we cannot exclude the possibility that it may occur.
Intriguingly, the pathogenicity of CoNS such as S. epidermidis does not rely on the production of phage-associated virulence factors, such as toxins found in S. aureus. Instead, these species are well provided with mechanisms that allow them to escape host defenses and to persist in unfavorable environments (48). Nevertheless, a pathogenicity island encoding toxins has been recently described (43).
We then reassessed the classification of the serine recombinases in the context of the phage classification. Phages with serine recombinase are found in 5 of the 9 clusters (1, 2, 3, 6, and 7), and these groups do not reflect the phylogeny of the protein (Fig. 5). This means that the classification of phages based on the sequence of the integrase gene previously reported (26) is strongly at odds with the classification based on the gene repertoire. Interestingly, clusters including phages encoding serine recombinases always include phages encoding tyrosine recombinases, This suggests that recombination processes often exchange the integrase gene, including radically different recombinase types.
The modular evolution of staphylococcal phage genomes determines the cluster specificity.
The mosaic structure of phage genomes reflects module exchange via horizontal transfer and recombination (10, 31, 37). In order to study the modular evolution of StB12, StB27, and StB20, their ORF contents were compared to the data set described above. For each predicted ORF, a family of homologous proteins was defined, and the relative proportions of homologs in the nine clusters were calculated (see Materials and Methods) (Fig. 7).
Fig 7.

Modular evolution of StB12, StB27, and StB20. The schematic representations of the genomes of StB12, StB27, and StB20 show the frequencies of homologs detected among the 9 clusters of phages. Functional modules are represented in different colors: red, lysogeny; yellow, DNA metabolism; green, DNA packaging and head; blue, tail; light pink, lysis. Purple boxes, structural components considered specific to a cluster; light-blue boxes, non-cluster-specific structural components.
StB12, StB27, and StB20 ORFs fall into two categories. ORFs belonging to the modules of lysogeny, DNA metabolism, and lysis are conserved among the different clusters (except for clusters 7 and 8). On the other hand, a large proportion of ORFs from the structural modules, i.e., DNA-packaging and head, as well as tail, modules, appear to be cluster specific (Fig. 7, purple boxes). Notable exceptions are the ORFs at the ends of the tail modules, which are conserved among the different clusters (Fig. 7, light-blue boxes). Interestingly, some tail end ORFs of StB12 and StB27 that are likely involved in host-phage interaction (e.g., putative tail-associated Zn carboxypeptidase) are not conserved within cluster 1 but are shared by non-S. aureus phages (phages in cluster 9 and the S. capitis phage in cluster 1). Acquisition of such ORFs might modify host specificity independently of any other structural components. Consistently, horizontal transfer of tail fiber genes among unrelated phages infecting a given host could be used by phages to alter their host specificity under selective pressure (29).
Thus, the differentiation of a Siphoviridae phage relies mainly on structural features. This indicates that within the overall similarities of virion structure, there are subtle variations that differentiate Staphylococcus Siphoviridae phages in distinct clusters. This is in accordance with a modular mechanism of evolution in which differentiation processes mainly rely on the exchange of a restricted number of genes or parts of genes within the structural module (8).
Concluding remarks.
At the time of the present work, representation of Staphylococcus phages was mainly restricted to phages infecting the pathogen S. aureus. We report here the identification and molecular characterization of three novel Siphoviridae phages isolated from CoNS. In addition, we propose a refined Staphylococcus phage classification (Fig. 6). This classification highlights the close evolutionary relationships between CoNS and S. aureus species and confirms the modular nature of phage genome evolution. It also raises important questions regarding lysogenic conversion of commensal strains to pathogenic strains at the level of virulence genes, as well as host specificity genes shuffling between “nonpathogenic” and “pathogenic” phages. Consistently, several studies have reported host range shifts in phages, a process that is efficiently improved by phage-bacterium coevolution (15, 20, 29, 30, 45). As S. aureus and CoNS share the same niche, we cannot exclude the possibility that such shifts might occur, resulting from the interplay between genetic alteration and environmental conditions. In a larger context, our data also show that classification of staphylococcal phages should favor grouping methods based on common protein repertoires and take into account phage modular architecture (55) instead of single-locus information, such as integrase sequences (26, 34).
Supplementary Material
ACKNOWLEDGMENTS
Work in the Deghorain, Smeesters, Drèze, and Van Melderen laboratory is supported by the European Space Agency (MISSEX project ESA AO-2004: Prodex C90255), the Fonds Jean Brachet, and the Van Buuren Foundation. M.D. and P.R.S. are Chargé de Recherches at the Fonds de la Recherche Scientifique, Belgium (FNRS). Work in the Bobay, Rocha, and Touchon laboratories is supported by the Institut Pasteur and the CNRS. Work in the Bousbata laboratory was supported by the Fonds Jean Brachet, the Van Buuren Fondation, and the “CIBLES” program of the Walloon Region. Work in the Perez-Morga laboratory is funded by the Belgian Fund for Scientific Research (FNRS). The CMMI is supported by the European Regional Development Fund and the Walloon Region.
M.D. and L.V.M. thank Ariane Toussaint and Bernard Hallet for stimulating discussions and Lena Demazy for technical help.
Published ahead of print 24 August 2012
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1. Ackermann HW. 2007. 5500 Phages examined in the electron microscope. Arch. Virol. 152: 227–243 [DOI] [PubMed] [Google Scholar]
- 2. Ackermann HW, Kropinski AM. 2007. Curated list of prokaryote viruses with fully sequenced genomes. Res. Microbiol. 158: 555–566 [DOI] [PubMed] [Google Scholar]
- 3. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J. Mol. Biol. 215: 403–410 [DOI] [PubMed] [Google Scholar]
- 4. Altschul SF, et al. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25: 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Anisimova M, Gascuel O. 2006. Approximate likelihood-ratio test for branches: a fast, accurate, and powerful alternative. Syst. Biol. 55: 539–552 [DOI] [PubMed] [Google Scholar]
- 6. Bae T, Baba T, Hiramatsu K, Schneewind O. 2006. Prophages of Staphylococcus aureus Newman and their contribution to virulence. Mol. Microbiol. 62: 1035–1047 [DOI] [PubMed] [Google Scholar]
- 7. Besemer J, Borodovsky M. 2005. GeneMark: web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res. 33: W451–W454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Botstein D. 1980. A theory of modular evolution for bacteriophages. Ann. N. Y. Acad. Sci. 354: 484–490 [DOI] [PubMed] [Google Scholar]
- 9. Brussow H, Canchaya C, Hardt WD. 2004. Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. 68: 560–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Canchaya C, Proux C, Fournous G, Bruttin A, Brussow H. 2003. Prophage genomics. Microbiol. Mol. Biol. Rev. 67: 238–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Casjens SR, Gilcrease EB. 2009. Determining DNA packaging strategy by analysis of the termini of the chromosomes in tailed-bacteriophage virions. Methods Mol. Biol. 502: 91–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Castresana J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17: 540–552 [DOI] [PubMed] [Google Scholar]
- 13. Christie GE, et al. 2010. The complete genomes of Staphylococcus aureus bacteriophages 80 and 80alpha—implications for the specificity of SaPI mobilization. Virology 407: 381–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Daniel A, Bonnen PE, Fischetti VA. 2007. First complete genome sequence of two Staphylococcus epidermidis bacteriophages. J. Bacteriol. 189: 2086–2100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14a. Darling AE, Mau B, Perna NT. 2010. Progressive Mauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5: e11147 doi:10.1371/journal.pone.0011147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Duffy S, Burch CL, Turner PE. 2007. Evolution of host specificity drives reproductive isolation among RNA viruses. Evolution 61: 2614–2622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Eddy SR. 2011. Accelerated profile HMM searches. PLoS Comput. Biol. 7: e1002195 doi:10.1371/journal.pcbi.1002195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32: 1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Enright AJ, Van Dongen S, Ouzounis CA. 2002. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 30: 1575–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Feng Y, et al. 2008. Evolution and pathogenesis of Staphylococcus aureus: lessons learned from genotyping and comparative genomics. FEMS Microbiol. Rev. 32: 23–37 [DOI] [PubMed] [Google Scholar]
- 20. Ferris MT, Joyce P, Burch CL. 2007. High frequency of mutations that expand the host range of an RNA virus. Genetics 176: 1013–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Freeman TC, et al. 2007. Construction, visualisation, and clustering of transcription networks from microarray expression data. PLoS Comput. Biol. 3: 2032–2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Garcia P, et al. 2009. Functional genomic analysis of two Staphylococcus aureus phages isolated from the dairy environment. Appl. Environ. Microbiol. 75: 7663–7673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gascuel O. 1997. BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Mol. Biol. Evol. 14: 685–695 [DOI] [PubMed] [Google Scholar]
- 24. Gillespie JJ, et al. 2011. PATRIC: the comprehensive bacterial bioinformatics resource with a focus on human-pathogenic species. Infect. Immun. 79: 4286–4298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Goerke C, et al. 2004. Increased frequency of genomic alterations in Staphylococcus aureus during chronic infection is in part due to phage mobilization. J. Infect. Dis. 189: 724–734 [DOI] [PubMed] [Google Scholar]
- 26. Goerke C, et al. 2009. Diversity of prophages in dominant Staphylococcus aureus clonal lineages. J. Bacteriol. 191: 3462–3468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Goerke C, Wirtz C, Fluckiger U, Wolz C. 2006. Extensive phage dynamics in Staphylococcus aureus contributes to adaptation to the human host during infection. Mol. Microbiol. 61: 1673–1685 [DOI] [PubMed] [Google Scholar]
- 28. Gutierrez D, Martinez B, Rodriguez A, Garcia P. 2012. Genomic characterization of two Staphylococcus epidermidis bacteriophages with anti-biofilm potential. BMC Genomics 13: 228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Haggard-Ljungquist E, Halling C, Calendar R. 1992. DNA sequences of the tail fiber genes of bacteriophage P2: evidence for horizontal transfer of tail fiber genes among unrelated bacteriophages. J. Bacteriol. 174: 1462–1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hall AR, Scanlan PD, Buckling A. 2011. Bacteria-phage coevolution and the emergence of generalist pathogens. Am. Nat. 177: 44–53 [DOI] [PubMed] [Google Scholar]
- 31. Hatfull GF. 2008. Bacteriophage genomics. Curr. Opin. Microbiol. 11: 447–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hoshiba H, et al. 2010. Isolation and characterization of a novel Staphylococcus aureus bacteriophage, phiMR25, and its therapeutic potential. Arch. Virol. 155: 545–552 [DOI] [PubMed] [Google Scholar]
- 33. Iandolo JJ, et al. 2002. Comparative analysis of the genomes of the temperate bacteriophages phi 11, phi 12 and phi 13 of Staphylococcus aureus 8325. Gene 289: 109–118 [DOI] [PubMed] [Google Scholar]
- 34. Kahankova J, et al. 2010. Multilocus PCR typing strategy for differentiation of Staphylococcus aureus siphoviruses reflecting their modular genome structure. Environ. Microbiol. 12: 2527–2538 [DOI] [PubMed] [Google Scholar]
- 35. Kalinina E, et al. 2007. A novel subfamily of mouse cytosolic carboxypeptidases. FASEB J. 21: 836–850 [DOI] [PubMed] [Google Scholar]
- 36. Kaneko J, Kimura T, Narita S, Tomita T, Kamio Y. 1998. Complete nucleotide sequence and molecular characterization of the temperate staphylococcal bacteriophage phiPVL carrying Panton-Valentine leukocidin genes. Gene 215: 57–67 [DOI] [PubMed] [Google Scholar]
- 37. Kwan T, Liu J, DuBow M, Gros P, Pelletier J. 2005. The complete genomes and proteomes of 27 Staphylococcus aureus bacteriophages. Proc. Natl. Acad. Sci. U. S. A. 102: 5174–5179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lee JS, Stewart PR. 1985. The virion proteins and ultrastructure of Staphylococcus aureus bacteriophages. J. Gen. Virol. 66: 2017–2027 [DOI] [PubMed] [Google Scholar]
- 39. Lee YD, Chang HI, Park JH. 2011. Genomic sequence of temperate phage TEM126 isolated from wild type S. aureus. Arch. Virol. 156: 717–720 [DOI] [PubMed] [Google Scholar]
- 40. Leplae R, Hebrant A, Wodak SJ, Toussaint A. 2004. ACLAME: a CLAssification of Mobile genetic Elements. Nucleic Acids Res. 32: D45–D49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lima-Mendez G, Toussaint A, Leplae R. 2011. A modular view of the bacteriophage genomic space: identification of host and lifestyle marker modules. Res. Microbiol. 162: 737–746 [DOI] [PubMed] [Google Scholar]
- 42. Ma XX, et al. 2008. Two different Panton-Valentine leukocidin phage lineages predominate in Japan. J. Clin. Microbiol. 46: 3246–3258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Madhusoodanan J, et al. 2011. An enterotoxin-bearing pathogenicity island in Staphylococcus epidermidis. J. Bacteriol. 193: 1854–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Matsuzaki S, et al. 2003. Experimental protection of mice against lethal Staphylococcus aureus infection by novel bacteriophage phi MR11. J. Infect. Dis. 187: 613–624 [DOI] [PubMed] [Google Scholar]
- 45. Meyer JR, et al. 2012. Repeatability and contingency in the evolution of a key innovation in phage lambda. Science 335: 428–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Narita S, et al. 2001. Phage conversion of Panton-Valentine leukocidin in Staphylococcus aureus: molecular analysis of a PVL-converting phage, phiSLT. Gene 268: 195–206 [DOI] [PubMed] [Google Scholar]
- 47. O'Flaherty S, et al. 2004. Genome of staphylococcal phage K: a new lineage of Myoviridae infecting gram-positive bacteria with a low G+C content. J. Bacteriol. 186: 2862–2871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Otto M. 2009. Staphylococcus epidermidis—the ‘accidental’ pathogen. Nat. Rev. Microbiol. 7: 555–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pantucek R, et al. 2004. Identification of bacteriophage types and their carriage in Staphylococcus aureus. Arch. Virol. 149: 1689–1703 [DOI] [PubMed] [Google Scholar]
- 50. Poliakov A, et al. 2008. Capsid size determination by Staphylococcus aureus pathogenicity island SaPI1 involves specific incorporation of SaPI1 proteins into procapsids. J. Mol. Biol. 380: 465–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Punta M, et al. 2012. The Pfam protein families database. Nucleic Acids Res. 40: D290–D301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rao VB, Feiss M. 2008. The bacteriophage DNA packaging motor. Annu. Rev. Genet. 42: 647–681 [DOI] [PubMed] [Google Scholar]
- 53. Rashel M, et al. 2008. Tail-associated structural protein gp61 of Staphylococcus aureus phage phi MR11 has bifunctional lytic activity. FEMS Microbiol. Lett. 284: 9–16 [DOI] [PubMed] [Google Scholar]
- 54. Rashel M, et al. 2008. A novel site-specific recombination system derived from bacteriophage phiMR11. Biochem. Biophys. Res. Commun. 368: 192–198 [DOI] [PubMed] [Google Scholar]
- 55. Rohwer F, Edwards R. 2002. The Phage Proteomic Tree: a genome-based taxonomy for phage. J. Bacteriol. 184: 4529–4535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rombel IT, Sykes KF, Rayner S, Johnston SA. 2002. ORF-FINDER: a vector for high-throughput gene identification. Gene 282: 33–41 [DOI] [PubMed] [Google Scholar]
- 56a. Rosenstein R, et al. 2009. Genome analysis of the meat starter culture bacterium Staphylococcus carnosus TM300. Appl. Environ. Microbiol. 75: 811–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sambrook J, Russell DW. 2006. Purification of bacteriophage lambda particles by isopycnic centrifugation through CsCl gradients. CSH Protoc. 2006: pii:pdb.prot3968 doi:10.1101/pdb.prot3968 [DOI] [PubMed] [Google Scholar]
- 58. Shevchenko A, Wilm M, Vorm O, Mann M. 1996. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 68: 850–858 [DOI] [PubMed] [Google Scholar]
- 59. Smeesters PR, et al. 2011. Characterization of a novel temperate phage originating from a cereulide-producing Bacillus cereus strain. Res. Microbiol. 162: 446–459 [DOI] [PubMed] [Google Scholar]
- 60. Smith MC, Brown WR, McEwan AR, Rowley PA. 2010. Site-specific recombination by phiC31 integrase and other large serine recombinases. Biochem. Soc. Trans. 38: 388–394 [DOI] [PubMed] [Google Scholar]
- 61. Snel B, Bork P, Huynen MA. 1999. Genome phylogeny based on gene content. Nat. Genet. 21: 108–110 [DOI] [PubMed] [Google Scholar]
- 62. Tallent SM, Langston TB, Moran RG, Christie GE. 2007. Transducing particles of Staphylococcus aureus pathogenicity island SaPI1 are comprised of helper phage-encoded proteins. J. Bacteriol. 189: 7520–7524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. von Eiff C, Peters G, Heilmann C. 2002. Pathogenesis of infections due to coagulase-negative staphylococci. Lancet Infect. Dis. 2: 677–685 [DOI] [PubMed] [Google Scholar]
- 64. Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. 2011. PHAST: a fast phage search tool. Nucleic Acids Res. 39: W347–W352 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.