

Abstract
Although it is vital that cells detect and respond to oxidative stress to allow adaptation and repair damage, the underlying sensing and signaling mechanisms that control these responses are unclear. Protein ubiquitinylation plays an important role in controlling many biological processes, including cell division. In Saccharomyces cerevisiae, ubiquitinylation involves a single E1 enzyme, Uba1, with multiple E2s and E3s providing substrate specificity. For instance, the conserved E2 Cdc34 ubiquitinylates many substrates, including the cyclin-dependent kinase inhibitor Sic1, targeting it for degradation to allow cell cycle progression. Here we reveal that, in contrast to other ubiquitin pathway E2 enzymes, Cdc34 is particularly sensitive to oxidative inactivation, through sequestration of the catalytic cysteine in a disulfide complex with Uba1, by levels of oxidant that do not reduce global ubiquitinylation of proteins. This Cdc34 oxidation is associated with (i) reduced levels of Cdc34-ubiquitin thioester forms, (ii) increased stability of at least one Cdc34 substrate, Sic1, and (iii) Sic1-dependent delay in cell cycle progression. Together, these data reveal that the differential sensitivity of a ubiquitin pathway E2 enzyme to oxidation is utilized as a stress-sensing mechanism to respond to oxidative stress.
INTRODUCTION
Reactive oxygen species (ROS), such as H2O2, arise as a by-product of aerobic metabolism and, also, from the exposure of cells to environmental oxidizing agents (15). Oxidative stress occurs when cells are exposed to increased levels of ROS, causing oxidative damage to components such as DNA, proteins, and lipids. This damage has been linked with diseases, including cancer, diabetes, and cardiovascular and neurodegenerative diseases, and also with ageing (2, 4, 15). Dividing cells respond to oxidative stress by inhibiting cell cycle progression and by inducing the expression of genes involved in repair and restoring redox balance. However, the mechanisms by which these responses to increased levels of ROS are coordinated and regulated are not clear.
In eukaryotes, the covalent modification of proteins by ubiquitinylation plays an important role in regulating the activity of a multitude of proteins or targeting them for degradation (for reviews, see references 17, 18, and 21). Consequently, ubiquitinylation regulates many fundamental processes, such as cell cycle progression. For example, the ubiquitinylation of key regulatory proteins, including cyclins and cyclin-dependent kinase (CDK) inhibitors, such as Sic1 in the yeast Saccharomyces cerevisiae, determines the timing of their degradation in the cell division cycle (29, 39, 40). The conjugation of ubiquitin to a substrate requires sequential transfer of ubiquitin from the catalytic cysteine residue of the E1 activating enzyme to the catalytic cysteine residue of the E2 conjugating enzyme and then, finally, to the target substrate through the action of E3 enzymes (ligases) (Fig. 1) (17, 18, 21). A single E1, Uba1, is responsible for ubiquitinylation in S. cerevisiae, but multiple E2s and E3s confer substrate specificity on ubiquitinylation (Fig. 1) (18).
Fig 1.

The ubiquitin pathway. The transfer of ubiquitin to the target protein requires the sequential formation of an E1∼ and an E2∼ubiquitin thioester involving the catalytic cysteine residue of each enzyme. With the subsequent function of a specific E3, ubiquitin is finally transferred to the target protein. In S. cerevisiae, a single E1, Uba1, transfers ubiquitin to proteins via 11 E2 enzymes and multiple E3 enzymes, which provide target specificity.
A key to understanding the effects of ROS on cells is the identification and characterization of signaling pathways that detect ROS and subsequently initiate the appropriate response (for reviews, see references 8 and 38). Several eukaryotic signaling pathways respond to ROS via reversible oxidation of cysteine-thiols in signaling proteins. For example, reversible oxidation of a catalytic cysteine residue in some tyrosine phosphatases regulates their activity (for reviews, see references 6 and 37). E1 and E2 enzymes in ubiquitin and ubiquitin-like (Ubl) modification pathways also utilize catalytic cysteine residues, providing a potential route by which they could be regulated by oxidation (17, 18, 21). Indeed, in response to H2O2, an intermolecular disulfide forms between the catalytic cysteine residue of the E1 and E2 enzymes in the sumoylation pathway, inhibiting global sumoylation in mammalian cells (5). The absence of any global reductions in protein ubiquitinylation under the same conditions suggested that H2O2-induced formation of E1/E2-containing disulfide complexes did not involve ubiquitin pathway enzymes (5). However, in contrast to the sumoylation pathway, which involves only one E2 enzyme, many E2s are utilized for ubiquitinylation, each targeting specific substrates (18). Thus, if only a few ubiquitin pathway E2 enzymes are more sensitive to oxidation, it is possible that the effects of inhibition of these enzymes on global ubiquitinylation would be masked by the influence of the majority of less-sensitive E2 enzymes.
Here, we have utilized the tractability of the model eukaryote S. cerevisiae to investigate the sensitivity of ubiquitin pathway enzymes to oxidation in response to H2O2 and a drug that oxidizes the antioxidant glutathione. Significantly, our data indicate that the differential sensitivity of ubiquitin pathway E2 enzymes to such oxidation is utilized to allow specific inhibition of an essential E2, Cdc34. Furthermore, our results suggest that the specific sensitivity of Cdc34 to nonlethal levels of oxidative stress leads to the stabilization of at least one Cdc34 substrate, the CDK inhibitor Sic1, and moreover, that this stabilization is associated with a transient delay in cell cycle progression. Together, these data suggest that oxidation of ubiquitin pathway enzymes is utilized to regulate cellular processes, such as the cell division cycle, in response to oxidative stress.
MATERIALS AND METHODS
Yeast strains and growth conditions.
The yeast strains used were derived either from W303-1a (MATa ade2-1 can1-100 his3-11 leu2-3,112 trp1-1 ura3) (36) or from BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) (TAP-tagged strains) (11). The W303-1a strains constructed were KD6 (MATa sic1::HIS3), KD79 (MATa CDC34-13Myc kanR), KD102 (MATa UBA1-3HA kanR), KD103 (MATa CDC34-13Myc kanR UBA1-3HA kanR), KD119 (MATa SIC1-13Myc kanR CDC34-13Myc kanR), and KD130 (MATa UBC1-13Myc kanR). Strains were normally grown at 30°C using yeast extract-peptone-dextrose (YPD) medium (35). For Gsh1 inhibition, cells were grown in synthetic dextrose (SD) medium with buthionine sulfoximine (BSO; Sigma) (14) added to a final concentration of 5 mM.
Yeast sensitivity tests.
Sensitivities to oxidative stress conditions were tested by using a 48-pronged replica plater instrument (Sigma) to spot 10-fold serial dilutions of exponentially growing cells onto YPD plates containing oxidizing agents as indicated in the figures.
Yeast strain construction.
Cdc34, Sic1, Uba1, and Ubc1 expressed from their normal chromosomal locus were epitope tagged at the C terminus by integration of a PCR-amplified cassette obtained using primer pair Cdc34tagF/Cdc34tagR, Sic1tagF/Sic1tagR, Uba1TagF/Uba1TagR, or Ubc1TagF/Ubc1TagR, respectively, with either pFA6a-13Myc-kanMX6 or pFA6a-3HA-kanMX6 as the template (24). Correct integration was confirmed by PCR using primers flanking the region of interest. The SIC1 gene was replaced by HIS3 by introduction of a PCR cassette obtained using the primer pair Sic1KOF/Sic1KOR, with the YDp-H plasmid as the template (3). Gene deletion was confirmed by PCR. The sequences of the oligonucleotide primers are listed in Table S1 in the supplemental material.
Plasmid construction.
pUba1-3HA was constructed by ligating an EcoRI-digested PCR product containing UBA1-3HA, generated using primers Uba1EcoRIF and Adh1tEcoRIR and KD102 genomic DNA as the template, into YEplac112 (12) digested with EcoRI. pUba1C600S-3HA was constructed by first producing two PCR products by using primer pairs Uba1EcoRIF/Uba1MutCR and Uba1MutCF/Adh1tEcoRIR with KD102 genomic DNA as the template. Overlapping PCR with these two PCR products created a cassette which was then digested with EcoRI and ligated into YEplac112 digested with EcoRI. The plasmid pHA-Ubiquitin was constructed by ligating a BamHI-digested PCR product, containing the sequence encoding 3 hemagglutinin (HA) epitope tags, 6 His residues, and ubiquitin and generated using primers BamH13HA6HISF and BamH1UbR with the plasmid pREP-6His-Ubiquitin as the template (gift from Caroline Wilkinson), into p416-MET25 (28) digested with BamHI. The sequences of the oligonucleotide primers are listed in Table S1 in the supplemental material.
Cell synchronization and cell cycle analyses.
Exponentially growing cells were blocked in G1 phase by adding 15 μg/ml α-factor for 1 h and then a further 5 μg/ml α factor for 1 h. Cells were released from the block by washing with prewarmed YPD medium. For DNA content analysis, 4 × 106 to 6 × 106 cells/ml were stained with propidium iodide (7) and a FACScan flow cytometer (Becton, Dickinson) was used to measure DNA content.
Hog1 phosphorylation assay.
Cells were lysed in ice-cold protein lysis solution (20 mM HEPES, pH 7.3, 350 mM NaCl, 10% glycerol, 0.1% Tween 20, 0.097 trypsin inhibitor unit/ml aprotinin, 2 μg/ml leupeptin, 10 mM β-mercaptoethanol, 2 μg/ml pepstatin A, 1 mM NaF, 2 mM Na3VO4) with ice-cold glass beads using a Mini-Beadbeater (Biospec Products). Protein concentrations were measured using the Bradford assay (Pierce). For Western blotting, proteins were separated by SDS-PAGE (see below) and transferred to Protan nitrocellulose membrane using a Bio-Rad mini-transfer apparatus, and the membrane was incubated with either anti-Skn7 antibody (27) or anti-phospho-p38 antibody (New England BioLabs). After incubation with secondary anti-rabbit horseradish peroxidase (HRP)-conjugated antibody (Sigma), proteins were visualized using the chemiluminescence detection system (Amersham Pharmacia Biotech).
Protein extraction, immunoprecipitation, and Western blotting.
Cells were lysed in nonreducing conditions (10% trichloroacetic acid) with ice-cold glass beads using a Mini-Beadbeater (Biospec Products), and the insoluble protein pellet washed with ice-cold acetone. Proteins were solubilized in resuspension buffer (100 mM Tris-HCl, pH 8, 1% SDS, 1 mM EDTA) and concentrations measured using the bicinchoninic acid (BCA) protein assay kit (Thermo Scientific). For the analysis of ubiquitin conjugates and for immunoprecipitation experiments, cells were lysed in ice-cold lysis solution (20 mM HEPES, pH 7.3, 350 mM NaCl, 10% glycerol, 0.1% Tween 20, 0.097 trypsin inhibitor unit/ml aprotinin, 2 μg/ml leupeptin, 2 μg/ml pepstatin A, 1 mM NaF, 2 mM Na3VO4) with ice-cold glass beads using a Mini-Beadbeater (Biospec Products). Protein concentrations were measured using the Bradford assay (Pierce). Proteins were immunoprecipitated by incubating extracts for 1 h at 4°C with agarose beads coupled with either anti-HA or anti-Myc antibody.
For Western blotting, proteins were separated by SDS-PAGE; typically, 10% SDS–PAGE gels were used to analyze low-molecular-weight (LMW) proteins and 6% SDS–PAGE gels to resolve high-molecular-weight (HMW) proteins/complexes. Prestained Fermentas standard protein markers (for LMW proteins) and Invitrogen's HiMark HMW protein standard (for HMW proteins/complexes) were included as appropriate to allow estimation of molecular weights. Following separation, proteins were transferred to Protan nitrocellulose membrane using a Bio-Rad mini-transfer apparatus and the membrane incubated with the appropriate primary antibody: anti-myc 9E10 (Sigma), anti-HA (Sigma), anti-CBP (Source BioScience LifeSciences), anti-Hog1 (Santa Cruz Biotechnology), or anti-Skn7 (27) antibody. After incubation with the relevant secondary antibody (either anti-mouse HRP-conjugated antibody [Sigma] or anti-rabbit HRP-conjugated antibody [Sigma]), proteins were visualized using the chemiluminescence detection system (Amersham Pharmacia Biotech).
RESULTS
Oxidative stress stimulates oxidation of specific ubiquitin pathway enzymes.
In yeast, a single E1 enzyme, Uba1, transfers ubiquitin to multiple E2 enzymes which, together with E3s, conjugate ubiquitin to specific target proteins (Fig. 1) (17, 18, 21). It has previously been shown that the catalytic cysteine residues of the E1 and E2 enzymes in the sumoylation pathway, Uba2 and Ubc9, respectively, are sensitive to oxidation in mammalian cells by a concentration of H2O2 that causes no reduction in overall levels of general protein ubiquitinylation (5). However, it was possible that only a subset of the many E2 enzymes in the ubiquitin pathway might be sensitive to oxidation. To test this hypothesis, we first examined whether Uba1 becomes oxidized in response to H2O2 by using yeast cells expressing HA epitope-tagged Uba1 (Uba1-HA) from the normal chromosomal locus (Fig. 2A). Although only a relatively small proportion of Uba1-HA was oxidized by H2O2, significantly, Western blot analysis revealed an HMW Uba1-containing complex when cells were exposed to increasing concentrations of H2O2 for 20 min (Fig. 2A, asterisk). The β-mercaptoethanol sensitivity of this complex is consistent with it containing a disulfide (Fig. 2C). The previous study in mammalian cells suggested that glutathione is involved in the reduction of the oxidized E1-E2 mixed disulfide in the sumoylation pathway (5). Hence, we also examined the effects of a drug that oxidizes glutathione, the thiol-oxidizing agent diamide (22), on Uba1 oxidation. Similar to H2O2, a relatively small proportion of Uba1 was detected in an HMW disulfide complex after treatment with diamide for 30 min (Fig. 2B and C, asterisk). Taken together, the stimulation of a predominant, HMW, Uba1-containing disulfide complex by H2O2 and diamide suggested that perhaps one specific ubiquitin pathway E2 enzyme is more sensitive to oxidation.
Fig 2.

Uba1 and Cdc34 form HMW complexes in response to oxidative stress conditions which do not decrease global levels of protein ubiquitinylation. (A to C) Western blot analyses detected an HMW Uba1-containing disulfide complex (*) formed in response to diamide or H2O2. Extracts were prepared under nonreducing conditions from cells (KD102) expressing 3HA epitope-tagged Uba1 (Uba1-HA) from the normal chromosomal locus that were either untreated or treated with the indicated concentrations of H2O2 or diamide (A and B) or 5 mM diamide or 2 mM H2O2 (C). In the experiment whose results are shown in panel C, samples were prepared with or without β-mercaptoethanol treatment (βm) prior to loading. (D) Western blot analyses indicated that Cdc34 but not other abundant ubiquitin pathway E2 enzymes forms a predominant HMW complex (*) after diamide treatment. Extracts were prepared under nonreducing conditions from BY4741-derived cells expressing TAP-tagged Ubc1, Ubc2/Rad6, Ubc3/Cdc34, Ubc4, Ubc6, or Ubc13 from their normal chromosomal locus; the cells were untreated or treated with 5 mM diamide. The mobility of each individual TAP-tagged E2 enzyme is indicated by an arrow and the labeling at the top of each panel. (E to G) Western blot analyses revealed that the levels of global protein ubiquitinylation are not decreased by treatment of cells with diamide or H2O2. (E) Protein-ubiquitin conjugates in cells expressing HA epitope-tagged ubiquitin were detected as a large collection of HMW bands. Extracts were prepared from wild-type (W303-1a) cells containing either p416-MET25 (vector) or pHA-Ubiquitin, expressing ubiquitin tagged with 3HA epitopes. (F and G) Extracts were prepared from wild-type (W303-1a) cells containing pHA-Ubiquitin treated with the indicated concentrations of H2O2 (F) or diamide (G). (E to G) Protein-ubiquitin conjugates and a band of the approximate size of epitope-tagged ubiquitin (HA-Ubi) are indicated. An example of ubiquitinylated protein whose levels were observed to decrease following exposure to oxidative stress (a) is indicated. (A to D, F, and G) Cells were treated with either diamide for 30 min or H2O2 for 20 min. (A to G) Western blots were probed with anti-HA (A to C and E to G), anti-CBP (D), and anti-Skn7 (loading control) (F and G) antibodies. Molecular masses (in kilodaltons) are shown to the left of panels.
Previous studies have shown that there are many E2 enzymes in the ubiquitin pathway in S. cerevisiae (Fig. 1), and 10 of these have been tagged with a tandem affinity purification (TAP) tag and expressed from their normal chromosomal locus (11). Several of these E2 enzymes are at very low abundance within cells (11), but we were able to detect the more-abundant tagged Ubc1, Ubc2/Rad6, Ubc3/Cdc34, Ubc4, Ubc6, and Ubc13 by Western blot analyses (Fig. 2D). Hence, to investigate whether any of these ubiquitin pathway E2 enzymes form an HMW complex in response to oxidative stress, cells expressing these individually TAP-tagged E2s were treated with 5 mM diamide, a concentration of diamide that stimulates formation of the Uba1 HMW disulfide complex (Fig. 2B). Excitingly, consistent with our hypothesis that a specific E2(s) is susceptible to oxidation, an HMW band was detected only in the lane containing Cdc34 after diamide treatment (Fig. 2D).
Consistent with the observation that only one ubiquitin pathway E2 enzyme formed an HMW complex, together with the finding that only a small proportion of Uba1 is oxidized, treatment of cells expressing HA epitope-tagged ubiquitin (Fig. 2E) with levels of H2O2 and diamide which lead to the formation of HMW Uba1 and Cdc34 complexes was not associated with a general decrease in the overall levels of ubiquitinylated proteins (Fig. 2F and G). Indeed, the general overall level of ubiquitinylation detected appeared to increase with oxidative stress. However, interestingly, a reduction in some specific protein ubiquitinylation was detected at higher levels of oxidative stress (Fig. 2F).
Cdc34 is more sensitive to diamide- and H2O2-induced oxidation than other E2 enzymes.
The data presented above suggested that Cdc34, but not other ubiquitin pathway E2 enzymes, forms a disulfide complex with Uba1. To explore this hypothesis further and to rule out any potential effect of the strain background (the Uba1-HA strain was derived from W303, while the Cdc34-TAP strain was derived from BY4741), Cdc34 and another ubiquitin pathway E2 enzyme, Ubc1, which has a similar abundance in cells and which did not show any evidence of oxidation when TAP tagged (11) (Fig. 2D), were tagged with 13Myc epitopes (Cdc34-Myc and Ubc1-Myc) and expressed from their normal chromosomal locus in the W303 strain background. Significantly, consistent with our analyses of the TAP-tagged strains, Western blot analysis of extracts from cells expressing Cdc34-Myc revealed an HMW Cdc34-containing band after cells were treated with either diamide or H2O2 (Fig. 3A and B, asterisk). Moreover, as predicted by our analysis of TAP-tagged Ubc1, analysis of extracts from cells expressing Ubc1-Myc revealed that HMW Ubc1-containing bands were barely detectable even at the highest levels of diamide or H2O2 (Fig. 3A and B). The sensitivity of the HMW Cdc34-containing band (asterisk) to the reducing agents β-mercaptoethanol and Tris(2-carboxyethyl)phosphine (TCEP) indicated that it was a disulfide-containing complex (Fig. 3C and D). Hence, taken together, these data indicate that Cdc34 is more susceptible to oxidation than Ubc1 at lower levels of oxidative stress, forming an HMW complex in different strain backgrounds and using different tags.
Fig 3.

Cdc34 is more sensitive than Ubc1 to oxidation by oxidative stress. (A to F) Western blot analyses demonstrated that Cdc34 is more sensitive to oxidation than another abundant E2, Ubc1, forming a predominant HMW disulfide complex (*) after H2O2 and diamide treatment. (A and B) Western blot analyses of extracts prepared from cells expressing either Cdc34-Myc (KD79) or Ubc1-Myc (KD130) from their normal chromosomal locus; cells were treated with the indicated concentrations of either diamide for 30 min (A) or H2O2 for 20 min (B). HMW Cdc34-containing complexes (*) and HMW Ubc1-containing complexes formed under these stress conditions (* and **) are indicated. (C to E) Western blot analyses of samples not treated or treated prior to loading with either β-mercaptoethanol (βm) or TCEP revealed that the stress-induced HMW form of Cdc34 (*) is a disulfide complex and identified the E2∼ubiquitin thioester forms of Cdc34 (Cdc34-MycUb) and Ubc1 (Ubc1-MycUb). Extracts were prepared from cells expressing Cdc34-Myc (KD79) that were either not treated or treated with 5 mM diamide for 30 min (C) or 2 mM H2O2 for 20 min (C and D) or from untreated cells expressing Ubc1-Myc (KD130) (E). (F) Western blot analysis indicated that Cdc34∼ubiquitin thioester forms start to decrease with a timing similar to that of the formation of the HMW Cdc34-containing disulfide complex (*). Extracts were prepared from cells expressing Cdc34-Myc (KD79) that were treated with 2 mM H2O2 for the indicated times. (A to F) All extracts were prepared under nonreducing conditions, and proteins were separated by SDS-PAGE and analyzed by Western blotting with anti-Myc antibodies. Molecular masses (in kilodaltons) are shown to the left of panels. (A, B, and F) Lighter exposures are shown (bottom panels) to demonstrate the effects of diamide and H2O2 on the E2∼ubiquitin thioester forms.
E2∼ubiquitin thioester forms are an active intermediate in the transfer of ubiquitin to substrates (Fig. 1). We next examined whether oxidative stress affected the formation of these Cdc34∼ and Ubc1∼ubiquitin thioester forms (1, 34). In extracts from unstressed cells in the absence of β-mercaptoethanol, Cdc34-Myc and Ubc1-Myc were present in two bands and were also found to have a much slower mobility than expected (Fig. 3A and B). Consistent with the prediction that the upper of these two bands represents the Cdc34∼ and Ubc1∼ubiquitin thioester forms (1, 34), both upper bands were sensitive to β-mercaptoethanol, which can reduce thioester bonds, but not to TCEP, which can reduce disulfide but not thioester bonds (Fig. 3D [compare first and second lanes with fifth and sixth lanes] and E). Although the basis for the slower-than-expected mobilities of the reduced Cdc34-Myc and Ubc1-Myc proteins is unclear, the epitope tagging did not affect either the growth under normal conditions of cells expressing either of the epitope-tagged proteins or the sensitivity of the cells to stress conditions (see Fig. S1 in the supplemental material). Interestingly, although there was some decrease in the levels of Ubc1∼ubiquitin thioester forms at the highest concentration of H2O2 tested, a marked decrease in the levels of Cdc34∼ubiquitin thioester forms was observed at concentrations of both H2O2 and diamide (Fig. 3A to C) where no effect on Ubc1∼ubiquitin thioester forms was detected (Fig. 3A and B). Moreover, consistent with formation of the Cdc34-containing disulfide complex causing the H2O2-induced decrease in Cdc34∼ubiquitin thioester forms, both events occurred with similar timing (Fig. 3F, compare darker- and lighter-exposure panels). Collectively, these data suggest that Cdc34 is sensitive to oxidation, lowering the levels of active Cdc34∼ubiquitin thioester intermediates, at concentrations of diamide and H2O2 which do not inhibit Ubc1.
Uba1 and Cdc34 form an oxidative stress-induced disulfide complex.
H2O2 induces the formation of an intermolecular disulfide bond between the catalytic cysteines of the sumoylation pathway E1 and E2 enzymes in mammalian cells (5). Hence, we next determined whether the oxidized forms of Uba1 and Cdc34 were also E1-E2 disulfides and, if so, whether this involved their catalytic cysteines. To facilitate this, cells expressing different combinations of untagged and epitope-tagged Uba1 and Cdc34 from their normal chromosomal locus were exposed to either diamide or H2O2 and the resulting disulfide complexes analyzed. The decreased mobility of the HMW Uba1-HA-containing complex in cells expressing Cdc34-Myc instead of untagged Cdc34 suggested that these HMW complexes did indeed contain both proteins (Fig. 4A and B, compare lanes 2 and 3 in panel A with panel B). Furthermore, using either anti-HA or anti-Myc antibodies, an HMW complex with the same mobility was detected in cells expressing both Uba1-HA and Cdc34-Myc (Fig. 4A, compare lanes 3 and 4). However, importantly, these conclusions were confirmed by Western blot analyses of extracts from H2O2-treated cells expressing both Uba1-HA and Cdc34-Myc, which revealed that stress-induced HMW Uba1-HA-containing complexes were enriched by immunoprecipitation with anti-Myc antibodies (Fig. 4C, rightmost lane) and, similarly, HMW Cdc34-Myc-containing complexes were enriched by immunoprecipitation with anti-HA antibodies (Fig. 4D, rightmost lane). Collectively, these data are consistent with the formation of oxidative stress-induced mixed disulfide complexes between Uba1 and Cdc34.
Fig 4.

Uba1 forms a disulfide complex with Cdc34. (A to D) Western blot analyses revealed that Uba1 and Cdc34 are components of the HMW disulfide complex induced by H2O2 and diamide. Extracts were prepared from cells expressing either Cdc34-Myc (KD79), Uba1-HA (KD102), or both Cdc34-Myc and Uba1-HA (KD103) from their normal chromosomal locus as indicated; cells were either not treated (C and D) or treated with 5 mM diamide for 30 min (A) or 2 mM H2O2 for 20 min (B to D). (B) Immunoprecipitated proteins were analyzed. (C and D) Whole-cell lysate (WCL) and immunopreciptiated were analyzed. (A to D) Proteins were prepared as described in Materials and Methods and separated by nonreducing SDS-PAGE. Antibodies used for immunoprecipitation (IP) or Western blotting (WB) were either anti-Myc or anti-HA antibodies as indicated. Cdc34-Myc, Uba1-HA, and HMW complexes containing both Cdc34 and Uba1 are indicated (arrows). Molecular masses (in kilodaltons) are shown to the left of panels. (C and D) For comparison, WCL equivalent to 7% (C) and 12% (D) of the lysates used for IP were analyzed.
Having established that both Uba1 and Cdc34 are present in this HMW disulfide complex, we next investigated the role of the catalytic cysteines in the formation of this complex. The activities of Cdc34 and Uba1 are essential, which prevented the construction of viable strains expressing mutant versions lacking their catalytic cysteine residues, Cys600 of Uba1 (16, 25) and Cys95 of Cdc34 (13). However, Cdc34 contains a single cysteine residue and, hence, the HMW Cdc34/Uba1 disulfide complex must involve this catalytic cysteine. Uba1 contains nine cysteine residues (and one cysteine residue in the 3HA epitopes) in addition to the catalytic Cys600. To determine whether Cys600 of Uba1 was involved in disulfide formation with Cdc34, we tested the ability of plasmid-expressed, HA epitope-tagged, wild-type Uba1-HA and the mutant Uba1C600S-HA (where Cys600 replaced with serine did not affect the stability of Uba1) (Fig. 5A) to compete with chromosomally expressed untagged wild-type Uba1 for disulfide complex formation with chromosomally expressed Cdc34-Myc. Importantly, Western blot analysis of extracts from H2O2-treated cells revealed that stress-induced HMW Cdc34-Myc-containing complexes were enriched by immunoprecipitation with anti-HA antibodies from cells expressing wild-type Uba1-HA but not from cells expressing mutant Uba1C600S-HA (Fig. 5B). Similarly, HMW complexes containing plasmid-expressed, wild-type Uba1-HA but not mutant Uba1C600S-HA were enriched by immunoprecipitation with anti-Myc antibodies (Fig. 5C). Thus, these data indicate that these oxidative stress conditions stimulate the formation of an HMW disulfide complex between the catalytic cysteine residues of Uba1 and Cdc34.
Fig 5.
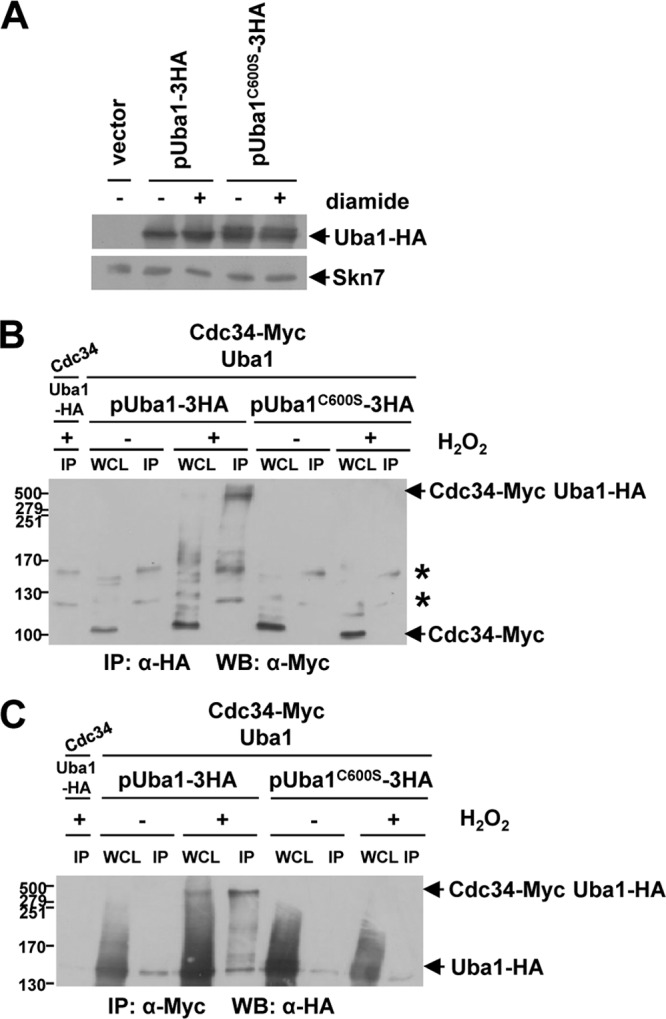
The catalytic cysteine residue of Uba1 is essential for disulfide complex formation with Cdc34. (A to C) Western blot analyses revealed that plasmid-expressed wild-type Uba1-HA but not Uba1C600S-HA can compete with chromosomally expressed untagged Uba1 for HMW complex formation with chromosomally expressed Cdc34-Myc. (A) Western blot analysis of extracts from wild-type (W303-1a) cells containing either YEplac112 (vector), pUba1-3HA, or pUba1C600S-3HA that were treated with 5 mM diamide for 0 (−) or 30 (+) min revealed that mutation of the catalytic cysteine residue, Cys600, of Uba1 did not decrease protein stability. Extracts were prepared under nonreducing conditions, and proteins were separated by nonreducing SDS-PAGE and analyzed with either anti-HA or anti-Skn7 (loading control) antibodies. (B and C) Immunoprecipitation (IP) and Western blot (WB) analyses of proteins extracted from cells (KD79) expressing Cdc34-Myc and untagged Uba1 from their normal chromosomal locus and containing either pUba1-3HA or pUba1C600S-3HA; cells were treated with 2 mM H2O2 for 0 (−) or 20 (+) min, and the results demonstrate that the catalytic cysteine residue of Uba1 (Cys600) is essential for HMW complex formation with Cdc34. Whole-cell lysate (WCL) and immunoprecipitated proteins, prepared as described in Materials and Methods, were separated by nonreducing SDS-PAGE and analyzed by Western blotting. Extracts from cells (KD102) expressing Uba1-HA and untagged Cdc34 from their normal chromosomal locus that were treated with 2 mM H2O2 for 20 min were included as a negative control (lane 1). The antibodies used for IP or WB were either anti-Myc or anti-HA antibody as indicated. (B) Two nonspecific bands (*) present in all IP lanes, including lane 1, which contains no Myc epitope-tagged proteins, are indicated. (A to C) Cdc34-Myc, Uba1-HA, Skn7, and HMW complexes containing both Cdc34-Myc and Uba1-HA are indicated (arrows). For comparison, WCL equivalent to 11% of the lysates used for IP were analyzed. Molecular masses (in kilodaltons) are shown to the left of panels.
Oxidative stress-induced cell cycle delay is associated with reversible oxidation of Cdc34.
The specificity of the ubiquitin pathway E2 enzyme Cdc34 to oxidation in response to oxidative stress (Fig. 2D and 3A and B) suggested that this may be important for the cellular response to this stress. Protein ubiquitinylation influences many fundamental cellular processes, including cell cycle progression. Furthermore, Cdc34 regulates the stability of many proteins via multiple E3s which determine substrate specificity (40). For example, the levels of one such Cdc34 substrate, the CDK inhibitor Sic1, vary during the cell cycle, peaking in early G1 phase where it inhibits Cdc28-Clb CDK complexes (9, 26, 33). Interestingly, exposure of an asynchronous culture of wild-type cells to diamide results in an accumulation of cells with a 1C (G1 phase) content of DNA (Fig. 6A). Different environmental stress conditions have been found to inhibit cell cycle progression. For example, recent work demonstrated that phosphorylation of Sic1 by the Hog1 stress-activated protein kinase, related to the mammalian Jun N-terminal protein kinase (JNK)/p38 stress kinases, is associated with increased stability of Sic1 and arrest in G1 phase of the cell cycle in response to osmotic stress (10). Hence, it was possible that Sic1 might play a role in the accumulation of cells in G1 phase in response to diamide. Indeed, in contrast to SIC1 wild-type cells, diamide-treated sic1Δ cells did not accumulate with a G1 phase content of DNA (Fig. 6A). Furthermore, consistent with an important role for Sic1 in maintaining viability under these conditions, sic1Δ cells were found to be more sensitive than SIC1 wild-type cells to diamide (Fig. 6B). However, in contrast to osmotic stress, we can find no evidence that diamide activates Hog1, suggesting that this Sic1 role is independent of regulation by Hog1 (Fig. 6C). To further examine the role of Sic1 in the response of cells to diamide, G1-phase-synchronized SIC1 and sic1Δ cells were treated with 2 mM diamide. Diamide-treated synchronized SIC1 wild-type cells were found to delay exit from G1 phase (Fig. 6D, compare first and second columns). However, in contrast, diamide induced a much shorter delay in cell cycle progression of sic1Δ cells than of SIC1 wild-type cells (Fig. 6D, compare first and second columns with third and fourth columns), further supporting a role for Sic1 in the response of cells to this oxidizing agent. These data suggested that Sic1 may be stabilized under different oxidative stress conditions. To examine this possibility, the stability of Myc epitope-tagged Sic1 (Sic1-Myc) expressed from the normal chromosomal locus was examined in G1-phase-synchronized cells in the presence or absence of 2 mM diamide. As expected, the levels of Sic1-Myc decreased rapidly as untreated synchronized cells progressed from G1 phase (Fig. 6E, left, compare the 20- and 40-min time points with the DNA content analyses below). However, Sic1-Myc persisted much longer in diamide-treated cells and, moreover, this is associated with the timing of the diamide-induced cell cycle delay (Fig. 6E, right, compare the time points with the DNA content analyses below). G1-phase-synchronized cells expressing Sic1-Myc were also treated with 0.5 mM H2O2 and, similar to diamide treatment, this was also found to delay exit from G1 phase compared with that of untreated cells (Fig. 6F, DNA content analyses). Moreover, Sic1-Myc persisted much longer in the H2O2-treated cells and was associated with the timing of the cell cycle delay (Fig. 6F). The levels of diamide (2 mM) and H2O2 (0.5 mM) used here that affect the levels of Sic1-Myc also oxidize Cdc34 (Fig. 3A and B) but do not decrease global ubiquitinylation (Fig. 2F and G). Hence, it was possible that the timing of the formation of the Uba1-Cdc34 disulfide complex may be linked to this oxidative stress-induced inhibition of cell cycle progression by influencing the ubiquitinylation/stability of downstream substrates, such as Sic1. Indeed, consistent with this model, we observed oxidation of Cdc34 at the first time point after the treatment of G1-phase-synchronized cells with diamide or H2O2 and, moreover, the timing of the disappearance of this Cdc34 oxidation is associated with the persistence of Sic1 and with the diamide- and H2O2-induced cell cycle delay (Fig. 6E and F, right, compare the time points with the DNA content analyses below). Although we cannot rule out the potential role of other mechanisms of regulation of Sic1 and/or of the potential role of regulation of other Cdc34 substrates in the cell cycle delay, the results are consistent with a model whereby the oxidation of Cdc34 influences the oxidant-induced cell cycle delay and adaptation to specific oxidative stress.
Fig 6.
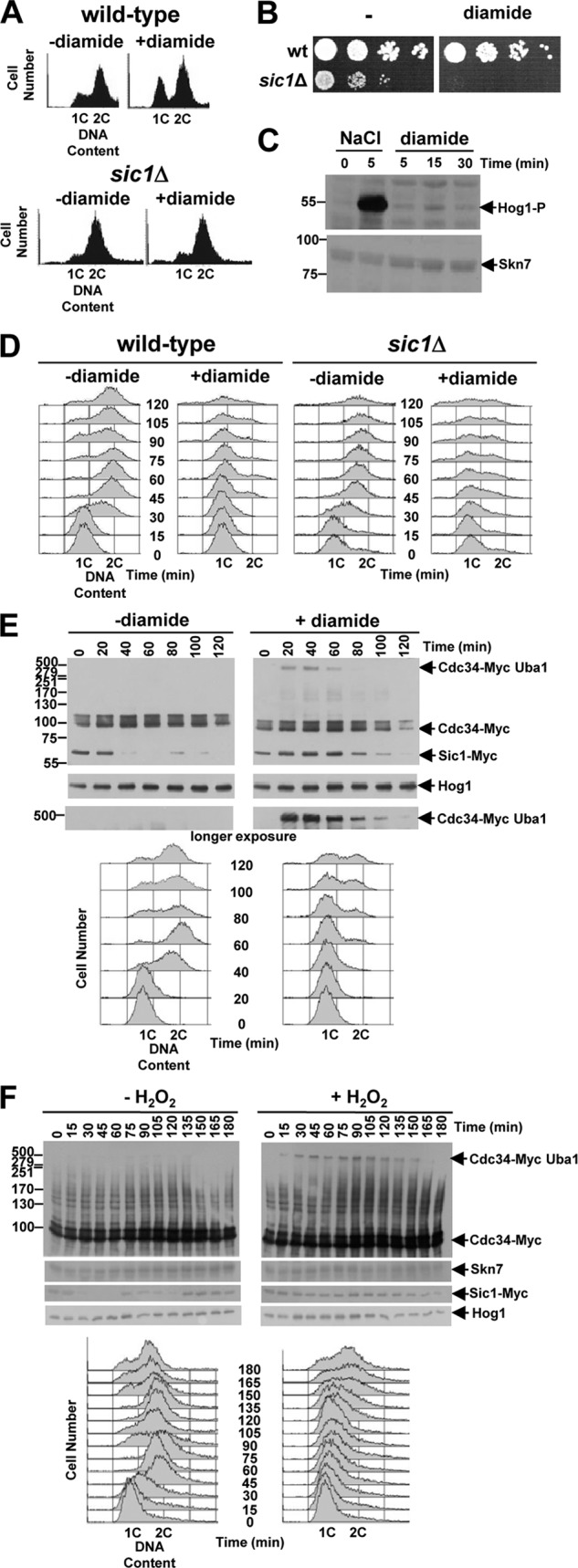
Oxidation of Cdc34 is associated with delays of cell cycle progression and stabilization of the Cdc34 substrate Sic1. (A to D) Sic1 is required for the response of cells to diamide. (A) DNA content analyses of asynchronous cultures of SIC1 wild-type (W303-1a) and sic1Δ (KD6) cells not treated or treated with 5 mM diamide for 2 h revealed that Sic1 is required for the accumulation of cells in G1 phase in response to diamide. (B) Cells lacking Sic1 display increased sensitivity to diamide. Tenfold serial dilutions of SIC1 wild-type (wt) cells (W303-1a) and cells lacking Sic1 (sic1Δ) (KD6) were spotted onto either YPD medium or YPD medium containing 1.75 mM diamide. Plates were incubated at 30°C for 2 days. (C) The Hog1 pathway is not activated by diamide-induced oxidative stress. Western blot analysis of protein extracts prepared from wild-type (W303-1a) cells treated with either 0.4 M NaCl or 1 mM diamide for the indicated times revealed that, in contrast to NaCl treatment, diamide treatment does not stimulate phosphorylation of Hog1. Anti-phospho-p38 antibodies were used to detect phosphorylation of Hog1 (Hog1-P), and anti-Skn7 (Skn7) antibodies were used to confirm loading. (D) Sic1 is essential for delay in G1 phase in response to diamide. DNA content analyses of SIC1 wild-type (W303-1a) and sic1Δ (KD6) cells synchronized in G1 phase with α-factor and then released into medium with or without 2 mM diamide is shown. (E and F) Western blot and DNA content analyses of cells expressing Cdc34-Myc and Sic1-Myc from their normal chromosomal locus that were synchronized in G1 phase with α-factor and then released into medium either with or without 2 mM diamide (E) or with or without 0.5 mM H2O2 (F) revealed diamide- and H2O2-induced formation of the HMW Cdc34-Myc-containing complex, delay of destabilization of Sic1-Myc, and transient delay of cell cycle progression. Bands with the expected mobilities of Cdc34-Myc, Sic1-Myc, and the Cdc34-Myc- and Uba1-containing HMW disulfide complex are indicated (arrows). Anti-Hog1 (Hog1) and anti-Skn7 (Skn7) antibodies confirmed loading. DNA content analyses performed at the indicated times on the cells used in the Western blot analyses revealed a diamide- and H2O2-induced delay in cell cycle progression that corresponded with the formation of the HMW Cdc34-Myc-containing complex and the persistence of Sic1-Myc. (C, E, and F) Molecular masses (in kilodaltons) are shown to the left of panels.
DISCUSSION
Here, we show that different E2 enzymes in the ubiquitin pathway are differentially sensitive to inactivation by oxidative stress. Moreover, our data suggest that the increased sensitivity of the E2 Cdc34 to inhibition by reversible oxidation is employed as a mechanism to regulate cellular responses to oxidative stress. We find that levels of diamide and H2O2 which do not decrease global levels of protein ubiquitinylation or affect the oxidation of many ubiquitin pathway E2 enzymes stimulate disulfide bond formation between the catalytic cysteines of the E2 enzyme Cdc34 and the E1 enzyme Uba1. Furthermore, oxidation of Cdc34 is associated with decreased levels of Cdc34∼ubiquitin thioester forms, delayed destabilization of at least one Cdc34 substrate, the CDK inhibitor Sic1, and a transient Sic1-dependent cell cycle delay.
Previous work in mammalian cells revealed that H2O2 induced the formation of a disulfide complex involving the catalytic cysteine residues of the E1 and E2 enzymes in the sumoylation pathway and, moreover, that this was associated with reduced levels of global sumoylation but not ubiquitinylation (5). Here, in agreement with the previous study, H2O2 treatment did not reduce global levels of ubiquitinylated proteins in S. cerevisiae (5) (Fig. 2F). However, we found that Cdc34 oxidation is associated with reduced levels of Cdc34∼ubiquitin thioesters and increased stability of Sic1, a Cdc34 substrate. Indeed, consistent with this effect, Uba1 displays specificity in the formation of disulfide complexes with one E2 enzyme, Cdc34, and, moreover, Cdc34 is more sensitive to oxidation than other ubiquitin pathway E2 enzymes, including those which have a cellular abundance similar to (Ubc1 and Ubc13) or higher than (Ubc4) that of Cdc34 (11) (Fig. 2D and 3A and B). The basis for the increased sensitivity of Cdc34 to oxidation is unknown. It seems unlikely that the relative sensitivity of each E2 enzyme to oxidation is linked to its cellular location. Indeed, Cdc34 appears to have a cellular distribution similar to that of many of the other E2 enzymes which do not display increased sensitivity (19). However, one possibility is that the catalytic cysteine residue of Cdc34 is inherently more sensitive to oxidation than those of the other E2 enzymes. The E1-E2 disulfide complex in the sumoylation pathway is also influenced by glutathione (5). Similarly, we find that both diamide, a thiol-oxidizing agent which oxidizes glutathione, and buthionine sulfoximine (BSO) (14), an inhibitor of gamma glutamylcysteine synthetase, the first enzyme in the glutathione synthesis pathway (encoded by GSH1 in S. cerevisiae), induce the formation of the Uba1-Cdc34 disulfide complex (Fig. 2 to 4 and 7). Moreover, the addition of reduced glutathione reverses the effects of BSO on Uba1-Cdc34 oxidation (Fig. 7, compare lanes 5 and 7). Thus, collectively, these results suggest that glutathione influences the formation of the Uba1-Cdc34 disulfide complex and the ubiquitinylation of downstream substrates of Cdc34.
Fig 7.
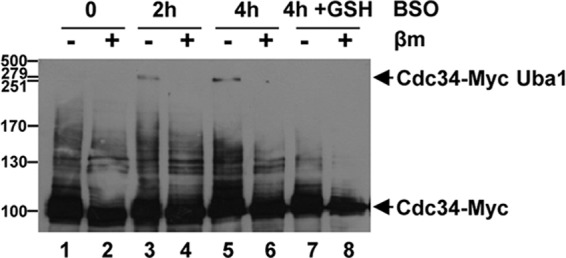
Inhibition of Gsh1 by BSO stimulates formation of the Uba1-Cdc34 disulfide complex. Western blot analysis of extracts prepared under nonreducing conditions from cells (KD79) expressing Cdc34-Myc revealed that treatment with 5 mM buthionine sulfoximine (BSO), an inhibitor of gamma glutamylcysteine synthetase (Gsh1 in S. cerevisiae), for 0, 2 and 4 h stimulated the formation of the Cdc34-Myc- and Uba1-containing HMW disulfide complexes (lanes 1 to 6). Lanes 7 and 8 contain extracts from cells that were exposed to BSO for 4 h (equivalent to lanes 5 and 6), except that 1 mM reduced glutathione (+GSH) was added 2 h after the addition of BSO. Thus, comparison of lanes 5 and 7 indicates that reduced GSH inhibits the formation of the disulfide complex. Proteins were separated by SDS-PAGE (with or without β-mercaptoethanol [βm] treatment prior to loading) and analyzed with anti-Myc antibodies. Cdc34-Myc and the Cdc34-Myc- and Uba1-containing HMW disulfide complexes are indicated (arrows). Molecular masses (in kilodaltons) are shown to the left.
Cdc34 is evolutionarily conserved between yeast and mammals (31) and is a potential oncogene in humans (32). In S. cerevisiae, Cdc34 regulates cell cycle progression through the ubiquitinylation of proteins such as the CDK inhibitor Sic1 (40) (Fig. 6). Sic1 is regulated by multiple mechanisms in the cell division cycle and in response to stress (10, 29, 39, 40). Here, we demonstrate that Sic1 is essential for viability when cells are exposed to diamide (Fig. 6B) and, moreover, that diamide induces a delay in early cell cycle progression by a Sic1-dependent mechanism (Fig. 6A and D). However, in contrast to osmotic stress, we find no evidence that diamide activates the Hog1 stress-activated protein kinase, suggesting that these diamide-induced cell cycle inhibitory mechanisms are Hog1 independent (Fig. 6C). Instead, our results suggest that Cdc34 oxidation contributes to the increased stabilization of Sic1 (Fig. 6E), which is at least one Cdc34 target that is important for the response of cells to diamide. We also find that Sic1 is stabilized in response to H2O2 and, significantly, that this corresponds to the timing of H2O2-induced cell cycle delay and Cdc34 oxidation (Fig. 6F). Although we cannot rule out the influence of other diamide- and/or H2O2-induced regulatory mechanisms on Sic1 stability or of the effects of the oxidation of Cdc34 on the ubiquitinylation of other substrates, our data suggest that the regulation of Sic1 by diamide and H2O2 is linked to Cdc34 oxidation-dependent inhibition of ubiquitinylation and, hence, degradation of Sic1 (Fig. 6). Interestingly, our analysis of sic1Δ cells also suggests the existence of an additional Sic1-independent mechanism(s) for oxidant-induced cell cycle delay (Fig. 6D), in which it is possible that other targets of Cdc34-directed ubiquitinylation could be involved. In our model, the cellular response to specific oxidative stresses involves the induced oxidation of one specific ubiquitin pathway E2 enzyme, Cdc34, blocking the catalytic cysteine residue, which as a consequence contributes to the inhibition of ubiquitinylation of a subset of proteins important for adaptation to the conditions (Fig. 8).
Fig 8.
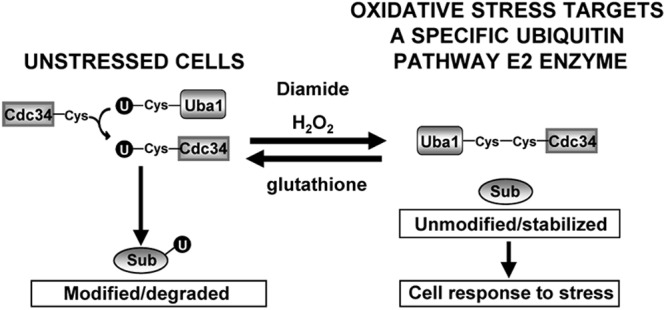
Model for the regulation of cellular responses to oxidative stress by Cdc34 oxidation. In our model, certain oxidative stress conditions promote thiol oxidation of a specific ubiquitin pathway E2 enzyme, Cdc34, which inhibits ubiquitinylation of downstream substrates (Sub). This inhibition results in cellular responses to oxidative stress, including, for example, a delay in cell cycle progression. In the model, reduced glutathione is coupled to Uba1 and Cdc34 oxidation via a negative feedback loop that reverses their oxidation, restoring normal regulation of downstream substrates when redox conditions have improved.
In addition to our studies in yeast and the study of the sumoylation pathway in mammalian cells (5), there is some evidence of disulfide complex formation in the ubiquitinylation (involving E1 enzyme) and neddylation (involving E2 enzyme) pathways in mammalian cells, although it is not clear in either case whether these disulfides are in fact E1-E2 disulfides (23, 30). Nevertheless, taken together, our work and that of others suggests that a key element in the regulation of specific cellular responses to ROS involves the oxidation of enzymes of several ubiquitin and Ubl modification pathways. Ubiquitin and Ubl modification play key roles in regulating diverse, fundamental cellular pathways. However, in contrast to other Ubl pathways, the ubiquitin pathway utilizes many E2 and E3 enzymes to determine target protein specificity (Fig. 1). The regulation of ubiquitinylation involves several mechanisms, including phosphorylation of individual substrates and/or enzymes in the ubiquitin pathway (20). Here, we have identified a mechanism by which the ubiquitinylation of specific substrates can be regulated by ROS that involves the relative sensitivities of ubiquitin pathway E2 enzymes to oxidation and inhibition.
ROS are generated as signaling effector molecules to regulate a multitude of processes in mammals and plants. The sensitivity of particular phosphotyrosine protein phosphatases to reversible inhibition by H2O2-induced oxidation has become established as a key mechanism mediating H2O2 signal transduction through changes in protein phosphorylation. Our data suggest that oxidative stress regulates cell cycle progression in yeast by directly inhibiting the activity of a specific ubiquitin pathway E2 enzyme and, thus, the ubiquitinylation of specific substrate(s). This raises the possibility that the relative sensitivities of ubiquitin pathway E2 enzymes to oxidation may also be important in mediating specific ROS signal transduction to regulate other cellular responses. For instance, it is possible that the inhibition of one E2 enzyme may also drive ubiquitin through other E2 enzyme(s). Indeed, our analysis of global ubiquitinylation suggests that certain proteins become increasingly ubiquitinylated in response to increasing concentrations of H2O2 (Fig. 2F). Determining the relative sensitivities to oxidation of particular ubiquitin pathway E2 enzymes in eukaryotes will be key to understanding the significance of redox-regulatory mechanisms in the control of protein ubiquitinylation.
Supplementary Material
ACKNOWLEDGMENTS
We thank Elizabeth Veal for many discussions, Marc Flanagan, Janet Quinn, Elizabeth Veal, and Caroline Wilkinson for comments on the manuscript, Chris Grant, David Lydall, and Caroline Wilkinson for reagents and strains, and Michelle Wray for technical assistance.
The research was supported by the Medical Research Council (K.S.D. and E.L.R.), the National Institute for Health Research (NIHR) Newcastle Biomedical Research Centre based at Newcastle upon Tyne Hospitals NHS Foundation Trust and Newcastle University (E.L.R.), and the Faculty of Medical Sciences Newcastle University (K.S.D. and B.A.M.).
The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, or the Department of Health.
Footnotes
Published ahead of print 4 September 2012
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1. Banerjee A, Gregori L, Xu Y, Chau V. 1993. The bacterially expressed yeast CDC34 gene product can undergo autoubiquitination to form a multiubiquitin chain-linked protein. J. Biol. Chem. 268: 5668–5675 [PubMed] [Google Scholar]
- 2. Beckman KB, Ames BN. 1997. Oxidative decay of DNA. J. Biol. Chem. 272: 19633–19636 [DOI] [PubMed] [Google Scholar]
- 3. Berben G, Dumont J, Gilliquet V, Bolle PA, Hilger F. 1991. The YDp plasmids: a uniform set of vectors bearing versatile gene disruption cassettes for Saccharomyces cerevisiae. Yeast 7: 475–477 [DOI] [PubMed] [Google Scholar]
- 4. Berlett BS, Stadtman ER. 1997. Protein oxidation in aging, disease, and oxidative stress. J. Biol. Chem. 272: 20313–20316 [DOI] [PubMed] [Google Scholar]
- 5. Bossis G, Melchior F. 2006. Regulation of SUMOylation by reversible oxidation of SUMO conjugating enzymes. Mol. Cell 21: 349–357 [DOI] [PubMed] [Google Scholar]
- 6. Cho SH, et al. 2004. Redox regulation of PTEN and protein tyrosine phosphatases in H2O2-mediated cell signaling. FEBS Lett. 560: 7–13 [DOI] [PubMed] [Google Scholar]
- 7. Corliss DA, White WE. 1981. Fluorescence of yeast vitally stained with ethidium bromide and propidium iodide. J. Histochem. Cytochem. 29: 45–48 [DOI] [PubMed] [Google Scholar]
- 8. D'Autréaux B, Toledano MB. 2007. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 8: 813–824 [DOI] [PubMed] [Google Scholar]
- 9. Donovan JD, Toyn JH, Johnson AL, Johnston LH. 1994. P40SDB25, a putative CDK inhibitor, has a role in the M/G1 transition in Saccharomyces cerevisiae. Genes Dev. 8: 1640–1653 [DOI] [PubMed] [Google Scholar]
- 10. Escoté X, Zapater M, Clotet J, Posas F. 2004. Hog1 mediates cell-cycle arrest in G1 phase by the dual targeting of Sic1. Nat. Cell Biol. 6: 997–1002 [DOI] [PubMed] [Google Scholar]
- 11. Ghaemmaghami S, et al. 2003. Global analysis of protein expression in yeast. Nature 425: 737–741 [DOI] [PubMed] [Google Scholar]
- 12. Gietz RD, Sugino A. 1988. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74: 527–534 [DOI] [PubMed] [Google Scholar]
- 13. Goebl MG, et al. 1988. The yeast cell cycle gene CDC34 encodes a ubiquitin-conjugating enzyme. Science 241: 1331–1335 [DOI] [PubMed] [Google Scholar]
- 14. Griffith OW, Meister A. 1979. Potent and specific inhibition of glutathione synthesis by buthionine sulfoximine (S-n-butyl homocysteine sulfoximine). J. Biol. Chem. 254: 7558–7560 [PubMed] [Google Scholar]
- 15. Halliwell B, Gutteridge JMC. 1999. Free radicals in biology and medicine. Oxford University Press, Oxford, United Kingdom [Google Scholar]
- 16. Hatfield PM, Vierstra RD. 1992. Multiple forms of ubiquitin-activating enzyme e1 from wheat: identification of an essential cysteine by in vitro mutagenesis. J. Biol. Chem. 267: 14799–14803 [PubMed] [Google Scholar]
- 17. Herrmann J, Lerman LO, Lerman A. 2007. Ubiquitin and ubiquitin-like proteins in protein regulation. Circ. Res. 100: 1276–1291 [DOI] [PubMed] [Google Scholar]
- 18. Hochstrasser M. 2000. Evolution and function of ubiquitin-like protein-conjugation systems. Nat. Cell Biol. 2: 153–157 [DOI] [PubMed] [Google Scholar]
- 19. Huh WK, et al. 2003. Global analysis of protein localization in budding yeast. Nature 425: 686–691 [DOI] [PubMed] [Google Scholar]
- 20. Hunter T. 2007. The age of crosstalk: phosphorylation, ubiquitination, and beyond. Mol. Cell 28: 730–738 [DOI] [PubMed] [Google Scholar]
- 21. Kerscher O, Felberbaum R, Hochstrasser M. 2006. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu. Rev. Cell Dev. Biol. 22: 159–180 [DOI] [PubMed] [Google Scholar]
- 22. Kosower NS, Kosower EM. 1995. Diamide: an oxidant probe for thiols. Methods Enzymol. 251: 123–133 [DOI] [PubMed] [Google Scholar]
- 23. Kumar A, et al. 2007. Commensal bacteria modulate cullin-dependent signaling via generation of reactive oxygen species. EMBO J. 26: 4457–4466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Longtine MS, et al. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14: 953–961 [DOI] [PubMed] [Google Scholar]
- 25. McGrath JP, Jentsch S, Varshavsky A. 1991. UBA1: an essential yeast gene encoding ubiquitin-activating enzyme. EMBO J. 10: 227–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mendenhall MD. 1993. An inhibitor of p34CDC28 protein kinase activity from Saccharomyces cerevisiae. Science 259: 216–219 [DOI] [PubMed] [Google Scholar]
- 27. Morgan BA, et al. 1997. The Skn7 response regulator controls gene expression in the oxidative stress response of the budding yeast Saccharomyces cerevisiae. EMBO J. 16: 1035–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mumberg D, Müller R, Funk M. 1994. Regulatable promoters of Saccharomyces cerevisiae: comparison of transcriptional activity and their use for heterologous expression. Nucleic Acids Res. 22: 5767–5768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nash P, et al. 2001. Multisite phosphorylation of a CDK inhibitor sets a threshold for the onset of DNA replication. Nature 414: 514–521 [DOI] [PubMed] [Google Scholar]
- 30. Obin M, et al. 1998. Redox regulation of ubiquitin-conjugating enzymes: mechanistic insights using the thiol-specific oxidant diamide. FASEB J. 12: 561–569 [DOI] [PubMed] [Google Scholar]
- 31. Plon SE, Leppig KA, Do HN, Groudine M. 1993. Cloning of the human homolog of the CDC34 cell cycle gene by complementation in yeast. Proc. Natl. Acad. Sci. U. S. A. 90: 10484–10488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Price GR, et al. 2006. Phenotype-directed analysis of genotype in early-onset, familial breast cancers. Pathology 38: 520–527 [DOI] [PubMed] [Google Scholar]
- 33. Schwob E, Bohm T, Mendenhall MD, Nasmyth K. 1994. The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition in S. cerevisiae. Cell 79: 233–244 [DOI] [PubMed] [Google Scholar]
- 34. Seufert W, McGrath JP, Jentsch S. 1990. UBC1 encodes a novel member of an essential subfamily of yeast ubiquitin-conjugating enzymes involved in protein degradation. EMBO J. 9: 4535–4541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sherman F, Fink GR, Hicks JB. 1986. Methods in yeast genetics. Cold Spring Harbor Press, Cold Spring Harbor, NY [Google Scholar]
- 36. Thomas BJ, Rothstein R. 1989. Elevated recombination rates in transcriptionally active DNA. Cell 56: 619–630 [DOI] [PubMed] [Google Scholar]
- 37. Tonks NK. 2005. Redox redux: revisiting PTPs and the control of cell signaling. Cell 121: 667–670 [DOI] [PubMed] [Google Scholar]
- 38. Veal EA, Day AM, Morgan BA. 2007. Hydrogen peroxide sensing and signaling. Mol. Cell 26: 1–14 [DOI] [PubMed] [Google Scholar]
- 39. Verma R, et al. 1997. Phosphorylation of Sic1p by G1 Cdk required for its degradation and entry into S phase. Science 278: 455–460 [DOI] [PubMed] [Google Scholar]
- 40. Willems AR, Schwab M, Tyers M. 2004. A hitchhiker's guide to the cullin ubiquitin ligases: SCF and its kin. Biochim. Biophys. Acta 1695: 133–170 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.