

Abstract
The expression of the pre-B cell receptor (BCR) is confined to the early stage of B cell development, and its dysregulation is associated with anomalies of B-lineage cells, including leukemogenesis. Previous studies suggested that the pre-BCR signal might trigger the autonomous termination of pre-BCR expression even before the silencing of pre-BCR gene expression to prevent sustained pre-BCR expression. However, the underlying mechanism remains ill defined. Here we demonstrate that the pre-BCR signal induces the expression of lysosome-associated protein transmembrane 5 (LAPTM5), which leads to the prompt downmodulation of the pre-BCR. While LAPTM5 induction had no significant impact on the internalization of cell surface pre-BCR, it elicited the translocation of a large pool of intracellular pre-BCR from the endoplasmic reticulum to the lysosomal compartment concomitantly with a drastic reduction of the level of intracellular pre-BCR proteins. This reduction was inhibited by lysosomal inhibitors, indicating the lysosomal degradation of the pre-BCR. Notably, the LAPTM5 deficiency in pre-B cells led to the augmented expression level of surface pre-BCR. Collectively, the pre-BCR induces the prompt downmodulation of its own expression through the induction of LAPTM5, which promotes the lysosomal transport and degradation of the intracellular pre-BCR pool and, hence, limits the supply of pre-BCR to the cell surface.
INTRODUCTION
B cell development in the bone marrow is characterized by the ordered production of Ig heavy (H) and light (L) chains (2). At the pre-B cell stage, L chains are not yet produced, and μH chains are covalently linked to surrogate L (SL) chains composed of VpreB and λ5 to form the pre-B cell receptor (BCR) in a noncovalent association with signal-transducing Igα-Igβ heterodimers (4, 16, 30). The pre-BCR does not function as a receptor for antigen recognition, unlike the BCR on mature B cells. Instead, the pre-BCR plays a crucial role in B cell development in the fetal liver and adult bone marrow in an antigen-independent manner. The deficiency of pre-BCR components or signaling modules such as BLNK (also known as BASH or SLP-65) results in impaired B cell differentiation at the pre-B cell stage in both humans and mice (7, 11). Pre-BCR expression is temporally confined to the large pre-B cell stage and is promptly downmodulated as cells differentiate toward the small pre-B cell stage, where L chains are produced (4, 17, 43). This must be tightly regulated, and dysregulated pre-BCR expression and signal transduction lead to impaired B cell development, the leukemogenesis of pre-B cells, and autoantibody production (9, 10, 15, 19, 24, 25, 35, 41).
The pre-BCR was reported previously to terminate its own expression through the induction of the transcriptional shutoff of the λ5 and VpreB genes (36). The BLNK-mediated pre-BCR signal upregulates the expression of the transcription factors interferon regulatory factor 4 (IRF4) and Aiolos, which in turn induce the silencing of SL chain gene expression (22, 40). In mice deficient for both IRF4 and IRF8, pre-B cells fail to downregulate the pre-BCR and therefore express higher pre-BCR levels than do wild-type (WT) pre-B cells (21). Similarly, BLNK-deficient pre-B cells fail to downregulate the pre-BCR, and the reconstitution of BLNK in these cells leads to the upregulation of Aiolos and pre-BCR downregulation (9, 26, 40, 45). Aiolos competes with early B-cell factor (EBF), an essential transcriptional activator of the λ5 gene, for binding to an overlapping region on the λ5 promoter, thereby silencing λ5 transcription (40). Intriguingly, some studies previously suggested that the downmodulation of the pre-BCR might be triggered by the pre-BCR signal even before the gene silencing of the SL chains (37, 45). However, the underlying mechanism remains to be investigated.
The pre-BCR complex is distinct from the BCR complex not only in its composition but also in the topology within the cell. Only a small fraction (∼2%) of the newly synthesized pre-BCR complex is transported to the cell surface, compared to over 90% of the BCR in mature B cells, even though the pre-BCR and BCR show comparable rates of synthesis and assembly of their components in the endoplasmic reticulum (ER) (3, 8). In accordance with this, most of the μH chains produced by pre-BCR-expressing cells show immature glycosylation, whereas those produced by BCR-expressing cells possess a Golgi-modified, fully mature form of polysaccharides. Thus, the vast majority of the pre-BCR complex is retained in the ER of pre-B cells, in contrast to the dominant expression of the BCR complex on the cell surface. These observations suggest that the regulation of pre-BCR metabolism may differ from that of BCR metabolism.
Lysosome-associated protein transmembrane 5 (LAPTM5) is a transmembrane protein that resides in late endosomes and lysosomes and is expressed preferentially in hematopoietic tissues (1, 38). LAPTM5 contains five membrane-spanning segments, three polyproline-tyrosine motifs, and a ubiquitin-interacting motif at its C terminus (33). LAPTM4, structurally related to LAPTM5, was shown previously to transport and sequestrate various small molecules, including nucleosides and antibiotics, into the lysosome, thereby protecting the cell from their harmful effects (5, 12, 13). Even though the exact function of LAPTM5 remains ill defined, recent studies demonstrated that LAPTM5 is involved in the regulation of antigen receptor expression on the surface of lymphocytes after antigen stimulation (31, 32).
In the present study, we explored the possible mechanism underlying the autonomous downregulation of the pre-BCR on pre-B cells at the protein level. We found that the BLNK-mediated pre-BCR signal induces LAPTM5 expression in pre-B cells. While the induction of LAPTM5 did not show any significant impact on the internalization of cell surface pre-BCR, it promoted the translocation of a large intracellular pool of pre-BCR complexes from the ER to the lysosomal compartment, leading to their lysosomal degradation. This shut off the supply of the pre-BCR to the cell surface, resulting in the downmodulation of surface pre-BCR. Importantly, pre-B cells in LAPTM5-deficient mice showed augmented pre-BCR expression levels, illustrating the importance of LAPTM5 in the autonomous downmodulation of the pre-BCR.
MATERIALS AND METHODS
Mice.
Mice deficient for the membrane exon of the μH chain gene (μMT mice) (20), RAG2-deficient mice (39), and LAPTM5-deficient mice (32) were bred and maintained under specific-pathogen-free conditions in our animal facility. All animal studies were approved by the Institutional Animal Care and Use Committee of Tokyo Medical and Dental University.
Cell lines and cell culture.
An interleukin-7 (IL-7)-independent BLNK-deficient pre-B cell line, BKO84 (45), and its transfectants were cultured in Iscove's modified Dulbecco's medium (IMDM) supplemented with 5% fetal calf serum (FCS), 2 mM l-glutamine, 1 mM sodium pyruvate, 0.1 mM nonessential amino acids, 100 U/ml penicillin-streptomycin, and 5 × 10−5 M 2-mercaptoethanol (2-ME) at 37°C in 5% CO2. The retroviral packaging cell line Plat-E (28) was cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FCS, 100 U/ml penicillin-streptomycin, 1 μg/ml puromycin (Sigma-Aldrich), and 10 μg/ml blasticidin (Gibco BRL). The BKO/BASH-ERtm cell line was established by infecting BKO84 cells with a retroviral vector encoding a fusion protein consisting of BLNK and a mutated hormone-binding domain of mouse estrogen receptor (45). The expression of functional BLNK was induced by the incubation of BKO/BASH-ERtm cells with 10 nM 4-hydroxytamoxifen (4-OHT; Sigma). The BKO/Ton-LAPTM5 cell line was generated by a retroviral infection of BKO cells with pRetroX-Tet-On-Advanced/BLAST and pRetroX-Tight-Pur/LAPTM5 (prepared as described below), followed by selection with 0.5 μg/ml puromycin and 25 μg/ml blasticidin. LAPTM5 expression was induced by the incubation of BKO/Ton-LAPTM5 cells with 2 μg/ml doxycycline. In some experiments, cells were incubated with 0.1 μM bafilomycin A1 (Sigma), 1 μM clasto-lactacystin β-lactone (Calbio Chem), or vehicle (dimethyl sulfoxide [DMSO]) alone as a control.
Construction of retroviral vectors and their infection.
A retroviral vector for μH chain expression, pMX-IRES-GFP/IgμH chain (μHHi and μHUn), was previously described (18). The μHHi and μHUn chains differ in their variable regions and their abilities to form the pre-BCR. The former but not the latter is capable of pairing with the SL chain. The retroviral vector used for doxycycline-induced expression, pRetroX-Tet-On-Advanced/Blast, was generated by replacing the neomycin-resistant gene in the pRetroX-Tet-On-Advanced/Neo retroviral vector (Clontech) with the blasticidin-resistant gene derived from the pCALBL5 vector. The LAPTM5 expression vectors pRetroX-Tight-Pur/LAPTM5 and pRetroX-Tight-Pur/Flag-LAPTM5 (WT or Δcyto [LAPTM5 ER retention mutant]) were created by inserting cDNAs encoding LAPTM5 and Flag-tagged LAPTM5 (WT or Δcyto), respectively, into the multiple-cloning site of the pRetroX-Tight-Pur retroviral vector (Clontech). The LAPTM5 Δcyto mutant includes amino acids (aa) 1 to 204 of LAPTM5 but lacks the cytoplasmic tail. The retroviral vector used for short hairpin RNA (shRNA) expression, MSCV-LMP8, was generated by replacing the green fluorescent protein (GFP) gene in MSCV-LMP (Open Biosystems) with the human CD8 gene (hCD8) from the pMX-IRES-huCD8 vector, which was a gift from T. Saito (Riken, Yokohama, Japan). The following sequences were selected: 5′-CCCTTCAAATAGCTTGCTTAAA (Laptm5sh-1) and (5′-GGTAAAGTGTCCTGTAGGTT (Laptm5sh-2), as targets of short hairpin RNA for the LAPTM5 knockdown; 5′-AAGCTCGAAACTGTTTGGTAA, for the Btk knockdown; and a scramble sequence, ATTCAATCGCTATCGCATACTA, as a negative control. Plat-E cells were transfected with the above-described vectors by using Fugene 6 (Roche). The culture supernatants were collected 48 h later and used to infect cells in the presence of 1 μg/ml Polybrene in 24-well plates. The retroviral infection of bone marrow pro-B cells was performed as previously described (18).
Quantitative RT-PCR analysis.
Total RNA was prepared from cells by using TRIzol (Invitrogen) or a total RNA extraction and purification kit (JenaBioscience). cDNAs were synthesized with ReverTra Ace (Toyobo), an oligo(dT) primer (Amersham), and random hexamer primers (Invitrogen). Quantitative reverse transcription (RT)-PCR was performed on a Stepone Plus PCR instrument (Applied Biosystems) with Fast SYBR green master mix (Applied Biosystems) and the following primer sets for various genes: sense primer TGCCGCACAGCCAGTTCATCA and antisense primer AGGACGGCAGAGCCACCTTGA for Laptm5, sense primer ACGTGGGAGAAGAGGCAGTA and antisense primer TCTGTCTTTCTGGTGGGGGA for Btk, and sense primer GTTGGATACAGGCCAGACTTTGTTG and antisense primer GAGGGTAGGCTGGCCTATAGGCT for Hprt.
Flow cytometric analysis and cell sorting.
Cells were stained with the following antibodies: fluorescein isothiocyanate (FITC)-labeled anti-CD19, anti-GL7, and anti-B220 antibodies; biotin-labeled anti-CD43 (S7) and anti-human CD8 antibodies; phycoerythrin (PE)-labeled anti-CD43 antibody (S7); and anti-IL-7Rα chain antibody, peridinin chlorophyll protein (PerCP)-Cy5.5-labeled anti-IgM antibody, and allophycocyanin-labeled anti-CD19 antibody and streptavidin. All of these reagents were purchased from BD PharMingen. The monoclonal antibody (MAb) specific for the pre-BCR (SL156) (44) was biotinylated in our laboratory. Stained cells were analyzed with a FACSCalibur or FACSCanto II system (BD Biosciences). In some experiments, GFP-positive (GFP+) cells were sorted with a FACSVantage or FACSAria II system (BD Biosciences). In some experiments, B220+ cells were isolated with B220-iMag beads (BD Biosciences).
Immunofluorescence microscopic analysis.
Cells were placed onto silane-coated glass slides, fixed with 4% paraformaldehyde for 10 min, and permeabilized with 0.1% Tween 20 in phosphate-buffered saline (PBS) for 10 min. After being blocked with 5% goat serum and 2% bovine serum albumin (BSA) in PBS, the cells were incubated for 1 h with rat anti-mouse LAMP1 antibody (1D4B; eBiosciences), biotinylated goat anti-IgμH Fab (Jackson ImmunoResearch), rabbit anti-LAPTM5 antibody (32), or mouse anti-Flag M2 antibody (Sigma), followed by Alexa Fluor 488-labeled streptavidin, Alexa Fluor 405-labeled anti-mouse IgG, Alexa Fluor 555-labeled anti-rat IgG, or Alexa Fluor 647-labeled anti-rabbit IgG (Molecular Probes). Single-plane images were acquired with an A1R confocal laser scanning microscope (Nikon Instech) equipped with a 60× oil objective lens and processed with NIS-Elements C software (Nikon Instech).
Immunoblot analysis.
Immunoprecipitation and immunoblotting were performed with the following antibodies: goat polyclonal anti-μH chain antibody (Jackson ImmunoResearch); rabbit polyclonal anti-Igα antibody; goat polyclonal anti-λ5 antibody (V-20; Santa Cruz); mouse anti-α-tubulin antibody (Sigma-Aldrich); mouse anti-protein disulfide isomerase (anti-PDI) antibody (Abcam); and rabbit polyclonal antibodies specific for Lyn (sc-15; Santa Cruz), extracellular signal-regulated kinase 1 (ERK1) (C-16; Santa Cruz), phosphorylated ERK1/2 (Cell Signaling Technology), Syk (Cell Signaling Technology), phosphorylated Syk (Cell Signaling Technology), and LAPTM5 (32).
Labeling and monitoring of pre-BCR on the cell surface.
To measure the rate of pre-BCR internalization from the cell surface, BKO/BASH-ERtm (or BKO/Ton-LAPTM5) cells were incubated with or without 4-OHT (or doxycycline) for 7 h, followed by the labeling of surface pre-BCR with biotinylated anti-μH Fab (10 μg/ml) at 4°C for 10 min. After washing, the labeled cells were further cultured with or without 4-OHT (or doxycycline) for the indicated time periods and then harvested and fixed with 1% paraformaldehyde to stop pre-BCR internalization. The levels of pre-BCR remaining on the cell surface at the indicated time points were measured by staining the cells with allophycocyanin-labeled streptavidin.
Statistical analysis.
Statistical analyses were performed by using a two-tailed unpaired Student t test. A P value of <0.05 was considered statistically significant.
RESULTS
The BLNK-mediated signal induces rapid pre-BCR downmodulation in a lysosome-dependent manner.
To explore the molecular mechanism underlying the autonomous termination of pre-BCR expression, we first employed a pre-BCR-expressing cell line, BKO/BASH-ERtm. This pre-B cell line was originally derived from BLNK-deficient mice and engineered to stably express a fusion protein consisting of BLNK and a mutant hormone-binding domain of mouse estrogen receptor so that pre-BCR signal transduction via BLNK can be turned on by the treatment of cells with 4-hydroxytamoxifen (4-OHT) (45). The surface pre-BCR expression level was reduced by 4-OHT treatment down to 20% of the control level by 24 h, whereas the surface expression levels of CD19 and CD43 remained unchanged (Fig. 1A; not all data shown), demonstrating that the BLNK-mediated pre-BCR signal selectively downregulated pre-BCR expression.
Fig 1.

The lysosomal pathway is involved in BLNK-mediated pre-BCR downmodulation. (A) BKO/BASH-ERtm cells were cultured with 4-OHT (filled histograms in top panels) or control ethanol (open histograms in top panels) for 24 h (top) or for the indicated time periods (bottom) and analyzed for the surface expression of pre-BCR and CD19. Dashed histograms in the top panels show control staining with an isotype-matched antibody. Bottom panels show the time course of the relative level (mean fluorescence intensity) of pre-BCR and CD19 expressions (means ± standard errors of the means; n = 4 for each time point) (∗, P < 0.05, ∗∗, P < 0.01), where the level of expression before the culture (0 h) is set as 100%. (B) BKO/BASH-ERtm cells were cultured with 4-OHT (filled bars) or ethanol (open bars) for 7 h and subjected to surface pre-BCR labeling with anti-μH Fab antibodies, followed by further culturing with 4-OHT (filled bars) or ethanol (open bars) for the indicated time periods after labeling. The relative expression level of labeled pre-BCR on the cell surface at each time point is shown (means ± standard errors of the means; n = 3 each), in that the level of expression before the start of measurements (0 min) is set as 100% for both experimental groups (with and without 4-OHT treatment, respectively) for easier comparisons of the rates of pre-BCR internalization between them. Note that as shown in panel A, the actual level of pre-BCR on 4-OHT-treated cells was ∼70 to 80% of that on untreated control cells before the start of the measurements (0 min), that is, 7 h after treatment with or without 4-OHT. (C) BKO/BASH-ERtm cells were cultured for 12 h with 4-OHT (filled bars) or ethanol (open bars) in the presence of the indicated reagents or vehicle (DMSO) alone and then analyzed for the expression of surface pre-BCR. The relative expression level of pre-BCR in each group is shown (means ± standard errors of the means; n = 3 each) (∗, P < 0.05), in that the level of expression on control cells cultured with ethanol is set as 100%. Data shown in panels A to C are representative of data from three or four independent experiments.
The BLNK-induced downmodulation of the pre-BCR could not be attributed to accelerated pre-BCR internalization from the cell surface, because 4-OHT treatment had no significant impact on pre-BCR internalization (Fig. 1B). Of note, the downmodulation of the pre-BCR was almost completely abolished by the treatment of cells with a lysosomal inhibitor, bafilomycin or NH4Cl, but not with a proteasome inhibitor, lactacystin (Fig. 1C), suggesting that the BLNK-mediated pre-BCR signal induces the downmodulation of the pre-BCR in a manner dependent on the function of the lysosome.
The BLNK-mediated pre-BCR signal induces the expression of a lysosomal protein, LAPTM5, that is crucial for the downmodulation of the pre-BCR.
We next compared the expression profiles of genes, particularly those associated with the lysosome, of 4-OHT-treated and control BKO/BASH-ERtm cells. RT-PCR analysis revealed that Laptm5 gene expression was upregulated by 4-OHT treatment (Fig. 2A), and immunoblot analysis further demonstrated the induction of LAPTM5 at the protein level (Fig. 2B). When shRNAs specific to Bruton's tyrosine kinase (Btk) were expressed in BKO/BASH-ERtm cells, the induction of Laptm5 by treatment with 4-OHT was significantly suppressed, in parallel with the reduction in the Btk expression level (Fig. 2C). Thus, the BLNK-mediated pre-BCR signal induces LAPTM5 expression via Btk. To explore the possible link between LAPTM5 and pre-BCR downmodulation, shRNAs specific for LAPTM5 were expressed in BKO/BASH-ERtm cells (Fig. 2D and E). shRNA expression showed no significant impact on the basal level of pre-BCR expression in control (vehicle-treated) BKO/BASH-ERtm cells (Fig. 2D). Importantly, the inhibition of 4-OHT-induced LAPTM5 expression by the shRNAs (Fig. 2E, bottom) resulted in the suppression of the 4-OHT-induced downmodulation of the pre-BCR (Fig. 2D and E, top). These results suggested that the BLNK-mediated pre-BCR signal induces the expression of LAPTM5, which in turn promotes the downmodulation of the pre-BCR.
Fig 2.

BLNK-induced LAPTM5 expression is crucial for pre-BCR downmodulation. (A) BKO/BASH-ERtm cells were cultured with 4-OHT (filled bars) or control ethanol (open bars) for the indicated time periods and subjected to RT-PCR analysis. The relative expression level of Laptm5 mRNA at each time point is shown (means ± standard errors of the means; n = 3 each) (∗∗, P < 0.01), in that the level of Laptm5 mRNA expression in untreated cells is set as 1. (B) Immunoblot analysis of LAPTM5 and α-tubulin proteins in BKO/BASH-ERtm cells that were cultured with 4-OHT for the indicated time periods. (C, top) BKO/BASH-ERtm cells were infected with an hCD8-expressing retroviral vector carrying Btk shRNA or an irrelevant control shRNA, and hCD8+ cells were subjected to RT-PCR analysis for the expression of Btk (top). The level of Btk expression in cells infected with control shRNA is set as 1 (means ± standard errors of the means; n = 3 each) (∗∗, P < 0.01). (Bottom) hCD8+ cells were cultured for 24 h with or without 4-OHT and subjected to RT-PCR analysis for the expression of Laptm5 mRNAs. The level of Laptm5 expression in cells that were infected with control shRNA and cultured without 4-OHT is set as 1 (means ± standard errors of the means; n = 3 each) (∗∗, P < 0.01). (D and E) BKO/BASH-ERtm cells were infected with an hCD8-expressing retroviral vector carrying Laptm5 shRNA (Laptm5sh-1 or Laptm5sh-2) or an irrelevant control shRNA. Two days after infection, the cells were cultured for 24 h with 4-OHT (filled bars) or the control (open bars) and were subjected to flow cytometric analysis for pre-BCR expression (representative profiles are shown in panel D, and all data are summarized at the top of panel E) and immunoblot analysis for LAPTM5 expression (bottom of panel E). The top of panel E shows the relative expression level of pre-BCR in each group (means ± standard errors of the means; n = 3 each) (∗∗, P < 0.01), in that the level of expression on control cells cultured with ethanol is set as 100%. Data shown in panels A to E are representative of data from three or four independent experiments.
We next examined whether the downmodulation of the pre-BCR can be achieved by LAPTM5 alone or in cooperation with other molecules induced by the BLNK-mediated pre-BCR signal. To this end, we established a cell line, designated BKO/Ton-LAPTM5, by engineering the BLNK-deficient pre-B cell line BKO84 so that the expression of LAPTM5 proteins can be induced by treatment with doxycycline (Fig. 3A). Doxycycline-induced LAPTM5 expression elicited no detectable tyrosine phosphorylation of the signal-transducing molecules Syk and ERK (Fig. 3B), demonstrating that the induction of LATPM5 by itself does not trigger pre-BCR signaling. Nevertheless, doxycycline-induced LAPTM5 expression led to the downmodulation of the pre-BCR but not CD19 on the cell surface, even in the absence of BLNK (Fig. 3C). Thus, among the BLNK-elicited events, LAPTM5 induction appears to be critically important for pre-BCR downmodulation.
Fig 3.

LAPTM5 expression elicits the downmodulation of the pre-BCR in the absence of BLNK. (A and B) BKO/Ton-LAPTM5 cells were cultured with doxycycline (Dox) for the indicated time periods or stimulated with pervanadate (PV) for 5 min and subjected to immunoblot analysis for the expressions of LATPM5 and Lyn (A) and Syk, ERK, and their phosphorylated forms (B). Note that in the sequential blotting, the bands of Syk and ERK in pervanadate-stimulated cells did not show up well due to strong signals for the phosphorylated proteins in the first blotting. (C) BKO/Ton-LAPTM5 cells were cultured with doxycycline (filled histograms) or control H2O (open histograms) for 24 h (top) or for the indicated time periods (bottom) and analyzed for the surface expression of pre-BCR and CD19 as described in the legend to Fig. 1A (means ± standard errors of the means; n = 4 for each time point) (∗∗, P < 0.01). Data shown in panels A to C are representative of three independent experiments.
LAPTM5 promotes the translocation of μH chains from the ER to the lysosomal compartment.
The induced expression of LAPTM5 in BKO/Ton-LAPTM5 cells did not show any significant impact on the rate of internalization of cell surface pre-BCR (Fig. 4A), as observed when the BLNK-mediated pre-BCR signal was turned on in BKO/BASH-ERtm cells (Fig. 1B). Moreover, no significant recycling of the internalized pre-BCR back to the cell surface was detected in BKO/Ton-LAPTM5 cells, regardless of LAPTM5 induction (data not shown). No recycling was also demonstrated in the pre-T cell receptor (TCR), a functional analogue of the pre-BCR in pre-T cells (6). To understand the mechanism underlying LAPTM5-mediated pre-BCR downmodulation, we next examined the intracellular localization of LAPTM5 and the pre-BCR by using confocal microscopy (Fig. 4B). In untreated BKO/Ton-LAPTM5 cells, most of the μH chains were detected in the ER, in accordance with data from a previous report (3). Notably, when LAPTM5 was inducibly expressed by doxycycline treatment, μH chains changed their intracellular localization and were colocalized with LAPTM5 proteins that were also colocalized with LAMP-1 proteins, a lysosomal marker (Fig. 4B). This LAPTM5-associated translocation of μH chains from the ER to the lysosomal compartment was also observed when BLNK was inducibly expressed in BKO/BASH-ERtm cells with 4-OHT (data not shown). Conversely, the localization of μH chains was shifted from the lysosomal compartment to the ER concomitantly with the upregulation of surface pre-BCR expression when LAPTM5 expression was knocked down in 70Z/3 cells, a pre-B cell line that constitutively expresses the pre-BCR and LAPTM5 (Fig. 4C to E).Of note, in 70Z/3 cells, μH chains were coprecipitated with LAPTM5 (Fig. 4F), demonstrating the association of these two proteins.
Fig 4.

LAPTM5 promotes the translocation of μH chains from the ER to the lysosomal compartment. (A) BKO/Ton-LAPTM5 cells were cultured with doxycycline (filled bars) or control H2O (open bars) for 7 h and subjected to surface pre-BCR labeling with anti-μH Fab antibodies, followed by further culturing with doxycycline (filled bars) or control H2O (open bars) for the indicated time periods after labeling. The relative expression of labeled pre-BCR on the cell surface at each time point is shown, as described in the legend to Fig. 1B (means ± standard errors of the means; n = 3 each). Representative data from two independent experiments are shown. (B) BKO/Ton-LAPTM5 cells were cultured with doxycycline or control H2O for 24 h. The cells were stained simultaneously for intracellular LAPTM5 (green), LAMP1 (red), and the μH chain (blue) and subjected to confocal microscopy analysis. Representative images from three independent experiments are displayed in pseudocolor processed with NIS-Elements software. Right panels show merges of all images overlaid with transmitted light. Scale bars indicate 5 μm. (C to F) 70Z/3 pre-B cells were infected with an hCD8-expressiong retroviral vector carrying Laptm5 shRNA (Laptm5sh-1) or an irrelevant control shRNA. (C) hCD8+-infected cells were subjected to immunoblot analysis for LAPTM5 expression to confirm knockdown by Laptm5sh-1. WB, Western blot. (D) hCD8+ cells infected with the Laptm5sh-1 vector (filled histogram) or the control vector (open histogram) were analyzed for surface pre-BCR expression. The dashed histogram indicates the staining profile with an isotype-matched control antibody. (E) hCD8+-infected cells were subjected to confocal microscopy analysis for the expression of the intracellular μH chain (green) and LAMP1 (red), as described above for panel B. (F) Cell lysates of hCD8+-infected cells were subjected to immunoprecipitation with anti-LAPTM5 antibody or control rabbit IgG, followed by immunoblotting (IB) with anti-μH chain or anti-LAPTM5 antibody. Filled and open arrowheads indicate the LAPTM5 band and the nonspecific one, respectively. As a control, the whole lysates (without immunoprecipitation) were subjected to immunoblotting with anti-μH chain antibody. Data shown in panels C to F are representative of data from at least three independent experiments. (G) The BKO/Ton-FlagLAPTM5 (WT) and BKO/Ton-FlagLAPTM5 (Δcyto) cell lines were cultured for 12 h with doxycycline (filled histograms) or control H2O (open histograms) and analyzed for their pre-BCR expression on the cell surface. Dashed lines show the staining profile of the isotype-matched control. (H) The BKO/Ton-FlagLAPTM5 (WT) and BKO/Ton-FlagLAPTM5 (Δcyto) cell lines were cultured for 12 h with doxycycline and subjected to confocal microscopy examination, as described above for panel B. Data shown in panels G and H are representative of data from three independent experiments.
Collectively, LAPTM5 appears to escort μH chains along the way from the ER to the lysosomal compartment. Indeed, the translocation of μH chains from the ER to the lysosomal compartment was not observed when a LAPTM5 ER retention mutant (Δcyto) lacking the cytoplasmic tail crucial for sorting to the lysosomal compartment (33) was inducibly expressed in the BKO84 cell line (Fig. 4H). Importantly, the downmodulation of surface pre-BCR was observed when the wild-type but not the mutant LAPTM5 proteins were expressed in the BKO84 cell line (Fig. 4G), suggesting that the lysosomal translocation of μH chains is essential for the downmodulation of surface pre-BCR.
LAPTM5 promotes the lysosomal degradation of the intracellular pool of pre-BCR.
We next examined the fate of μH chains that were subjected to lysosomal translocation upon BLNK-mediated LAPTM5 expression. Western blot analyses revealed that in 4-OHT-treated BKO/BASH-ERtm cells, LAPTM5 proteins were inducibly produced, while the amounts of pre-BCR components, including the μH chain, λ5 proteins, and Igα, were drastically reduced compared with levels in the ethanol (EtOH)-treated controls (Fig. 5A). The Laptm5 gene knockdown inhibited the reduction in the levels of the pre-BCR proteins in 4-OHT-treated cells (Fig. 5A). Intriguingly, the reduction in the levels of μH chain proteins in 4-OHT-treated cells was observed not only for their mature form (cell surface type) but also for their immature form (ER retention type) (Fig. 5A). This was also the case when LAPTM5 was inducibly expressed in BKO/Ton-LAPTM5 cells (Fig. 5B and C), indicating that LAPTM5 is involved in the massive pre-BCR-induced reduction of the intracellular pool of pre-BCR. Because the amount of protein disulfide isomerase (PDI), a typical ER-resident protein, remained unaffected by LAPTM5 expression (Fig. 5A and B), it is unlikely that LAPTM5 nonselectively induced the reduction of the bulk of the ER-resident proteins. Notably, treatment with bafilomycin abolished the LAPTM5-induced reduction in the levels of μH chains (Fig. 5C). These results suggest that LAPTM5 promotes the translocation of pre-BCR complexes from the ER to the lysosomal compartment, leading to the lysosomal degradation of the intracellular pool of pre-BCR and, hence, limiting the new supply of pre-BCR to the cell surface.
Fig 5.

LAPTM5 promotes lysosomal degradation of the intracellular pool of pre-BCR. (A) BKO/BASH-ERtm cells infected with an hCD8-expressing retroviral vector carrying Laptm5 shRNA (Laptm5sh-1) or an irrelevant control shRNA were cultured for 24 h with 4-OHT or control ethanol, as described in the legend to Fig. 2D and E, and subjected to immunoblot analysis for the detection of the indicated proteins. Open and filled arrowheads indicate mature and immature forms of μH chains, respectively. Data shown are representative of data from three independent experiments. (B) BKO/Ton-LAPTM5 cells were cultured for 24 h with doxycycline or control H2O and subjected to immunoblot analysis for the detection of the indicated proteins. (C) BKO/Ton-LAPTM5 cells were cultured with doxycycline or control H2O for 12 h, and bafilomycin or vehicle (DMSO) was included in the culture medium during the last 4-h period, followed by immunoblot analysis, as described above for panel B. Data shown in panels B and C are representative of data from five independent experiments.
Crucial role for LAPTM5 in the downmodulation of the pre-BCR in primary pre-B cells.
We next explored whether pre-BCR-mediated LAPTM5 induction followed by the lysosomal translocation and degradation of the intracellular pre-BCR pool observed for the pre-B cell lines indeed recapitulates the mechanism underlying the autonomous downmodulation of the pre-BCR in primary pre-B cells. We previously showed that the cross-linking of Igβ on pro-B cells mimics pre-BCR signaling, and the in vivo administration of anti-Igβ MAb induces pro-B-to-pre-B cell differentiation even in the absence of μH chains (23, 29). The treatment of μH chain-deficient μMT mice with anti-Igβ antibody induced the upregulation of Laptm5 mRNA in B220+ pro-B cells, along with the upregulated expression of a differentiation marker, CD25 (Fig. 6A), suggesting that the pre-BCR signal can induce LAPTM5 expression in primary pre-B cells. Indeed, the level of Laptm5 mRNA was greatly upregulated when μH chains capable of forming pre-BCR (μHHi) but not those incapable of forming pre-BCR (μHUn) were expressed in pro-B cells isolated from μMT mice (Fig. 6B). LAPTM5 proteins were microscopically detected to be colocalized with LAMP-1 when μHHi proteins were expressed in primary pro-B cells (Fig. 6C). In contrast, the expression of μHUn proteins failed to induce LAPTM5 expression, indicating that the formation of pre-BCR but not the expression of the μH chain by itself can elicit LAPTM5 expression. In accordance with the observations for the cell lines (Fig. 4), μHHi proteins were detected in the lysosomal compartment as being colocalized with LAPTM5 and LAMP-1, whereas μHUn proteins were detected in the ER compartment (Fig. 6C). Importantly, surface pre-BCR expression was augmented (approximately 2-fold in terms of the mean fluorescence intensity) when LAPTM5-specific shRNAs were expressed in μHHi-expressing pro-B cells (Fig. 6D). Furthermore, GL7+ pre-B cells freshly isolated from LAPTM5-deficient mice expressed significantly higher (approximately 2.5-fold) levels of surface pre-BCR than did those from wild-type mice, while the levels of CD19 and GL7 expression were comparable (Fig. 7; not all data shown). These results clearly demonstrated that LAPTM5 induced by pre-BCR formation plays an important role in the autonomous downmodulation of the pre-BCR in vivo. Of note, in the presence of LAPTM5, the level of surface pre-BCR expression was higher in pro-B cells expressing μHHi (Fig. 6D) than in freshly isolated pre-B cells (Fig. 7), even though the extents of pre-BCR upregulation in the absence of LAPTM5 were comparable (∼2- to 2.5-fold). This can be explained by the facts that the former cells express a selected μH chain (μHHi), which confers the highest level of pre-BCR on pro-B cells (18), and were cultured with IL-7, which kept cells alive and also increased the pre-BCR expression level.
Fig 6.

Crucial role for LAPTM5 in pre-BCR downmodulation in primary pre-B cells. (A) μMT mice were left untreated or treated with an intraperitoneal injection of anti-Igβ antibody or control Ig. The anti-Igβ antibody-mediated stimulation of pro-B cells was confirmed by the increased CD25 expression levels among the B220+ bone marrow pro-B cells on day 7 postinjection (top), in that the level of CD25 expression on pro-B cells isolated from untreated mice is set as 1. The top panel shows the relative expression level of Laptm5 mRNA in B220+ bone marrow pro-B cells isolated 1 day after injection, where the level of Laptm5 mRNA in cells from untreated mice is set as 1. Data shown are the means ± standard errors of the means (n = 3) and are representative of two independent experiments (∗, P < 0.05; ∗∗, P < 0.01). (B) B220+ pro-B cells freshly isolated from bone marrow of μMT mice were infected with a GFP-expressing retroviral vector encoding the μH chains capable (μHHi) or incapable (μHUn) of forming pre-BCR or with a control vector, cultured with a low dose (10 ng/ml) of IL-7 in vitro for 24 h, and analyzed for the expressions of surface pre-BCR (n = 5 each) (top) and Laptm5 mRNA (n = 3 each) (bottom) in CD19+ GFP+ cells. The level of expression in cells infected with the control vector is set as 1. Data shown are the means ± standard errors of the means and are representative of three independent experiments (∗∗, P < 0.01). (C) B220+ pro-B cells freshly isolated from bone marrow of RAG2-deficient mice were infected with the vectors and then cultured as described above for panel B. GFP+ cells were intracellularly stained and subjected to examination by confocal microscopy, as described in the legend to Fig. 4B. (D) Pro-B cells from μMT mice were first infected with an hCD8-expressing retroviral vector encoding Laptm5 shRNA (Laptm5sh-1) or a control shRNA vector and subsequently infected with a GFP-expressing retroviral vector encoding the μHHi chain. One day after the second infection, the cells were cultured with 10 ng/ml of IL-7 for 24 h and then analyzed for the surface expression of pre-BCR on GFP(μH)+ hCD8(Laptm5 shRNA)+ cells (filled histograms) and GFP(μH)+ hCD8(control shRNA)+ cells (open histograms), as shown in the left panel. All the data are summarized in the right panel, which shows the relative expression level of pre-BCR (means ± standard errors of the means; n = 3), where the expression level on GFP(μH)− hCD8(control shRNA)+ cells is set as 1. Data shown in panels C and D are representative of data from three independent experiments.
Fig 7.
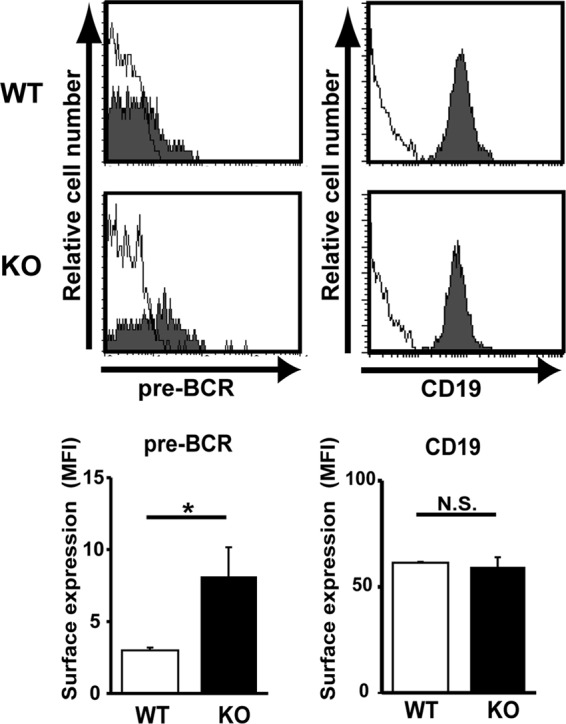
Augmented pre-BCR expression on large pre-B cells in LAPTM5-deficient mice. Bone marrow cells isolated from wild-type (WT) and Laptm5−/− (knockout [KO]) mice were stained for pre-BCR, CD19, CD43, and GL7 on the cell surface. Filled histograms in the top panels show representative staining profiles for pre-BCR and CD19 expressed on large pre-B cells (CD19+ CD43+ GL7+), while open histograms indicate control staining with an isotype-matched control antibody. All the data are summarized in the panels, which show the mean fluorescence intensity (MFI) values (means ± standard errors of the means; n = 4 each) for pre-BCR and CD19 staining (∗, P < 0.05; N.S., not significant).
DISCUSSION
Previous studies reported that the pre-BCR autonomously terminates its expression through the BLNK-mediated induction of the transcriptional silencing of the SL chain genes (22, 36, 40). In the present study, we have identified a novel mechanism for promoting prompt pre-BCR termination at the protein level. The BLNK-mediated pre-BCR signal induced the expression of LAPTM5, which in turn promoted the topological change of the intracellular pool of pre-BCR complexes, leading to their lysosomal degradation. The LAPTM5 deficiency resulted in the augmentation of pre-BCR expression on pre-B cells. Thus, the temporal expression of the pre-BCR at the early stage of B cell development is tightly guaranteed by the autonomous termination of pre-BCR expression via the LAPTM5-mediated degradation of existing pre-BCR proteins, in addition to SL chain gene silencing.
LAPTM5 is structurally related to LAPTM4, a lysosomal protein that is widely expressed in many cell types in mammals, insects, and nematodes. LAPTM4 was shown previously to transport and sequestrate various small molecules, including nucleosides and antibiotics, into the lysosome, thereby protecting the cell from their harmful effects (5, 12, 13). This finding suggests that LAPTM5 may also function as an intracellular transporter to the lysosome. In the present study, we demonstrated that the expression of wild-type LAPTM5 but not its ER retention mutant promotes the translocation of the large pre-BCR pool from the ER to the lysosomal compartment, indicating that LAPTM5 escorts the pre-BCR from the ER to the lysosome. On the other hand, LAPTM5 expression did not accelerate the internalization of the pre-BCR from the cell surface (Fig. 1B and 4A) and showed little or no inhibitory effect on pre-BCR recycling back to the cell surface or the transcription of any pre-BCR components (data not shown). Moreover, LAPTM5 expression did not stimulate pre-BCR signaling. Collectively, LAPTM5 appears to induce the prompt downmodulation of surface pre-BCR expression, mainly by accelerating the lysosomal transport and degradation of the large pool of intracellular pre-BCR and, hence, limiting the new supply of pre-BCR to the cell surface.
It was demonstrated previously that LAPTM5 in T cells targets the homodimer of CD3zeta but not other components of the TCR for degradation (32). In contrast, our data in the present study suggest that LAPTM5 in pre-B cells promotes the degradation of the whole complex of the pre-BCR. Although the exact mechanism of the interaction between LAPTM5 and the pre-BCR remains to be determined, it is possible that LAPTM5 associates with the pre-BCR via the Igα-Igβ heterodimer, a functional analogue of CD3. Alternatively, LAPTM5 may interact indirectly with the pre-BCR and CD3zeta through a chaperone-like molecule that recognizes a particular structure shared by these molecules.
Autophagy is a crucial clearance mechanism that prevents the accumulation of protein aggregates and abnormal organelles (27, 46). Recent studies demonstrated that the overexpression of LAPTM5 and LAPTM4b in a neuroblastoma cell line and a retinal pigment epithelial cell line, respectively, results in an altered autophagic process (14, 42). In the present study, we found that bafilomycin or NH4Cl efficiently inhibited LAPTM5-induced pre-BCR downmodulation and degradation, whereas neither 3-methyladenine, an inhibitor of phosphatidylinositol 3-kinase (PI3K), nor the shRNA-mediated knockdown of Atg5 showed any significant effect on pre-BCR metabolism (data not shown). Thus, the autophagic process does not seem to be significantly involved in LAPTM5-mediated pre-BCR degradation. Ubiquitin-dependent proteasomal degradation is involved in the downmodulation of the pre-TCR, a functional analogue of the pre-BCR in pre-T cells (34). Pre-TCR degradation is reduced in pre-T cells by treatment with proteasome inhibitors or the expression of dominant negative c-Cbl, a ubiquitin ligase. In accordance with this, the surface expression level of the pre-TCR on immature thymocytes is increased in c-Cbl-deficient mice. Thus, c-Cbl-mediated ubiquitination followed by proteasomal degradation is important for pre-TCR downmodulation. In contrast, we found that the BLNK- and LAPTM5-mediated downmodulation of the pre-BCR was not counteracted by proteasome inhibitor treatment (Fig. 1C) or the knockdown of c-Cbl (data not shown). Therefore, the mode of pre-BCR degradation does not seem to be identical to that of the pre-TCR, although it remains to be determined whether LAPTM5 has some contribution to the downmodulation of the pre-TCR.
In the present study, we found that the expression of the Laptm5 gene is upregulated through the pre-BCR signal in pre-B cells, unlike in mature B and T cells, in which it is transiently downregulated upon the cross-linking of antigen receptors (32). Although it remains to be clarified what makes the difference in Laptm5 gene regulation between pre-B cells and mature lymphocytes, this differential gene regulation appears to be reasonable, given the fact that pre-BCR expression is transient and should be terminated, while BCR and TCR expression returns to the basal level following their downregulation upon cross-linking. The steady-state level of the antigen receptor on mature lymphocytes remains unchanged regardless of the presence or absence of LAPTM5 (31, 32), in contrast to the augmented pre-BCR expression on LAPTM5-deficient pre-B cells, suggesting that LAPTM5 is not the major contributor to the basal-level degradation of antigen receptors. The cross-linking of the antigen receptors induces their downmodulation, regardless of the presence or absence of LAPTM5. However, the level of the antigen receptors after the cross-link is higher on LAPTM5-deficient lymphocytes than on wild-type lymphocytes, particularly during the phase of recovery to the basal level. Therefore, it is thought that LAPTM5 facilitates the lysosomal sorting of the antigen receptors that are internalized from the cell surface after cross-linking, leading to their degradation in the lysosomes. Notably, the vast majority of the pre-BCR produced by pre-B cells is retained in the ER, in sharp contrast to the dominant expression of the BCR on the cell surface of mature B cells. We demonstrated in the present study that pre-BCR-mediated LAPTM5 induction in pre-B cells has a great impact on the catabolism of intracellular pre-BCR rather than cell surface pre-BCR.
In conclusion, we demonstrated in the present study that the pre-BCR signal induces LAPTM5 expression, which in turn promotes the lysosomal transport and degradation of the intracellular pre-BCR pool. This regulation at the protein level, together with the transcriptional silencing of the SL chain genes, ensures the obligatory downmodulation of the pre-BCR at the early stage of B cell development.
ACKNOWLEDGMENTS
We thank T. Kitamura for providing pMX-IRES-GFP and Plat-E, T. Saito for pMX-IRES-hCD8, T. Kurosaki for anti-Igα antibody, and Y. Makishima for her excellent assistance with cell sorting.
This work was supported by grant-in-aid 23790530 to Y. Kawano from the Japanese Ministry of Education, Culture, Sports, Science, and Technology.
Footnotes
Published ahead of print 4 September 2012
REFERENCES
- 1. Adra CN, et al. 1996. LAPTM5: a novel lysosomal-associated multispanning membrane protein preferentially expressed in hematopoietic cells. Genomics 35: 328–337 [DOI] [PubMed] [Google Scholar]
- 2. Alt FW, et al. 1984. Ordered rearrangement of immunoglobulin heavy chain variable region segments. EMBO J. 3: 1209–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brouns GS, de Vries E, Neefjes JJ, Borst J. 1996. Assembled pre-B cell receptor complexes are retained in the endoplasmic reticulum by a mechanism that is not selective for the pseudo-light chain. J. Biol. Chem. 271: 19272–19278 [DOI] [PubMed] [Google Scholar]
- 4. Burrows PD, et al. 2002. The transient expression of pre-B cell receptors governs B cell development. Semin. Immunol. 14: 343–349 [DOI] [PubMed] [Google Scholar]
- 5. Cabrita MA, Hobman TC, Hogue DL, King KM, Cass CE. 1999. Mouse transporter protein, a membrane protein that regulates cellular multidrug resistance, is localized to lysosomes. Cancer Res. 59: 4890–4897 [PubMed] [Google Scholar]
- 6. Carrasco YR, Navarro MN, Toribio ML. 2003. A role for the cytoplasmic tail of the pre-T cell receptor (TCR) alpha chain in promoting constitutive internalization and degradation of the pre-TCR. J. Biol. Chem. 278: 14507–14513 [DOI] [PubMed] [Google Scholar]
- 7. Conley ME, Rohrer J, Rapalus L, Boylin EC, Minegishi Y. 2000. Defects in early B-cell development: comparing the consequences of abnormalities in pre-BCR signaling in the human and the mouse. Immunol. Rev. 178: 75–90 [DOI] [PubMed] [Google Scholar]
- 8. Fang T, Smith BP, Roman CA. 2001. Conventional and surrogate light chains differentially regulate Ig mu and Dmu heavy chain maturation and surface expression. J. Immunol. 167: 3846–3857 [DOI] [PubMed] [Google Scholar]
- 9. Flemming A, Brummer T, Reth M, Jumaa H. 2003. The adaptor protein SLP-65 acts as a tumor suppressor that limits pre-B cell expansion. Nat. Immunol. 4: 38–43 [DOI] [PubMed] [Google Scholar]
- 10. Hayashi K, Yamamoto M, Nojima T, Goitsuka R, Kitamura D. 2003. Distinct signaling requirements for Dmu selection, IgH allelic exclusion, pre-B cell transition, and tumor suppression in B cell progenitors. Immunity 18: 825–836 [DOI] [PubMed] [Google Scholar]
- 11. Herzog S, Reth M, Jumaa H. 2009. Regulation of B-cell proliferation and differentiation by pre-B-cell receptor signalling. Nat. Rev. Immunol. 9: 195–205 [DOI] [PubMed] [Google Scholar]
- 12. Hogue DL, Ellison MJ, Young JD, Cass CE. 1996. Identification of a novel membrane transporter associated with intracellular membranes by phenotypic complementation in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 271: 9801–9808 [DOI] [PubMed] [Google Scholar]
- 13. Hogue DL, Kerby L, Ling V. 1999. A mammalian lysosomal membrane protein confers multidrug resistance upon expression in Saccharomyces cerevisiae. J. Biol. Chem. 274: 12877–12882 [DOI] [PubMed] [Google Scholar]
- 14. Inoue J, et al. 2009. Lysosomal-associated protein multispanning transmembrane 5 gene (LAPTM5) is associated with spontaneous regression of neuroblastomas. PLoS One 4: e7099 doi:10.1371/journal.pone.0007099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jumaa H, et al. 2003. Deficiency of the adaptor SLP-65 in pre-B-cell acute lymphoblastic leukaemia. Nature 423: 452–456 [DOI] [PubMed] [Google Scholar]
- 16. Karasuyama H, Rolink A, Melchers F. 1996. Surrogate light chain in B cell development. Adv. Immunol. 63: 1–41 [DOI] [PubMed] [Google Scholar]
- 17. Karasuyama H, et al. 1994. The expression of Vpre-B/lambda 5 surrogate light chain in early bone marrow precursor B cells of normal and B cell-deficient mutant mice. Cell 77: 133–143 [DOI] [PubMed] [Google Scholar]
- 18. Kawano Y, Yoshikawa S, Minegishi Y, Karasuyama H. 2006. Pre-B cell receptor assesses the quality of IgH chains and tunes the pre-B cell repertoire by delivering differential signals. J. Immunol. 177: 2242–2249 [DOI] [PubMed] [Google Scholar]
- 19. Keenan RA, et al. 2008. Censoring of autoreactive B cell development by the pre-B cell receptor. Science 321: 696–699 [DOI] [PubMed] [Google Scholar]
- 20. Kitamura D, Rajewsky K. 1992. Targeted disruption of mu chain membrane exon causes loss of heavy-chain allelic exclusion. Nature 356: 154–156 [DOI] [PubMed] [Google Scholar]
- 21. Lu R, Medina KL, Lancki DW, Singh H. 2003. IRF-4,8 orchestrate the pre-B-to-B transition in lymphocyte development. Genes Dev. 17: 1703–1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ma S, Pathak S, Trinh L, Lu R. 2008. Interferon regulatory factors 4 and 8 induce the expression of Ikaros and Aiolos to down-regulate pre-B-cell receptor and promote cell-cycle withdrawal in pre-B-cell development. Blood 111: 1396–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maki K, Nagata K, Kitamura F, Takemori T, Karasuyama H. 2000. Immunoglobulin beta signaling regulates locus accessibility for ordered immunoglobulin gene rearrangements. J. Exp. Med. 191: 1333–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Martensson IL, Almqvist N, Grimsholm O, Bernardi AI. 2010. The pre-B cell receptor checkpoint. FEBS Lett. 584: 2572–2579 [DOI] [PubMed] [Google Scholar]
- 25. Martin DA, Lu L, Cascalho M, Wu GE. 2007. Maintenance of surrogate light chain expression induces developmental delay in early B cell compartment. J. Immunol. 179: 4996–5005 [DOI] [PubMed] [Google Scholar]
- 26. Meixlsperger S, et al. 2007. Conventional light chains inhibit the autonomous signaling capacity of the B cell receptor. Immunity 26: 323–333 [DOI] [PubMed] [Google Scholar]
- 27. Mizushima N, Komatsu M. 2011. Autophagy: renovation of cells and tissues. Cell 147: 728–741 [DOI] [PubMed] [Google Scholar]
- 28. Morita S, Kojima T, Kitamura T. 2000. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 7: 1063–1066 [DOI] [PubMed] [Google Scholar]
- 29. Nagata K, et al. 1997. The Ig alpha/Igbeta heterodimer on mu-negative proB cells is competent for transducing signals to induce early B cell differentiation. Immunity 7: 559–570 [DOI] [PubMed] [Google Scholar]
- 30. Ohnishi K, Melchers F. 2003. The nonimmunoglobulin portion of lambda5 mediates cell-autonomous pre-B cell receptor signaling. Nat. Immunol. 4: 849–856 [DOI] [PubMed] [Google Scholar]
- 31. Ouchida R, Kurosaki T, Wang JY. 2010. A role for lysosomal-associated protein transmembrane 5 in the negative regulation of surface B cell receptor levels and B cell activation. J. Immunol. 185: 294–301 [DOI] [PubMed] [Google Scholar]
- 32. Ouchida R, et al. 2008. A lysosomal protein negatively regulates surface T cell antigen receptor expression by promoting CD3zeta-chain degradation. Immunity 29: 33–43 [DOI] [PubMed] [Google Scholar]
- 33. Pak Y, Glowacka WK, Bruce MC, Pham N, Rotin D. 2006. Transport of LAPTM5 to lysosomes requires association with the ubiquitin ligase Nedd4, but not LAPTM5 ubiquitination. J. Cell Biol. 175: 631–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Panigada M, et al. 2002. Constitutive endocytosis and degradation of the pre-T cell receptor. J. Exp. Med. 195: 1585–1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pappu R, et al. 1999. Requirement for B cell linker protein (BLNK) in B cell development. Science 286: 1949–1954 [DOI] [PubMed] [Google Scholar]
- 36. Parker MJ, et al. 2005. The pre-B-cell receptor induces silencing of VpreB and lambda5 transcription. EMBO J. 24: 3895–3905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schebesta M, Pfeffer PL, Busslinger M. 2002. Control of pre-BCR signaling by Pax5-dependent activation of the BLNK gene. Immunity 17: 473–485 [DOI] [PubMed] [Google Scholar]
- 38. Scott LM, Mueller L, Collins SJ. 1996. E3, a hematopoietic-specific transcript directly regulated by the retinoic acid receptor alpha. Blood 88: 2517–2530 [PubMed] [Google Scholar]
- 39. Shinkai Y, et al. 1992. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J. rearrangement. Cell 68: 855–867 [DOI] [PubMed] [Google Scholar]
- 40. Thompson EC, et al. 2007. Ikaros DNA-binding proteins as integral components of B cell developmental-stage-specific regulatory circuits. Immunity 26: 335–344 [DOI] [PubMed] [Google Scholar]
- 41. van Loo PF, Dingjan GM, Maas A, Hendriks RW. 2007. Surrogate-light-chain silencing is not critical for the limitation of pre-B cell expansion but is for the termination of constitutive signaling. Immunity 27: 468–480 [DOI] [PubMed] [Google Scholar]
- 42. Vergarajauregui S, Martina JA, Puertollano R. 2011. LAPTMs regulate lysosomal function and interact with mucolipin 1: new clues for understanding mucolipidosis type IV. J. Cell Sci. 124: 459–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang YH, et al. 2002. Differential surrogate light chain expression governs B-cell differentiation. Blood 99: 2459–2467 [DOI] [PubMed] [Google Scholar]
- 44. Winkler TH, Rolink A, Melchers F, Karasuyama H. 1995. Precursor B cells of mouse bone marrow express two different complexes with the surrogate light chain on the surface. Eur. J. Immunol. 25: 446–450 [DOI] [PubMed] [Google Scholar]
- 45. Yamamoto M, et al. 2006. BASH—novel PKC-Raf-1 pathway of pre-BCR signaling induces kappa gene rearrangement. Blood 108: 2703–2711 [DOI] [PubMed] [Google Scholar]
- 46. Yang Z, Klionsky DJ. 2010. Eaten alive: a history of macroautophagy. Nat. Cell Biol. 12: 814–822 [DOI] [PMC free article] [PubMed] [Google Scholar]