

Abstract
Ribonucleotide reductase (RNR) and deoxycytidylate deaminase (dCMP deaminase) are pivotal allosteric enzymes required to maintain adequate pools of deoxyribonucleoside triphosphates (dNTPs) for DNA synthesis and repair. Whereas RNR inhibition slows DNA replication and activates checkpoint responses, the effect of dCMP deaminase deficiency is largely unknown. Here, we report that deleting the Schizosaccharomyces pombe dcd1+ dCMP deaminase gene (SPBC2G2.13c) increases dCTP ∼30-fold and decreases dTTP ∼4-fold. In contrast to the robust growth of a Saccharomyces cerevisiae dcd1Δ mutant, fission yeast dcd1Δ cells delay cell cycle progression in early S phase and are sensitive to multiple DNA-damaging agents, indicating impaired DNA replication and repair. DNA content profiling of dcd1Δ cells differs from an RNR-deficient mutant. Dcd1 deficiency activates genome integrity checkpoints enforced by Rad3 (ATR), Cds1 (Chk2), and Chk1 and creates critical requirements for proteins involved in recovery from replication fork collapse, including the γH2AX-binding protein Brc1 and Mus81 Holliday junction resolvase. These effects correlate with increased nuclear foci of the single-stranded DNA binding protein RPA and the homologous recombination repair protein Rad52. Moreover, Brc1 suppresses spontaneous mutagenesis in dcd1Δ cells. We propose that replication forks stall and collapse in dcd1Δ cells, burdening DNA damage and checkpoint responses to maintain genome integrity.
INTRODUCTION
The accurate duplication of a eukaryotic genome demands abundant supplies of deoxyribonucleoside triphosphates (dNTPs), which are the building blocks of DNA. Much of the burden for providing both ample and balanced pools of dNTPs falls to two allosteric enzymes: ribonucleotide reductase (RNR) and deoxycytidylate deaminase (dCMP deaminase) (22, 31, 40). RNR plays essential roles in the de novo biosynthesis of all four dNTPs required for DNA synthesis, while dCMP deaminase is specifically involved in the production of dTTP (Fig. 1). Whereas the physiological consequences of RNR defects have been investigated in great detail (9, 15, 47), the effects of dCMP deaminase deficiency are much less well understood, despite its presumptive key roles in efficient genome duplication and in influencing the outcomes of nucleoside-based antitumor and antiviral therapies (24, 25, 34).
Fig 1.
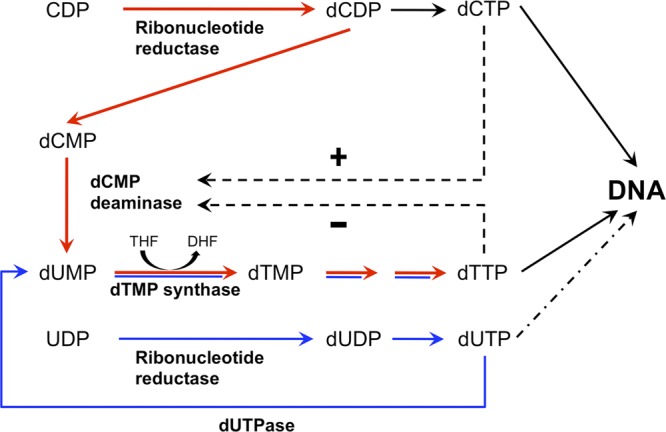
Simplified scheme of de novo dTMP synthesis. De novo synthesis of dCDP is catalyzed by ribonucleotide reductase. Conversion of dCMP to dUMP is catalyzed by dCMP deaminase, which is positively regulated by dCTP (+) and inhibited by dTTP (−). Red arrows indicate the major dTTP synthesis pathway via dCMP deaminase, and blue arrows indicate the minor dTTP synthesis pathway via dUTPase.
Investigations of dCMP deaminase in a genetically tractable organism have been carried out with the budding yeast Saccharomyces cerevisiae (29, 38). As predicted, null mutations of the DCD1 dCMP deaminase gene significantly increase dCTP and decrease dTTP pools, resulting in an ∼125-fold increase in the dCTP/dTTP ratio. Surprisingly, these dNTP pool imbalances do not reduce growth rates or have other obvious effects but rather modestly increase mutagenesis rates (29). In contrast, chemical or mutational inhibition of RNR slows DNA replication and activates checkpoint responses. Thus, RNR and dCMP deaminase deficiencies have quite different effects in Saccharomyces cerevisiae.
The fission yeast Schizosaccharomyces pombe has networks of genome protection mechanisms, ranging from highly specialized lesion repair enzymes to checkpoint mechanisms that coordinate multiple DNA damage responses (60). These include master checkpoint regulators such as the protein kinase Rad3 (ATR in mammals, Mec1 in S. cerevisiae). Rad3 phosphorylates many substrates, including the C terminus of histone H2A in chromatin flanking DNA lesions (45). Phosphohistone H2A (here γH2A), which is analogous to γH2AX in mammals (6), provides a multikilobase recruitment platform for genome protection factors, notably Mdc1 in mammals and Crb2 in fission yeast (14, 27, 55, 59). Recently, we discovered that γH2A also recruits Brc1, a protein with six BRCT domains, via docking of the phosphorylated C terminus of γH2A with the C-terminal pair of BRCT domains in Brc1 (65). This interaction is conserved in S. cerevisiae Rtt107 and mammalian PTIP, which are also 6-BRCT domain proteins (36, 67). Brc1 is important for survival of collapsed replication forks (2). Here, we report that Brc1 and other genome protection proteins are critical for survival in the absence of dCMP deaminase, revealing that unbalanced dNTP pools in the dCMP deaminase mutant profoundly affect DNA replication and repair.
MATERIALS AND METHODS
Strains and genetic methods.
Strains used in this study were made using standard techniques (17) and are listed in Table 1. The complete deletion of the dcd1+ open reading frame was made using pFA6a-KanMX6 as the template and the primers Dcd1 Forward (5′-TGGGGAGATAATCAAACTGAAATACGTTTCTTAAGCTTAAGTAGCTTTGTTTGATTTATTCAGCTGAAGCTTCGTACGCT-3′) and Dcd1 Reverse (5′-ATAAAGATATTAAAATCGTATTTTCTGCAATCAAAAAGAGCGGTAATCTAAAAAGTAAGTCTAGTGGATCTGATATCATC-3′). The PCR product was transformed into the wild-type strain (h− leu1-32) cells by the lithium acetate protocol (43), and disruption was verified by PCR. Tetrad analysis was performed to construct double mutants and verified by PCR.
Table 1.
S. pombe strains used in this study
| Strain | Genotype | Source |
|---|---|---|
| AS237 | h−leu1-32 | This study |
| PR110 | h+leu1-32 ura4-D18 | Laboratory stock |
| AS137 | h−leu1-32 brc1::hphMX6 | This study |
| AS304 | h−leu1-32 dcd1::kanMX6 | This study |
| AS351 | h−leu1-32 ura4-D18 dcd1::kanMX6 | This study |
| AS281 | h−leu1-32 dcd1::kanMX6 brc1::hphMX6 | This study |
| KS1452 | h−leu1-32 ura4-D18 ade6-704 chk1Δ::ura4+ | S. Forsburg |
| AS517 | h+leu1-32 ura4-D18 ade6-704 dcd1::kanMX6 chk1Δ::ura4+ | This study |
| NB2117 | h−leu1-32 ura4-D18 cds1Δ::ura4+ | Laboratory stock |
| KT2752 | h−leu1-32 ura4-D18 cds1-T8A | Laboratory stock |
| AS809 | h+leu1-32 ura4-D18 dcd1::kanMX6 cds1-T8A | This study |
| TM2937 | h−leu1-32 ura4-D18 ade6-M216 his3-D1 rad3::ura4 | Laboratory stock |
| NR1593 | h−leu1-32 ura4-D18 his3-237 rad1::ura4 | Laboratory stock |
| KT2785 | h−leu1-32 ura4-D18 mrc1::ura4 | Laboratory stock |
| AS364 | h−leu1-32 ddb1::hphMX6 | This study |
| AS355 | h−leu1-32 ssb3::hphMX6 | This study |
| AS329 | h−leu1-32 dcd1::kanMX6 ssb3::hphMX6 | This study |
| AS646 | h−leu1-32 swi1::kanMX6 | This study |
| AS738 | h−leu1-32 dcd1::kanMX6 swi1::kanMX6 | This study |
| SP896 | h−leu1-32 ura4-D18 nse5::ura4+ | M. Boddy |
| AS743 | h−leu1-32 ura4-D18 dcd1::kanMX6 nse5::ura4+ | This study |
| AS637 | h+leu1-32 mus81::natMX6 | This study |
| AS860 | h+leu1-32 dcd1::kanMX6 mus81::natMX6 | This study |
| PS2386 | h−leu1-32 ura4-D18 rhp51::ura4+ | Laboratory stock |
| AS661 | h+leu1-32 ura4-D18 dcd1::kanMX6 rhp51::ura4 | This study |
| OL4590 | h−leu1-32 ura4-D18 ctp1::hphMx6 | Laboratory stock |
| AS664 | h+leu1-32 ura4-D18 dcd1::kanMX6 ctp1::HphMX6 | This study |
| LLD3427 | h−ura4-D18 leu1-32 chk1-9myc-2HA6His::ura4 | Laboratory stock |
| AS546 | h−ura4-D18 leu1-32 dcd1::kanMX6 chk1-9myc-2HA6His::ura4 | This study |
| NB2118 | h−ura4-D18 leu1-32 cds1-2HA6His::ura4 | Laboratory stock |
| AS536 | h−ura4-D18 leu1-32 dcd1::kanMX6 cds1-2HA6His::ura4 | This study |
| EM4889 | h+leu1-32 ura4-D18 rad11-GFP::hphMX6 | Laboratory stock |
| AS673 | h+leu1-32 ura4-D18 rad11-GFP::hphMX6 dcd1::kanMX6 | This study |
| YYY4215 | h+leu1-32 rad22-YFP::kanMX6 | Laboratory stock |
| AS701 | h+leu1-32 rad22-YFP::kanMX6 dcd1::kanMX6 | This study |
| TMN3510 | h+leu1-32::[hENT1 leu1+]ura4-D18 ade6-M210 his7-366::[hsv-tk his7+] | S. Forsburg |
| AS873 | h+leu1-32 ura4-D18 ade6-M210 his7-366::[hsv-tk his7+] | This study |
| AS915 | h+leu1-32 ura4-D18 ade6-M210 his7-366::[hsv-tk his7+]dcd1::kanMX6 | This study |
Survival assays.
For DNA damage sensitivity assays, cells were grown in YES (yeast extract, glucose, and supplements) medium to log phase, plated as 10-fold serial dilutions, and treated with indicated amounts of hydroxyurea (HU), camptothecin (CPT), methyl methanesulfonate (MMS), 4-nitroquinoline N-oxide (4-NQO), and bleomycin. For UV treatment, cells were serially diluted onto YES plates and irradiated using a Stratagene UV source. Cell survival was determined after 3 to 4 days at 30°C.
Determination of dNTP and NTP pools.
At a density of 0.4 × 107 to 0.5 × 107 cells/ml, ∼3.70 × 108 (as determined by the optical density at 600 nm [OD600]) cells were harvested by filtration through 25-mm white AAWP nitrocellulose filters (0.8 mm; Millipore AB, Solna, Sweden). To determine the exact number of cells, the cells were also counted in a hemocytometer. The filters were immersed in 700 μl of ice-cold extraction solution (12% [wt/vol] trichloroacetic acid, 15 mM MgCl2) in Eppendorf tubes. The following steps were carried out at 4°C. The tubes were vortexed for 30 s, incubated for 15 min, and vortexed again for 30 s. The filters were removed, and the 700-μl supernatants were collected after centrifugation at 20,000 × g for 1 min and added to 800 μl of ice-cold Freon-trioctylamine mixture (10 ml of 99% pure Freon [1,1,2-trichlorotrifluoroethane; Aldrich, Sigma-Aldrich Sweden AB, Stockholm, Sweden] and 2.8 ml of >99% pure trioctylamine [Fluka, Sigma-Aldrich Sweden AB, Stockholm, Sweden]). The samples were vortexed and centrifuged for 1 min at 20,000 × g. The aqueous phase was collected and added to 700 μl of ice-cold Freon-trioctylamine mixture. Volumes of 475 and 47.5 μl of the aqueous phase were collected. The 475-μl aliquots of the aqueous phase were pH adjusted with 1 M NH4HCO3 (pH 8.9), loaded on boronate columns (Affi-Gel 601; Bio-Rad), and eluted with 50 mM NH4HCO3, pH 8.9, 15 mM MgCl2 to separate dNTPs and NTPs. The eluates with purified dNTPs were adjusted to pH 3.4 with 6 M HCl, separated on a Partisphere SAX-5 high-performance liquid chromatography (HPLC) column (4.6 by 12.5 cm; PolyLC Inc., Columbia, MD), and quantified using a UV-2075 Plus detector (Jasco, Mölndal, Sweden). Nucleotides were isocratically eluted using 0.36 M ammonium phosphate buffer (pH 3.4; containing 2.5% [vol/vol] acetonitrile). The 47.5-μl aliquots of the aqueous phase were adjusted to pH 3.4 and used to quantify NTPs by HPLC in the same way.
Nitrogen starvation and release.
Cells were grown to late-log growth phase in Edinburg minimal medium (EMM), harvested, and washed twice in EMM lacking nitrogen (EMM-N). Starvation and G1 arrest were achieved by resuspending cells in EMM-N to a volume equal to that of the original culture and grown overnight at 25°C. Nitrogen was restored to half of the culture by transfer to a fresh flask and refeeding with an equal volume of EMM plus appropriate supplements and sampled every hour.
Flow cytometry.
Cells were fixed in 70% ethanol and then treated with 0.1 mg/ml RNase A in 10 mM EDTA, pH 8.0, for at least 2 h at 37°C to eliminate RNA. Cells were stained with 1 μM SYTOX Green, sonicated, and analyzed using a FACSCalibur (Becton, Dickinson). Data analysis was carried out with Cell Quest software.
Immunoblots.
For Cds1 and Chk1 shifts, whole-cell extracts were prepared from exponentially growing cells in standard NP-40 lysis buffer (50 mM Tris [pH 8.0], 150 mM NaCl, 2.5 mM EDTA, 0.002% NP-40, 50 mM NaF, protease inhibitor tablet [Complete Mini; Roche]). Protein amounting to ∼100 mg was resolved by SDS-PAGE using 10% gels with an acrylamide/bisacrylamide ratio of 99:1. Proteins were transferred to nitrocellulose membranes, blocked with 5% milk in Tris-buffered saline with 0.05% Tween, and probed with antihemagglutinin (12C5) antibody (Roche).
Microscopy.
Cells were photographed using a Nikon Eclipse E800 microscope equipped with a Photometrics Quantix charge-coupled device (CCD) camera and IPLab Spectrum software. All fusion proteins were expressed at their own genomic locus. Rad52(Rad22)-yellow fluorescent protein (YFP)- and Rad11-green fluorescent protein (GFP)-expressing strains were grown in EMM until mid-log phase for focus quantification assays. Quantification was performed by scoring 500 or more nuclei from three independent experiments.
Mutagenesis assay.
A mutation frequency assay was carried out as described previously (26). Yeast strains used in this assay contain the wild-type ura4+ gene at their endogenous locus. The strains were first grown on EMM plates without uracil to select for cells harboring wild-type ura4+. For analysis of spontaneous mutations, the ura4+ cells were plated on minimal medium containing uracil to allow accumulation of mutations. Colonies (≥11) were grown to late log phase in EMM containing 0.1 mg/ml uracil. Each culture (1 ml) was plated onto EMM agar plates containing 1 mg/ml 5-fluoro-orotic acid (FOA), 0.1 mg/ml uracil. FOA-resistant colonies (FOAr) were counted after 7 to 10 days of growth. Total cells were determined by plating 100 μl of a 10−5 dilution onto EMM plates with 0.1 mg/ml uracil. Each assay was repeated at least 3 times. Mutation rates were calculated by fluctuation analysis using the Lea-Coulson method of the median by using the FALCOR program (20).
RESULTS
Brc1 is crucial in the absence of dCMP deaminase.
Brc1 is required for survival of DNA-damaging agents that disrupt DNA replication (57). To gain new insights into Brc1, we carried out a genetic interaction screen known as epistatic mini-array profiles (E-MAP) (52) in which brc1Δ was combined with deletions of ∼3,000 nonessential genes (28). The complete results of the screen will be described elsewhere; here, we focus on the negative genetic interaction between Brc1 and SPBC2G2.13c, which encodes a predicted dCMP deaminase that converts dCMP into dUMP, leading to the production of dTTP (Fig. 1). As SPBC2G2.13c is the ortholog of S. cerevisiae DCD1, we adopted the name dcd1+, but it should be noted that dcd1+ is also the obsolete name for pim1+, which encodes a Ran GDP/GTP exchange factor (63). We confirmed the genetic interaction between dcd1Δ and brc1Δ by creating and testing a new dcd1Δ null allele, which caused a modest growth defect that was substantially enhanced when combined with brc1Δ (Fig. 2, untreated). The dcd1Δ cells were mildly sensitive to 4-nitroquinoline N-oxide (4-NQO), which is consistent with a recent screen of the haploid deletion collection (11). The dcd1Δ cells were less sensitive to 4-NQO than brc1Δ cells, but they were noticeably more sensitive to hydroxyurea (HU), which inhibits DNA replication by inactivating RNR, and bleomycin, which causes double-strand breaks (DSBs). Unlike brc1Δ cells, the dcd1Δ mutant was largely resistant to the effects of the topoisomerase I inhibitor camptothecin (CPT) and the DNA alkylating agent methyl methanesulfonate (MMS), which cause replication fork stalling and collapse. Both dcd1Δ and brc1Δ mutants were modestly sensitive to UV. Double mutant dcd1Δ brc1Δ cells appeared to be highly sensitive to many types of DNA-damaging agents. The negative genetic interactions between dcd1Δ and brc1Δ mutations imply that Dcd1 and Brc1 independently act in genome maintenance pathways that are partially complementary.
Fig 2.
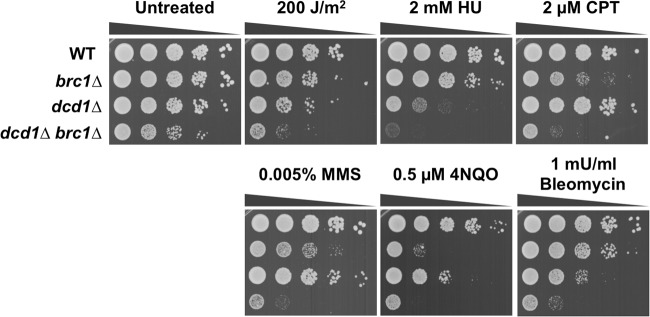
Genetic interaction between Dcd1 and Brc1. Tenfold serial dilutions of cells were exposed to the indicated DNA-damaging agents, and plates were incubated at 30°C for 3 to 4 days.
dCMP deaminase is required to maintain balanced pools of dNTPs.
We used high-performance liquid chromatography to measure the cellular levels of dNTPs in log-phase cultures of wild-type and dcd1Δ cells (32). In the dcd1Δ mutant the dTTP concentration decreased 4-fold while that of dCTP increased 30-fold (Fig. 3). These data are consistent with dCMP deaminase diverting dNTP synthesis from dCTP to dTTP (Fig. 1) and with evidence that thymidylate depletion increases dCTP pools due to the lack of the feedback inhibition by dTTP on RNR (33). Smaller changes in dATP (2.5-fold increase) and dGTP (∼2-fold decrease) were also detected in the dcd1Δ mutant (Fig. 3).
Fig 3.
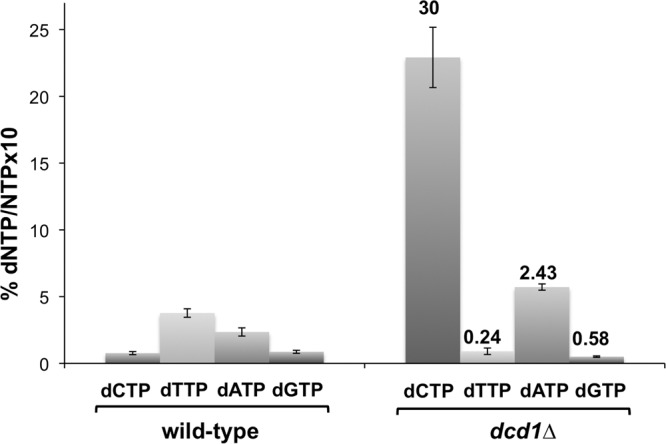
dCMP deaminase deficiency imbalances dNTPs pools. dNTP/NTP ratios are represented as means ± SD. Numbers above the bars indicate fold increase or fold decrease relative to the wild type.
Different DNA replication effects caused by dCMP deaminase deficiency versus reduction of RNR.
We used flow cytometry to measure DNA content in log-phase dcd1Δ cultures (54). Whereas the haploid wild type had a single ∼2C DNA peak because G1 is very short and completion of S phase coincides with separation of daughter cells, the dcd1Δ mutant displayed a large enrichment of cells with a <2C DNA content, with a prominent peak indicating a cell cycle delay in G1 or early S phase (Fig. 4). To investigate this defect further, we monitored DNA content in cells synchronously released from a G1-phase arrest imposed by nitrogen starvation (see Materials and Methods). Upon nitrogen refeeding, progression into S phase in the dcd1Δ cells was delayed 1 additional hour relative to the wild type. This analysis also revealed that dcd1Δ cells were unable to efficiently arrest in G1 phase when nitrogen starved, although very few cells appeared to arrest in S phase (Fig. 4). This dcd1Δ mutant phenotype may arise from an inability to complete S phase in a timely manner before nitrogen starvation halts growth, perhaps coupled with DNA damage checkpoint activation.
Fig 4.
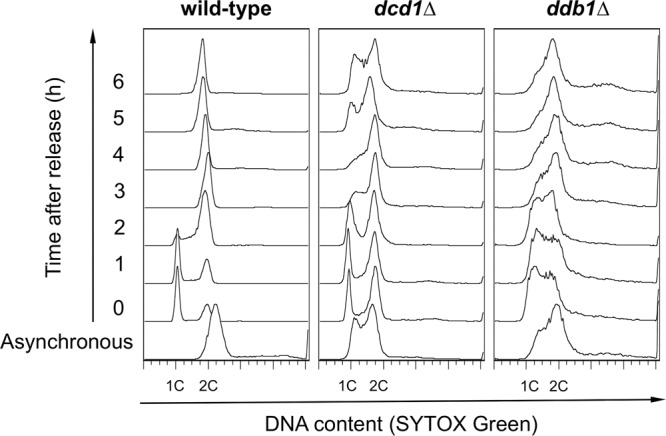
dcd1+ deletion slows progression through S phase. Flow cytometer analysis of wild-type, dcd1Δ, and ddb1Δ strains. Cells were arrested in G1 phase by nitrogen starvation for 16 h at 25°C, released from the block at 30°C in EMM containing nitrogen, and sampled every hour.
We performed a similar experiment with a ddb1Δ mutant, which is unable to degrade the RNR inhibitor protein Spd1 and has a more balanced ∼3-fold decrease in all dNTP pools (23). Compared to dcd1Δ, this mutant displayed a less pronounced early S phase peak, but it had many more cells with an intermediate DNA content when starved of nitrogen. These effects are typical of cells treated with HU. The recovery kinetics from nitrogen starvation appeared to be similar in the two mutants. These data indicated that dCMP deficiency and failure to relieve inhibition of RNR have quite different effects on DNA replication.
Suppression of dCMP deaminase deficiency by expression of Herpes simplex virus thymidine kinase.
Yeast cells are unable to salvage thymidine as they lack a thymidine kinase gene, therefore they completely rely on the de novo pathway to generate dTTP (Fig. 1) (19). A question raised by our studies is whether the phenotypes of dcd1Δ cells can be rescued by providing an alternative pathway for producing dTTP. We addressed this question by expressing the Herpes simplex virus thymidine kinase gene (hsv-tk), allowing conversion of thymidine to dTMP, which then enters the pathway of dTTP conversion (21, 58). Expression of hsv-tk in dcd1Δ cells suppressed their growth defect and restored resistance to UV, HU, 4NQO, and bleomycin (Fig. 5A). It also restored the flow cytometry profile of DNA content to a nearly normal pattern, with just a small number of cells in S phase (Fig. 5B).
Fig 5.
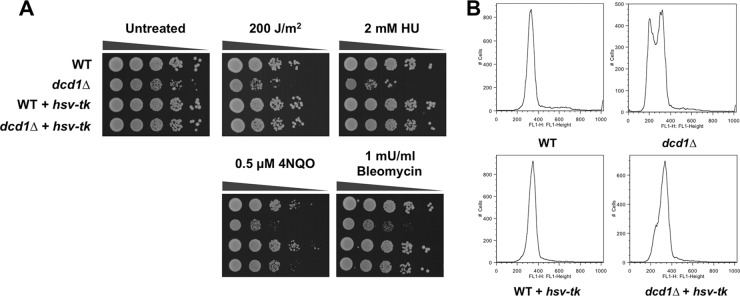
Suppression of dcd1Δ sensitivity phenotypes by expressing a herpes simplex virus thymidine kinase gene (hsv-tk). (A) Tenfold serial dilutions of cells were exposed to the indicated DNA-damaging agents, and plates were incubated at 30°C for 3 to 4 days. (B) Flow cytometer analysis of asynchronous, exponentially growing cultures of wild-type and dcd1Δ strains with or without expression of the hsv-tk gene.
Replication stress and DNA damage checkpoints are crucial in the absence of dCMP deaminase.
Replication fork stalling activates the master checkpoint kinase Rad3 (ATR), which in turn activates the downstream effector kinase Cds1, which is required for recovery from replication fork arrest (51, 68, 69). We crossed dcd1Δ cells to rad3Δ and cds1Δ cells to assess the importance of the replication checkpoint. Tetrad analysis revealed that dcd1Δ was synthetically lethal with rad3Δ and cds1Δ. We also found that dcd1Δ was synthetically lethal with mrc1Δ (Table 2). Mrc1 is a checkpoint mediator protein required for activation of Cds1 by Rad3 (1, 62). To further probe the genetic interactions between Dcd1 and Cds1, we crossed dcd1Δ cells with the hypomorphic cds1-T8A mutant, which partially decreases Cds1 kinase activity and causes mild sensitivity to HU and other genotoxins that impair DNA replication (61). We observed an enhanced growth defect in the dcd1Δ cds1-T8A double mutant, which was further accentuated in cells treated with UV, HU, CPT, or MMS (Fig. 6C).
Table 2.
Summary of genetic interactions involving dcd1Δa
| Allele | Function | Treatment |
||||
|---|---|---|---|---|---|---|
| Untreated | UV | HU | CPT | MMS | ||
| brc1Δ | BRCT domain protein | Yes | YES | YES | YES | YES |
| rad3Δ | ATR checkpoint kinase | SL | ||||
| chk1Δ | DNA damage checkpoint kinase | YES | YES | YES | YES | YES |
| cds1Δ | DNA replication checkpoint kinase | SL | ||||
| cds1.T8A | DNA replication checkpoint kinase mutant | Yes | YES | YES | YES | YES |
| mrc1Δ | Mediator of replication checkpoint 1 | SL | ||||
| rad1Δ | Checkpoint clamp complex protein | SL | ||||
| ddb1Δ | Damaged DNA binding protein | SL | ||||
| ssb3Δ | DNA replication factor A subunit | YES | YES | YES | YES | YES |
| swi1Δ | Replication fork protection complex subunit | YES | YES | YES | YES | YES |
| nse5Δ | Smc5-6 complex non-SMC subunit | YES | YES | YES | YES | YES |
| mus81Δ | Holliday junction resolvase subunit | YES | YES | YES | YES | YES |
| rhp51Δ | RecA family recombinase | YES | YES | YES | YES | YES |
| ctp1Δ | CtIP-related endonuclease | Yes | YES | YES | YES | YES |
Double mutants were assessed for growth in the absence or presence of specified genotoxins. YES, strong negative interaction; Yes, negative interaction; SL, synthetically lethal.
Fig 6.

Replication and DNA damage checkpoint responses in dcd1Δ cells. (A) After HU treatment, Cds1 is phosphorylated in control and dcd1Δ cells, as indicated by the appearance of a slow-mobility species. Cds1 undergoes activating phosphorylation in dcd1Δ cells in the absence of damage. (B) After CPT and MMS treatment, Chk1 is phosphorylated in control cells, as indicated by the appearance of a slow-mobility species. Chk1 undergoes activating phosphorylation in untreated dcd1Δ cells. (C) Phenotype of dcd1Δ cells in combination with the cds1-T8A mutant. (D) Phenotype of dcd1Δ cells in combination with chk1Δ. Tenfold serial dilutions of cells were exposed to the indicated DNA-damaging agents, and plates were incubated at 30°C for 3 to 4 days.
These data established that the Rad3-Mrc1-Cds1 signaling pathway is essential in dcd1Δ cells, suggesting that replication stalling in these cells activates the replication stress checkpoint. To address this question, we compared Cds1 phosphorylation in wild-type and dcd1Δ cells. Treatment of wild-type cells with HU triggers replication arrest and Cds1 activation as assessed by the appearance of a retarded electrophoretic mobility form of Cds1 (Fig. 6A). In dcd1Δ cells, phospho-Cds1 was detected even in the absence of HU treatment (Fig. 6A). These data indicated that Cds1 is activated in each cell cycle in the dcd1Δ mutant, which explains why Cds1 is essential in these cells.
Replication fork collapse creates one-ended DSBs that trigger Rad3 to activate Chk1, which is required to delay the onset of mitosis when DNA remains damaged after the completion of S phase. A strong negative genetic interaction was observed in dcd1Δ chk1Δ cells (Fig. 6D, untreated), which was enhanced in the presence of UV, HU, CPT, and MMS (Fig. 6D). These data suggested that increased spontaneous DNA damage in dcd1Δ cells activates Chk1. Indeed, immunoblot assays that detect phospho-Chk1 confirmed that Chk1 is activated even in the absence of genotoxin treatment in dcd1Δ cells (Fig. 6B). Taken together, these data strongly indicate that abnormal dNTP pools in dcd1Δ cells lead to replication fork stalling and collapse.
Increased RPA and Rad52 foci in dcd1Δ cells.
The Chk1 activation in dcd1Δ cells, and the strong negative genetic interaction between dcd1Δ and chk1Δ, suggested that unbalanced dNTP pools in dcd1Δ cells lead to increased spontaneous DNA damage. To test this proposition, we monitored Rad11-GFP foci in dcd1Δ cells. Rad11 is the S. pombe ortholog of RPA1, which is a subunit of replication protein A (RPA), which is the major single-strand DNA (ssDNA)-binding protein in eukaryotic cells (48, 66). We observed a large increase in cells with Rad11-GFP foci in the dcd1Δ strain (∼35%) compared to the wild type (∼9%), indicating increased replication fork stalling and/or spontaneous DNA damage (Fig. 7A). We also monitored Rad52-YFP foci in dcd1Δ cells. Rad52 (also called Rad22) is essential for DSB repair by homologous recombination (HR). Many mutants with genome maintenance defects have increased numbers of Rad52 foci (13, 41, 46). About 9% of wild-type cells had spontaneous Rad52-YFP foci, whereas this percentage increased 2.3-fold (∼21%) in dcd1Δ cells (Fig. 7B). These findings suggested that dcd1Δ cells suffer increased rates of spontaneous DNA damage.
Fig 7.
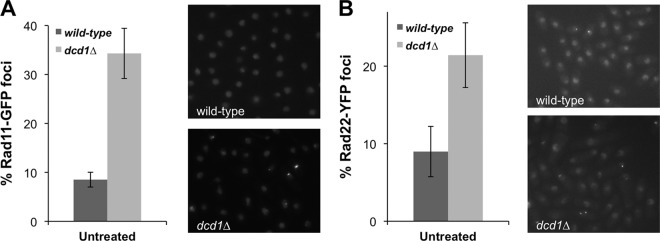
Rad11Rpa1 and Rad52 (Rad22) foci are increased in dcd1Δ cells. Rad11-GFP (A) and Rad22-YFP (B) form nuclear foci that increased in number in the absence of Dcd1. Cells expressing endogenous Rad11-GFP or Rad22-YFP in a wild-type and dcd1Δ background were cultured in minimal medium at 25°C until mid-log phase. Foci were scored in three independent experiments, and the mean values are represented. Error bars correspond to standard deviations of the means.
Pathways required for recovery from replication fork collapse are crucial in the absence of Dcd1.
These data suggested that dcd1Δ cells suffer increased rates of replication fork collapse, leading to formation of single-strand DNA and DNA repair foci and activation of the replication stress and DNA damage checkpoints. To further explore these effects, we performed a genetic epistasis analysis of dcd1Δ cells with genes involved in different pathways of DNA replication and repair. Double mutant strains were assessed for growth in dilution series on rich growth medium in the absence or presence of different genotoxins (Fig. 8). The data are summarized in Table 2. Deletion of the dcd1+ gene is lethal in the absence of Rad1, which is a component of the 9-1-1 checkpoint clamp involved in the replication stress and DNA damage checkpoints (49). There was also a synthetic lethal interaction between dcd1Δ and ddb1Δ. As noted above, Ddb1 is required for the degradation of Spd1, which is an inhibitor of RNR (23). We also detected strong negative interactions with mutations that eliminate Ssb3, which is a small nonessential subunit of RPA (8); Swi1, a component with Swi3 of the replication fork protection complex (FPC), which stabilizes replication forks in a configuration that is recognized by replication checkpoint sensors (46); Nse5, a nonessential subunit of the Smc5-6 complex, which is crucial for recovery from replication-associated DNA damage (7, 50); Mus81, which associates with Eme1 to form a heterodimeric endonuclease that resolves Holliday junctions and is required for recovery from collapsed replication forks and resolution of crossovers in meiosis (4, 12, 18); and Rhp51, the Rad51 ortholog required for most types of HR repair (44). We also detected a weaker negative genetic interaction with Ctp1, which is an HR repair factor required for the initiation of resection of DSBs prior to repair (35, 37).
Fig 8.

Genetic interactions involving Dcd1 and components required for recovery from replication fork collapse. Phenotypes of dcd1Δ in combination with ssb3Δ (A), swi1Δ (B), nse5Δ (C), and mus81Δ (D). Tenfold serial dilutions of cells were exposed to the indicated DNA-damaging agents, and plates were incubated at 30°C for 3 to 4 days.
All these data suggest that dCMP deaminase deficiency leads to replication fork stalling and collapse, necessitating requirements for genome maintenance pathways that stabilize replication forks and carry out the repair of collapsed replication forks.
Brc1 protects genome integrity in dcd1Δ cells.
The phenotypes of dcd1Δ cells strongly suggest that dCMP deaminase is required for efficient DNA replication and repair, with the consequences of a dCMP deaminase defect being increased genetic instability. To directly test whether deletion of dcd1+ increases the spontaneous mutation rate, we used a mutation rate assay that detects mutations inactivating the ura4+ gene which confer resistance to FOA as described previously (26). Surprisingly, despite the acute dNTP pool imbalance in dcd1Δ cells, the mutation rate was unaffected (Table 3).
Table 3.
Effect of dCMP deaminase deficiency on spontaneous mutation ratea
| Strain | Mutation frequencyb (per 108/generation) | CI | Ratioc |
|---|---|---|---|
| Wild type | 3.9986 | 5.25–2.36 | 1.0 |
| rad2Δ | 41.1053 | 68.64–27.55 | 10.3 |
| dcd1Δ | 4.2736 | 6.11–3.22 | 1.1 |
| brc1Δ | 2.8495 | 3.78–1.74 | 0.7 |
| dcd1Δ brc1Δ | 15.1217 | 26.91–7.81 | 3.8 |
Mutation rates are the averages from three or more experiments. CI is the 95% confidence interval (10−8 per cell division).
Mutation rates were calculated by using the Lea-Coulson method of the median (20).
Ratio is relative to the wild type.
From these data, we hypothesized that other genome protection factors are important for reducing the spontaneous mutation rate in dcd1Δ cells. As a first step to investigate this possibility, we examined the role of Brc1. Whereas brc1Δ, like dcd1Δ, did not increase the spontaneous mutation rate, the brc1Δ dcd1Δ double mutant displayed an ∼4-fold increase in spontaneous FOA-resistant colonies (Table 3). From these results, we conclude that Brc1 is required to maintain genome integrity in the cells lacking dCMP deaminase.
DISCUSSION
dCMP deaminase converts dCMP to dUMP, which is the nucleotide substrate for thymidylate synthase that is required for dTTP synthesis. Its activity is allosterically regulated by the ratio of dCTP to dTTP, with dCTP as an activator and dTTP as an inhibitor (Fig. 1). With this central role in deoxyribonucleotide metabolism, dCMP deaminase should be crucial for efficient DNA replication and repair and for maintaining genome integrity. However, studies of S. cerevisiae suggest that eliminating dCMP deaminase has mild consequences, with the principal phenotype being a small increase in spontaneous mutagenesis (29). This mild phenotype contrasts with the profound consequences of decreasing RNR activity by chemical or genetic means. In this study, we have investigated dCMP deaminase in fission yeast, where we find that deleting the predicted dCMP deaminase gene increases dCTP ∼30-fold and decreases dTTP ∼4-fold. Expansion of the dCTP pool is likely due to the allosteric properties of RNR, which is regulated by dTTP but not dCTP, hence dCDP production was not diminished (33). Similar dNTP imbalances have been reported in other species (30, 39, 42, 64). Our key discovery is that dNTP imbalance in dcd1Δ cells delays progression in early S phase and creates critical requirements for key genome maintenance factors. These data establish that provision of balanced pools of dNTPs by dCMP deaminase is crucial for genome stability in fission yeast, in contrast to the situation in S. cerevisiae.
In many respects, the phenotypes of fission yeast dcd1Δ cells parallel the effects of decreased RNR activity. Notably, our studies revealed that dcd1Δ is synthetically lethal with mutations that delete Rad3, Cds1, and Mrc1, which are all critical for survival of HU treatment. Moreover, we found that dcd1Δ has a strong negative genetic interaction with cds1-T8A, which partially decreases Cds1 activity (61). The evidence indicating higher activation of Cds1 during the cell cycle in dcd1Δ cells supports these genetic data. These findings establish that the dcd1Δ mutant creates a requirement for a replication stress checkpoint. The dNTP imbalance in the dcd1Δ mutant, especially the low levels of dTTP, likely limits DNA synthesis and causes replication fork stalling, which leads to activation of the Rad3-Mrc1-Cds1 replication stress checkpoint. Also, this fork stalling may lead to replication fork collapse, triggering a DNA damage checkpoint, which explains why there is a strong negative genetic interaction between dcd1Δ and chk1Δ. Supporting this idea, dcd1Δ cells displayed higher activation of Chk1 in cycling cells and showed an elevated number of RPA and Rad52 foci, suggesting high rates of spontaneous DNA damage.
Indeed, dcd1Δ had negative genetic interactions with a large number of genes involved in genome protection, including Brc1, Ssb3, Mus81, Nse5, and Swi1. The negative genetic interaction with brc1Δ is noteworthy because Brc1 has an important role in promoting recovery from replication fork collapse (57, 65). The requirement for Mus81 in dcd1Δ cells is particularly informative, as besides its essential meiotic function, Mus81 is also required for recovery from a site-specific broken replication fork (4, 5, 53). It is thought that Mus81, in a heterodimeric complex with Eme1, cleaves Holliday junctions that are formed during reestablishment of a broken replication fork by homology-directed repair.
Although there are many parallels in the effects of dCMP deaminase and RNR deficiency in fission yeast, our data also suggest there are clear differences. In particular, we note that DNA content profiling of dcd1Δ cells reveals a prominent ∼1C DNA peak that indicates a cell cycle delay in late G1 or early S phase (Fig. 4). In contrast, cells with reduced RNR activity (e.g., ddb1Δ mutant or HU-treated wild-type cells) typically display a broader distribution of cells in S phase with DNA contents between 1C and 2C. An even more striking difference was observed in the DNA content profiles of nitrogen-starved dcd1Δ and ddb1Δ cells, in which the latter had many more cells arrested in S phase. These data suggest that dNTP pool imbalance in dcd1Δ cells inhibits entry into S phase, or, once having entered S phase and triggered a replication checkpoint response, they have more biochemical options for providing adequate supplies of dTTP to complete S phase. These options may operate more effectively in budding yeast, which could explain the mild phenotypes of dcd1Δ mutants in S. cerevisiae. Alternatively, the DNA polymerases in budding yeast may work more effectively with unbalanced pools of dNTPs.
The different phenotypes of dcd1Δ mutants in budding yeast and fission yeast suggest there are considerable variations in other species and cell types. In studies that predated molecular genetic characterizations, mouse S49 cells defective for dCMP deaminase had nearly normal doubling times with a somewhat prolonged S phase (16), whereas V79 hamster fibroblasts lacking dCMP deaminase had almost twice the generation time of a control (3). We speculate that the viability of these cell lines has depended on the activation of checkpoint and DNA repair mechanisms, as we have observed with fission yeast.
Mouse S49 dCMP deaminase-deficient cells (16) exhibited a 10- to 20-fold increase in spontaneous mutation rates (64). Qualitatively similar results were observed in budding yeast, in which a dcd1Δ mutant had a 2.7-fold increase in spontaneous mutation rates (29), and in Escherichia coli infected with a dCMP deaminase deletion mutant of phage T4, in which mutagenesis increased 10- to 1,000-fold (56). The E. coli study observed a specific increase in AT→GC transition mutation. On the other hand, most of the mutations in the yeast study were GC→CG transversion mutations, an event that is not straightforwardly expected from the dNTP pool imbalance in these cells. These data indicate that mutagenesis induced by a DNA precursor pool imbalance involves factors other than concentration-dependent competition between nucleotides that are correctly and incorrectly base paired to a template base. In contrast, in a dCMP deaminase-defective hamster V79 cell line, the same dNTP imbalance had only a small effect on the spontaneous mutation rate (10). In our study, we found that the spontaneous mutation rate was unaffected in S. pombe dcd1Δ cells. However, protection against spontaneous mutations in S. pombe dcd1Δ cells depended on at least Brc1 and probably other genome maintenance factors. As seen for the viability of dCMP deaminase mutants, it is likely that mutation rates are kept in check by multiple DNA repair mechanisms, and these may play as important roles in mammalian cells as they do in fission yeast. In this regard, it will be interesting in future experiments to assess the spectrum of mutations in brc1Δ dcd1Δ cells.
Deoxycytidine analogs such as gemcitabine (2′,2′-difluorodeoxycytidine) have strong antineoplastic activities that have led to their widespread use in clinical settings for treatment of solid tumors, including non-small-cell lung cancer, pancreatic cancer, bladder cancer, and breast cancer (24, 25, 34). The anticancer activity of gemcitabine is affected by metabolic inactivation through a pathway requiring dCMP deaminase. Thus, individual variation in dCMP deaminase activity likely influences the therapeutic response to treatment with gemcitabine, as well as to related nucleosides used in antiviral therapies. Moreover, dCMP deaminase may play an important role in tumors developing resistance to gemcitabine, which is a major problem in the clinic. The discovery of strong genetic interactions involving dCMP deaminase and genome maintenance factors in fission yeast, most of which are conserved in humans, may open up new opportunities for improving the therapeutic activities of deoxycytidine analogs.
ACKNOWLEDGMENTS
We thank Oliver Limbo for expert advice and assistance.
S.S. was supported by the Wenner-Gren Foundations. This work was funded by the Swedish Foundation for Strategic Research, the Swedish Research Council, and the Swedish Cancer Society (to A.C.); QB3@UCSF and NIH grants GM084448, GM084279, GM081879, and GM098101 (to N.J.K.); and NIH grants GM59447 and CA77325 (to P.R.).
Footnotes
Published ahead of print 27 August 2012
REFERENCES
- 1.Alcasabas AA, et al. 2001. Mrc1 transduces signals of DNA replication stress to activate Rad53. Nat. Cell Biol. 3:958–965 [DOI] [PubMed] [Google Scholar]
- 2.Bass KL, Murray JM, O'Connell MJ. 2012. Brc1-dependent recovery from replication stress. J. Cell Sci. 125:2753–2764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bianchi V, Pontis E, Reichard P. 1987. Regulation of pyrimidine deoxyribonucleotide metabolism by substrate cycles in dCMP deaminase-deficient V79 hamster cells. Mol. Cell. Biol. 7:4218–4224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boddy MN, et al. 2001. Mus81-Eme1 are essential components of a Holliday junction resolvase. Cell 107:537–548 [DOI] [PubMed] [Google Scholar]
- 5.Boddy MN, et al. 2000. Damage tolerance protein Mus81 associates with the FHA1 domain of checkpoint kinase Cds1. Mol. Cell. Biol. 20:8758–8766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonner WM, et al. 2008. GammaH2AX and cancer. Nat. Rev. Cancer 8:957–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bustard DE, et al. 2012. During replication stress Non-Smc-Element 5 is required for Smc5/6 complex functionality at stalled forks. J. Biol. Chem. 287:11374–11383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cavero S, Limbo O, Russell P. 2010. Critical functions of Rpa3/Ssb3 in S-phase DNA damage responses in fission yeast. PLoS Genet. 6:e1001138 doi:10.1371/journal.pgen.1001138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cerqueira NM, Fernandes PA, Ramos MJ. 2007. Ribonucleotide reductase: a critical enzyme for cancer chemotherapy and antiviral agents. Rec. Pat. Anticancer Drug Discov. 2:11–29 [DOI] [PubMed] [Google Scholar]
- 10.Dare E, Zhang LH, Jenssen D, Bianchi V. 1995. Molecular analysis of mutations in the hprt gene of V79 hamster fibroblasts: effects of imbalances in the dCTP, dGTP and dTTP pools. J. Mol. Biol. 252:514–521 [DOI] [PubMed] [Google Scholar]
- 11.Deshpande GP, et al. 2009. Screening a genome-wide S. pombe deletion library identifies novel genes and pathways involved in genome stability maintenance. DNA Repair 8:672–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doe CL, Ahn JS, Dixon J, Whitby MC. 2002. Mus81-Eme1 and Rqh1 involvement in processing stalled and collapsed replication forks. J. Biol. Chem. 277:32753–32759 [DOI] [PubMed] [Google Scholar]
- 13.Du LL, Nakamura TM, Moser BA, Russell P. 2003. Retention but not recruitment of Crb2 at double-strand breaks requires Rad1 and Rad3 complexes. Mol. Cell. Biol. 23:6150–6158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Du LL, Nakamura TM, Russell P. 2006. Histone modification-dependent and -independent pathways for recruitment of checkpoint protein Crb2 to double-strand breaks. Genes Dev. 20:1583–1596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elledge SJ, Zhou Z, Allen JB. 1992. Ribonucleotide reductase: regulation, regulation, regulation. Trends Biochem. Sci. 17:119–123 [DOI] [PubMed] [Google Scholar]
- 16.Eriksson S, Skog S, Tribukait B, Jaderberg K. 1984. Deoxyribonucleoside triphosphate metabolism and the mammalian cell cycle. Effects of thymidine on wild-type and dCMP deaminase-deficient mouse S49 T-lymphoma cells. Exp. Cell Res. 155:129–140 [DOI] [PubMed] [Google Scholar]
- 17.Forsburg SL, Rhind N. 2006. Basic methods for fission yeast. Yeast 23:173–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gaillard PH, Noguchi E, Shanahan P, Russell P. 2003. The endogenous Mus81-Eme1 complex resolves Holliday junctions by a nick and counternick mechanism. Mol. Cell 12:747–759 [DOI] [PubMed] [Google Scholar]
- 19.Grivell AR, Jackson JF. 1968. Thymidine kinase: evidence for its absence from Neurospora crassa and some other micro-organisms, and the relevance of this to the specific labelling of deoxyribonucleic acid. J. Gen. Microbiol. 54:307–317 [DOI] [PubMed] [Google Scholar]
- 20.Hall BM, Ma CX, Liang P, Singh KK. 2009. Fluctuation analysis CalculatOR: a web tool for the determination of mutation rate using Luria-Delbruck fluctuation analysis. Bioinformatics 25:1564–1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hodson JA, Bailis JM, Forsburg SL. 2003. Efficient labeling of fission yeast Schizosaccharomyces pombe with thymidine and BUdR. Nucleic Acids Res. 31:e134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hofer A, Crona M, Logan DT, Sjoberg BM. 2012. DNA building blocks: keeping control of manufacture. Crit. Rev. Biochem. Mol. Biol. 47:50–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holmberg C, et al. 2005. Ddb1 controls genome stability and meiosis in fission yeast. Genes Dev. 19:853–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jansen RS, Rosing H, Schellens JH, Beijnen JH. 2011. Deoxyuridine analog nucleotides in deoxycytidine analog treatment: secondary active metabolites? Fundam. Clin. Pharmacol. 25:172–185 [DOI] [PubMed] [Google Scholar]
- 25.Jordheim LP, Dumontet C. 2007. Review of recent studies on resistance to cytotoxic deoxynucleoside analogues. Biochim. Biophys. Acta 1776:138–159 [DOI] [PubMed] [Google Scholar]
- 26.Kai M, Taricani L, Wang TS. 2006. Methods for studying mutagenesis and checkpoints in Schizosaccharomyces pombe. Methods Enzymol. 409:183–194 [DOI] [PubMed] [Google Scholar]
- 27.Kilkenny ML, et al. 2008. Structural and functional analysis of the Crb2-BRCT2 domain reveals distinct roles in checkpoint signaling and DNA damage repair. Genes Dev. 22:2034–2047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim DU, et al. 2010. Analysis of a genome-wide set of gene deletions in the fission yeast Schizosaccharomyces pombe. Nat. Biotechnol. 28:617–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kohalmi SE, Glattke M, McIntosh EM, Kunz BA. 1991. Mutational specificity of DNA precursor pool imbalances in yeast arising from deoxycytidylate deaminase deficiency or treatment with thymidylate. J. Mol. Biol. 220:933–946 [DOI] [PubMed] [Google Scholar]
- 30.Kohalmi SE, Kunz BA. 1993. Mutational specificity of thymine nucleotide depletion in yeast. Mutat. Res. 289:73–81 [DOI] [PubMed] [Google Scholar]
- 31.Kumar D, et al. 2011. Mechanisms of mutagenesis in vivo due to imbalanced dNTP pools. Nucleic Acids Res. 39:1360–1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kumar D, Viberg J, Nilsson AK, Chabes A. 2010. Highly mutagenic and severely imbalanced dNTP pools can escape detection by the S-phase checkpoint. Nucleic Acids Res. 38:3975–3983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kunz BA. 1982. Genetic effects of deoxyribonucleotide pool imbalances. Environ. Mutagen. 4:695–725 [DOI] [PubMed] [Google Scholar]
- 34.Lamba JK. 2009. Genetic factors influencing cytarabine therapy. Pharmacogenomics 10:1657–1674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Langerak P, Mejia-Ramirez E, Limbo O, Russell P. 2011. Release of Ku and MRN from DNA ends by Mre11 nuclease activity and Ctp1 is required for homologous recombination repair of double-strand breaks. PLoS Genet. 7:e1002271 doi:10.1371/journal.pgen.1002271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li X, et al. 2012. Structure of C-terminal tandem BRCT repeats of Rtt107 protein reveals critical role in interaction with phosphorylated histone H2A during DNA damage repair. J. Biol. Chem. 287:9137–9146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Limbo O, et al. 2007. Ctp1 is a cell-cycle-regulated protein that functions with Mre11 complex to control double-strand break repair by homologous recombination. Mol. Cell 28:134–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liskay RM, Wheeler LJ, Mathews CK, Erdeniz N. 2007. Involvement of deoxycytidylate deaminase in the response to S(n)1-type methylation DNA damage in budding yeast. Curr. Biol. 17:R755–R757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maley F, Maley GF. 1990. A tale of two enzymes, deoxycytidylate deaminase and thymidylate synthase. Prog. Nucleic Acid Res. Mol. Biol. 39:49–80 [DOI] [PubMed] [Google Scholar]
- 40.Mathews CK. 2006. DNA precursor metabolism and genomic stability. FASEB J. 20:1300–1314 [DOI] [PubMed] [Google Scholar]
- 41.Meister P, et al. 2003. Nuclear factories for signalling and repairing DNA double strand breaks in living fission yeast. Nucleic Acids Res. 31:5064–5073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mollgaard H, Neuhard J. 1978. Deoxycytidylate deaminase from Bacillus subtilis. Purification, characterization, and physiological function. J. Biol. Chem. 253:3536–3542 [PubMed] [Google Scholar]
- 43.Moreno S, Klar A, Nurse P. 1991. Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol. 194:795–823 [DOI] [PubMed] [Google Scholar]
- 44.Muris DF, et al. 1993. Cloning the RAD51 homologue of Schizosaccharomyces pombe. Nucleic Acids Res. 21:4586–4591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakamura TM, Du LL, Redon C, Russell P. 2004. Histone H2A phosphorylation controls Crb2 recruitment at DNA breaks, maintains checkpoint arrest, and influences DNA repair in fission yeast. Mol. Cell. Biol. 24:6215–6230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Noguchi E, Noguchi C, Du LL, Russell P. 2003. Swi1 prevents replication fork collapse and controls checkpoint kinase Cds1. Mol. Cell. Biol. 23:7861–7874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nordlund P, Reichard P. 2006. Ribonucleotide reductases. Annu. Rev. Biochem. 75:681–706 [DOI] [PubMed] [Google Scholar]
- 48.Parker AE, Clyne RK, Carr AM, Kelly TJ. 1997. The Schizosaccharomyces pombe rad11+ gene encodes the large subunit of replication protein A. Mol. Cell. Biol. 17:2381–2390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Parrilla-Castellar ER, Arlander SJ, Karnitz L. 2004. Dial 9-1-1 for DNA damage: the Rad9-Hus1-Rad1 (9-1-1) clamp complex. DNA Repair 3:1009–1014 [DOI] [PubMed] [Google Scholar]
- 50.Pebernard S, Wohlschlegel J, McDonald WH, Yates JR, III, Boddy MN. 2006. The Nse5-Nse6 dimer mediates DNA repair roles of the Smc5-Smc6 complex. Mol. Cell. Biol. 26:1617–1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rhind N, Russell P. 2000. Chk1 and Cds1: linchpins of the DNA damage and replication checkpoint pathways. J. Cell Sci. 113:3889–3896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roguev A, Wiren M, Weissman JS, Krogan NJ. 2007. High-throughput genetic interaction mapping in the fission yeast Schizosaccharomyces pombe. Nat. Methods 4:861–866 [DOI] [PubMed] [Google Scholar]
- 53.Roseaulin L, et al. 2008. Mus81 is essential for sister chromatid recombination at broken replication forks. EMBO J. 27:1378–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sabatinos SA, Forsburg SL. 2009. Measuring DNA content by flow cytometry in fission yeast. Methods Mol. Biol. 521:449–461 [DOI] [PubMed] [Google Scholar]
- 55.Sanders SL, Arida AR, Phan FP. 2010. Requirement for the phospho-H2AX binding module of Crb2 in double-strand break targeting and checkpoint activation. Mol. Cell. Biol. 30:4722–4731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sargent RG, Mathews CK. 1987. Imbalanced deoxyribonucleoside triphosphate pools and spontaneous mutation rates determined during dCMP deaminase-defective bacteriophage T4 infections. J. Biol. Chem. 262:5546–5553 [PubMed] [Google Scholar]
- 57.Sheedy DM, et al. 2005. Brc1-mediated DNA repair and damage tolerance. Genetics 171:457–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sivakumar S, Porter-Goff M, Patel PK, Benoit K, Rhind N. 2004. In vivo labeling of fission yeast DNA with thymidine and thymidine analogs. Methods 33:213–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sofueva S, Du LL, Limbo O, Williams JS, Russell P. 2010. BRCT domain interactions with phospho-histone H2A target Crb2 to chromatin at double-strand breaks and maintain the DNA damage checkpoint. Mol. Cell. Biol. 30:4732–4743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stracker TH, Usui T, Petrini JH. 2009. Taking the time to make important decisions: the checkpoint effector kinases Chk1 and Chk2 and the DNA damage response. DNA Repair 8:1047–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tanaka K, Boddy MN, Chen XB, McGowan CH, Russell P. 2001. Threonine-11, phosphorylated by Rad3 and atm in vitro, is required for activation of fission yeast checkpoint kinase Cds1. Mol. Cell. Biol. 21:3398–3404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tanaka K, Russell P. 2001. Mrc1 channels the DNA replication arrest signal to checkpoint kinase Cds1. Nat. Cell Biol. 3:966–972 [DOI] [PubMed] [Google Scholar]
- 63.Toone WM, et al. 1998. Regulation of the fission yeast transcription factor Pap1 by oxidative stress: requirement for the nuclear export factor Crm1 (Exportin) and the stress-activated MAP kinase StyI/Spc1. Genes Dev. 12:1453–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weinberg G, Ullman B, Martin DW., Jr 1981. Mutator phenotypes in mammalian cell mutants with distinct biochemical defects and abnormal deoxyribonucleoside triphosphate pools. Proc. Natl. Acad. Sci. U. S. A. 78:2447–2451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Williams JS, et al. 2010. γH2A binds Brc1 to maintain genome integrity during S-phase. EMBO J. 29:1136–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wold MS. 1997. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem. 66:61–92 [DOI] [PubMed] [Google Scholar]
- 67.Yan W, et al. 2011. Structural basis of γH2AX recognition by human PTIP BRCT5-BRCT6 domains in the DNA damage response pathway. FEBS Lett. 585:3874–3879 [DOI] [PubMed] [Google Scholar]
- 68.Yin L, Locovei AM, D'Urso G. 2008. Activation of the DNA damage checkpoint in mutants defective in DNA replication initiation. Mol. Biol. Cell 19:4374–4382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zegerman P, Diffley JF. 2009. DNA replication as a target of the DNA damage checkpoint. DNA Repair 8:1077–1088 [DOI] [PubMed] [Google Scholar]