

Abstract
Processing bodies (PBs, or P bodies) are cytoplasmic granules involved in mRNA storage and degradation that participate in the regulation of gene expression. PBs concentrate nontranslated mRNAs and several factors involved in mRNA decay and translational repression, including the eukaryotic translation initiation factor 4E-transporter (4E-T). 4E-T is required for PB assembly, but little is known about the molecular mechanisms that regulate its function. Here, we demonstrate that oxidative stress promotes multisite 4E-T phosphorylation. We show that the c-Jun N-terminal kinase (JNK) is targeted to PBs in response to oxidative stress and promotes the phosphorylation of 4E-T. Quantitative mass spectrometry analysis reveals that JNK phosphorylates 4E-T on six proline-directed sites that are required for the formation of the 4E-T complex upon stress. We have developed an image-based computational method to quantify the size, number, and density of PBs in cells, and we find that while 4E-T is required for steady-state PB assembly, its phosphorylation facilitates the formation of larger PBs upon oxidative stress. Using polysomal mRNA profiling, we assessed global and specific mRNA translation but did not find that 4E-T phosphorylation impacts translational control. Collectively, these data support a model whereby PB assembly is regulated by a two-step mechanism involving a 4E-T-dependent assembly stage in unstressed cells and a 4E-T phosphorylation-dependent aggregation stage in response to stress stimuli.
INTRODUCTION
The regulation of mRNA turnover plays an essential role in modulating gene expression (16, 29). In eukaryotes, the bulk of mRNAs undergoes decay by pathways that are initiated by poly(A)-tail shortening, commonly known as deadenylation, an event that decreases mRNA stability and translation efficiency (16). Deadenylation is an important step for all major paths of mRNA decay in mammalian cells, including AU-rich element (ARE)-mediated decay (AMD), nonsense-mediated decay (NMD), and decay mediated by microRNAs (miRNAs) (44). Deadenylated mRNAs are degraded either through the exosome in the 3′-to-5′ direction or through decapping and subsequent degradation by the exoribonuclease Xrn1 in the 5′-to-3′ direction (16, 29). In many cases, the transit of an mRNA to a repressed state is associated with its recruitment to processing bodies (PBs, or P bodies), which are cytoplasmic foci containing the enzymatic machinery involved in mRNA deadenylation, decapping, and degradation (28).
PBs are highly dynamic structures, and their assembly is tightly linked with the availability of free cytoplasmic mRNAs. Translation inhibition, which leads to the release of mRNAs from polysomes, increases the number and size of PBs (38), and conversely, limiting the pool of free cytoplasmic mRNAs using cycloheximide disrupts PB formation (1). The assembly of PBs is thought to be orchestrated by protein-protein interactions among components and depends on oligomerization domains found in factors such as NOT1, Dcp2, or LSm4 (31). Depletion of proteins involved in mRNA decapping (RCK, LSm1-7, and Hedls) and deadenylation (the CCR4-CAF1-NOT1 complex) strongly decreases PB assembly (13), suggesting that mRNAs must undergo these processes to form PBs. In contrast to stress granules (SGs), which contain stalled 48S translation preinitiation complexes (3), PBs contain only one translation initiation factor, the eukaryotic initiation factor 4E (eIF4E) (32). eIF4E binds to the 5′ cap of mRNAs and, in addition to its role in translation, has been suggested to target transcripts to PBs in collaboration with the eIF4E-binding protein 4E-transporter (4E-T) (1, 14). 4E-T is required for the localization of eIF4E to PBs, and its depletion was found to strongly decrease PB assembly (1, 14). Several large-scale proteomics studies have shown that 4E-T becomes phosphorylated on multiple residues (10, 26), but the regulation and function of these phosphorylation events with regard to PB assembly remain unknown.
Unlike SGs, PBs are found in unstressed cells, but their assembly can be modulated by levels of reactive oxygen species (ROS) (28). Indeed, oxidative stress, which can be induced by arsenite treatment, increases the number and size of PBs in cells (23). This response was shown to be independent of eIF2α phosphorylation, a necessary event in SG assembly (23), suggesting that additional pathways must contribute to PB assembly. Oxidative stress leads to the activation of the mitogen-activated protein kinase (MAPK) pathways (4), which include the c-Jun N-terminal kinase (JNK) (40). The mechanisms underlying JNK activation in response to oxidative stress are not well-defined but may involve upstream kinases that respond to oxidative modifications, such as the apoptosis signal-regulating kinase 1 (ASK1) (25). It has been proposed that oxidative stress also inhibits a JNK phosphatase, thus contributing to sustained JNK activation in response to arsenite treatment (5). A recent study has proposed a role for JNK in the phosphorylation of the PB component Dcp1 (35), suggesting that JNK may participate in PB assembly. An unresolved issue at the moment is the significance of the increase in PB size, as aggregation of messenger ribonucleoproteins (mRNPs) into microscopically visible PBs has been shown not to be required for mRNA degradation and translational repression (9). However, as aggregation into mRNP granules is an evolutionarily conserved feature of eukaryotic cells, it is anticipated to have some role (8).
Here, we show that JNK interacts with and phosphorylates 4E-T on proline-directed sites in response to oxidative stress. We found that JNK-mediated phosphorylation of 4E-T promotes complex formation and 4E-T localization to PBs, suggesting a new molecular mechanism by which stress signaling modulates the assembly of PBs.
MATERIALS AND METHODS
DNA constructs.
The plasmid encoding human hemagglutinin (HA)-tagged 4E-T was kindly provided by Nahum Sonenberg (McGill University, Montreal, Quebec, Canada) and described previously (11). Myc-tagged 4E-T was generated by subcloning the coding region of 4E-T from pACTAG-HA-4E-T into pcDNA3-6myc. The human 4E-T mutant S6A was generated using the QuikChange methodology (Stratagene, La Jolla, CA). The vectors encoding Flag MKK7-JNK1 (constitutively activated form of JNK1 [CA] and the kinase-inactive mutant [KD]) were obtained from Addgene (reference numbers 19726 and 19730), and the plasmid encoding HA-JNK1 was a kind gift from John Blenis (Harvard Medical School, Boston, MA).
Antibodies.
Antibodies targeted against 4E-T were purchased from Abcam (Cambridge, United Kingdom). Anti-HA, -myc, -Flag, -tubulin, and -phospho-extracellular signal-regulated kinase 1/2 (ERK1/2) monoclonal antibodies were purchased from Sigma-Aldrich (Oakville, Ontario, Canada). Antibodies targeted against phospho-eIF2α, phospho-JNK (Thr183/Tyr185), phospho-p38 (Thr180/Tyr182), and poly(A)-binding protein (PABP) were purchased from Cell Signaling Technologies (Beverly, MA). Antibodies targeted against Dcp1 were purchased from Abnova. All secondary horseradish peroxidase (HRP)-conjugated antibodies used for immunoblotting were purchased from Chemicon (Temecula, CA).
Cell culture and transfection.
HEK293, U2OS, and HeLa cells were maintained at 37°C in Dulbecco's modified Eagle's medium (DMEM) with 4.5 g/liter glucose supplemented with 10% fetal bovine serum (FBS) and antibiotics. HEK293 cells were transfected with calcium phosphate as described previously (34). U2OS and HeLa cells were transfected with Fugene 6 (Roche) according to the manufacturer's instructions. Cells were grown for 48 h after transfection and were pretreated with AS601245 (20 μM), SP600125 (20 μM), PD184352 (10 to 50 μM), SB203580 (10 to 50 μM), or N-acetyl-l-cysteine (NAC; 5 to 20 mM) for 30 to 45 min. Cells were stimulated with either sodium arsenite (500 μM), anisomycin (10 μM), clotrimazole (20 μM), carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP; 1 μM), emetine (20 μg/ml), or cycloheximide (50 μg/ml) for 30 to 60 min before being harvested.
RNA interference (RNAi) and viral infections.
For small interfering RNA (siRNA)-mediated knockdown of 4E-T, predesigned Dicer-substrate siRNA duplexes against the 3′ untranslated region (UTR) of human 4E-T (HSC.RNAI.N019843.4.10) were purchased from Integrated DNA Technologies (Coralville, IA). U2OS cells were transfected using Oligofectamine (Invitrogen) and 25 nM siRNA per six-well plate. Retroviruses were produced using the pMSCV-GFP vector system to stably overexpress ectopic human wild-type (wt) 4E-T or the S6A mutant in HEK293 cells. 293GPG cells were transfected using calcium phosphate and 30 μg of pMSCV-GFP 4E-T wild type or S6A mutant. At 4 days posttransfection, the retroviral supernatant was collected and filtered (0.45-μm-pore-size filter) for use. Host HEK293 cells were infected in the presence of 4 μg/ml Polybrene. After two passages postinfection, positive pools were selected by fluorescence-activated cell sorting with green fluorescent protein (GFP).
Immunoprecipitations and immunoblotting.
Cell lysates were prepared as previously described (34). Briefly, cells were washed with ice-cold phosphate-buffered saline (PBS) and lysed in 10 mM K3PO4, 1 mM EDTA, 5 mM EGTA, 10 mM MgCl2, 50 mM β-glycerophosphate, 0.5% Nonidet P-40, 0.1% Brij 35, 0.1% deoxycholic acid, 1 mM sodium orthovanadate (Na3VO4), and 1 mM phenylmethylsulfonyl fluoride with a Complete protease inhibitor cocktail tablet (Roche). For immunoprecipitations, cell lysates were incubated with the indicated antibody or control IgG (Santa Cruz Biotechnology, Santa Cruz, CA) for 2 h, followed by a 1-h incubation with protein A-Sepharose CL-4B beads (GE Healthcare). Immunoprecipitates were washed thrice in lysis buffer, and beads were eluted and boiled in 2× reducing sample buffer (5× reducing sample buffer is 60 mM Tris-HCl [pH 6.8], 25% glycerol, 2% SDS, 14.4 mM 2-mercaptoethanol, and 0.1% bromophenol blue). For λ-phosphatase assay, endogenous 4E-T immunoprecipitates were washed twice in lysis buffer and incubated for 1 h at 30°C with λ-phosphatase (New England BioLabs, Beverly, MA) in λ-phosphatase buffer. The reaction was stopped by adding 2× reducing sample buffer. Eluates and total cell lysates were subjected to 10 to 12% SDS-PAGE, and resolved proteins were transferred onto polyvinylidene difluoride (PVDF) membranes for immunoblotting.
Protein phosphotransferase assays.
For JNK assays, beads from Flag-MKK7-JNK1 immunoprecipitations were washed twice in lysis buffer and twice in kinase buffer (25 mm Tris-HCl, pH 7.4, 10 mm MgCl2, 5 mm β-glycerophosphate). Kinase assays were performed with immunopurified full-length myc-tagged 4E-T wt or S6A mutant as the substrates under linear assay conditions. Assays were performed for 10 min at 30°C in kinase buffer supplemented with 5 μCi of [γ-32P]ATP. All samples were subjected to SDS-PAGE, and incorporation of radioactive 32P label was determined by autoradiography using a Fuji PhosphorImager with ImageQuant software.
Tryptic digestion, TMT labeling, and mass spectrometry analysis.
Following SDS-PAGE separation with Coomassie staining, bands corresponding to 4E-T were excised and digested in gel with sequencing-grade trypsin (Promega, Madison, WI) as described previously (37). Labeling with six-plex tandem mass tag (TMT6) reagents (Thermo Scientific) was accomplished as published previously with minimal modifications (39). Labeled peptides were then mixed and underwent C18 solid-phase extraction as described previously (30). Finally, combined samples were resuspended in 5% acetonitrile–5% formic acid for mass spectrometry (MS) analysis. All liquid chromatography (LC)-MS experiments were performed on an LTQ-Velos-Orbitrap apparatus (Thermo Fisher Scientific) equipped with a Famos autosampler (LC Packings) and an Agilent 1100 binary high-pressure liquid chromatography pump (Agilent Technologies) essentially as described previously (39). The LTQ-Velos-Orbitrap was operated in data-dependent mode, collecting high-resolution Orbitrap MS/MS spectra after higher-energy C-trap dissociation for the top 10 most intense ions following each survey scan collected in the Orbitrap.
Peptide, protein, and phosphorylation site identification.
Following acquisition, individual MS/MS spectra were assigned to peptides using the Sequest program (42). To maximize phosphorylation site identifications, a two-stage database searching strategy was employed. In the first stage, all MS/MS spectra were searched against a database containing all protein sequences from the human International Protein Index database (version 3.6) in forward and reverse orientations as well as sequences of common contaminants. Initial searches were performed with the following parameters: 25-ppm precursor ion tolerance; 0.02-Da product ion tolerance; fully tryptic digestion with up to two missed cleavages; static modifications Cys alkylation (+57.021464) and TMT labeling of Lys and peptide N termini (+229.162932); and dynamic modifications Met oxidation (+15.994915) and phosphorylation of Ser, Thr, and Tyr (+79.966330). The target-decoy approach (12) was then used to distinguish correct and incorrect peptide identifications using linear discriminant analysis based on several parameters, including Xcorr, dCn′, peptide length, precursor ion mass error, numbers of missed cleavages, peptide length, and charge state (20). After filtering to an initial 1% peptide-level false discovery rate (FDR), peptides were then assembled into proteins, and proteins were scored and filtered to a final protein FDR of 1% (20).
In the second stage, relaxed parameters were used to match additional MS/MS spectra against a filtered database. Sequest was again employed, but this time the database was filtered to include only forward and reverse sequences for proteins that were identified in stage one. Stage two search parameters included 3.1-Da precursor ion tolerance; 0.02-Da product ion tolerance; no enzyme specificity; static modifications Cys alkylation (+57.021464) and TMT labeling of Lys and peptide N termini (+229.162932); and dynamic modifications Met oxidation (+15.994915) and phosphorylation of Ser, Thr, and Tyr (+79.966330). The resulting peptides were again filtered via linear discriminant analysis, but this time the number of tryptic ends was included as an additional feature and peptide mass errors were corrected to account for occasional incorrect monoisotopic mass assignments.
To evaluate phosphorylation site localization, all phosphopeptides matching 4E-T were scored using the Ascore algorithm (2) and peptides were grouped according to the phosphorylation sites that they contained. A minimum Ascore of 13 was required for phosphorylation site localization (P < 0.05), and phosphorylation site assignments were manually validated to ensure reliability.
Phosphorylation site quantification.
Relative quantification of each peptide was accomplished on the basis of the intensities observed for all six reporter ions from high-resolution Orbitrap MS/MS spectra, after correcting for batch-specific isotopic enrichments of each TMT reagent. Each peptide was required to have a minimum isolation specificity of 0.75 (39) and a summed reporter ion intensity of at least 500 with no more than four missing reporter ions. Individual sites were quantified on the basis of the summed reporter ion intensities for all matching peptides. Nonphosphorylated peptides matching 4E-T were combined to estimate unmodified protein abundance. Quantitative profiles for all phosphorylation sites were normalized to account for slight changes in 4E-T abundance. Finally, analysis of variance (ANOVA) was used to identify statistically significant, site-specific changes in protein phosphorylation. Within each experiment, all P values were adjusted to account for multiple-hypothesis testing via the method of Hochberg and Benjamini (18).
Confocal microscopy.
For immunofluorescence analyses, 1.5 × 105 U2OS cells or 1 × 105 HeLa cells were seeded in six-well plates containing coverslips. Cells were treated as indicated, washed in PBS, and fixed in 3.7% formaldehyde for 15 min at room temperature. Cells were permeabilized for 7 min in PBS containing 0.2% Triton X-100 and blocked for 45 min in PBS containing 10% FBS (blocking buffer). Cells were incubated for 2.5 h with the indicated primary antibodies diluted in blocking buffer, followed by 1 h of incubation with Alexa Fluor 488- or 555-conjugated anti-mouse or -rabbit secondary antibodies (Invitrogen) diluted in blocking buffer. Cells were stained for 2 min with DAPI (4′,6′-diamidino-2-phenyindole) at 1 μg/ml, and coverslips were mounted on slides with Prolong gold (Invitrogen). Immunofluorescence microscopy images from single confocal sections (0.7 μm) were acquired at room temperature with an LSM 510 Meta laser scanning confocal microscope (Zeiss) using a Plan Apochromat 63× oil-immersion objective. All images were acquired using identical parameters.
Image-based quantification of PB characteristics.
PB characteristics were measured using custom image analysis software written for MATLAB (MathWorks, Natick, MA) in two steps: image segmentation and data analysis. Cells were detected using nonspecific background fluorescence in the GFP channel by identifying local clusters of pixels with intensities significantly above the noise level. To separate touching cells, a watershed transform was applied using nuclei that had been segmented in the DAPI channel using Otsu's algorithm as seeds. PBs were detected similarly to cells: by identifying local clusters of pixels in the cytoplasm with intensities significantly above the noise level (at least 2 noise standard deviations), after subtracting the background, as estimated via a 3- by 3-μm local median filter.
We characterized PBs by their density and their fluorescence intensity. We chose to use PB density (number of PBs divided by the cell area) as a measure for the quantity of PBs, since the number of PBs correlates with cell area. The peak fluorescence intensity of PBs above the background is used as a measure for the amount of 4E-T localized in PBs and, consequently, as a proxy for PB size. This measure is more robust than, for example, total fluorescence intensity within the PB. Since PBs are mostly subresolution features, variations in their size are reflected in variations in fluorescence intensity rather than in the area occupied by their image.
Cell-to-cell variations in expression levels were corrected on the basis of the assumptions that higher expression would equally increase both the localized (peak PB intensity) and diffuse (background intensity) 4E-T signal and that the different genetic and pharmacological experimental settings affect only the relation between the localized and the diffuse 4E-T signal but not their reaction to differences in expression level. Consequently, we applied a robust linear fit of the form PI(k,x) = s · BI(k,x) + i(x) to the intensity measurements for each PB, where PI(k,x) is the peak fluorescence intensity of the kth PB k of experiment x, BI(k,x) is the local cellular background fluorescence around PB k, s is the slope, which is assumed to be the same for all experimental conditions, and i(x) is the intercept, which is considered to be different for each experimental condition x. To eliminate the effect of expression level, we subtracted s · BI(k,x) from each peak intensity measurement.
Overall recruitment of 4E-T into PBs can occur either via an increase in the size of the PB or via assembly into new PBs. Thus, we define “PB agglomeration” as the total amount of 4E-T localized to PBs, which is calculated as the product of PB density and peak fluorescence intensity. The uncertainty of PB agglomeration is calculated from the uncertainties of PB density and peak fluorescence intensity via Gaussian error propagation.
Polysomal mRNA profiling.
Sucrose gradient velocity sedimentation was employed to isolate the polysomal fractions. Ten minutes before collection, 100 μg/ml cycloheximide was added to the culture medium. Cells were washed in ice-cold PBS supplemented with 100 μg/ml cycloheximide and collected in polysome lysis buffer (PLB; 15 mM Tris [pH 7.4], 250 mM NaCl, 15 mM MgCl2, 1% Triton X-100, 100 μg/ml cycloheximide, 1 mM dithiothreitol, 400 U/ml RNaseOut [Invitrogen], and protease inhibitors), as described elsewhere (33). Samples were centrifuged at 10,000 × g for 10 min at 4°C. The resulting supernatant was layered on a 20 to 50% linear sucrose gradient and centrifuged in a Beckman SW41Ti rotor at 92,000 × g for 3 h at 4°C. Following centrifugation, the A254 was continuously monitored and recorded using a Gradient Station IP instrument (Biocomp, Fredericton, NB, Canada) attached to a UV-MII spectrophotometer (GE Healthcare). Polysomal fractions were collected, and RNA was extracted using an RNeasy minikit (Qiagen).
Real-time quantitative PCR (qPCR).
Total RNA was reverse transcribed using a cDNA reverse transcription kit (Applied Biosystems, Carlsbad, CA), as described by the manufacturer. Gene expression levels of endogenous controls glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and beta-actin (ActB) were determined using prevalidated TaqMan gene expression assays (Applied Biosystems), and the levels of the survivin, c-myc, vascular endothelial growth factor A (VEGFA), and ornithine decarboxylase (ODC1) transcripts were determined using assays designed with the Universal Probe Library (UPL) from Roche.
RESULTS
4E-T localizes to PBs and becomes phosphorylated in response to oxidative stress.
Several high-throughput phosphoproteomics studies have shown that 4E-T can be phosphorylated on several residues, but the biological significance of these modifications is unknown (10, 26). To determine if conditions that promote PB assembly result in 4E-T phosphorylation, we treated HEK293 cells with arsenite, which is frequently used to induce PB assembly (22), and we evaluated cellular responses via indirect immunofluorescence microscopy and biochemistry. Validating the effect of arsenite in U2OS cells transfected with HA-tagged 4E-T confirmed that at steady state, 4E-T localized mainly within discrete foci in the cytoplasm that were also labeled by anti-Dcp1 antibodies, which is indicative of PBs (Fig. 1A). As expected, arsenite treatment increased both the number and the size of PBs. Arsenite treatment also induced the formation of SGs, as shown by the labeling of cytoplasmic foci with anti-PABP antibodies (Fig. 1B). Notably, none of the 4E-T puncta overlapped the PABP-positive granules, indicating that 4E-T did not localize within other types of cytoplasmic RNA granules, such as SGs. To determine if arsenite treatment altered the electrophoretic properties of 4E-T, we analyzed endogenous 4E-T by SDS-PAGE. Interestingly, we found that arsenite treatment induced a robust mobility shift in 4E-T, whereas no effect was seen in response to the known SG inducers clotrimazole and FCCP (Fig. 1C) (22). To determine if the mobility shift in 4E-T was due to phosphorylation, we immunoprecipitated endogenous 4E-T and treated immunoprecipitates with λ-phosphatase. We found that phosphatase treatment completely abolished the 4E-T mobility shift (Fig. 1D), indicating that arsenite treatment promotes 4E-T phosphorylation.
Fig 1.

4E-T becomes phosphorylated in response to oxidative stress. (A) U2OS cells growing in serum were transfected with HA-tagged 4E-T. Subcellular localization of endogenous Dcp1 (P-body component, green) and HA-tagged 4E-T (red) was determined by indirect immunofluorescence. The colocalization of these factors appears yellow in the merged image. Nuclei were stained with DAPI (blue). (B) U2OS cells stably expressing HA-tagged 4E-T were grown in the presence of serum and treated for 45 min with arsenite (0.5 mM). Endogenous PABP (stress granule component, green) and HA-tagged 4E-T (red) were analyzed by immunofluorescence. Higher-magnification views of boxed areas are shown in insets at the bottom right and display 4E-T and Dcp1 colocalization (A) and the distinct localization of 4E-T and PABP (B). Bars, 10 μm. (C) HEK293 cells were treated with arsenite (0.5 mM) for 45 min and clotrimazole (mitochondrial stress inducer, 20 μM) or FCCP (metabolic stress inducer, 1 μM) for 60 min. The phosphorylation of 4E-T was assayed by mobility shift. Induction of oxidative stress by arsenite was verified by monitoring the phosphorylation of eIF2α on Ser51. (D) Endogenous 4E-T was immunoprecipitated (IP) from HEK293 cells treated with arsenite. Immunoprecipitates were treated with λ-phosphatase (λ-PPase), and 4E-T phosphorylation was assayed as described for panel C. (E) HEK293 cells were incubated for 45 min with the antioxidant agent N-acetyl-l-cysteine (NAC) at the indicated concentrations prior to arsenite treatment. 4E-T phosphorylation was assayed as described for panel C.
One of the main molecular mechanisms of action of arsenite is the induction of oxidative stress (19). To address whether oxidative stress was required for 4E-T phosphorylation, cells were pretreated with increasing concentrations of the antioxidant NAC. Phosphorylation of 4E-T induced by arsenite was efficiently prevented by the presence of NAC (Fig. 1E), indicating that oxidative stress is a prerequisite for 4E-T phosphorylation. Together, these results demonstrate that 4E-T becomes phosphorylated in response to oxidative stress, which correlates with the induction of PBs.
JNK phosphorylates 4E-T in vitro and in vivo.
We next sought to identify candidate kinases responsible for 4E-T phosphorylation upon arsenite treatment. Given that arsenite promotes the activation of all three conventional MAPK pathways (Fig. 2A) (19), we tested whether pretreatment of cells with MAPK pathway inhibitors would affect 4E-T phosphorylation (Fig. 2B). To assess the role of the ERK1/2 and p38MAPK pathways, we pretreated HEK293 cells with increasing concentrations of the inhibitors PD184352 and SB203580, respectively, before cell stimulation experiments. Inhibition of MEK1/2 (Fig. 2C) and p38MAPK (Fig. 2D) did not affect the mobility shift of 4E-T induced by arsenite, indicating that these pathways do not contribute to 4E-T phosphorylation. In contrast, treatment of cells with two different JNK inhibitors (SP600125 and AS601245) efficiently blocked the mobility shift of 4E-T induced by arsenite (Fig. 2E and F), suggesting that JNK is required for 4E-T phosphorylation. To address whether JNK activity is sufficient to stimulate 4E-T phosphorylation in the absence of stress stimuli, we transfected HEK293 cells with an MKK7-JNK1 fusion protein that exhibits constitutive JNK activity (24). Transient expression of this construct (CA) strongly enhanced the electrophoretic mobility shift of 4E-T, whereas no effect was seen with the kinase-inactive MKK7-JNK1 allele (KD) (Fig. 3A). To further demonstrate the involvement of JNK in 4E-T phosphorylation, we used anisomycin, which activates JNK in the absence of oxidative stress. Consistent with JNK being required for 4E-T phosphorylation, we found that anisomycin treatment also induced a mobility shift in 4E-T (Fig. 3B). Next, we investigated whether 4E-T is a direct JNK substrate by performing in vitro kinase assays with immunopurified proteins. While no incorporation of 32P label was seen in purified 4E-T incubated with control beads or the kinase-inactive MKK7-JNK1 (KD), we found that activated MKK7-JNK1 (CA) robustly increased 32P incorporation (∼9-fold) in purified 4E-T (Fig. 3C). Taken together, these results demonstrate that JNK directly phosphorylates 4E-T both in vitro and in vivo.
Fig 2.

4E-T is a target of JNK signaling. (A) HEK293 cells were treated with arsenite (0.5 mM) for the indicated times. Activation of JNK, p38, and ERK was determined by immunoblotting, and 4E-T phosphorylation was determined by mobility shift assay. (B) Schematic representation of the agonist and pharmacological inhibitors used in this study. (C to F) Cells were pretreated for 30 min with PD184352 (C), SB203580 (D), SP600125 (E), or AS601245 (F) at the indicated concentrations prior to arsenite treatment. The phosphorylation of 4E-T was analyzed by mobility shift assay.
Fig 3.

JNK phosphorylates 4E-T in vitro and in vivo. (A) Cells were cotransfected with myc-tagged 4E-T and Flag-tagged MKK7-JNK1 in constitutively activated (CA) or kinase-inactive (KD) forms. 4E-T phosphorylation was analyzed by mobility shift assay. (B) JNK activation was induced by anisomycin treatment (10 μM) for the indicated times, and 4E-T phosphorylation was analyzed as described for panel A. (C) HEK293T cells were transfected with Flag-tagged MKK7-JNK1 (CA or KD) or myc-tagged 4E-T. Immunoprecipitated myc-tagged 4E-T from serum-starved cells was incubated with immunopurified Flag-tagged MKK7-JNK1 in a kinase reaction with [γ-32P]ATP. The resulting samples were subjected to SDS-PAGE, and the dried Coomassie-stained gel was autoradiographed. The histogram shows the n-fold increase of 4E-T phosphorylation normalized to the condition where JNK was not transfected.
JNK interacts and colocalizes with 4E-T upon oxidative stress.
Our results indicate that JNK directly phosphorylates 4E-T, suggesting that both proteins physically interact in cells. To explore this possibility, we expressed HA-tagged JNK1 and myc-tagged 4E-T in HEK293 cells and coimmunoprecipitated them with anti-HA antibodies. At steady state, 4E-T and JNK did not appear to interact in cells; however, upon arsenite treatment, we observed a strong, time-dependent interaction between the two proteins (Fig. 4A). Notably, similar results were found when performing the reverse coimmunoprecipitation (Fig. 4B). Having found that JNK interacts with and directly phosphorylates 4E-T, we investigated whether arsenite treatment could promote their colocalization to PBs. Immunofluorescence of HA-tagged JNK1 in untreated U2OS cells resulted in uniform staining throughout the cytoplasm and the nucleus, whereas endogenous 4E-T was enriched in PBs (Fig. 4C, top). The anti-4E-T antibody was found to be specific, as cells transfected with an siRNA targeting 4E-T did not display specific staining (see Fig. S1 in the supplemental material). Upon arsenite treatment, a significant proportion of JNK appeared to relocalize to distinct cytoplasmic foci that overlapped 4E-T puncta (Fig. 4C, bottom). This raises the possibility that JNK promotes the phosphorylation of 4E-T within PBs. Together, these results suggest that JNK interacts with 4E-T and is targeted to PBs in response to oxidative stress.
Fig 4.

JNK interacts and colocalizes with 4E-T upon induction of oxidative stress. (A) HEK293 cells were cotransfected with HA-tagged JNK1 and myc-tagged 4E-T and treated with arsenite (0.5 mM) for the indicated times. Associated 4E-T was assayed within HA-JNK immunoprecipitates by immunoblotting. (B) As described for panel A, except that associated JNK was assayed within myc 4E-T immunoprecipitates by immunoblotting. (C) U2OS cells were transfected with HA-tagged JNK1, left untreated (top), or treated with arsenite (0.5 mM) for 45 min (bottom). Endogenous 4E-T (red) and HA-tagged JNK1 (green) were immunostained. Nuclei were stained by DAPI (blue). Arrowheads show examples of colocalized puncta. Bar, 10 μm.
Identification of 4E-T phosphorylation sites regulated by JNK.
Next, we sought to identify JNK-regulated phosphorylation sites in 4E-T using a quantitative MS-based approach. HEK293 cells were transfected with myc-tagged 4E-T and either the constitutively activated form of JNK1 (CA) or the kinase-inactive mutant (KD). Immunoprecipitated 4E-T was then isolated via SDS-PAGE, digested in gel with trypsin, and labeled with TMT6s. Samples were then combined and analyzed by liquid chromatography-assisted tandem MS, and the relative abundance of each phosphopeptide was measured across experimental conditions (Fig. 5A). Using this approach, we identified a total of 34 phosphorylation sites, from which 6 showed significant changes (ANOVA; FDR < 5%) between the activated and inactive JNK conditions (Fig. 5B). Identification of these residues as phosphorylation sites (Ser301, Ser374, Ser513, Ser587, Ser693, and Ser752) was obtained by MS/MS sequencing, as depicted by annotated high-resolution MS/MS spectra from representative tryptic peptides (see Fig. S2 in the supplemental material). These phosphorylation sites are located in the central portion of 4E-T (Fig. 5C; see Fig. S3 in the supplemental material) and reside within proline-directed sequences (S/T-P) characteristic of JNK phosphorylation motifs (Fig. 5B; see Fig. S3 in the supplemental material). To validate that JNK phosphorylates these sites in vivo, we performed site-directed mutagenesis to replace all six serine residues with unphosphorylatable alanines in a single mutant (S6A). Notably, mutation of these sites completely abolished the electrophoretic mobility shift in 4E-T induced by the constitutive activation of JNK (Fig. 6A) and significantly reduced the mobility shift induced by arsenite treatment (Fig. 6B), indicating that JNK phosphorylates these residues in cells. To further validate that these sites are directly phosphorylated by JNK, we performed in vitro kinase assays using constitutively activated MKK7-JNK1 (CA) and immunopurified wt 4E-T or the S6A mutant as the substrates. Mutation of all six residues significantly inhibited JNK-mediated 32P label incorporation in immunopurified full-length 4E-T (Fig. 6C), confirming that JNK predominantly phosphorylates 4E-T at these residues.
Fig 5.

Identification of 4E-T phosphorylation sites regulated by JNK. (A) Outline of the quantitative phosphoproteomic work flow developed for phosphopeptide quantitation in HEK293 cells using the TMT technology. Cells were cotransfected with myc-tagged 4E-T and either empty vector, constitutively activated JNK1, or inactivated JNK1. Immunoprecipitated 4E-T from cell extracts was resolved by SDS-PAGE and digested in gel with trypsin prior to TMT6 labeling and mass spectrometry analysis. (B) MS/MS analysis revealed six phosphorylation sites in 4E-T which are differentially regulated between the activated (CA) and inactivated (KD) JNK conditions. The six serine residues (Ser301, Ser374, Ser513, Ser587, Ser693, and Ser752) reside within the S/T-P consensus JNK phosphorylation motif. The data were normalized to the empty vector condition. Error bars indicate the standard deviation (SD) of duplicate samples (for all changes, FDR was <0.05). Sites of phosphorylation within each tryptic peptide are indicated by a # symbol immediately following the modified residue, with numbering based on the sequence found in the International Protein Index (IPI00291800.1). (C) Schematic representation of the identified phosphorylation sites within 4E-T. NES, nuclear export signal; NLS, nuclear localization signal. The gray areas represent regions with high Q/N contents.
Fig 6.

JNK phosphorylates 4E-T on six serine residues. (A) HEK293 cells were cotransfected with wt 4E-T or the S6A mutant (S301A, S374A, S513A, S587A, S693A, S752A) and activated JNK1. The phosphorylation of 4E-T was analyzed by mobility shift assay. (B) Cells were transfected with wt or S6A 4E-T and treated with arsenite (0.5 mM) for 45 min. 4E-T phosphorylation was assayed as described for panel A. (C) HEK293T cells were cotransfected with wt or S6A 4E-T and activated JNK1. Immunoprecipitated myc-tagged 4E-T from serum-starved cells was incubated with immunopurified Flag-tagged MKK7-JNK1 (CA) in a kinase reaction with [γ-32P]ATP. The resulting samples were subjected to SDS-PAGE, and the dried Coomassie-stained gel was autoradiographed. The histogram shows the n-fold increase of 4E-T phosphorylation normalized to the condition where JNK was not transfected.
JNK promotes 4E-T complex assembly upon oxidative stress.
PB assembly has been shown to be partly mediated through oligomerization of its components (9). To determine whether 4E-T forms higher-molecular-weight complexes in mammalian cells, we used immunoprecipitation of differentially tagged 4E-T proteins. HEK293 cells were cotransfected with plasmids coding for myc- and HA-tagged forms of 4E-T, and coimmunoprecipitated proteins were analyzed by immunoblotting. At steady state, relatively low levels of HA-tagged 4E-T coimmunoprecipitated with myc-tagged protein, indicating that differentially tagged proteins can assemble in the same complex (Fig. 7A). Arsenite treatment dramatically increased coimmunoprecipitation of the two tagged forms of 4E-T in a time-dependent manner, regardless of whether we pulled down myc- or HA-tagged protein (Fig. 7A and B), suggesting that oxidative stress promotes 4E-T complex assembly. To test whether 4E-T complex formation further correlated with the assembly of PBs, we treated HEK293 cells with cycloheximide or emetine, each of which disrupts PBs by decreasing the pool of cytoplasmic mRNAs (38). Importantly, both compounds strongly decreased the ability of 4E-T to form complexes (Fig. 7C), indicating that coimmunoprecipitation of differentially tagged proteins correlates with PB assembly. Furthermore, we found that treatment of cells with NAC decreased the ability of 4E-T to form complexes (Fig. 7D), suggesting that oxidative stress is required for 4E-T complex formation.
Fig 7.

JNK contributes to 4E-T complex assembly upon oxidative stress. (A) HEK293 cells were transfected with wt HA and myc-tagged 4E-T and treated with arsenite (0.5 mM) for the indicated times. Associated HA-tagged 4E-T was assayed within myc 4E-T immunoprecipitates by immunoblotting. (B) Same as described for panel A, except that myc-tagged 4E-T was assayed within HA-tagged 4E-T immunoprecipitates by immunoblotting. (C to E) Same as described for panel A, except that cells were treated with cycloheximide (50 μg/ml) or emetine (20 μg/ml) for 60 min (C), NAC (10 mM) for 45 min prior to arsenite treatment (D), and SP600125 (20 μM) or AS601245 (20 μM) for 30 min prior to arsenite treatment (E). (F) Cells were transfected with wt myc-tagged 4E-T and HA-tagged wt or JNK-nonphosphorylatable forms of 4E-T and treated with arsenite (0.5 mM) for 45 min. Associated HA-tagged 4E-T within myc 4E-T immunoprecipitates was assayed by immunoblotting.
To investigate the molecular mechanism of 4E-T complex assembly, we tested whether phosphorylation of 4E-T by JNK was required for complex assembly. Indeed, we found that coimmunoprecipitation of differentially tagged 4E-T proteins from arsenite-treated cells was impaired by both JNK inhibitors (SP600125 or AS601245) (Fig. 7E), suggesting that 4E-T phosphorylation may regulate 4E-T complex formation. To explore this possibility, we compared wt 4E-T with the S6A mutant in coimmunoprecipitation experiments. While arsenite treatment strongly induced the coimmunoprecipitation of wt 4E-T with itself, we found that the S6A mutant was significantly impaired in complex formation (Fig. 7F). Together, these results indicate that oxidative stress-mediated JNK activation promotes the assembly of 4E-T in higher-molecular-weight complexes. Moreover, we found that 4E-T phosphorylation promotes complex assembly, suggesting a potential mechanism by which arsenite induces PB assembly.
4E-T phosphorylation promotes PB assembly in response to oxidative stress.
Based on our in vitro and in vivo results, we hypothesized that JNK-mediated phosphorylation of 4E-T was involved in PB assembly. To test this hypothesis, we measured PB assembly using a quantitative image-based assay. Since PB assembly in response to oxidative stress could result in an increase in either PB number or PB size, we initially characterized PB assembly via the total amount of 4E-T present in PBs relative to the area of the cell, which we defined as the product of PB size and density. To validate our assay, we measured the effects of arsenite and cycloheximide treatments in U2OS cells (Fig. 8A), the latter of which was expected to eliminate PBs (36). Indeed, our assay reported that the amount of 4E-T per area (number of PBs) was significantly increased by arsenite treatment (∼60%, P < 10−58) and significantly decreased by cycloheximide treatment (∼10-fold, P < 10−48) (Fig. 8B), which validated our quantitative approach. Next, we assessed the role of JNK in PB assembly and 4E-T localization by treating cells with JNK inhibitors (AS601245 and SP600125) prior to arsenite treatment. Notably, we found that JNK inhibition significantly impaired PB assembly (Fig. 8C), as seen by the decrease of 4E-T accumulation to these structures following arsenite treatment (∼25%, P < 10−23). These results demonstrate that JNK activity is required to stimulate the assembly of PBs in response to oxidative stress.
Fig 8.

4E-T phosphorylation promotes PB assembly in response to oxidative stress. (A) U2OS cells growing in serum were treated with cycloheximide (50 μg/ml) for 1 h or arsenite (0.5 mM) for 45 min. Endogenous 4E-T was immunostained and analyzed by confocal microscopy (top). The images were processed using the software Matlab (bottom). Cell shapes were visualized by the fluorescence of the background cytoplasm, and the cell outline was traced (white lines). PBs were detected and circled (red line) on the basis of the endogenous 4E-T immunofluorescence. The number of PBs per cell and their associated intensity were reported as explained in Materials and Methods. Higher-magnification views of boxed areas are shown in the insets at the bottom right and display the recognition of PBs by the informatics program. Bar, 10 μm. (B) Data generated for panel A were analyzed to quantify the total amount of endogenous 4E-T accumulating in PBs from cells left untreated and treated with either cycloheximide (CHX) or arsenite (ARS). Error bars indicate the standard error of the mean (SEM). *, P < 0,001. a.u., arbitrary units. (C) U2OS cells were incubated with AS601245 (20 μM) or SP600125 (20 μM) for 30 min prior to treatment with arsenite. Endogenous 4E-T was immunostained and confocal images were processed as described for panel A. The amount of 4E-T localizing in PBs was determined as described for panel B. Error bars indicate the standard error of the mean. *, P < 0,001. (D) HeLa cells were transfected with wt or S6A mutant 4E-T and treated with arsenite. HA-tagged 4E-T was immunostained, and the localization of 4E-T within PBs was determined as described for panels A and B, except that 4E-T localization in PBs was normalized to its expression levels prior to quantification. Error bars indicate the standard error of the mean. *, P < 0,001. The number of cells analyzed for each condition is indicated below the bar graphs in panels B to D. (E) Representation of the distribution of wt and S6A HA-tagged 4E-T from the data in panel D. The average PB intensities were plotted over the PB densities for wt and S6A 4E-T in untreated and arsenite-treated cells. Arrows show the change in the accumulation of 4E-T in PBs upon arsenite treatment. Error bars indicate the standard error of the mean. *, P < 0,001. pix, number of pixels.
JNK-mediated phosphorylation of 4E-T is required for 4E-T complex formation. To test whether 4E-T phosphorylation is involved in PB nucleation or growth of existing PBs, we transfected wt 4E-T or the S6A mutant in HeLa cells and quantified the localization of transfected proteins in PBs. Using our quantitative image-based assay (Fig. 8A), we found that compared to wt 4E-T, arsenite did not efficiently promote the accumulation of the S6A mutant to PBs (∼30%, P < 10−15) (Fig. 8D). While we found that wt 4E-T and the S6A mutant were similarly localized to PBs in resting cells, our results indicate that the S6A mutant accumulated preferentially in small PBs upon oxidative stress (Fig. 8E). Indeed, while wt 4E-T was associated with an increased number (reflected by PB density) and size (reflected by PB intensity) of PBs in response to arsenite, we found that the S6A mutant was severely impaired in its ability to mediate PB size increases (P < 10−25) (Fig. 8E). These results suggest that 4E-T phosphorylation may not be necessary for the nucleation of PBs but, rather, may be involved in the growth of preexisting PBs in response to oxidative stress.
4E-T phosphorylation does not affect global protein synthesis or the loading of eIF4E-sensitive transcripts on polysomes.
Our results indicate that JNK-mediated 4E-T phosphorylation promotes the assembly of PBs, but whether these changes have an impact on translational control remains unknown. To address this, we first tested the impact of 4E-T phosphorylation on global translation rates by analyzing polysome profiles from stable HEK293 cell lines expressing either wt 4E-T or the S6A mutant treated with increasing concentrations of arsenite (0, 100, and 500 μM). As shown in Fig. 9A, we found that arsenite treatment strongly suppressed polysome formation, which coincided with a robust increase in the 80S ribosomal subunit peak. Although our results indicate that arsenite treatment promotes 4E-T phosphorylation, we did not see differences in arsenite-mediated translational suppression between cells expressing wt 4E-T and cells expressing the S6A mutant. These results indicate that 4E-T phosphorylation does not affect global translation rates.
Fig 9.

Expression of the S6A mutant of 4E-T does not affect global protein synthesis or loading of eIF4E-sensitive transcripts on polysomes. (A) Polysome profiles of HEK293 cells stably expressing HA-tagged 4E-T wild type (blue) or S6A (red) treated for 45 min with increasing doses of arsenite. Cell extracts were size fractionated by centrifugation through sucrose density gradients (20 to 50%). The absorbance (Abs) at 254 nm is shown as a function of sedimentation. The 40S, 60S, and 80S ribosomal subunits and polysomes are indicated. (B) Polysomal mRNAs from HEK293 cells stably expressing HA-tagged 4E-T wild type (blue) or S6A (red) were collected in three fractions corresponding to the light, medium, and heavy polysomes. (C) The expression levels of ODC1, survivin, VEGFA, and c-myc transcripts were analyzed by quantitative PCR, and the data were normalized to those for GAPDH and ActB from each polysomal fraction as well as the input. Data are expressed as the fold enrichment relative to the input. (D and E) Same as for panels B and C, except that cells were treated with 50 μM arsenite.
The translation of certain mRNAs containing structured elements in their 5′ UTR, such as those encoding VEGF, survivin, ODC1, and c-myc, is known to be more dependent on eIF4E (7). To assess whether 4E-T phosphorylation specifically regulates the translation of these transcripts, we measured their association with three polysomal fractions (low, medium, and high) using real-time qPCR (Fig. 9B). Total and polysomal mRNA was extracted from cells expressing wt 4E-T or the S6A mutant, and the abundances of the ODC1, VEGFA, survivin, and c-myc transcripts relative to those of control mRNAs (ActB and GAPDH) were evaluated. As shown in Fig. 9C, we did not find significant differences in the relative abundance of these transcripts in cells expressing wt 4E-T compared to the S6A mutant. Furthermore, we repeated these experiments by extracting polysomal mRNA from control and arsenite-treated cells to specifically address the role of 4E-T phosphorylation (Fig. 9D). We repeated the qPCR analyses of two eIF4E-sensitive transcripts (ODC1 and survivin) but did not find significant differences in arsenite-mediated translational suppression (Fig. 9E).
Altogether, our findings are consistent with an overall model whereby JNK, by mediating the phosphorylation of 4E-T, promotes the accumulation of 4E-T in larger PBs (Fig. 10). These results support the idea that PBs are dynamic structures regulated in part by the JNK signaling pathway in response to stress stimuli. Our data also indicate that PB assembly into microscopically visible structures does not affect global mRNA translation rates.
Fig 10.
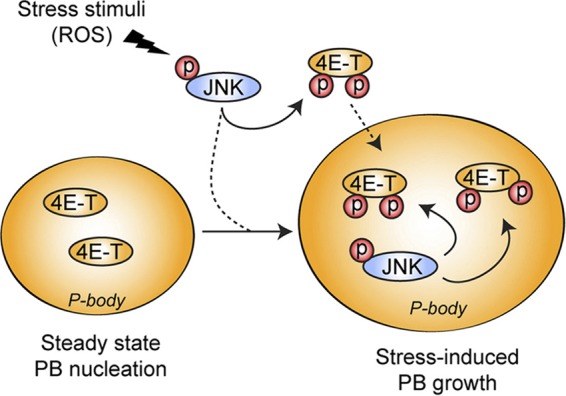
Schematic representation of the role of 4E-T phosphorylation in PB assembly. Upon oxidative stress, activated JNK localizes to PBs, where it interacts with and phosphorylates 4E-T. The phosphorylation of 4E-T mediated by JNK enhances its accumulation in PBs and participates in the increased assembly of PBs upon oxidative stress. JNK can then regulate the formation of PBs by phosphorylating its substrate, 4E-T.
DISCUSSION
PBs are dynamic aggregates of specific mRNAs and proteins that play important roles in mRNA storage and degradation. Despite being evolutionarily conserved, little is known about the regulatory mechanisms that participate in their steady-state assembly and induction in stressed cells. In this study, we show that JNK signaling regulates PB assembly by phosphorylating one of its components, the eIF4E-binding protein 4E-T. In particular, our data demonstrate that activation of JNK by oxidative stress promotes the formation of large PB foci. JNK directly phosphorylates 4E-T on several residues, and these phosphorylation events were found to promote the assembly of in vitro complexes containing 4E-T and to increase the size of PBs. These observations indicate that 4E-T might function as an important scaffold upon which PBs are assembled and suggest that JNK phosphorylation may therefore be required for the formation of larger PBs.
The assembly of PBs has been proposed to occur in two discrete stages (15). In the first step, mRNAs associate with the protein constituents of PBs to form ribonucleoprotein (RNP) monomers. Several PB proteins likely contribute to the translationally repressed state of these mRNAs (6, 15), as was suggested for 4E-T, which presumably competes with translation initiation factors for binding to eIF4E (14). The PB monomers then aggregate in the second stage to form the larger cytoplasmic foci that are microscopically visible. While 4E-T may be involved in the formation of RNP monomers, our data indicate that it mainly participates in the second step of this model by specifically promoting the formation of larger aggregate structures. At the moment, the function of these large aggregates is unknown, but these structures were not found to be required for mRNA degradation and translational repression (9). Consistent with this, our results indicate that 4E-T phosphorylation and, thus, PB assembly in response to stress do not affect global translation rates or the translation of transcripts previously shown to be more dependent on eIF4E (7). While we cannot exclude the possibility that certain cell types are more sensitive to changes in the size of PBs, our data suggest that assembly of PBs in large aggregates may participate in other unresolved cellular functions.
4E-T is part of a subset of PB factors, including Lsm1, RCK/p54, and CCR4, which are required for the accumulation of each other in PBs (1). It is not known what makes any of these factors concentrate in cytoplasmic foci, but at a minimum, this seems to require RNA (38). Many Saccharomyces cerevisiae PB components, including LSm4, CCR4, and NOT1, contain Q/N (glutamine/asparagine)-rich regions that have the potential to form aggregates similar to the Q/N-rich domains found in prion proteins (9, 17, 31). The average Q/N content in the eukaryotic proteome is 7.7 per 80-mer (27), and many PB components contain at least one region that exceeds this average (31). Such domains were shown to participate in protein aggregation and accumulation to PBs (31) and are also found in proteins prone to aggregation, such as huntingtin (27). It was also observed that PB components contain high numbers of proline residues in or just downstream of these Q/N-rich regions (31). Proline-rich regions often form extended and flexible regions that facilitate protein-protein interactions and are often regulated by phosphorylation (41). Binding via these proline-rich domains is generally not very specific but can be both very rapid and strong (21). Our data suggest that 4E-T participates in the aggregation stage of PB assembly, and interestingly, we found that 4E-T contains two regions with higher than average Q/N contents (11 and 14 per 80-mer, respectively) (Fig. 5C; see Fig. S4 in the supplemental material). We also found that these regions are enriched in proline residues (see Fig. S4 in the supplemental material), suggesting that they may participate in 4E-T protein aggregation. While this model is suggested by our data, more experimentation will be required to characterize the 4E-T interaction network.
Accumulating evidence indicates that several stress-activated signaling pathways directly regulate PB assembly. In S. cerevisiae, the protein kinase Ste20 was recently shown to promote phosphorylation of the decapping factor Dcp2, which increased Dcp2 accumulation in PBs upon stress (43). In mammalian cells, the decapping cofactor Dcp1 was found to be phosphorylated by JNK in response to interleukin-1 (IL-1) stimulation (35). While Dcp1 does not seem to be required for PB assembly (36), expression of a phosphomimetic mutant was shown to disrupt PBs, suggesting a possible antagonistic role of JNK in response to IL-1 stimulation (35). We found that JNK activity is required for PB assembly in response to oxidative stress, suggesting that PBs are likely subject to multiple regulatory mechanisms that affect mRNP maturation. Our data are consistent with JNK affecting the second step of PB assembly, by specifically promoting the formation of larger aggregate structures. Indeed, expression of the S6A mutant resulted in the presence of smaller but more numerous PBs in response to oxidative stress, suggesting that JNK-mediated 4E-T phosphorylation regulates the final aggregation of these smaller structures. Interestingly, many of the JNK phosphorylation sites are located within or in proximity to the Q/N-rich regions (see Fig. S4 in the supplemental material), suggesting that 4E-T phosphorylation may affect its ability to form larger aggregates. These phosphorylation events may be a rapid and reversible mechanism to promote PB assembly in response to different types of stress stimuli.
In closing, we propose a model whereby 4E-T regulates the second stage of PB assembly in unstressed cells. We found that JNK regulates 4E-T function by facilitating the aggregation stage in response to stress stimuli. Together, these results demonstrate an important role for 4E-T in PB assembly and underscore the role of JNK in this regulation.
Supplementary Material
ACKNOWLEDGMENTS
We deeply appreciate all the members of our laboratories for their insightful discussions and comments on the manuscript and the data. We also specifically acknowledge Chris Jahns (HMS Taplin Biological Mass Spectrometry Facility) for assistance with preparation of in-gel digests.
This work was supported by grants from the Canadian Cancer Society Research Institute (700878), the Cancer Research Society (F121153), and the Canadian Institutes of Health Research (MOP123408) (to P.P.R.), as well as the U.S. National Institutes of Health (to S.P.G.; HG3456). P.P.R. holds a Canada Research Chair in Signal Transduction and Proteomics. M.C. holds a doctoral studentship from the Fonds de la Recherche en Santé du Québec (FRSQ). IRIC core facilities are supported by the FRSQ.
We have no conflict of interest to declare.
Footnotes
Published ahead of print 10 September 2012
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1. Andrei MA, et al. 2005. A role for eIF4E and eIF4E-transporter in targeting mRNPs to mammalian processing bodies. RNA 11:717–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Beausoleil SA, Villen J, Gerber SA, Rush J, Gygi SP. 2006. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 24:1285–1292 [DOI] [PubMed] [Google Scholar]
- 3. Buchan JR, Parker R. 2009. Eukaryotic stress granules: the ins and outs of translation. Mol. Cell 36:932–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cargnello M, Roux PP. 2011. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 75:50–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cavigelli M, et al. 1996. The tumor promoter arsenite stimulates AP-1 activity by inhibiting a JNK phosphatase. EMBO J. 15:6269–6279 [PMC free article] [PubMed] [Google Scholar]
- 6. Coller J, Parker R. 2005. General translational repression by activators of mRNA decapping. Cell 122:875–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. De Benedetti A, Graff JR. 2004. eIF-4E expression and its role in malignancies and metastases. Oncogene 23:3189–3199 [DOI] [PubMed] [Google Scholar]
- 8. Decker CJ, Parker R. 2012. P-bodies and stress granules: possible roles in the control of translation and mRNA degradation. Cold Spring Harbor Perspect. Biol. 4(9):pii=a012286. doi:10.1101/cshperspect.a012286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Decker CJ, Teixeira D, Parker R. 2007. Edc3p and a glutamine/asparagine-rich domain of Lsm4p function in processing body assembly in Saccharomyces cerevisiae. J. Cell Biol. 179:437–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dephoure N, et al. 2008. A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. U. S. A. 105:10762–10767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dostie J, Ferraiuolo M, Pause A, Adam SA, Sonenberg N. 2000. A novel shuttling protein, 4E-T, mediates the nuclear import of the mRNA 5′ cap-binding protein, eIF4E. EMBO J. 19:3142–3156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Elias JE, Gygi SP. 2007. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4:207–214 [DOI] [PubMed] [Google Scholar]
- 13. Eulalio A, Behm-Ansmant I, Izaurralde E. 2007. P bodies: at the crossroads of post-transcriptional pathways. Nat. Rev. Mol. Cell Biol. 8:9–22 [DOI] [PubMed] [Google Scholar]
- 14. Ferraiuolo MA, et al. 2005. A role for the eIF4E-binding protein 4E-T in P-body formation and mRNA decay. J. Cell Biol. 170:913–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Franks TM, Lykke-Andersen J. 2008. The control of mRNA decapping and P-body formation. Mol. Cell 32:605–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Garneau NL, Wilusz J, Wilusz CJ. 2007. The highways and byways of mRNA decay. Nat. Rev. Mol. Cell Biol. 8:113–126 [DOI] [PubMed] [Google Scholar]
- 17. Gilks N, et al. 2004. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol. Biol. Cell 15:5383–5398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hochberg Y, Benjamini Y. 1990. More powerful procedures for multiple significance testing. Stat. Med. 9:811–818 [DOI] [PubMed] [Google Scholar]
- 19. Huang C, Ke Q, Costa M, Shi X. 2004. Molecular mechanisms of arsenic carcinogenesis. Mol. Cell. Biochem. 255:57–66 [DOI] [PubMed] [Google Scholar]
- 20. Huttlin EL, et al. 2010. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143:1174–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kay BK, Williamson MP, Sudol M. 2000. The importance of being proline: the interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J. 14:231–241 [PubMed] [Google Scholar]
- 22. Kedersha N, Anderson P. 2007. Mammalian stress granules and processing bodies. Methods Enzymol. 431:61–81 [DOI] [PubMed] [Google Scholar]
- 23. Kedersha N, et al. 2005. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J. Cell Biol. 169:871–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lei K, et al. 2002. The Bax subfamily of Bcl2-related proteins is essential for apoptotic signal transduction by c-Jun NH(2)-terminal kinase. Mol. Cell. Biol. 22:4929–4942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Matsuzawa A, Ichijo H. 2008. Redox control of cell fate by MAP kinase: physiological roles of ASK1-MAP kinase pathway in stress signaling. Biochim. Biophys. Acta 1780:1325–1336 [DOI] [PubMed] [Google Scholar]
- 26. Mayya V, et al. 2009. Quantitative phosphoproteomic analysis of T cell receptor signaling reveals system-wide modulation of protein-protein interactions. Sci. Signal. 2:ra46 doi:10.1126/scisignal.2000007 [DOI] [PubMed] [Google Scholar]
- 27. Michelitsch MD, Weissman JS. 2000. A census of glutamine/asparagine-rich regions: implications for their conserved function and the prediction of novel prions. Proc. Natl. Acad. Sci. U. S. A. 97:11910–11915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Parker R, Sheth U. 2007. P bodies and the control of mRNA translation and degradation. Mol. Cell 25:635–646 [DOI] [PubMed] [Google Scholar]
- 29. Parker R, Song H. 2004. The enzymes and control of eukaryotic mRNA turnover. Nat. Struct. Mol. Biol. 11:121–127 [DOI] [PubMed] [Google Scholar]
- 30. Rappsilber J, Ishihama Y, Mann M. 2003. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 75:663–670 [DOI] [PubMed] [Google Scholar]
- 31. Reijns MA, Alexander RD, Spiller MP, Beggs JD. 2008. A role for Q./N-rich aggregation-prone regions in P-body localization. J. Cell Sci. 121:2463–2472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rhoads RE. 2009. eIF4E: new family members, new binding partners, new roles. J. Biol. Chem. 284:16711–16715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Romeo Y, et al. 16 July 2012. RSK regulates activated BRAF signalling to mTORC1 and promotes melanoma growth. Oncogene [Epub ahead of print.] doi:10.1038/onc.2012.312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J. 2004. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc. Natl. Acad. Sci. U. S. A. 101:13489–13494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rzeczkowski K, et al. 2011. c-Jun N-terminal kinase phosphorylates DCP1a to control formation of P bodies. J. Cell Biol. 194:581–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sheth U, Parker R. 2003. Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science 300:805–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shevchenko A, Wilm M, Vorm O, Mann M. 1996. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 68:850–858 [DOI] [PubMed] [Google Scholar]
- 38. Teixeira D, Sheth U, Valencia-Sanchez MA, Brengues M, Parker R. 2005. Processing bodies require RNA for assembly and contain nontranslating mRNAs. RNA 11:371–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ting L, Rad R, Gygi SP, Haas W. 2011. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods 8:937–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Weston CR, Davis RJ. 2007. The JNK signal transduction pathway. Curr. Opin. Cell Biol. 19:142–149 [DOI] [PubMed] [Google Scholar]
- 41. Williamson MP. 1994. The structure and function of proline-rich regions in proteins. Biochem. J. 297(Pt 2):249–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yates JR, III, Eng JK, McCormack AL, Schieltz D. 1995. Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal. Chem. 67:1426–1436 [DOI] [PubMed] [Google Scholar]
- 43. Yoon JH, Choi EJ, Parker R. 2010. Dcp2 phosphorylation by Ste20 modulates stress granule assembly and mRNA decay in Saccharomyces cerevisiae. J. Cell Biol. 189:813–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zheng D, et al. 2008. Deadenylation is prerequisite for P-body formation and mRNA decay in mammalian cells. J. Cell Biol. 182:89–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.