

Abstract
Deregulation of transcription arising from mutations in key signaling pathways is a hallmark of cancer. In melanoma, the most aggressive and lethal form of skin cancer, the Brn-2 transcription factor (POU3F2) regulates proliferation and invasiveness and lies downstream from mitogen-activated protein kinase (MAPK) and Wnt/β-catenin, two melanoma-associated signaling pathways. In vivo Brn-2 represses expression of the microphthalmia-associated transcription factor, MITF, to drive cells to a more stem cell-like and invasive phenotype. Given the key role of Brn-2 in regulating melanoma biology, understanding the signaling pathways that drive Brn-2 expression is an important issue. Here, we show that inhibition of phosphatidylinositol 3-kinase (PI3K) signaling reduces invasiveness of melanoma cells in culture and strongly inhibits Brn-2 expression. Pax3, a transcription factor regulating melanocyte lineage-specific genes, directly binds and regulates the Brn-2 promoter, and Pax3 expression is also decreased upon PI3K inhibition. Collectively, our results highlight a crucial role for PI3K in regulating Brn-2 and Pax3 expression, reveal a mechanism by which PI3K can regulate invasiveness, and imply that PI3K signaling is a key determinant of melanoma subpopulation diversity. Together with our previous work, the results presented here now place Brn-2 downstream of three melanoma-associated signaling pathways.
INTRODUCTION
A major challenge to effective anticancer therapy is tumor cell population heterogeneity. Different subpopulations of cells with substantially different biological properties coexist within tumors (24, 35). While some heterogeneity will be genetic and consequently irreversible, a further level of complexity is imposed by dynamic nongenetic heterogeneity driven by the cellular microenvironment. The ability of cancer cells to adapt their phenotype in response to environmental cues is exemplified in the epithelial to mesenchymal transition that characterizes a switch from a noninvasive to invasive phenotype (23). Moreover, reversible switching between cell phenotypes is likely to underpin the generation of therapeutically resistant cancer stem cells that are proposed to self renew, seed, and maintain tumors and provides a reservoir of therapeutically resistant cells (25, 51). Understanding the signals and microenvironmental cues that mediate phenotype switching is a key issue.
In melanoma, recent evidence suggests that cells switch between a minimum of three phenotypes (26): cells expressing hallmarks of differentiation, including the manufacture of the pigment melanin; cells with the potential for proliferative tumorigenesis; and an invasive, therapeutically resistant, slow-proliferating stem cell-like phenotype. The different subpopulations may be defined by the activity of the so-called melanocyte master regulator, the microphthalmia-associated transcription factor, Mitf. Low-Mitf-expressing cells are G1 arrested and stem cell like with tumor-initiating potential and are highly invasive, while Mitf-positive cells either proliferate or differentiate into G1-arrested pigment-producing cells depending on posttranslational modifications or level of expression (1, 5, 6, 8, 9). Accordingly, transcription profiling of 86 melanoma cell lines revealed two phenotypes: slow proliferating with high invasive potential, low Mitf expression, and high tumor growth factor beta (TGF-β) signaling; and rapidly proliferating with higher Mitf expression and low invasive potential (27). Importantly, the stable phenotype of cell lines in culture is reversible in xenograft tumors (26).
A key to understanding how different melanoma subpopulations can be generated is the identification of key regulators of Mitf expression, especially those that may suppress Mitf transcription to generate invasive stem-like cells. One such factor is the POU domain transcription factor Brn-2 (POU3F2) (11, 19, 30), whose expression lies downstream from two melanoma-associated signaling pathways: the mitogen-activated protein kinase (MAPK) cascade (21), including receptor tyrosinase kinases, NRAS, BRAF, and MEK, that is constitutively active via genetic lesions in the majority of, if not all, melanomas (15, 33), and the Wnt/β-catenin pathway (20), which has been implicated in melanocyte immortality and proliferation (16, 44). Importantly, Brn-2, which is frequently overexpressed in melanoma (17, 48), can repress Mitf (19), leading to either increased proliferation if expressed in melanocytes (20) or increased invasiveness in melanoma (19). Moreover, the key role played by Brn-2 in promoting invasiveness in melanoma was also highlighted when it was shown to mediate repression of the cGMP phosphodiesterase PDE5 in response to BRAF signaling (2). Strikingly, although Brn-2 and Mitf are coexpressed in cell lines, they mark two distinct subpopulations of melanoma cells in tumors (19, 49). Consistent with this, recent evidence obtained using real-time intravital imaging of melanoma syngeneic tumors engineered to express a Brn-2 promoter-green fluorescent protein (GFP) reporter has revealed that high levels of Brn-2 promoter activity identifies invasive melanoma cells and confirms that melanoma cells self renew in vivo and switch phenotypes from stem cell like (invasive) to proliferative/differentiated (frequently) and back (infrequently) (42). These data highlight the close relationship between stem cell-like properties and invasiveness and indicate that a major contribution to phenotype switching in vivo is the cellular microenvironment. The Brn-2 promoter therefore responds to signals that generate the invasive subpopulation of melanoma cells that in humans is responsible for seeding metastases that are heterogeneous. The identification of the signals that target the Brn-2 promoter will therefore provide important clues as to the signaling pathways that operate in vivo to trigger a switch between different melanoma phenotypes. Here, we show that in addition to being regulated by BRAF and β-catenin, the Brn-2 promoter is controlled by Pax3 and that both Pax3 and Brn-2 expression is downregulated by inhibitors that target phosphatidylinositol 3-kinase (PI3K) signaling. Collectively the results provide a mechanistic link between PI3K signaling and invasiveness in melanoma.
MATERIALS AND METHODS
Cell cultures and reagents.
501mel, Skmel28, and A375 human melanoma cell lines and the B16F10 mouse melanoma cell line (here referred to as B16) were maintained in RPMI 1640 (Gibco BRL, Invitrogen) supplemented with 10% fetal bovine serum (Biosera). Cells were grown at 37°C with 5% CO2. Where indicated, cells were treated for 24 h with specific inhibitors of all classes of PI3K (LY294002; 25 μM; Sigma) or class I only (GDC-0941; 1 μM; Selleck), MEK (U0126; 20 μM; Sigma), or dimethylsulfoxide (DMSO) alone.
Proliferation assay.
A 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay was performed using Thiazol Blue Tetrazolium Bromide (Sigma). Cells were seeded in a 96-well plate, and the MTT solution (1 mg/ml in medium) was added to the wells and left for 2 h at 37°C. At different time points the medium was removed, the cells were lysed in 100 μl of DMSO, and the absorbance was read at 560 nm.
Brn-2 luciferase assays.
The Brn-2 promoter–luciferase reporter has been described already (20, 21). Cells were harvested 36 h after transfection of 100 ng of promoter-reporter construct with or without plasmid expressing activators or empty expression vector DNA with FuGENE6 (Roche) and assayed for firefly luciferase activity (Promega). The point mutations in the Pax3 binding sites were introduced according to the DpnI-based QuikChange site-directed mutagenesis methodology (Stratagene, La Jolla, CA) using the following primers: 5′-TATCCACTGGGATCAAAGGGCGCAGAGCCCGGGGGAGGGGGTGGA-3′ and 5′-TCCACCCCCTCCCCCGGGCTCTGCGCCCTTTGATCCCAGTGGATA-3′.
siRNA.
Small interfering RNA (siRNA)-mediated downregulation of Pax3 was achieved with the following Pax3-specific sequences: 5′-CCACAUCCGCCACAAGAUCTT-3′ and 5′-GAUCUUGUGGCGGAUGUGGTT-3′, with the control being 5′-UUCUCCGAACGUGUCACGUTT-3′ and 5′-ACGUGACACGUUCGGAGAATT-3′. siRNA was transfected into cells with Lipofectamine (Invitrogen) per the manufacturer's instructions. Cells were harvested 3 days posttransfection.
Lentivirus infection.
A lentiviral vector (pCSII EF1alpha) containing the sequence encoding Pax3 and mCherry fluorescent protein separated by a 2A self-cleaving peptide was used to establish the stable cell line SKmel28 mCherry-Pax3. Virus was produced by transient transfection into Phoenix cells with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions together with vectors encoding Gag/Pol, Rev, and the vesicular stomatitis virus (VSV) envelope. Virus-containing supernatant was harvested at 48 and 72 h posttransfection, passed through a 0.45-μm filter, and concentrated by ultracentrifugation at 70,000 × g for 2 h. The pellet was resuspended in Hanks balanced salt solution (HBSS) and supplemented with 2 μg/ml Polybrene (Millipore), and target cells were incubated with concentrated virus for 5 min, after which fresh medium was added.
Infection efficiency was measured by flow cytometry using a FACSCantoII (BD Biosciences) and determined as >80% (data not shown).
Reverse transcription-quantitative PCR (RT-qPCR).
Total RNA was extracted from melanoma cells by using the RNA isolation kit (Invitrogen). RNA was treated with DNase for 1 h at 37°C, and 500 ng was reverse transcribed. The resulting cDNA was used to detect mRNA abundance with the following primers: 5′-ATGTGCAAGCTGAAGCCTTT-3′ and 5′-CTCACCACCTCCTTCTCCAG-3′ for human and mouse Brn-2, 5′-CCGACTTGGAGAGGAAGGA-3′ and 5′-CATCTGATTGGGGTGCTGA-3′ for human Pax3, 5′-ACCCACTCCTCCACCTTTGA-3′ and 5′-CATACCAGGAAATGAGCTTGACAA-3′ for human glyeraldehyde-3-phosphate dehydrogenase (GAPDH), 5′-TGGCTTTTCAACCATCTCATTCC-3′ and 5′-AGAGTGCTCCGACAGCTGGTAT-3′ for mouse Pax3, and 5′-GCATGGCCTTCCGTGTTC-3′ and 5′-ATGTCATCATACTTGGCAGGTTTC-3′ for mouse GAPDH. Reactions were done in SYBR green mix and analyzed using a Corbett rotor-gene 6000. Relative mRNA levels were calculated using the comparative threshold cycle (CT) method.
Western blot analysis.
Cells were lysed in Laemmli buffer in reducing conditions, and whole-cell extracts were subjected to 10% polyacrylamide SDS-PAGE. Proteins were then transferred onto polyvinylidene difluoride (PVDF) membranes (Amersham Biosciences). Membranes were blocked with 5% nonfat milk in phosphate-buffered saline containing 0.1% Tween 20 and probed with appropriate primary antibodies (mouse monoclonal anti-Brn-2 [Biogenes], mouse monoclonal anti-Pax3 [Developmental Studies Hybridoma Bank, University of Iowa], mouse monoclonal anti-MITF variant M [Dako], rabbit polyclonal anti-β-catenin [Abcam], rabbit polyclonal anti-phospho-S6 ribosomal protein [Ser235/236; Cell Signaling], rabbit monoclonal anti-S6 ribosomal protein [Cell Signaling], rabbit monoclonal anti-phospho-Akt [Ser473; Cell Signaling], rabbit polyclonal anti-Akt [Cell Signaling], rabbit polyclonal anti-p44/42 [T202/Y204; Cell Signaling], and rabbit polyclonal anti-ERK2 [Santa Cruz]) for 1 h at room temperature and then overnight at 4°C. Proteins were detected using anti-mouse or anti-rabbit immunoglobulin coupled to horseradish peroxidase (Bio-Rad) and visualized with the ECL detection kit (Amersham Biosciences).
Band shift assays.
The band shift assays for Pax3 were performed in a 20-μl final volume containing 25 mM HEPES, pH 7.9, 10% glycerol, 150 mM KCl, 1 μg of dGdC, 10 μg of bovine serum albumin (BSA), 5 mM dithiothreitol (DTT), and the protein of interest. The Pax3 paired domain and homeo domain was expressed as a histidine fusion (Pax3-PDHD-His) (12) (a gift from Alan Underhill) and Lef1 protein as a glutathione S-transferase (GST) fusion and purified. Unlabeled oligonucleotides specific for the Brn-2 promoter were used in the competition assays, and the Brn-2 (or Tyrp-1 promoter as a control)-specific oligonucleotides were labeled with 32P and used as probes.
Chromatin immunoprecipitation assays.
Chromatin immunoprecipitation assays were performed with 2 μg of mouse monoclonal anti-Pax3 (DSHB, University of Iowa), anti-Lef1 (Santa Cruz), anti-RNA polymerase II (PolII; Santa Cruz), or normal immunoglobulin G (Upstate Biotechnology) using the μMAC technology (μMAC column; Miltenyi Biotec). After immunoprecipitation and reverse cross-linking, DNA was phenol-chloroform extracted and samples were analyzed by semiquantitative PCR for 35 cycles or quantitative PCR for 45 cycles, taking care that the PCR was in the log phase of amplification. The primers used for PCR were 5′-GGGTACAGCTCTGCACCAAT-3′ (−277 to +178 relative to the transcription start site [TSS]) for the mouse Brn-2 promoter, 5′-GTAGCTCTGCGCCAATCAGT-3′ and 5′-GGACTGAGCGCTCCGGTTAAA-3′ (−276 to +157 relative to the TSS) for the human Brn-2 promoter containing the potential Pax3 binding site, 5′-AAAGAGCATGACTCCTTGTTA-3′ and 5′-CTCGCTCTATCGCTGGTTTTG-3′ (−1806 to −1647 relative to the TSS) for a region of the human Brn-2 promoter located upstream of the potential Pax3 binding site, and 5′-GAAAAGCCCGCACCAACCAT-3′ and 5-GCTAAGTTTAGCCTGCCTGG-3′ (−900 to −742 relative to TSS) for the human GAPDH promoter.
Wound-healing assay.
501mel, Skmel28, and B16F10 melanoma cells were seeded onto 24-well plates. After 48 h, a wound was formed by scraping the cells with a 200-μl tip. The monolayers of cells were then washed three times with phosphate-buffered saline (PBS) to remove floating cells and incubated in fresh complete medium containing either PI3K inhibitors or DMSO. Photographs were taken every 15 min for 72 h at the same position of the wounds at ×10 magnification using a Nikon microscope and Metamorph software.
Invasion assays.
Quantitative invasion assays were performed using a cell invasion assay kit (Chemicon). Melanoma cell suspensions in serum-free medium were added to the invasion chamber and allowed to migrate through an extracellular matrix for 24 to 72 h. The invading cells present on the lower surface of the membrane were stained and counted under the microscope.
Immunofluorescence microscopy.
Immunofluorescence was performed as described previously (6) with a 1:100 dilution of rabbit polyclonal anti-phospho-S6 ribosomal protein (Ser235/236) (Cell Signaling), mouse monoclonal anti-Brn-2 (Biogenes), and rabbit anti-β-catenin (Abcam) and with a 1:300 dilution of appropriate Alexa Fluor dye-conjugated antibodies (Invitrogen). Nuclei were counterstained using 4′,6-diamidino-2-phenylindole (DAPI), and slides were mounted with Vectashield mounting media for fluorescence (Vector laboratories).
RESULTS
Inhibition of PI3K inhibits melanoma invasiveness.
Although Brn-2 expression is upregulated by MAPK signaling (21), the fact that it exhibits heterogeneous expression within human tumors (19) in which all cells would be expected to bear activating mutations in BRAF or NRAS suggests that other signaling pathways regulate its promoter. The notion that Brn-2-positive cells may be subject to additional microenvironment-regulated signals is reinforced by the observation that in xenograft tumors derived from melanoma cell lines that coexpress Brn-2 and MITF, cells segregate in vivo into two mutually exclusive populations expressing either Brn-2 or MITF (49), and that within human tumors Brn-2 and MITF expression is mutually exclusive (19). One clue to the identity of a Brn-2-activating signaling pathway comes from the fact that MITF-low, Brn-2-high cells are more invasive than their MITF-high, Brn-2-low counterparts (19, 42). Signals that promote invasiveness might be expected to upregulate Brn-2, leading to downregulation of MITF and triggering an invasive phenotype as a consequence. In this respect, increasing evidence suggests that activation of the PI3K pathway promotes invasiveness in melanoma (38) and the transition from a radial to a vertical growth phase (22, 34). We therefore investigated the possibility that inhibition of PI3K signaling could diminish invasive behavior in melanoma cells in culture and downregulate Brn-2 expression.
Melanoma cells were treated with the PI3K inhibitor LY294002, and invasive capacity compared to that of untreated cells was assessed using a Boyden chamber Matrigel assay. Treatment of SKmel28 cells with LY294002 for 24 h led to a 3- to 4-fold decrease in the numbers of cells passing through the membrane (Fig. 1A). Similar results were obtained using 3 other melanoma cell lines (Lu1205, B16, and Colo858 [not shown]). These results are consistent with PI3K signaling promoting invasiveness, as has been observed previously (39, 56). Similarly, treatment of SKmel28, 501mel, and B16 with LY294002 inhibited wound healing in a scratch-wound assay (Fig. 1B to D) that measures the amount of time it takes for confluent cells to fill a wound in the monolayer. In this assay, wound healing results from a combination of polarized cell migration and proliferation. Although PI3K inhibition reduced proliferation in Skmel28 cells (Fig. 1E), which have been reported to have a PTEN mutation (54), as well as 501mel and B16 cells (data not shown), which might account in part for any delay in wound healing, these experiments were performed over a 10-h period, shorter than the doubling time of the cell lines used. As such, we conclude that the major effect of LY294002 in delaying wound healing in the melanoma cell lines is a consequence of inhibition of cell migration.
Fig 1.

LY294002 treatment decreases invasion and migration of melanoma cell lines. (A) Skmel28 invasive melanoma cells were treated with 20 μM LY294002. After 24 h, the number of LY294002-treated invading cells was counted and compared to the control (DMSO). Columns represent means from three experiments; bars indicate SD; *, P < 0.05. (B to D) LY294002 treatment decreases the wound-healing ability of the SKmel28 (B), B16 (C), and 501mel (D) melanoma cell lines in a scratch/wound assay. (E) Proliferation MTT assay on Skmel28 cells over 48 h in the presence or absence of PI3K inhibitors. Values represent means of optical density values measured at 560 nm ± standard deviations (SD).
PI3K signaling is required for Brn-2 expression.
To investigate whether LY294002 could also regulate Brn-2 expression, which we (19) and others (2) have previously shown to promote invasiveness, 501mel, SKmel28, and A375 melanoma cells were treated with LY294002, and the expression of Brn-2 was assessed by Western blotting. As a control we used U0126, a MEK inhibitor, since we have shown previously that Brn-2 is strongly regulated by MAPK signaling downstream from both a receptor tyrosine kinase and BRAF (21). The results reveal that while U0126 diminished Brn-2 expression as expected, treatment with LY294002 led to an even more robust inhibition of Brn-2 levels in all three cell lines tested (Fig. 2A). The effect of LY294002 on Brn-2 expression was not mediated by off-target effects on MAPK signaling, since we observed no effect on phospho-ERK levels (data not shown; also see Fig. 6B). Moreover, the effects appear to be specific for PI3K, since we observed a similar effect using a distinct PI3K inhibitor GDC0941 (Fig. 2B) where phosphorylation of ribosomal S6 protein was used as a measure of the effectiveness of the inhibitors used.
Fig 2.

PI3K inhibition decreases Brn-2 protein expression. (A) Western blot of 501mel, SKmel28, and A375 melanoma cells treated with LY294002 (25 μM) for 24 h. MEK inhibition (U0126; 10 μM) was used as a control, and Erk2 was used to ensure equal loading. (B) Western blot performed on SKmel28 cells treated with two different PI3K inhibitors (LY294002 at 25 μM or GDC-0941 at 10 μM) for 24 h using anti-Brn-2 and anti-p-S6 antibodies. β-Catenin was used as a loading control. (C) Relative expression of Brn-2 mRNA as determined using quantitative real-time RT-PCR from the indicated melanoma cell lines. Values were normalized to those for GAPDH and ratios were established compared to DMSO treatment. (D) Schematic of the Brn-2 promoter showing the binding site for Lef1 (top). Shown is a luciferase assay of 501mel and SKmel28 cells transfected with a Brn-2-luciferase reporter and treated with LY294002 or DMSO. Columns represent means from three experiments; bars indicate SD; *, P < 0.05. (E) PI3K inhibition does not affect β-catenin cellular localization. Immunofluorescence performed on SKmel28 treated with LY294002, GDC-0941, or DMSO (Ctl) using a monoclonal mouse anti-β-catenin (green), a polyclonal goat anti-Brn2, and a polyclonal rabbit anti-p-S6 antibody. Nuclei were counterstained using DAPI.
Fig 6.

Pax3 is implicated in melanoma cell migration and invasiveness. (A) Relative Brn-2, Pax3, and Mitf mRNA levels of 501mel and SKmel28 cells were determined by RT-qPCR. Results represent the means from three independent experiments with error bars indicating standard deviations. (B) Western blot of 501mel, B16, and Skeml28 cells after 24 h of treatment with LY294002 using anti-Brn-2 and anti-Pax3 antibodies. Anti-p-S6 was used as a control of LY294002 efficiency, and total S6 (tS6) and Erk2 were used for the loading control. p-Erk was used for PI3K inhibition specificity. (C) Pax3 knockdown decreases the wound-healing ability of the B16 cell line in a scratch/wound assay. (D) Pax3 knockdown decreases invasiveness potential of B16 cells. After 24 h, the number B16ShPax3 clone 1 invading cells was counted and compared to the control (parental cell line). (E) Brn-2 expression is sustained upon LY294002 exposure when Pax3 is overexpressed. Brn-2 and Pax-3 expression analysis was assessed by Western blotting of Skmel28 parental or Skmel28 cells expressing mCherry-Pax3 protein. Anti-p-S6 was used as a control of LY294002 efficiency, and total S6 (tS6) and Erk2 were used for the loading control. Quantification was achieved using ImageJ software. (F) LY294002 does not decrease invasion of Skmel28 melanoma cells when Pax3 is overexpressed. After 24 h, the number of Skmel28 parental cells or those expressing mCherry-Pax3 treated with LY294002 was counted and compared to the control (DMSO). Columns represent means from three experiments; bars indicate SD; *, P < 0.05.
The inhibition of Brn-2 levels following LY294002 treatment was likely due to a decrease in Brn-2 transcription, since quantitative RT-PCR analysis using 501mel and Skmel28 cells showed a diminution in Brn-2 mRNA expression in both cell lines (Fig. 2C). The analysis of Brn-2 promoter activity using a well-characterized Brn-2-luciferase reporter (20, 21) showed a strong repression following LY294002 treatment in a luciferase assay (Fig. 2D), again implying that inhibition of PI3K signaling targets Brn-2 transcription.
The effect of LY294002 presumably was mediated via regulation of a transcription factor(s) able to recognize the Brn-2 promoter. To date, only β-catenin/Lef1 has been shown to regulate Brn-2 expression (20). However, no obvious increase in β-catenin levels (Fig. 2B) or in nuclear β-catenin (Fig. 2E) was observed following treatment of cells with either LY294002 or GDC0941, suggesting that another factor was involved. Phosphorylation of ribosomal S6 protein was again used as a control for the effectiveness of the PI3K inhibitors.
Pax3 directly regulates the Brn-2 promoter.
One key regulator of the melanocyte lineage is Pax3 (31), which not only regulates MITF expression (52) but also plays a key role in preventing differentiation of melanocyte stem cells (32). Although little is known as to how Pax3 is regulated, it is widely expressed in melanomas (37, 43), and we therefore asked whether Pax3 also regulates Brn-2 promoter activity. Cotransfection of a Pax3 expression vector together with the Brn-2-luciferase reporter into B16, SKmel28, or 501mel melanoma cells led to up to 5-fold activation of luciferase activity in all three cell lines (Fig. 3A). Consistent with Pax3 regulating Brn-2 expression, Western blotting revealed that siRNA-mediated depletion of Pax3 led to decreased Brn-2 expression in 501mel cells (Fig. 3B). To confirm the role of Pax3 in mediating Brn-2 expression, we established two independent stable cell lines depleted for Pax3 using a plasmid (pGeneClip) carrying a short hairpin RNA (shRNA) specific for both mouse and human Pax3, and we verified that Pax3 mRNA (Fig. 3C) and protein (Fig. 3D) were reduced. shRNA-mediated depletion of Pax3 in B16 cells also decreased Brn-2 mRNA (Fig. 3C). The effect of Pax3 on Brn-2 mRNA expression was likely to be direct, since preliminary chromatin immunoprecipitation (ChIP) from B16 cells followed by semiquantitative PCR (Fig. 3E) revealed endogenous Pax3 bound to the Brn-2 promoter, using IgG and Lef1 as negative and positive controls, respectively. This conclusion was confirmed using qPCR/ChIP from both Skmel28 (Fig. 3F) or 501mel cells (Fig. 3G), which revealed substantial enrichment of Pax3 at the Brn-2 promoter compared to an unrelated upstream region.
Fig 3.
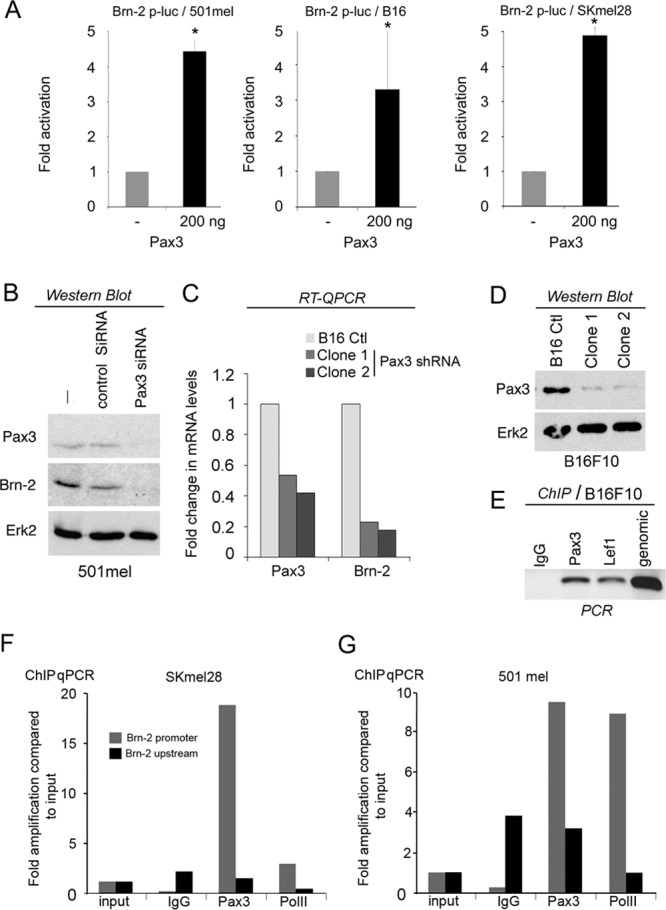
Pax3 regulates Brn-2 transcription. (A) Luciferase assay of B16, SKmel28, and 501mel cells transfected with a Brn-2-luciferase reporter together with a Pax3 expression vector or control vector as indicated. Data from a representative experiment are presented as fold changes compared to the control (set to 1). Columns represent means from four experiments; bars indicate SD; *, P < 0.05. (B) Western blot using anti-Brn-2 or anti-Pax3 antibodies of 501mel transfected with an siRNA of Pax3 or scrambled (control) as indicated. Erk2 was used as a loading control. (C) Relative expression of Pax3 and Brn-2 mRNA as determined using RT-qPCR from two independent clones of B16F10 depleted for Pax3 using shRNA compared to the WT B16F10. Values were normalized to GAPDH and are shown as fold changes. (D) Western blot of Pax3 protein in parental and shPax3 B16-expressing clones 1 and 2. ERK was used as a loading control. (E) Chromatin immunoprecipitation (ChIP) of B16 cells using an anti-Pax3 or anti-Lef1 antibody as a control. Amplification of the Brn-2 promoter was detected by semiquantitative PCR using primers specific for the Brn-2 promoter. (F and G) ChIP of Pax3 and RNA PolII followed by qPCR at the Brn-2 promoter or an unrelated region 1.8 kb upstream using IgG as a control in the indicated cell lines.
To confirm whether Pax3 could directly bind the Brn-2 promoter, we performed in vitro electrophoretic mobility shift assays (EMSA). To this end, we used a bacterially expressed Pax3 DNA binding domain, comprising the paired and homeo domains (PDHD) as a His-tagged protein (12), together with radiolabeled MSEu (melanocyte-specific element) from the Tyrp1 promoter that we have previously shown to represent a high-affinity Pax3 binding site (18, 55). The preliminary results showed that Pax3 bound the MSEu probe (Fig. 4A) and that the complex formed was specific, as seen with the supershift obtained with the anti-His antibody. Since Pax3 binding sites are highly variable, it is consequently difficult to identify putative binding sites on the basis of primary DNA sequence alone. We therefore performed an EMSA using Pax3 and the labeled MSEu with competitors corresponding to a series of unlabeled oligonucleotides spanning the entire proximal Brn-2 promoter and leader sequence from −265 to +168 (Fig. 4B). The results using 250 ng of competitor indicated that Pax3 could bind preferentially to some sequences, e.g., oligonucleotide 2, and less well to others (data not shown). To define better which region of the Brn-2 promoter was preferentially bound, we performed a series of EMSAs using a smaller amount of competitors (30 ng) against labeled MSEu (data not shown) and observed that Pax3 binding was preferentially competed for by oligonucleotide 2, which contains the LEF/Tcf recognition site, and less well by oligonucleotide 10, which lies in the 5′ untranslated leader sequence. Binding to the other competitors was significantly less efficient.
Fig 4.

Pax3 directly binds the Brn-2 promoter. (A) Band shift using the MSEu probe from the Tyrp1 promoter together with bacterially expressed and purified Pax3-PDHD-His tagged with or without α-His antibody. (B) Schematic of the Brn-2 promoter showing the Tcf/Lef1 binding site. The lines under the Brn-2 promoter represent the locations of 10 oligonucleotides used as probes or competitors in our band shift assays. (C) Band shift assay using an excess of probe (oligonucleotide 10) together with bacterially expressed and purified Pax3-PDHD-His. Thirty nanograms of the indicated competitors corresponding to a region of the proximal promoter of Brn-2 was used. The described Tcf/Lef1 binding site is highlighted in blue. (D) Binding of Pax3 to the probe oligonucleotide 2s was challenged using 30 ng of the indicated competitors. Blue, Lef1 binding site; red, mutated bases of the potential Pax3 binding site. (E) Binding of Pax3 to the probe oligonucleotide 2 was challenged using 30 ng of the indicated competitors. (F) Band shift using bacterially purified GST-Lef1 together with oligonucleotide 2s and 30 ng of unlabeled oligonucleotide 2s corresponding to the probe and mutant 1, 11, and 12 as competitors. (G) Binding of Pax3 to the probe oligonucleotide 2 was challenged using 30 ng of the indicated competitors. (H) Binding of Pax3 to WT oligonucleotide 2s or Mut1 probes as indicated. (I) Scheme representing the Pax3 binding sites (yellow) adjacent to Lef1 (blue) on the Brn-2 promoter. The bases shared by both Pax3 and Lef1 are represented in green.
To enhance the sensitivity of the DNA binding assay, we next used low-affinity oligonucleotide 10 as a probe, with Pax3 binding being confirmed using anti-His tag antibody. For these results only the bound DNA is shown, but probe was in excess in all experiments. The results (Fig. 4C) revealed that Pax3 was competed for by the unlabeled oligonucleotide 10, corresponding to the probe, but better was competed for by either oligonucleotide 2 or a short version, oligonucleotide 2s, lacking 23 bases at the 5′ end of oligonucleotide 2, consistent with the results obtained using the MSEu probe. Significantly, both oligonucleotide 2 and oligonucleotide 2s competed better for Pax3 binding than the well-characterized MSEu binding site. Additional competitors revealed that Pax3 was unable to bind sequences at the 5′ or 3′ ends of oligonucleotide 2, competitors 2c and 2d, respectively, while oligonucleotides 2a and 2b bound as well as oligonucleotide 10 and the MSEu. These results indicated that Pax3 binding was located close to the previously identified Lef/Tcf binding site that confers responsiveness to β-catenin.
To map better the Pax3 binding site, we used oligonucleotide 2s as a probe together with a series of unlabeled mutated competitors. The results (Fig. 4D) showed that Pax3 binding was efficiently competed for by mutants 2, 3, and 4, marginally less well by mutant 5, and poorly by either mutant 1 or mutant 6. To verify that the 4 bases replaced in mutant 1 were all necessary for Pax3 recognition, we replaced those 4 bases one by one (mutants 7, 8, 9, and 10). The importance of those 4 bases for Pax3 binding was revealed by the weak competition by each of the single-base-substituted competitors (Fig. 4E) compared to the binding observed with oligonucleotide 2 or oligonucleotide 2s.
We next checked whether Lef1 binding was altered when the Pax3 binding site was disrupted, with the ultimate aim of making a mutant promoter in which Pax3, but not Lef1, binding was impaired. To this end, we performed a band shift assay using purified Lef1 protein (20) together with labeled oligonucleotide 2s and showed (Fig. 4F) that while unlabeled oligonucleotide 2s was able to compete efficiently for Lef1 binding, mutant 1 failed to bind Lef1. Since we were interested in disrupting only the Pax3 binding site without affecting Lef1 recruitment, we checked two other oligonucleotides mutated toward the 5′ end of the Lef1 binding site (11 and 12 [Fig. 4E shows their sequences]) for their ability to compete. Both mutant oligonucleotides were able to compete with the oligonucleotide 2s probe for Lef1 binding. However, when using Pax3 to bind an oligonucleotide 2s probe (Fig. 4G), only mutant 12 was able to compete as efficiently as oligonucleotide 2s, while mutant 11 exhibited reduced binding to Pax3. The poor binding of Pax3 to the mutant 1 oligonucleotide revealed by competition assay was confirmed by direct binding, in which the ability of Pax3 to recognize a mutant 1 probe was substantially reduced compared to that of the wild-type (WT) oligonucleotide 2s probe (Fig. 4H).
Since we observed a difference in Pax3 binding of oligonucleotide 2 compared to that of 2a and 2b (Fig. 4C), which lack some bases on the 3′ end of the Lef1 binding site, and mutation within the CAAT motif (mutant 5) did not compete as well as the WT oligonucleotide 2s (Fig. 4D), we concluded that the Pax3 binding site was composed of a core element, AAAT, immediately 5′ to the Lef/Tcf binding site, and a secondary motif, CAAT, as schematized in Fig. 4I.
To investigate the contribution of this bipartite Pax3 binding site to Brn-2 expression, we introduced point mutations that disrupted Pax3 binding (Fig. 5A), while retaining binding by Lef1, based on the results of the in vitro DNA binding assays. Consistent with the motif identified in vitro mediating Pax3 responsiveness, mutation of the Pax3 binding site led to reduced activation in a Brn-2 reporter luciferase assay by ectopically expressed Pax3 (Fig. 5B). Note that the band shift assays also identified a downstream Pax3 binding site (oligonucleotide 10), although the contribution of this site to Brn-2 expression was not investigated further owing to the technical barriers associated with mutating a site within the 5′ untranslated region that could affect RNA stability or translation. Given the proximity of the Lef1 and Pax3 binding sites, we could envisage the possibility that the two factors cooperate for regulating Brn-2 expression, especially since Lef1 and Pax3 have been reported to interact (10). However, in cotransfection/luciferase reporter assays, we were unable to detect any synergy between Lef1 and Pax3 in regulating Brn-2 promoter activity (not shown).
Fig 5.
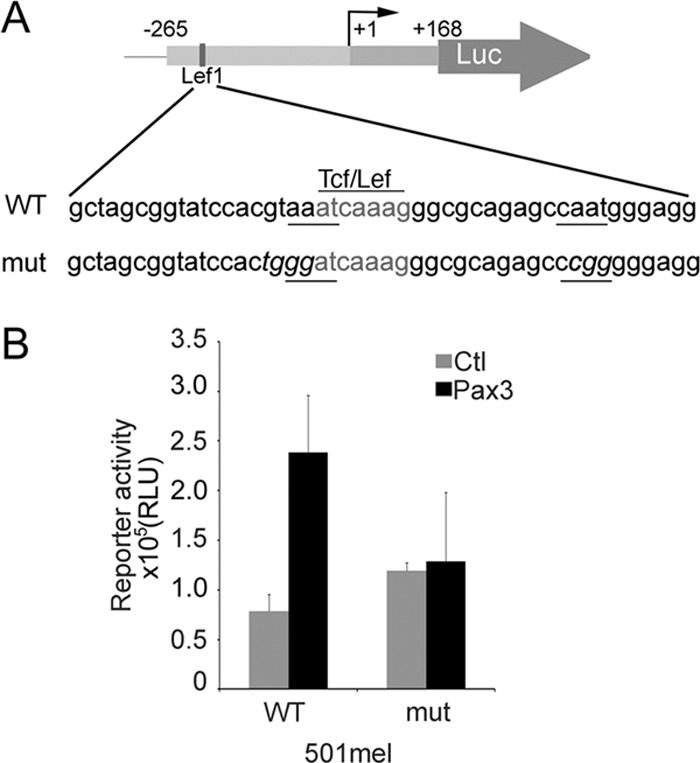
(A) Scheme representing the WT Brn-2 promoter reporter construct with the Lef/Tcf site in gray, the two putative Pax3 binding element underlined, and the mutated bases represented in italics. (B) Luciferase assay was performed on 501mel cells to confirm the loss of responsiveness of the Brn-2 promoter mutated for the Pax3 binding site after transfection of Pax3 or an empty vector (Ctl). Values reported are means from 2 independent experiments performed in triplicate; error bars represent SD.
PI3K and MAP signaling are required for Pax3 expression.
The data so far indicate, first, that PI3K inhibition decreases Brn-2 expression, and second, that Pax3 represents a novel activator of the Brn-2 promoter. We next asked whether the effects of LY294002 are mediated by Pax3. Analysis of Brn-2, Pax3, and Mitf mRNA levels by RT-qPCR in 501mel and Skmel28 cells revealed that LY294002 treatment led to a substantial reduction in Brn-2 mRNA (Fig. 2C and 6A) as well as Pax3 mRNA expression (Fig. 6A). In contrast, Mitf mRNA levels were marginally upregulated in 501mel cells and downregulated only around 15% in SKmel28 cells upon LY294002 treatment. Note that effects on regulation of MITF are difficult to interpret since its promoter is regulated both by Pax3 (52), positively (53) and negatively (19) by Brn-2, and by other factors, such as Sox10 (32a) and Lef/Tcf/β-catenin (47). However, consistent with the effect of PI3K inhibition on Pax3 RNA expression, Western blotting of 501mel, Skmel28, and B16 melanoma cells showed a downregulation of Pax3 and Brn-2 expression upon LY294002 treatment (Fig. 6B). Western blotting of p-S6, used as a marker for PI3K pathway activity, indicated that the inhibitor was working effectively, and p-ERK was used to show that this pathway was not affected by the LY294002 treatment. Total S6 and total ERK were used as loading controls. In addition we also used U0126, a MEK inhibitor that blocks MAPK signaling, as a positive control for downregulation of Brn-2; we have previously identified MAPK signaling downstream from activated BRAF as a key regulator of Brn-2 expression, though in our previous study we did not identify any transcription factor able to mediate the effect of MAPK signaling on the Brn-2 promoter (21). While U0126 reduced Brn-2 expression strongly in 501mel cells, the effect was reduced in B16 cells and was not apparent in the Skmel28 cell line. Interestingly, the effect of U0126 on Brn-2 was mirrored by an effect on Pax3 expression, with a substantial downregulation of Pax3 being observed in the presence of U0126 in 501mel cells, a minor effect in B16 cells, and no effect in Skmel28 cells. Thus, while additional experiments would be required to determine precisely how MAPK signaling can regulate Pax3 protein levels, it is clear that Pax3 may contribute to the regulation of Brn-2 expression by MAPK signaling. Combining LY294002 and U0126 provided some additive effect on Pax3 expression in 501mel cells, but in the other two cell lines the effect of the drug combination was not substantially greater than that of LY294002 alone.
Although the role of Brn-2 in invasion is well characterized, the effect of Pax3 was less clear. If Pax3 mediates the effect of PI3K on Brn-2 and invasiveness, then knockdown of Pax3 should also inhibit cell invasiveness. We therefore examined the behavior of B16 cells depleted for Pax3 (Fig. 3) and tested their migration and invasion capacities. B16 shPax3 cells showed delayed wound-healing compared to parental B16 (Fig. 6C), meaning that like Brn-2, Pax3 expression tends to favor cell mobility. The effect of Pax3 reduction was not restricted to cell migration, as invasiveness was also strongly decreased (Fig. 6D). These results suggested that Pax3 knockdown, like Brn-2 depletion and PI3K inhibition, affect cellular mobility/invasiveness in a similar manner. To further substantiate that PI3K signaling regulates invasiveness via Pax3, we also used a lentivirus to stably overexpress an mCherry-Pax3 fusion protein in invasive Skmel28 cells. Western blot analysis using an anti-Pax3 antibody showed that mCherry-Pax3 was highly overexpressed and partially cleaved (Fig. 6E). Importantly, while Brn-2 protein was downregulated upon LY294002 treatment in the parental Skmel28 cells (Fig. 2 and 6B), compared to the parental cells the overexpression of Pax3 increased Brn-2 expression, and LY294002 only reduced Brn-2 expression to the level observed in untreated control cells. The fact that such substantial overexpression of Pax3 did not lead to a greater elevated expression of Brn-2 most likely is related to the fact that Pax3, like many transcription factors, can both activate and repress transcription, and that Pax3 can activate transcription efficiently only over a narrow band of protein concentrations (7, 28, 32). Nevertheless, the fact that in the presence of LY294002 cells expressing mCherry-Pax3 expressed levels of Brn-2 similar to those of the parental untreated cells gave us an opportunity to ask whether the invasive capacity of these cells was affected. Importantly, the inhibition of the invasive capacity of the parental cells by LY294002 was abrogated by Pax3 overexpression (Fig. 6F). Thus, Pax3 appears to promote melanoma invasiveness downstream from PI3K signaling, at least in part via its capacity to regulate Brn-2.
DISCUSSION
Anticancer therapies will need to take into account the moving target represented by the ability of subpopulations of cells within tumors to reversibly switch their identities and consequently their biological properties. Deciphering how the combination of signals originating in the tumor microenvironment, superimposed on deregulation of signal transduction pathways by genetic lesions, affects cell identity is therefore a major challenge. In melanoma, subpopulation identity appears to be largely controlled by the activity and expression of the MITF transcription factor (6, 8). Since Brn-2 emerged as a key regulator of MITF expression (19, 49, 53), it is attracting increasing attention as a major factor in melanoma progression. Moreover, its role in promoting invasiveness has also been emphasized by the observation that Brn-2 represses the cGMP phosphodiesterase PDE5 (2). In the long term, the identification of signaling pathways that regulate Brn-2 expression, or its switch from activator to repressor, may reveal potential opportunities for therapeutic intervention aimed at eliminating MITF-low stem-like cells. Here, we have shown that Brn-2 is regulated by Pax3 and that expression of both factors is controlled by PI3K signaling. We also reveal that in some melanoma cell lines, Pax3 expression is also regulated by MAPK signaling, perhaps accounting in part for our previous observations that showed that BRAF signaling promotes Brn-2 expression (21). Indeed, although not directly addressed here, the positive regulation of MITF expression in cell lines by BRAF may not be mediated solely by Brn-2 (53), since Pax3 is also a key regulator of MITF expression (52).
Pax3 plays a well-established role in the development and homeostasis of the melanocyte lineage. In part, the impact of Pax3 in development arises through its ability to promote expression of MITF, and Pax3-deficient mice exhibit pigmentation defects as a result (31). In addition, Pax3 can also block terminal differentiation (32), a mechanism that also may contribute to the uncontrolled cell growth and loss of terminal differentiation in melanomas where Pax3 is commonly expressed (37, 43). Similarly, Pax3 activates expression of TGF-β2 during development (36), and TGF-β signaling promotes melanoma invasiveness and a switch from a proliferative to an invasive phenotype (26, 27). Although in our work we have revealed Pax3 as an activator of Brn-2 expression, other studies have shown that Pax3 may act as a transcriptional repressor via interaction with the KAP-1 corepressor or GRG4/TLE4 (7, 28, 32). Presumably, the capacity of Pax3 to repress or activate transcription will be determined by posttranslational modifications, though at present the nature of the signals that mediate the Pax3 repressor-activator switch are unknown. The identification of such signals will be important to determine whether Pax3 represses or activates Brn-2 in vivo, which in turn will have an impact on MITF and melanoma subpopulation identity.
The importance of the identification of Pax3 as a novel regulator of Brn-2 expression is underlined by our observations that expression of both Pax3 and Brn-2 are regulated via PI3K. During recent years, deregulation of the PI3K pathway has been increasingly recognized as a major contributor to cancer. In melanomas, activation of the PI3K pathway can act in concert with the RAS/RAF pathway to promote melanoma tumorigenesis and metastasis (14, 40). The PI3K signaling pathway is often upregulated in melanoma (3) through activating mutations of the PIK3CA gene encoding the p110α catalytic subunit (13, 41, 45), by loss-of-function mutations in PTEN, which have been detected in 10 to 30% of melanomas (50), by epigenetic silencing of PTEN expression (57), and by activation of AKT3 (46). The ability of PI3K to regulate expression of Brn-2, a transcription factor that can downregulate MITF expression to promote an invasive phenotype in vitro and in vivo (2, 4, 19, 42, 48), fits nicely with a role for PI3K signaling in promoting highly aggressive melanomas in a BRAF, Pten−/− mouse melanoma model (14). However, it should also be noted that PI3K signaling can also activate MITF expression in vitro (29), perhaps via the ability of Brn-2 to activate MITF expression in cultured cells. It will be instructive to examine the expression of Brn-2 in vivo, melanomas bearing defined combinations of activating mutations in BRAF and PI3K, or a loss of Pten. Finally, the fact that Brn-2 is now placed downstream from BRAF, β-catenin, and PI3K, three different melanoma-associated signaling pathways, highlights its likely importance in melanoma progression.
ACKNOWLEDGMENTS
We thank Alan Underhill for the bacterial Pax3 expression vector.
This work was supported by the Ludwig Institute for Cancer Research, La Ligue Nationale Contre le Cancer Comité de l'Oise, and a Wellcome Trust Fellowship to H.M.S.
Footnotes
Published ahead of print 17 September 2012
REFERENCES
- 1. Arnheiter H. 2010. The discovery of the microphthalmia locus and its gene, Mitf. Pigment Cell Melanoma Res. 23:729–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Arozarena I, et al. 2011. Oncogenic BRAF induces melanoma cell invasion by downregulating the cGMP-specific phosphodiesterase PDE5A. Cancer Cell 19:45–57 [DOI] [PubMed] [Google Scholar]
- 3. Aziz SA, et al. 2009. Phosphatidylinositol-3-kinase as a therapeutic target in melanoma. Clin. Cancer Res. 15:3029–3036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boyle GM, et al. 2011. Melanoma cell invasiveness is regulated by miR-211 suppression of the BRN2 transcription factor. Pigment Cell Melanoma Res. 24:525–537 [DOI] [PubMed] [Google Scholar]
- 5. Carreira S, et al. 2005. Mitf cooperates with Rb1 and activates p21Cip1 expression to regulate cell cycle progression. Nature 433:764–769 [DOI] [PubMed] [Google Scholar]
- 6. Carreira S, et al. 2006. Mitf regulation of Dia1 controls melanoma proliferation and invasiveness. Genes Dev. 20:3426–3439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chalepakis G, Goulding M, Read A, Strachan T, Gruss P. 1994. Molecular basis of splotch and Waardenburg Pax-3 mutations. Proc. Natl. Acad. Sci. U. S. A. 91:3685–3689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cheli Y, et al. 2011. Mitf is the key molecular switch between mouse or human melanoma initiating cells and their differentiated progeny. Oncogene 30:2307–2318 [DOI] [PubMed] [Google Scholar]
- 9. Cheli Y, Ohanna M, Ballotti R, Bertolotto C. 2010. Fifteen-year quest for microphthalmia-associated transcription factor target genes. Pigment Cell Melanoma Res. 23:27–40 [DOI] [PubMed] [Google Scholar]
- 10. Christova T, Mojtahedi G, Hamel PA. 2010. Lymphoid enhancer factor-1 mediates loading of Pax3 to a promoter harbouring lymphoid enhancer factor-1 binding sites resulting in enhancement of transcription. Int. J. Biochem. Cell Biol. 42:630–640 [DOI] [PubMed] [Google Scholar]
- 11. Cook AL, Sturm RA. 2008. POU domain transcription factors: BRN2 as a regulator of melanocytic growth and tumourigenesis. Pigment Cell Melanoma Res. 21:611–626 [DOI] [PubMed] [Google Scholar]
- 12. Corry GN, Underhill DA. 2005. Pax3 target gene recognition occurs through distinct modes that are differentially affected by disease-associated mutations. Pigment Cell Res. 18:427–438 [DOI] [PubMed] [Google Scholar]
- 13. Curtin JA, Stark MS, Pinkel D, Hayward NK, Bastian BC. 2006. PI3-kinase subunits are infrequent somatic targets in melanoma. J. Investig. Dermatol. 126:1660–1663 [DOI] [PubMed] [Google Scholar]
- 14. Dankort D, et al. 2009. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat. Genet. 41:544–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Davies H, et al. 2002. Mutations of the BRAF gene in human cancer. Nature 417:949–954 [DOI] [PubMed] [Google Scholar]
- 16. Delmas V, et al. 2007. Beta-catenin induces immortalization of melanocytes by suppressing p16INK4a expression and cooperates with N-Ras in melanoma development. Genes Dev. 21:2923–2935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Eisen T, Easty DJ, Bennett DC, Goding CR. 1995. The POU domain transcription factor Brn-2: elevated expression in malignant melanoma and regulation of melanocyte-specific gene expression. Oncogene 11:2157–2164 [PubMed] [Google Scholar]
- 18. Galibert M-D, Yavuzer U, Dexter TJ, Goding CR. 1999. Pax3 and regulation of the melanocyte-specific TRP-1 promoter. J. Biol. Chem. 274:26894–26900 [DOI] [PubMed] [Google Scholar]
- 19. Goodall J, et al. 2008. Brn-2 represses microphthalmia-associated transcription factor expression and marks a distinct subpopulation of microphthalmia-associated transcription factor-negative melanoma cells. Cancer Res. 68:7788–7794 [DOI] [PubMed] [Google Scholar]
- 20. Goodall J, et al. 2004. Brn-2 expression controls melanoma proliferation and is directly regulated by beta-catenin. Mol. Cell. Biol. 24:2915–2922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Goodall J, et al. 2004. The Brn-2 transcription factor links activated BRAF to melanoma proliferation. Mol. Cell. Biol. 24:2923–2931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Govindarajan B, et al. 2007. Overexpression of Akt converts radial growth melanoma to vertical growth melanoma. J. Clin. Investig. 117:719–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144:646–674 [DOI] [PubMed] [Google Scholar]
- 24. Heppner GH. 1984. Tumor heterogeneity. Cancer Res. 44:2259–2265 [PubMed] [Google Scholar]
- 25. Hoek K, Goding CR. 2010. Cancer stem cells versus phenotype switching in melanoma. Pigment Cell Melanoma Res. 23:746–759 [DOI] [PubMed] [Google Scholar]
- 26. Hoek KS, et al. 2008. In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res. 68:650–656 [DOI] [PubMed] [Google Scholar]
- 27. Hoek KS, et al. 2006. Metastatic potential of melanomas defined by specific gene expression profiles with no BRAF signature. Pigment Cell Res. 19:290–302 [DOI] [PubMed] [Google Scholar]
- 28. Hsieh MJ, Yao YL, Lai IL, Yang WM. 2006. Transcriptional repression activity of PAX3 is modulated by competition between corepressor KAP1 and heterochromatin protein 1. Biochem. Biophys. Res. Commun. 349:573–581 [DOI] [PubMed] [Google Scholar]
- 29. Khaled M, et al. 2003. Microphthalmia associated transcription factor is a target of the phosphatidylinositol-3-kinase pathway. J. Investig. Dermatol. 121:831–836 [DOI] [PubMed] [Google Scholar]
- 30. Kobi D, et al. 2010. Genome-wide analysis of POU3F2/BRN2 promoter occupancy in human melanoma cells reveals Kitl as a novel regulated target gene. Pigment Cell Melanoma Res. 23:404–418 [DOI] [PubMed] [Google Scholar]
- 31. Kubic JD, Young KP, Plummer RS, Ludvik AE, Lang D. 2008. Pigmentation PAX-ways: the role of Pax3 in melanogenesis, melanocyte stem cell maintenance, and disease. Pigment Cell Melanoma Res. 21:627–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lang D, et al. 2005. Pax3 functions at a nodal point in melanocyte stem cell differentiation. Nature 433:884–887 [DOI] [PubMed] [Google Scholar]
- 32a. Lee M, Goodall J, Verastegui C, Ballotti R, Goding CR. 2000. Direct regulation of the microphthalmia promoter by Sox10 links Waardenburg-Shah syndrome (WS4)-associated hypopigmentation and deafness to WS2. J. Biol. Chem. 275:37978–37983 [DOI] [PubMed] [Google Scholar]
- 33. Lopez-Bergami P. 2011. The role of mitogen- and stress-activated protein kinase pathways in melanoma. Pigment Cell Melanoma Res. 24:902–921 [DOI] [PubMed] [Google Scholar]
- 34. Madhunapantula SV, Robertson GP. 2009. The PTEN-AKT3 signaling cascade as a therapeutic target in melanoma. Pigment Cell Melanoma Res. 22:400–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Marusyk A, Polyak K. 2010. Tumor heterogeneity: causes and consequences. Biochim. Biophys. Acta 1805:105–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mayanil CS, et al. 2006. Regulation of murine TGFbeta2 by Pax3 during early embryonic development. J. Biol. Chem. 281:24544–24552 [DOI] [PubMed] [Google Scholar]
- 37. Medic S, Rizos H, Ziman M. 2011. Differential PAX3 functions in normal skin melanocytes and melanoma cells. Biochem. Biophys. Res. Commun. 411:832–837 [DOI] [PubMed] [Google Scholar]
- 38. Michailidou C, et al. 2009. Dissecting the roles of Raf- and PI3K-signalling pathways in melanoma formation and progression in a zebrafish model. Dis. Model Mech. 2:399–411 [DOI] [PubMed] [Google Scholar]
- 39. Monterrubio M, Mellado M, Carrera AC, Rodriguez-Frade JM. 2009. PI3Kgamma activation by CXCL12 regulates tumor cell adhesion and invasion. Biochem. Biophys. Res. Commun. 388:199–204 [DOI] [PubMed] [Google Scholar]
- 40. Nogueira C, et al. 2010. Cooperative interactions of PTEN deficiency and RAS activation in melanoma metastasis. Oncogene 29:6222–6232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Omholt K, Krockel D, Ringborg U, Hansson J. 2006. Mutations of PIK3CA are rare in cutaneous melanoma. Melanoma Res. 16:197–200 [DOI] [PubMed] [Google Scholar]
- 42. Pinner S, et al. 2009. Intravital imaging reveals transient changes in pigment production and Brn2 expression during metastatic melanoma dissemination. Cancer Res. 69:7969–7977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Plummer RS, et al. 2008. PAX3 expression in primary melanomas and nevi. Mod. Pathol. 21:525–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rubinfeld B, et al. 1997. Stabilization of beta-catenin by genetic defects in melanoma cell lines. Science 275:1790–1792 [DOI] [PubMed] [Google Scholar]
- 45. Shull AY, et al. 2012. Novel somatic mutations to PI3K pathway genes in metastatic melanoma. PLoS One 7:e43369 doi:10.1371/journal.pone.0043369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stahl JM, et al. 2004. Deregulated Akt3 activity promotes development of malignant melanoma. Cancer Res. 64:7002–7010 [DOI] [PubMed] [Google Scholar]
- 47. Takeda K, et al. 2000. Induction of melanocyte-specific microphthalmia-associated transcription factor by Wnt-3a. J. Biol. Chem. 275:14013–14016 [DOI] [PubMed] [Google Scholar]
- 48. Thomson JA, et al. 1995. The brn-2 gene regulates the melanocytic phenotype and tumorigenic potential of human melanoma cells. Oncogene 11:690–700 [PubMed] [Google Scholar]
- 49. Thurber AE, et al. 2011. Inverse expression states of the BRN2 and MITF transcription factors in melanoma spheres and tumour xenografts regulate the NOTCH pathway. Oncogene 30:3036–3048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tsao H, Zhang X, Benoit E, Haluska FG. 1998. Identification of PTEN/MMAC1 alterations in uncultured melanomas and melanoma cell lines. Oncogene 16:3397–3402 [DOI] [PubMed] [Google Scholar]
- 51. Visvader JE, Lindeman GJ. 2008. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat. Rev. Cancer 8:755–768 [DOI] [PubMed] [Google Scholar]
- 52. Watanabe A, Takeda K, Ploplis B, Tachibana M. 1998. Epistatic relationship between Waardenburg syndrome genes MITF and PAX3. Nat. Genet. 18:283–286 [DOI] [PubMed] [Google Scholar]
- 53. Wellbrock C, et al. 2008. Oncogenic BRAF regulates melanoma proliferation through the lineage specific factor MITF. PLoS One 3:e2734 doi:10.1371/journal.pone.0002734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wu H, Goel V, Haluska FG. 2003. PTEN signaling pathways in melanoma. Oncogene 22:3113–3122 [DOI] [PubMed] [Google Scholar]
- 55. Yavuzer U, Goding CR. 1994. Melanocyte-specific gene expression: role of repression and identification of a melanocyte-specific factor, MSF. Mol. Cell. Biol. 14:3494–3503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yoon SO, Shin S, Lee HJ, Chun HK, Chung AS. 2006. Isoginkgetin inhibits tumor cell invasion by regulating phosphatidylinositol 3-kinase/Akt-dependent matrix metalloproteinase-9 expression. Mol. Cancer Ther. 5:2666–2675 [DOI] [PubMed] [Google Scholar]
- 57. Zhou XP, et al. 2000. Epigenetic PTEN silencing in malignant melanomas without PTEN mutation. Am. J. Pathol. 157:1123–1128 [DOI] [PMC free article] [PubMed] [Google Scholar]