

Abstract
Fun30 is a Swi2/Snf2 homolog in budding yeast that has been shown to remodel chromatin both in vitro and in vivo. We report that Fun30 plays a key role in homologous recombination, by facilitating 5′-to-3′ resection of double-strand break (DSB) ends, apparently by facilitating exonuclease digestion of nucleosome-bound DNA adjacent to the DSB. Fun30 is recruited to an HO endonuclease-induced DSB and acts in both the Exo1-dependent and Sgs1-dependent resection pathways. Deletion of FUN30 slows the rate of 5′-to-3′ resection from 4 kb/h to about 1.2 kb/h. We also found that the resection rate is reduced by DNA damage-induced phosphorylation of histone H2A-S129 (γ-H2AX) and that Fun30 interacts preferentially with nucleosomes in which H2A-S129 is not phosphorylated. Fun30 is not required for later steps in homologous recombination. Like its homolog Rdh54/Tid1, Fun30 is required to allow the adaptation of DNA damage checkpoint-arrested cells with an unrepaired DSB to resume cell cycle progression.
INTRODUCTION
Chromatin structure and modifications play central roles in the ability of eukaryotic cells to sense and repair chromosomal breaks. Both the Snf5 subunit of the Swi2/Snf2 chromatin remodeler and the Swi2/Snf2 homolog Rad54 are required in the repair of a double-strand break (DSB) by gene conversion. In the well-studied case of yeast mating-type (MAT) gene switching, induced by site-specific HO endonuclease cleavage, Rad51-mediated strand invasion of the heterochromatic HML donor sequence depends on Snf5 (5). Strand invasion can occur without Rad54, but the necessary displacement of histones, apparently to facilitate the initiation of new DNA synthesis from the 3′ end of the invading strand, fails to occur (24, 59). Rad54 and two related proteins, Rdh54/Tid1 and Uls1, have also been implicated in displacing Rad51 from nonspecific associations with double-stranded DNA to allow the recombinase protein, which is not present in abundance, to bind to single-stranded DNA (ssDNA) that is produced by exonucleases degrading the DSB ends in a 5′-to-3′ direction (9). In addition, the RSC chromatin remodeling complexes have been shown to facilitate repair (5, 51, 55).
In budding yeast, as in mammals, the most immediate and notable alteration in response to DNA damage is the phosphorylation of histone H2A (H2AX in mammals), known as γ-H2AX, which covers about 50 kb of DNA around a single DSB (30, 56). In mammals, γ-H2AX serves to recruit many DNA repair factors, such as 53BP1, and the absence of this modification markedly reduces recombinational repair of DSBs between sister chromatids (3, 19, 69). In budding yeast, the spreading of γ-H2AX is required for the establishment of damage-induced cohesion between sister chromatids; thus, in a strain carrying the histone H2A-S129A mutation, which prevents phosphorylation, sister chromatid repair is reduced (58, 62). The presence of γ-H2AX also extends the time that cells remain arrested prior to anaphase by the DNA damage checkpoint (28). The prolongation of arrest involves the spreading of γ-H2AX across the centromere of the damaged chromosome and the activation of the spindle assembly checkpoint (17).
Very little is known about how γ-H2AX is removed from a damaged region after repair is complete. The histones are dephosphorylated by the Pph3 phosphatase, but this apparently occurs only after the histones have been displaced from DNA (28). γ-H2AX is not simply rapidly turned over, as inactivating the Mec1 (ATR) and Tel1 (ATM) checkpoint kinases responsible for the modification does not lead to a rapid displacement of γ-H2AX (30). Presumably this displacement requires the agency of a chromatin remodeler, but to date the identity of this protein has not been established. Experiments have ruled out a role for Arp4, a common subunit of the Ino80, Swr1, and NuA4 chromatin remodelers (J.-A. Kim and J. E. Haber, unpublished observation). Similarly, we have shown that neither the Asf1 nor CAF-1 histone chaperone is involved in removing γ-H2AX (29). However, Asf1 and CAF-1 are apparently involved in the re-establishment of chromatin after a DSB is repaired, and in the absence of both Asf1 and the CAF-1 subunit Cac1, repair at the DNA level is complete but cells fail to turn off the DNA damage checkpoint and thus fail to resume cell cycle progression (29).
When the DSB cannot be readily repaired, as a result of deletion of the HML and HMR donor sequences that are normally used to repair an HO-induced DSB at MAT, cells remain arrested by the Mec1-dependent DNA damage checkpoint for 12 to 15 h, but then they adapt, turning off the checkpoint signals (35). We and other labs have identified several adaptation-defective mutations. Adaptation fails in the absence of the PP2C phosphatases Ptc2 and Ptc3, which are needed to dephosphorylate Rad53 (Chk2), or in the absence of casein kinase II, which in turn is needed to phosphorylate Ptc2 (22, 37, 61). The DNA helicase Srs2 is also required for adaptation, possibly in its role to displace Rad51 or other damage-associated proteins from DNA after repair (64, 67). Among the adaptation-defective mutations is the chromatin remodeler Rdh54/Tid1 (36), but the reason for its defect is unknown. In contrast, there is no adaptation defect when Rad54 is absent.
In most cases, we do not know why cells fail to adapt. Initially we postulated that increased 5′-to-3′ resection generated a stronger signal that kept the checkpoint active (35). Thus, cells failed to adapt when the amount of resection was doubled or when a cell had two unrepaired DSBs. Permanent arrest was suppressed by deleting Mre11, which lowered the rate of resection but still allowed the establishment of the checkpoint (35). The idea that the rate of resection might regulate the persistence of checkpoint arrest was supported by the demonstration that short deoxyoligonucleotides—the presumed products of a helicase-endonuclease that resects DNA ends—stimulate the checkpoint response in Xenopus extracts (27). However, there now does not seem to be a clear correlation between resection rate and adaptation in budding yeast. For example, in contrast to deletion of Mre11, deletion of Sae2, which also slows resection, is adaptation defective (11, 12). Indeed, as we show below, deletion of the Sgs1 (BLM) helicase responsible for generating deoxyoligonucleotides in yeast proves to be itself adaptation defective.
For several reasons, then, we became interested in the possible role of another Swi2/Snf2 homolog, Fun30, in both repair and checkpoint signaling. Fun30 has recently been shown to be an ATPase that can facilitate transfer of H2A-H2B dimers and sliding of nucleosomes in vitro (1). Cells lacking Fun30 are viable, although they exhibit synthetic lethality with defects in ORC proteins, which are required to initiate new DNA synthesis (13, 50). In addition, fun30Δ cells show a mild weakening of heterochromatin structure at the HMR locus (68) and in maintaining budding yeast's centromere function (18). Fun30 also affects the distribution of the histone H2A variant, Htz1, across the genome (18). The Schizosaccharomyces pombe homolog of Fun30 (Fft3) has also been shown to play a role in maintaining heterochromatin in silenced regions (57).
We wished to know whether Fun30 would be required for any steps in homologous recombination, whether it might be responsible for the removal of γ-H2AX from chromatin after DNA had been repaired, or whether it might play a role in DNA damage signaling. We report that Fun30 does have a distinctive role in homologous recombination, namely, in facilitating 5′-to-3′ resection of DNA ends. Fun30 acts in both 5′-to-3′ resection pathways represented by Exo1 and Sgs1, apparently by interacting with and presumably remodeling nucleosomes around the DSB. We found that resection is accelerated about 2-fold in a strain lacking the γ-H2AX modification and that Fun30 interacts less strongly in vitro with histones carrying a phosphomimetic H2A-S129E. We also report that cells lacking Fun30 are adaptation defective, much like Rdh54/Tid1 cells.
MATERIALS AND METHODS
Yeast strains.
All yeast strains used in this study are described in Table 1. Deletions of open reading frames (ORFs) were carried out using single-step-PCR-mediated transformation of yeast colonies as described in reference 65 or introduced by genetic crosses. Primer sequences are available on request. FUN30 wild-type and fun30(K603R)-containing plasmids were a kind gift from Patrick Varga-Weisz and are described in further detail in the work of Neves-Costa et al. (50). The 2μ pGAL-EXO1 plasmid was also described in an earlier publication (34).
Table 1.
Yeast strains used in this study
| Strain | Genotype |
|---|---|
| JKM179 | MATα hoΔ hmlΔ::ADE1 hmrΔ::ADE1 ade1-100 leu2,3-112 lys5 trp1::hisG ura3-52 ade3::GAL::HO |
| JKM139 | JKM179 isogenic, MATa |
| YJK17 | MATα hoΔ hmlΔ::ADE1 hmrΔ::ADE1 arg5,6Δ::HPH::MATa-inc ade1-100 leu2,3-112 lys5 trp1::hisG ura3-52 ade3::GAL::HO |
| YMV80 | hoΔ hmlΔ::ADE1 mataΔ::hisG hmrΔ::ADE1 his4::[NAT-leu2(Asp718-SalI)] leu2::HOcs ade3::GAL::HO ade1 lys5 ura3-52 |
| YMV45 | hoΔ hmlΔ:: ADE1 mataΔ::hisG hmrΔ:: ADE1 leu2::leu2(Asp718-SalI)-URA3-pBR322-HOcs ade3::GAL::HO ade1 lys5 ura3-52 trp1 |
| JRL346 | mata::HOcsΔ::hisG ura3Δ851 trp1Δ63 sup53Δ::leu2Δ::NATMX hmlΔ::hisG hmrΔ::ADE3 ade3::GAL::HO can1,1-1446::HOcs::HPH::ΔAVT2 ykl215c::leu2::hisG::can1Δ1-289 |
| VE1a | JKM179 isogenic, fun30Δ::KANMX |
| VE2a | YJK17 isogenic, fun30Δ::KANMX |
| VE3a | YMV80 isogenic, fun30Δ::KANMX |
| VE5 | JKM179 isogenic, chk1Δ::URA3 fun30Δ::KANMX |
| YAA2 | JKM179 isogenic, chk1Δ::KANMX |
| VE27 | JKM179 isogenic, pGAL::EXO1 LEU2 |
| VE28 | VE1a isogenic, pGAL::EXO1 LEU2 |
| VE53 | JKM179 isogenic, tel1Δ::TRP1 fun30Δ::KANMX |
| YFD122 | JKM139 isogenic, mre11Δ::hisG′ |
| VE22 | JKM139 isogenic, mre11Δ::hisG′ fun30Δ::KANMX |
| YFD318 | JKM139 isogenic, tel1Δ::TRP1 |
| VE51 | VE3a isogenic, pFUN30-FLAG URA3 |
| VE52 | VE3a isogenic, pFUN30(K603R)-FLAG URA3 |
| VE70 | JRL346 isogenic, fun30Δ::KANMX |
| YMT004 | YMV80 isogenic, rad51Δ::HPHMX |
| YMT005 | YMV80 isogenic, fun30Δ::KANMX rad51Δ::HPHMX |
| YMT016 | YMV45 isogenic, fun30Δ::KANMX |
| VE73 | tNS2548 isogenic, pGAL::EXO1 LEU2 |
| YFD1081 | JKM139 isogenic, tid1Δ::URA3 |
| VE74 | JKM139 isogenic, tid1Δ::URA3 pGAL::EXO1 LEU2 |
| VE13 | VE1a isogenic, pFA1 URA3 |
| VE14 | VE1a isogenic, pFA3 URA3 |
| JY395 | JKM179 isogenic, ptc2Δ::URA3 ptc3Δ::KANMX bar1Δ::TRP1 |
| VE75 | JKM179 isogenic, ptc2Δ::URA3 ptc3Δ::KANMX bar1Δ::TRP1 pGAL::EXO1 LEU2 |
| VE80 | VE1a isogenic, pFUN30-3X-FLAG URA3 |
| tNS2540 | JKM179 isogenic, fun30Δ::KANMX exo1Δ::TRP1 |
| tNS2542 | JKM179 isogenic, exo1Δ::TRP1 |
| tNS2546 | JKM179 isogenic, fun30Δ::KANMX sgs1Δ::URA3 |
| tNS2548 | JKM179 isogenic, sgs1Δ::URA3 |
| R726 | JKM139 isogenic, hta1-S129A hta2-S129A |
| SD090 | JKM179 isogenic, fun30Δ::KANMX hta1-S129A hta2-S129A |
| tNS2569 | JKM139 isogenic, HOcs::HPHMX inserted into Chr 6 |
| tNS2575 | JKM139 isogenic, HOcs::HPHMX inserted into Chr 6 fun30Δ::KANMX |
| tNS2577 | JKM139 isogenic, HOcs::HPHMX and HMRα::LEU2 on Chr 6 |
| tNS2580 | JKM139 isogenic, HOcs::HPHMX and HMRα::LEU2 on Chr 6 fun30Δ::KANMX |
| tNS2592 | JKM139 isogenic, matΔ::KLURA3 and HMRα::LEU2 on Chr 6 |
| tNS2594 | JKM139 isogenic, matΔ::KLURA3 and HMRα::LEU2 on Chr 6 fun30Δ::KANMX |
| YWH503 | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 FUN30-3Flag-KANMX4 |
Viability and adaptation assays.
Viability and adaptation assays were carried out as previously described (28, 32, 38). For viability assays, the relevant strains were grown overnight in yeast extract-peptone (YEP)-lactate medium and then spread onto YEPD and YEP-Gal plates in triplicate to obtain roughly 100 to 150 cells per plate. The survival rate is calculated by dividing the number of cells that grew on YEP-Gal plates by those that grew on YEPD.
Nonhomologous-end joining (NHEJ) was assayed by measuring the proportion of MATα cells lacking HML and HMR that could survive continuous expression of HO endonuclease by misaligned rejoining of the 4-bp 3′-overhanging HO-cut DSB ends as described previously (48).
Adaptation assays were carried out using the protocol described by Dotiwala et al. (16). Strains were grown either on YEP-EG plates or in liquid dropout medium containing raffinose, if a plasmid had to be maintained, and were then plated onto YEP-Gal plates. Single, unbudded G1 cells were micromanipulated using a dissection needle into a grid pattern containing 50 to 100 cells in each experiment. The percentage of cells adapted at any time point was calculated by diving the number of cells that were beyond the 2-cell (G2/M) arrest stage by the total number of cells arranged.
ChIP.
Chromatin immunoprecipitation (ChIP) of γ-H2AX was carried out as described in reference 56 using an antibody against γ-H2AX purchased from ABCAM(15083). ChIP against Fun30 was carried out using a strain expressing a C-terminally FLAG-tagged version of the protein, transcribed at the normal FUN30 locus, as described by Chen et al. (8). Anti-FLAG antibody (M2) was purchased from Sigma.
Western blotting.
Western blotting was carried out using the trichloroacetic acid (TCA) protocol described by Pellicioli et al. (53). Protein lysates were separated on 6% sodium dodecyl sulfate polyacrylamide-gel electrophoresis (SDS-PAGE) gels, transferred to PVDF membranes, and blotted with anti-RAD53 antibodies generously provided by M. Foaini (IFOM, Milan, Italy).
qPCR resection assay.
Cells were grown to approximately 5 × 106 cells/ml in YEP-lactate and induced with 2% galactose. Cells were harvested at intervals, and DNA was extracted by vortexing with glass beads and phenol. For quantitative PCR (qPCR), dilutions of DNA from the 0-h time point prior to galactose induction were used for calibration samples, and values for loci at various distances from the DSB were measured relative to these samples. Similarly, several qPCR values were acquired and averaged for an independent locus (PHO5) and used for normalization. All values were also normalized to the 0-h time point. Sequences of the PCR primers are provided in the supplemental material. Resection rates were calculated by examining each curve on the plot of distance versus PCR signal and determining the time and distance when the PCR value reached 0.7 (calculated by interpolating between the nearest data points). All of the distance and time values were then analyzed by linear regression to calculate the rate. For many strains we observed an increase in the resection rates by the 8-h and 10-h time points, as evident by the increased slope in the distance-versus-time graphs. This may be due to the late activity of another factor, such as a protein, or due to a change in chromatin state. In view of this, we confined our analysis to the first 6 h of the time course, since this is the period that is most relevant to DSB repair processes.
Southern blotting.
Southern blot analysis was performed as previously described (26). Briefly, genomic DNA was purified and digested with the appropriate restriction enzyme (PstI for YMV45 derivatives, Asp718 for YMV80 derivatives, and EcoRI for YJK17 derivatives). Genomic DNA was also isolated from a colony that grew on a YEP-galactose (YEP-Gal) plate (a survivor) for quantification of repair. For YMV80 derivatives, the membranes were first probed with the U2 flanking repeat. Membranes were later washed and reprobed with a probe from a locus that does not participate in the reaction (GSY1 locus) to provide a loading control. Bands were normalized to the loading control and then to the survivor lane that has a repair product (100% repair).
Microscopy.
For assessing nuclear division in cells, we fixed cells using 70% ethanol at various time points and stained the nucleus using DAPI (4′,6′-diamidino-2-phenylindole) as previously described (16). Images were acquired using a Nikon Eclipse E600 microscope.
In vitro affinity measurements of nucleosome binding.
The yeast Fun30 protein was purified by anti-FLAG immunoaffinity purification from YWH501, in which three copies of the FLAG epitope tag were fused to the C terminus of Fun30 (W. H. Wu, unpublished data). Procedures for one-step anti-FLAG immunoaffinity purification were as described in reference 66. Yeast whole-cell extracts of YWH503 were incubated with anti-FLAG M2 agarose beads (Sigma) for 4 h at 4°C, followed by six washes with 0.5 M KCl. The Fun30 protein was eluted with the FLAG peptide.
The mutation for the S129A or S129E substitution in histone H2A was introduced into the HTA1 gene by site-directed mutagenesis (Agilent). Recombinant yeast histone purification, histone octamer reconstitution, and nucleosome array assembly were performed as described in reference 47. Dynabeads M-280 (Invitrogen) coupled to 1.5 μg of pBlueScript SK(−) DNA were used as the template. The amounts of wild-type and mutant octamers used were normalized for the H2B histone by Western blotting using anti-H2B (a gift from the laboratory of Carl Wu). The nucleosome array binding assay was performed essentially as described previously (66). Immobilized nucleosome arrays (150 ng DNA equivalents) were incubated with 0.8 pmol of purified Fun30 protein for 1 h at room temperature, followed by three 0.3 M KCl washes. The bound Fun30 protein was eluted with SDS-PAGE sample buffer and detected by Western blotting using anti-FLAG antibody (Sigma).
RESULTS
HO-induced gene conversion is not impaired in fun30Δ strains.
We first examined the role of Fun30 in the two major pathways of homologous recombination induced by the site-specific HO endonuclease, gene conversion and break-induced replication (BIR). In strain YJK17, a DSB induced at the MATα locus on chromosome 3 (Chr 3) can be repaired by ectopic gene conversion from a MATa-inc donor situated on Chr 5 (Fig. S1A) (28, 29). There was no significant difference between the viability of the fun30Δ strain and that of the wild type after induction of HO from a galactose-inducible promoter (Fig. 1A). Moreover, Southern blot analysis of the kinetics of repair also showed no significant difference when Fun30 was ablated (Fig. 1B).
Fig 1.
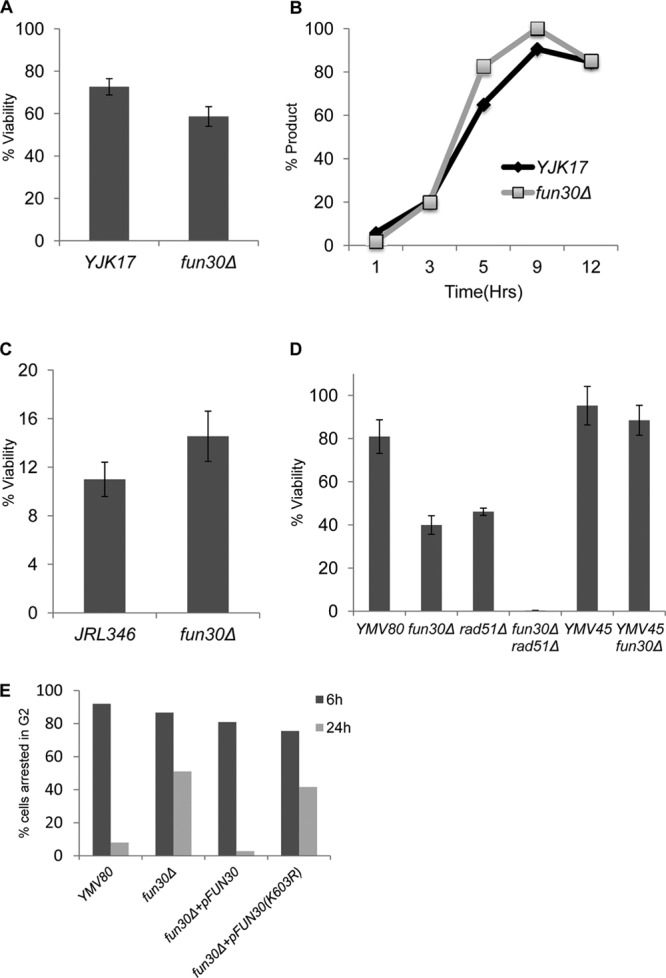
FUN30 does not affect interchromosomal gene conversion, break-induced replication, or single-strand annealing. (A) Viability of ectopic recombination strain YJK17 and its fun30Δ derivative on YEP-galactose plates relative to YEPD plates (see Materials and Methods). (B) Quantification of gene conversion kinetics based on Southern blot analysis. (C) Viability in BIR strain JRL346. (D) Viability in the SSA/BIR strains YMV80 and YMV45. (E) Microcolony assay of YMV80 strains harboring wild-type FUN30 or fun30 ATPase-defective mutant on plasmids. Data are means and standard errors of the means (SEM) from at least three independent experiments.
We then examined BIR in the strain JRL346 (38, 40), in which a 3′ truncation of the CAN1 gene lies adjacent to an HO cleavage site and is marked with the hygromycin resistance gene (CA-HOcs-HPH) (see Fig. S1C in the supplemental material). A donor sequence which carries 1 kb of overlapping homologous sequences and the 3′ end of the gene (AN1) was inserted on Chr 11. The DSB can be repaired by BIR to restore the CAN1 gene, making the cells canavanine sensitive, and to create a nonreciprocal translocation that results in the loss of hygromycin resistance as well as the terminal, nonessential portion of Chr 5. Deletion of FUN30 had no effect on the viability of BIR events (Fig. 1C). Similar to observations with the wild-type BIR strain, 94.4% of the fun30Δ colonies were both Cans and HPHs (data not shown). The absence of Canr HPHs colonies indicates that there were few de novo telomere additions (see Discussion). We conclude that Fun30 does not play an important role in strand invasion or later steps in homologous recombination.
Loss of Fun30 affects 5′-to-3′ resection and delays single-strand annealing.
Another important pathway of DSB repair is single-strand annealing (SSA). We examined SSA using two well-characterized strains, YMV45 and YMV80 (64). In YMV45, the flanking homologous sequences are separated by 5 kb; repair and viability were equivalently efficient in both wild-type and fun30Δ strains (Fig. 1D and data not shown). However, when we examined repair in YMV80, in which one of the flanking repeats of part of the LEU2 gene (U2) is separated from the DSB by 25 kb (see Fig. S1B in the supplemental material), we found that viability of the fun30Δ derivative was reduced to about 40%, which is consistent with the observation that only 40% of fun30Δ cells can efficiently repair the DSB, as measured by a Southern blot (Fig. 1D and 2A). Our previous work had established that repair in strain YMV80 can occur either by SSA or by BIR, with similar kinetics. To determine if one of these pathways was specifically disabled in the fun30Δ mutant, we knocked out RAD51, which is required for BIR but is not required for SSA (26). We discovered that the fun30Δ rad51Δ strain was nearly inviable, and product formation was almost completely abolished (Fig. 1D and 2A). In addition, the persistence of the HO-cut DNA was prolonged, as would be expected if 5′-to-3′ resection was reduced. These data suggested that Fun30 might play a role in regulating 5′-to-3′ resection and that a significant delay in resection prevented SSA between such distant flanking homologies, as has been shown for other mutations that slow resection (12).
Fig 2.

fun30Δ forces cells to undergo BIR in YMV80. (A) GAL::HO was induced at 0 h, and DNA was harvested at the time points shown. The Southern blot shows the repair kinetics in wild-type (YMV80), fun30Δ, rad51Δ, and fun30Δ rad51Δ strains using a probe specific to the repeated sequence, U2 (see Materials and Methods for details). (B) Quantification of the blots presented in panel A.
Neves-Costa et al. (50) found that a mutation in the Walker A motif of the ATPase binding domain of Fun30 rendered it inactive in a gene silencing assay. We asked whether the ATPase mutant was also required for resection and SSA. First, we monitored YMV80 fun30Δ cells microscopically 6 and 24 h after HO induction on YEP-Gal plates. Consistent with the viability data, at 24 h approximately half of the cells remained arrested at the dumbbell stage indicative of G2/M arrest (Fig. 1E). When the strain was transformed with a plasmid carrying wild-type FUN30, it fully complemented the repair defect of the fun30Δ strain; nearly 100% of cells escaped the initial G2/M arrest. In contrast, in strains transformed with a plasmid carrying the fun30(K603R) ATPase mutation, cells continued to behave as the null mutant, with 40% of the cells permanently arrested as large budded cells.
Fun30 is required for normal 5′-to-3′ resection of DSB ends.
To assess the effect of Fun30 on resection, we monitored the fate of DSB ends in strain JKM179, in which a DSB was created at MATα and the normal donor sequences that would repair the break were deleted (35, 48). We measured the progress of resection by a quantitative PCR (qPCR) assay by isolating DNA at different time intervals after inducing the DSB and using primer pairs that created 100- to 300-bp amplicons at many sites on both sides of the DSB. As shown in Fig. 3A, one can follow the progressive reduction in PCR amplification over many hours. Resection removes only one strand of the PCR template, so the signal is reduced to about half of the initial amplification, although with more time, further loss of signal can be seen. We converted the data to rate measurements by plotting the time and distance it took for the PCR signals to reach 70% of their initial value (Fig. 3D, E, and F). The rate of resection over the first 6 h proved to be about 4.0 kb/h (Table 2), which is very similar to previous measurements (20, 46, 71). In the absence of Fun30, however, there was a marked reduction in resection (Fig. 3A) to a rate that was approximately 1/3 that of the wild type (1.2 kb/h) (Table 2 and Fig. 3D).
Fig 3.

Quantitation of DNA resection by qPCR. (A to C) PCR products were amplified using primers located at different distances from the DSB. Template DNA was isolated from GAL::HO-induced cells at the times shown. All values were normalized to that for an independent locus on Chr 2 and then normalized to that for the 0-h time point. Resection was measured by qPCR in wild-type and fun30Δ strains (A), sgs1Δ and sgs1Δ fun30Δ strains (B), and exo1Δ and exo1Δ fun30 strains (C). (D to F) Resection rates were calculated by examining each curve and determining the time and distance when the PCR signal fell to 0.70. These values were plotted on time-versus-distance graphs, and the slopes (rates) were determined by linear regression analysis. PCR data using primers to the right of the DSB from one time course are illustrated using wild-type (WT) and fun30Δ (D), sgs1Δ (E), and exo1Δ (F) strains.
Table 2.
Resection rates for mutants
| Strain genotype | Rate of DNA resection (kb/h)a |
|---|---|
| Wild type | 4.0 ± 0.7b |
| fun30Δ | 1.2 ± 0.1c |
| sgs1Δ | 3.0 ± 0.2b |
| sgs1Δ fun30Δ | 1.8 ± 0.7b |
| exo1Δ | 2.1 ± 0.4c |
| fun30Δ exo1Δ | 1.2 ± 0.2b |
| H2A-S129A | 6.8 |
| fun30Δ H2A-S129A | 2.4 |
5′-to-3′ resection rates from an HO-induced unrepaired DSB in strain JKM179 and its derivatives were measured as described in the legend to Fig. 3 and in Materials and Methods. Resection rates were calculated from qPCR values as described in the text.
Average of three time courses (± standard deviation).
Average of two time courses (± range).
There are two major 5′-to-3′ resection pathways that act once the MRX complex and Sae2 have initiated resection close to the DSB ends: the exonuclease Exo1 and the helicase-endonuclease complex containing Sgs1-Top3-Rmi1 and Dna2 (21, 46, 71). The sgs1Δ and exo1Δ deletions each cause a partial reduction in resection, while sgs1Δ exo1Δ eliminates nearly all resection. We asked if fun30Δ was epistatic with one or both of these pathways. We confirmed that the sgs1Δ mutant had a reduced rate of resection—about 3.0 kb/h (Fig. 3B and Table 2)—but found that the sgs1Δ fun30Δ double mutant had an even greater reduction in resection (1.8 kb/h), similar to that of the strain carrying fun30Δ alone (Fig. 3B and Table 2). This suggested that Fun30 plays a role in the remaining Exo1-mediated resection. When we compared exo1Δ and exo1Δ fun30Δ, we saw a similar epistatic effect, with exo1Δ reducing resection to 2.1 kb/h and the rate in the double mutant being 1.2 kb/h (Table 2). Thus, Fun30 functions in both resection pathways, as would be expected if Fun30 facilitates resection ahead of the exonucleases by altering nucleosome structure.
As a further exploration of the role of Fun30 in resection, we asked if Fun30 might be especially important if resection had to cross a heterochromatic sequence with strongly positioned nucleosomes. However, we first needed to determine the degree to which Fun30 might affect HMR chromatin structure, as Fun30 has been reported to slightly weaken the silencing of a mutated HMR locus (68). Thus, the HO cleavage site that is normally inaccessible in HMR might become open to HO cleavage in the absence of Fun30. To address this question, we inserted a cloned HMRα sequence (30) into the middle of the left arm of Chr 6 in a strain lacking the normal HML, MAT, and HMR sites. We determined that the normally inaccessible heterochromatic HO cleavage site within the HMR locus was not accessible in the fun30Δ mutant, by measuring the loss of amplification of a PCR product that spans the cleavage site. Cutting within HMR was not observed in either the wild type or fun30Δ strain (Fig. 4C), indicating that HMR was largely protected. The presence of a cleavable HO cutting site within HMR was confirmed by adding the SIR2 inhibitor nicotinamide to desilence HMR (4) prior to inducing HO (Fig. 4C). HO clearly cuts within the desilenced HMR, although less efficiently than what we generally observe at the unsilenced MAT locus (24, 59).
Fig 4.

HMRα was inserted into Chr 6 at a position 3 kb from an HO cut site to examine whether fun30Δ had an effect on DNA resection through heterochromatin. (A) HMRα labeled with LEU2 was inserted near a HOcs marked with HPHMX. The HMRα X, Y, and Z sequences are flanked by the E and I silencers. LEU2 contains the ORF plus flanking sequences. The MATa sequences contained in the HOcs site are oriented in inverted orientation with respect to HMRα. (B) qPCR measurements as described for Fig. 3 were made of strains containing the HOcs on Chr 6 with and without HMR. DNA resection measurements showing the effect of fun30Δ on these strains are also shown. (C) A PCR product spanning the HO recognition site in HMR was used to measure the amount of HO cutting within heterochromatic DNA in a strain containing HMR on Chr 6 and lacking any other HO cut sites. No cutting was observed in either the wild type or the fun30Δ strain. These strains were also incubated in the presence of 5 mM nicotinamide for 2 h to desilence HMR and thereby confirm the presence of a cleavable HO cut site.
We then asked if resection through heterochromatin was dependent on Fun30. We inserted an HO cleavage site 3 kb centromere proximal to HMR on Chr 6 (Fig. 4A) and then examined resection as it progressed into the silent locus (Fig. 4B, compare top and bottom panels). The resection profile shown in Fig. 4B indicates that in a wild-type strain, there is a small delay in resection to the left of the DSB where HMR is located. This is evident from the asymmetry in resection on the left and right sides of the DSB that was not observed in the absence of HMR. This is the first evidence that heterochromatin imposes a tangible impediment to resection of DSB ends. Deletion of FUN30 caused a reduction in overall resection that makes it difficult to discern whether there was a small decrease in resection in the 3-kb region embracing HMR compared to the wild-type strain. However, it is apparent that fun30Δ does not significantly increase the barrier to resection.
γ-H2AX plays a role in governing resection that may involve Fun30.
If Fun30 interacts with nucleosomes near a DSB, it should encounter γ-H2AX. We wondered if this chromatin modification affects the rate of resection and if Fun30 specifically responds to this chromatin modification. We first measured resection in a donorless strain (R726), in which both histone H2A genes carry the S129A mutation that eliminates γ-H2AX formation (62). We report that γ-H2AX noticeably slows 5′-to-3′ resection, so that the rate is increased 1.7-fold in a strain carrying histone H2A-S129A mutations, from 4 kb/h to 6.8 kb/h (Fig. 5B and Table 2). We then investigated how fun30Δ affected this increased resection. The rate of resection in the fun30Δ H2A-S129A mutant was 2.4 kb/h (Fig. 5C and Table 2). Thus, the reduction was roughly proportional to that seen in wild-type cells; that is, fun30Δ caused a 3-fold drop in resection in both wild-type and H2A-S129A strains, but the rate in the fun30Δ H2A-S129A strain was still twice that seen in the fun30Δ strain. We interpret these data as indicating that Fun30 plays an important role in nucleosome remodeling to facilitate resection but that this activity is not solely dependent on the γ-H2AX modification.
Fig 5.

DNA resection in H2A-S129 and H2A-S129A strains. (A) Quantities of DNA before and after induction were measured by qPCR using primers located at different distances from the DSB, as described for Fig. 3. DNA resection was quantitated in wild-type, H2A-S129A, and fun30Δ H2A-S129A strains. (B) The time and distance at which the PCR signal fell to 0.70 were determined for each curve and plotted on a distance-versus-time graph. The fun30Δ deletion causes resection to become slower in a H2A-S129A strain.
Fun30 is recruited to a DSB.
The effect of Fun30 on resection implies that it should associate with a DSB. We created a FLAG-tagged derivative of Fun30 in the donorless strain JKM179 and induced an unrepairable DSB, as described above. Figure 6A shows that Fun30 is significantly recruited to a region about 2 kb from each side of a DSB within 1 h and that the ChIP signal is enriched 30-fold by 2 h. We conclude that Fun30's role in resection involves its direct association with DNA near the DSB ends.
Fig 6.
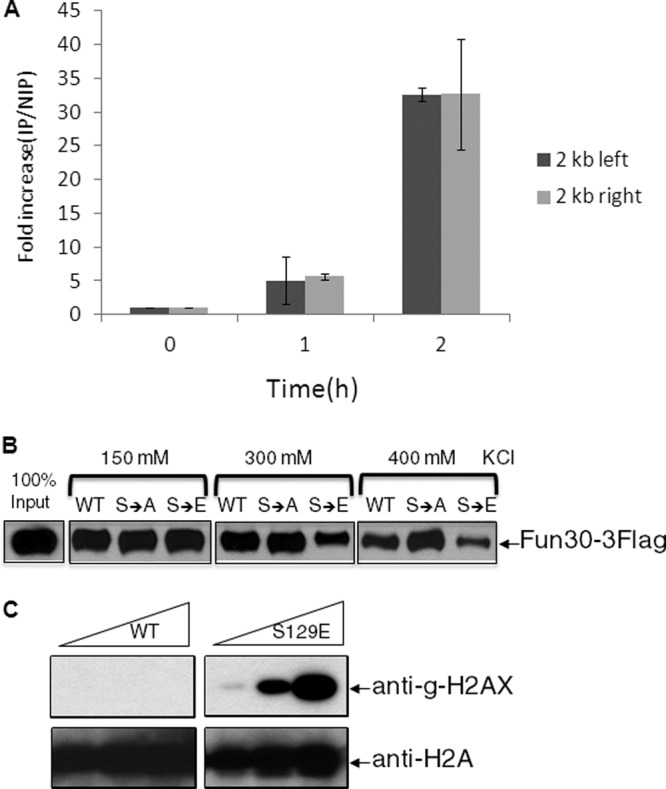
Fun30 is recruited to a DSB in vivo and has lower affinity for γ-H2AX than for unphosphorylated H2A. (A) Chromatin immunoprecipitation of Fun30 modified by a FLAG epitope at its C terminus, after induction of an unrepaired DSB in strain JKM179. The increase of IP signal relative to input DNA is shown. Error bars reflect ranges from two independent experiments. (B) Purified-FLAG-tagged Fun30 was incubated with immobilized nucleosome arrays at different salt concentrations. (C) Anti-γ-H2AX antibody recognizes the phosphomimetic form of histone H2A.
The interaction of Fun30 with nucleosomes in vitro is influenced by modifications at H2A-S129.
Fun30 has previously been shown to bind and remodel nucleosomes in vitro (1). To examine if Fun30's interaction with nucleosomes was influenced by γ-H2AX, we assembled nucleosome arrays on an immobilized linear pBlueScript SK(−) DNA, using reconstituted wild-type-H2A-, H2A-S129A-, or H2A-S129E-containing histone octamers. The assembled wild-type and mutant nucleosome arrays showed similar micrococcal-nuclease-digested DNA ladders with size increments of approximately 150 bp (see Fig. S2 in the supplemental material). The result indicates that most of the DNA is assembled into nucleosomes packaged with either wild-type or mutant histone octamers.
We then examined interactions between Fun30 and immobilized wild-type-H2A-, H2A-S129A-, or H2A-S129E-containing nucleosome arrays. We found that Fun30 was associated with the wild-type nucleosome array in the presence of 0.15, 0.3, and 0.4 M KCl (Fig. 6B). In contrast, while Fun30 showed binding to the S129E nucleosome array in 0.15 M KCl, this binding was substantially reduced in 0.3 and 0.4 M KCl (Fig. 6B). Compared to its binding to wild-type nucleosomes, Fun30 showed equal or stronger binding to a S129A-containing nucleosome array under the same conditions. We have confirmed that, in vitro, H2A-S129E is phosphomimetic, as it reacts with antibodies against γ-H2AX (Fig. 6C). The observation that the phosphomimetic H2A-S129E inhibits the binding of Fun30 to nucleosomes indicates that Fun30 binds preferentially to H2A over γ-H2AX.
Fun30 does not facilitate the removal of γ-H2AX after DSB repair.
In the ectopic gene conversion strain YJK17, described earlier, the induction of a DSB (29) results in the extensive spreading of γ-H2AX around the DSB. This modification persists until after the break is repaired and then it is removed, apparently by H2A-H2B exchange or displacement, prior to its being dephosphorylated by Pph3 (28, 29). How γ-H2AX is removed from chromatin is not yet known, but it is likely that some chromatin remodeler carries out this activity. Because Fun30 has the ability to exchange H2A-H2B dimers in vitro (1), we hypothesized that Fun30 could have a role in promoting the removal of γ-H2AX. To analyze the persistence of γ-H2AX in the 30-kb region on either side of the DSB, we used an antibody against γ-H2AX to carry out chromatin immunoprecipitation (ChIP) using strains YJK17 and its fun30Δ derivative. As seen in Fig. 7, induction of HO robustly promoted γ-H2AX formation, with the maximum signal peaking at 2 h for all the distances tested. We also note that the fold increase in γ-H2AX was similar in wild-type and fun30Δ strains; for example, at 20 kb from a DSB, the ChIP signal increased approximately 30-fold in both cases. By 3 h, we detected loss of γ-H2AX signal 10 kb away from the break site, a time that correlates with the appearance of the repair product (28) (Fig. 1B). By 7 h and 9 h, we saw loss of γ-H2AX at 20 and 30 kb from the repaired DSB, respectively. In the fun30Δ derivative, there is a peak of γ-H2AX phosphorylation by 2 to 3 h followed by a decrease in ChIP signal at 5 h, which decreases to wild-type levels by 12 h (Fig. 7). As noted above, the kinetics of product formation in the fun30Δ mutant is indistinguishable from that in the wild type (Fig. 1B). Thus, the removal of γ-H2AX after DSB repair is complete and does not uniquely depend on Fun30.
Fig 7.
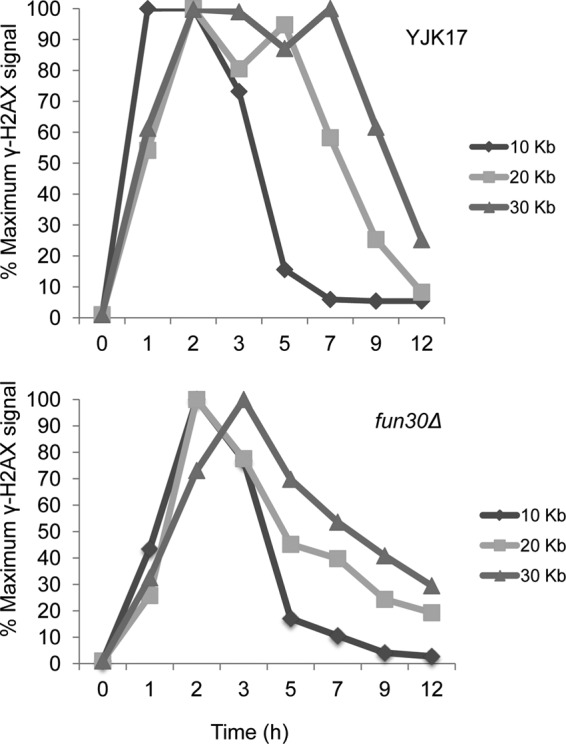
Fun30 is not required for the removal of γ-H2AX after DSB repair. γ-H2AX was quantitated by ChIP at 10 kb, 20 kb, and 30 kb to the right of the DSB at MAT in the ectopic recombination strain YJK17 and in its fun30Δ derivative. γ-H2AX ChIP values were measured at the indicated times after HO induction and graphed as percentages of the maximum ChIP value for each locus.
Fun30 is required for checkpoint adaptation to an unrepaired DSB.
In strain JKM179, in which there is a galactose-induced DSB but no homologous donor sequences, only a very small fraction of cells (∼0.2%) can repair the DSB by nonhomologous-end joining (35). Thus, nearly all cells respond to the DSB by activating the DNA damage checkpoint and arresting as large budded cells. This Mec1-dependent arrest persists for 12 to 15 h, after which cells adapt and resume cell cycle progression even though the DSB persists (53). Strain JKM179 and its fun30Δ derivative were grown on YEP-glycerol, and then single unbudded G1 cells were micromanipulated on YEP-Gal plates. By 8 h, 90% appeared as large budded cells, which is characteristic of checkpoint-mediated G2/M arrest (Fig. 8A). By 24 h, roughly 90% of wild-type cells adapted and formed microcolonies of 3 to 8 cells. In contrast, 90% of fun30Δ cells remained arrested as large budded cells (Fig. 8A). This adaptation defect is similar to that seen with the tid1Δ mutant previously (36).
Fig 8.

FUN30 is required for adaptation to a single, irreparable DNA DSB. (A) Single cells of wild-type, fun30Δ, and tid1Δ strains were plated on YEP-Gal plates and monitored at 8 h and 24 h. The percentage of cells adapted at any time point was calculated from the number of cells that progressed beyond the two-cell stage. Data are means and SEM from at least three independent experiments. (B) Wild-type and fun30Δ strains were stained with DAPI 24 h after galactose induction. The percentages of 150 G2/M-arrested cells that had a single nucleus are provided. (C) chk1Δ but not tel1Δ suppressed the adaptation defect of fun30Δ. (D) fun30Δ and tid1Δ prolong RAD53 hyperphosphorylation. Cells of the indicated strains were collected after galactose induction at the time points indicated. Proteins were extracted, and Western blotting was carried out using anti-Rad53 antibody. The arrows indicate the phosphorylated and unphosphorylated forms of Rad53.
We noted that the time to complete a cell cycle of a fun30Δ strain lacking the HO cleavage site but treated in the same fashion (4.3 h) was substantially longer than that of an isogenic wild-type cell (2.6 h) (data not shown). This result is consistent with previous reports of delayed cell cycle progression in fun30 mutants (50). To eliminate the possibility that the fun30 mutants are able to adapt and complete nuclear division but unable to rebud in the presence of DNA damage, we quantified the percentage of binucleate versus mononucleate cells by DAPI staining at 24 h. As shown in Fig. 8B, 85% of fun30Δ cells remain arrested as mononucleate cells, indicating that these cells are unable to resume mitotic progression; again, this result is similar to that seen when Rdh54/Tid1 is absent (36).
To confirm that the failure to adapt depends on the Mec1 (ATR)-mediated DNA damage checkpoint, we showed that a fun30Δ chk1Δ double mutant suppressed the adaptation defect of fun30Δ. Cells carrying chk1Δ arrest for approximately 12 h, about 80% as long as wild-type cells (17), and fun30Δ chk1Δ cells resume cell cycle progression only after a similar delay. Normally, the Tel1 (ATM) kinase does not play an important role in the DNA damage checkpoint, although its absence slightly shortens checkpoint arrest (23). In addition, a Tel1-Mre11-dependent checkpoint has been described (63); this complex is recruited to minimally resected or blunt DNA ends. Given that fun30Δ mutants are profoundly impaired in both resection and adaptation, we decided to investigate the interaction of Fun30 with both Mre11 and Tel1. Deletion of Mre11 suppressed the adaptation defect of fun30Δ, but deletion of TEL1 did not (Fig. 8C). These results are similar to what has been seen with tid1Δ, in that chk1Δ suppresses its permanent arrest whereas tel1Δ does not (36). We conclude that Fun30 is required for adaptation in response to a single irreparable DSB and that this defect is dependent on a functional checkpoint response which is Mec1 and Mre11 dependent but Tel1 independent.
In the case of some adaptation-defective mutants (e.g., ino80Δ strains), the inability to turn off the DNA damage checkpoint correlates with the failure to remove the histone variant Htz1 from the region around the DSB (52). Indeed, the adaptation defect of ino80Δ cells is suppressed by deleting either HTZ1 or SWR1 (the histone chaperone which is responsible for the deposition of Htz1). We also note that Fun30 appears to interact with Htz1, based on the finding that there is a global increase in Htz1 across the genome, notably near centromeres, in the absence of Fun30 (18). We deleted HTZ1 and monitored adaptation in a fun30Δ strain. Deletion of HTZ1 had no effect on the adaptation defect of fun30Δ cells (see Fig. S3 in the supplemental material).
Induction of an unrepairable DSB leads to the robust phosphorylation of the Rad53 (Chk2) kinase, and this kinase is then dephosphorylated at the time of adaptation (53). In adaptation-defective mutants, such as tid1Δ, yku70Δ, ptc2Δ ptc3Δ, and cdc5-ad mutants, Rad53 remains in a hyperphosphorylated state (37, 53). Figure 8D shows Western blots of Rad53 in wild-type, fun30Δ, and tid1Δ cells after induction of a DSB. We found that the absence of Fun30 prolonged Rad53 hyperphosphorylation compared to that in the wild type, apparently to a somewhat lesser extent than seen in tid1Δ cells (Fig. 8D). Hence, the failure to adapt correlates with an inability to turn off the Rad53-mediated checkpoint arrest, and as we showed above, prolonged checkpoint arrest depends on Chk1.
We also determined whether the ATPase mutant was required for adaptation. As shown in Fig. 8C, a plasmid carrying wild-type FUN30 was able to complement the adaptation defect of a fun30Δ strain, whereas the ATPase mutant was unable to restore adaptation. The ATPase mutation is dominant negative, as it caused a failure to adapt when the plasmid carrying the fun30(K603R) mutation is present in a wild-type cell (data not shown).
Overexpression of EXO1 suppresses the adaptation defects of both fun30Δ and sgs1Δ mutants.
As we mentioned in the introduction, it is now unclear whether the rate of resection or, more specifically, the rate of generating deoxyoligonucleotides during resection is required to maintain checkpoint arrest. One hypothesis that we wished to address more specifically was that the helicase-exonuclease Sgs1-Top3-Rmi1/Dna2, by producing deoxyoligonucleotides, might promote prolonged checkpoint arrest, whereas the exonuclease Exo1, producing mononucleotides, would not have this effect. Were this the case, we might expect that deleting SGS1 would result in a shorter arrest after a DSB triggered the DNA damage checkpoint, while exo1Δ might in fact result in a longer arrest if Sgs1 were the only resection activity. But in fact, the sgs1Δ strain proved to be adaptation defective, whereas exo1Δ reduced adaptation to about 50% (Fig. 9). Thus, the persistence of arrest in budding yeast does not seem to be related to the production of deoxyoligonucleotides, and again, mutants with slowed resection can be adaptation defective (sgs1Δ, fun30Δ, and sae2Δ mutants) or not (mre11Δ and rad50Δ mutants). The sgs1Δ fun30Δ and exo1Δ fun30Δ double mutants are no more adaptation defective than the fun30Δ mutant (Fig. 9).
Fig 9.
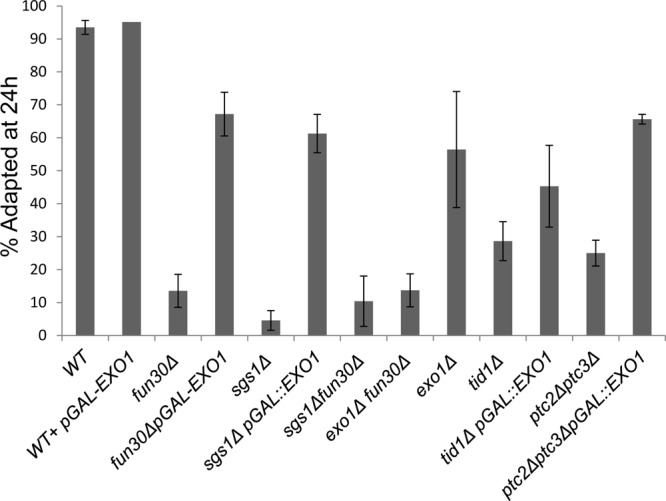
sgs1Δ is adaptation defective and overexpression of EXO1 suppresses the adaptation defect of fun30Δ and sgs1Δ. This assay was carried out as described in Fig. 8A except that strains harboring plasmids were pregrown in appropriate selective media overnight containing raffinose prior to the experiment and then micromanipulated onto YEP-Gal plates. Error bars reflect ranges from two independent experiments.
Given that there is a group of slow-resecting mutants that fail to adapt, one can imagine that in fun30Δ strains, resection or a resection by-product does not reach some threshold required to turn on adaptation. In this context, overexpressing EXOI might suppress the adaptation defect. We transformed fun30Δ and sgs1Δ strains with a 2μ plasmid that overexpresses EXO1 under the control of a galactose-inducible promoter, so that EXO1 is overexpressed at the same time as the unrepairable DSB is induced. Wild-type cells transformed with the same plasmid showed no major changes in the adaptation phenotype (Fig. 9), whereas the adaptation defects of fun30Δ and sgs1Δ were suppressed by overexpressing EXO1. In contrast, overexpressing EXO1 in another adaptation mutant, the tid1Δ mutant, had no significant effect on adaptation, although, unexpectedly, it did suppress the effect of ptc2Δ ptc3Δ. To assess whether EXO1 overexpression suppressed the fun30Δ resection defect, we measured resection by qPCR as described earlier. EXO1 overexpression caused a higher level of resection in both the wild-type and fun30Δ strains (see Fig. S4 in the supplemental material).
Fun30 also affects camptothecin sensitivity.
Finally, we report that the absence of Fun30 in exo1Δ and sgs1Δ strains exacerbates their sensitivity to the genotoxic agent camptothecin (see Fig. S5 in the supplemental material). However, in contrast to this mutation's epistatic effect on resection, the fun30Δ single mutant was only mildly sensitive to camptothecin, and the sgs1Δ fun30Δ and exo1Δ fun30Δ mutants were each much more sensitive than the strains with sgs1Δ or exo1Δ alone. This result suggests that the camptothecin sensitivity of these strains cannot be ascribed to their resection defects alone. The more severe effect of fun30Δ in the double mutants may reflect defects in turning off the DNA damage checkpoint, especially in the absence of either Sgs1 or Exo1.
Deletion of FUN30 increases the rate of nonhomologous-end joining.
Nonhomologous-end joining (NHEJ) is most efficient when 5′-to-3′ resection of DSB ends is very limited (2, 15, 25). We assayed NHEJ by plating MATα GAL::HO cells lacking the HML and HMR donor sequences (strain JKM179) on YEP-Gal plates so that HO endonuclease is constantly present and cells can grow into colonies only if they rejoin the ends by misalignment of the bases in the 4-bp 3′ overhanging complementary HO-cut ends so that HO cannot cleave the modified site (31, 48). This process depends on DNA ligase 4, the yeast Ku proteins, and the MRX complex, which not only controls the initial resection of DSB ends but also can hold DSB ends together for end joining (14, 48). As shown in Fig. S6 in the supplemental material, fun30Δ results in a 5-fold increase in NHEJ. This increase is similar to the ∼5-fold increase seen in G1-arrested cells, where resection is blocked by the lack of CDK1 activity (25), or when resection is impaired by deletion of SAE2 or mutation of the nuclease domain of Mre11 (33). The increase in NHEJ in the fun30Δ mutant is Ku dependent (see Fig. S6), suggesting that Fun30 acts in the classic NHEJ pathway rather than the Ku-independent microhomology-mediated end-joining (MMEJ) pathway (33, 41). These results suggest that Fun30 may act quite early in the resection process as well as controlling the rate of extensive resection.
DISCUSSION
We have investigated the roles played by the chromatin remodeler Fun30 in various aspects of DSB repair and checkpoint responses. Despite having been shown to facilitate H2A-H2B dimer exchange in vitro, Fun30 does not appear to be responsible for the displacement of γ-H2AX-containing dimers from chromatin surrounding a DSB after the break has been repaired. But Fun30 does play a highly significant role in facilitating 5′-to-3′ resection from the ends of a DSB. In the absence of Fun30, the rate of resection drops about 3-fold. This residual resection is not slowed further by deleting either Sgs1 or Exo1, suggesting that Fun30 acts to facilitate nucleosome remodeling in both major resection pathways but that some resection can occur without Fun30. Importantly, Fun30 binds near DSB ends, supporting the idea that it directly interacts with nucleosomes to facilitate resection. We also found that γ-H2AX impairs resection, so that a strain expressing only H2A-S129A promotes a 1.7-fold increase in resection. Interestingly, Fun30 binds less strongly to nucleosomes containing the phosphomimetic H2A-S129E mutation than to wild-type or H2A-S129A-containing nucleosomes. Together these findings suggest that Fun30 is restrained in promoting resection through chromatin containing γ-H2AX.
fun30Δ has a very low rate of 5′-to-3′ resection of DSB ends, though not as low as the rate of removal of the two major resection activities encoded by EXO1 and SGS1. We have shown previously that, in the BIR assay, a sgs1Δ exo1Δ derivative displayed a marked increase in survivors compared to the wild type, and this increase resulted from the appearance of a new class of events, in which telomere sequences had been added near the end of the DSB in 50% of the survivors (39). De novo telomere addition is apparently facilitated both by the very slow resection of the ends, so that there is very little single-stranded DNA (ssDNA), and by the failure to activate the (ssDNA-dependent) Mec1-dependent DNA damage checkpoint (10, 39). Mec1 activation results in the activation of Pif1 helicase, an antagonist of telomerase (44), so that new telomere formation is inhibited (49, 70). Resection was not completely eliminated, however, as new telomeres were added up to 6 kb centromere proximal to the site of the DSB. However, in the case of fun30Δ, more than 94% of the repair events proved to be BIR, with few new telomere additions, even though resection was impaired. One clear difference is that in the sgs1Δ exo1Δ mutant, the DNA damage checkpoint is not strongly activated, whereas in the fun30Δ strain, the DNA damage checkpoint is fully activated, and thus Pif1 would be activated to prevent new telomere addition. Interestingly, even in the mre11Δ fun30Δ mutant, in which resection should be reduced even further, the checkpoint is activated (cells arrest for 8 h), although arrest is not permanent.
During the course of our work we learned that similar results had been obtained by Chen et al. (7) and Costelloe et al. (4a), who showed that deleting FUN30 had a much more profound effect on extensive resection than ablations of other chromatin remodelers, such as RSC, INO80, SWR1, CHD1, and Rad54. However, fun30lΔ does not fully eliminate continuing resection compared to sgs1Δ exo1Δ, and indeed, overexpressing Exo1 overrides the resection defect of fun30Δ. Hence, other chromatin remodelers may play a role in facilitating Exo1's progression through chromatin.
Previous work has shown that fun30Δ partially weakens the silencing of HMR (50). Here we show that this weakening of silencing does not lead to the exposure of the HO cleavage site within HMR, even when HMR is inserted far from a telomere, where silencing is also weakened (60). We also found that heterochromatin slows resection slightly but is not an absolute barrier to resection and, moreover, that the absence of Fun30 does not absolutely prevent resection through such a region. We noted that since heterochromatic regions such as HMR are refractory to γ-H2AX phosphorylation (30), it is possible that any decrease in resection imposed by highly positioned nucleosomes is offset by an increase in affinity for Fun30 in a region lacking γ-H2AX.
Another significant role of FUN30 is in turning off the DNA damage checkpoint in response to an unrepaired DSB. Thus, two Swi2/Snf2 homologs—Fun30 and Tid1/Rdh54—both play similar roles in regulating adaptation, but the tid1Δ mutation does not result in the same resection defects as fun30Δ. Our finding that a sgs1Δ strain is adaptation defective removes any link between maintenance of the checkpoint signal and the extent of resection or the production of novel degradation products, at least in budding yeast. We had advanced the idea that deoxyoligonucleotides produced by a helicase-endonuclease (such as Sgs1-Dna2) might be required to maintain the checkpoint (27). This idea was supported by data obtained with Xenopus and mammalian cells indicating that deoxyoligonucleotides stimulated the DNA damage checkpoint and were bound by the Mre11-Rad50-Nbs1 complex (27). It is intriguing therefore that mre11Δ suppresses fun30Δ's adaptation defect. But how checkpoint adaptation is regulated remains unclear. Several hypotheses can still be entertained. First, it might be that some key signaling protein that binds near DSB ends needs to be displaced and this fails to happen in several mutants, so that the signaling persists or is intensified. Second, it is possible that some of these mutations increase the signaling from Chk1, which normally plays a modest role in the damage/arrest response. An alternative idea is that there still is a connection to resection, reflecting a competition between the exonucleases and the binding of the 9-1-1 DNA damage clamp that binds to a resected 5′ end (42, 43). Further experiments will be needed to sort out whether these mutants converge on a single cause of prolonging arrest or if they cause arrest by activating different signals.
Finally, we note that knockdown of the mammalian homolog of FUN30, SMARCAD1, displays many of the silencing defects seen in fun30Δ mutants (54). Knockdown of SMARCAD1 in HeLa cells results in a global increase in histone H3 and H4 acetylation and a decrease in H3K9 methylation. These results suggest that SMARCAD1 is required for the maintenance of silent heterochromatin, which is similar to Fun30's role in budding yeast. Interestingly a proteome-wide screen identified SMARCAD1 as being phosphorylated upon DNA damage (45), which has also been demonstrated for FUN30 (6). Indeed, knockdown of SMARCAD1 inhibits DNA end-resection from DSBs (4a). Therefore, it would be highly informative to investigate whether SMARCAD1 depletion in human cells leads to the same DNA damage defects we observe in budding yeast.
Supplementary Material
ACKNOWLEDGMENTS
We thank Wei Wang for help with quantitation of the histone octamers, Carl Wu for anti-H2B antibodies and histone expression plasmids, Sarah Dykstra for constructing the H2A S129A fun30Δ strain, Patrick Varga-Weisz for Fun30 plasmids and for communicating results prior to publication, Marco Foaini for Rad53 antibody, Grzgorz Ira and Betrand Llorente for communicating results, and the Haber lab and Carl Wu for discussions.
This research was supported by NIH grants GM20056 and GM61766 to J.E.H. and NCI grant 1K22CA122453 to W.-H.W.
Footnotes
Published ahead of print 24 September 2012
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1. Awad S, Ryan D, Prochasson P, Owen-Hughes T, Hassan AH. 2010. The Snf2 homolog Fun30 acts as a homodimeric ATP-dependent chromatin-remodeling enzyme. J. Biol. Chem. 285:9477–9484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aylon Y, Kupiec M. 2005. Cell cycle-dependent regulation of double-strand break repair: a role for the CDK. Cell Cycle 4:259–261 [PubMed] [Google Scholar]
- 3. Bassing CH, et al. 2002. Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc. Natl. Acad. Sci. U. S. A. 99:8173–8178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bitterman KJ, Anderson RM, Cohen HY, Latorre-Esteves M, Sinclair DA. 2002. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J. Biol. Chem. 277:45099–45107 [DOI] [PubMed] [Google Scholar]
- 4a. Costelloe T, et al. 2012. The yeast Fun30 and human SMARCAD1 chromatin remodellers promote DNA end resection. Nature 489:581–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chai B, Huang J, Cairns BR, Laurent BC. 2005. Distinct roles for the RSC and Swi/Snf ATP-dependent chromatin remodelers in DNA double-strand break repair. Genes Dev. 19:1656–1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen SH, Albuquerque CP, Liang J, Suhandynata RT, Zhou H. 2010. A proteome-wide analysis of kinase-substrate network in the DNA damage response. J. Biol. Chem. 285:12803–12812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen X, et al. 2012. The Fun30 nucleosome remodeler promotes resection of DNA double-strand break ends. Nature 489:576–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen X, et al. 2011. Cell cycle regulation of DNA double-strand break end resection by Cdk1-dependent Dna2 phosphorylation. Nat. Struct. Mol. Biol. 18:1015–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chi P, et al. 2011. Analyses of the yeast Rad51 recombinase A265V mutant reveal different in vivo roles of Swi2-like factors. Nucleic Acids Res. 39:6511–6522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chung WH, Zhu Z, Papusha A, Malkova A, Ira G. 2010. Defective resection at DNA double-strand breaks leads to de novo telomere formation and enhances gene targeting. PLoS Genet. 6:e1000948 doi:10.1371/journal.pgen.1000948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Clerici M, Mantiero D, Lucchini G, Longhese MP. 2006. The Saccharomyces cerevisiae Sae2 protein negatively regulates DNA damage checkpoint signalling. EMBO Rep. 7:212–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Clerici M, Mantiero D, Lucchini G, Longhese MP. 2005. The Saccharomyces cerevisiae Sae2 protein promotes resection and bridging of double strand break ends. J. Biol. Chem. 280:38631–38638 [DOI] [PubMed] [Google Scholar]
- 13. Costanzo M, Baryshnikova A, Myers CL, Andrews B, Boone C. 2011. Charting the genetic interaction map of a cell. Curr. Opin. Biotechnol. 22:66–74 [DOI] [PubMed] [Google Scholar]
- 14. Daley JM, Palmbos PL, Wu D, Wilson TE. 2005. Nonhomologous end joining in yeast. Annu. Rev. Genet. 39:431–451 [DOI] [PubMed] [Google Scholar]
- 15. Daley JM, Wilson TE. 2005. Rejoining of DNA double-strand breaks as a function of overhang length. Mol. Cell. Biol. 25:896–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dotiwala F, Haase J, Arbel-Eden A, Bloom K, Haber JE. 2007. The yeast DNA damage checkpoint proteins control a cytoplasmic response to DNA damage. Proc. Natl. Acad. Sci. U. S. A. 104:11358–11363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dotiwala F, Harrison JC, Jain S, Sugawara N, Haber JE. 2010. Mad2 prolongs DNA damage checkpoint arrest caused by a double-strand break via a centromere-dependent mechanism. Curr. Biol. 20:328–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Durand-Dubief M, et al. 2012. SWI/SNF-like chromatin remodeling factor Fun30 supports point centromere function in S. cerevisiae. PLoS Genet. 8:e1002974 doi:10.1371/journal.pgen.1002974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fernandez-Capetillo O, et al. 2002. DNA damage-induced G2-M checkpoint activation by histone H2AX and 53BP1. Nat. Cell Biol. 4:993–997 [DOI] [PubMed] [Google Scholar]
- 20. Fishman-Lobell J, Rudin N, Haber JE. 1992. Two alternative pathways of double-strand break repair that are kinetically separable and independently modulated. Mol. Cell. Biol. 12:1292–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gravel S, Chapman JR, Magill C, Jackson SP. 2008. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 22:2767–2772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guillemain G, et al. 2007. Mechanisms of checkpoint kinase Rad53 inactivation after a double-strand break in Saccharomyces cerevisiae. Mol. Cell. Biol. 27:3378–3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Harrison JC, Haber JE. 2006. Surviving the breakup: the DNA damage checkpoint. Annu. Rev. Genet. 40:209–235 [DOI] [PubMed] [Google Scholar]
- 24. Hicks WM, Yamaguchi M, Haber JE. 2011. Real-time analysis of double-strand DNA break repair by homologous recombination. Proc. Natl. Acad. Sci. U. S. A. 108:3108–3115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ira G, et al. 2004. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 431:1011–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jain S, et al. 2009. A recombination execution checkpoint regulates the choice of homologous recombination pathway during DNA double-strand break repair. Genes Dev. 23:291–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jazayeri A, Balestrini A, Garner E, Haber JE, Costanzo V. 2008. Mre11-Rad50-Nbs1-dependent processing of DNA breaks generates oligonucleotides that stimulate ATM activity. EMBO J. 27:1953–1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Keogh MC, et al. 2006. A phosphatase complex that dephosphorylates gammaH2AX regulates DNA damage checkpoint recovery. Nature 439:497–501 [DOI] [PubMed] [Google Scholar]
- 29. Kim JA, Haber JE. 2009. Chromatin assembly factors Asf1 and CAF-1 have overlapping roles in deactivating the DNA damage checkpoint when DNA repair is complete. Proc. Natl. Acad. Sci. U. S. A. 106:1151–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim JA, Kruhlak M, Dotiwala F, Nussenzweig A, Haber JE. 2007. Heterochromatin is refractory to gamma-H2AX modification in yeast and mammals. J. Cell Biol. 178:209–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kramer KM, Brock JA, Bloom K, Moore JK, Haber JE. 1994. Two different types of double-strand breaks in Saccharomyces cerevisiae are repaired by similar RAD52-independent, nonhomologous recombination events. Mol. Cell. Biol. 14:1293–1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee JH, Paull TT. 2004. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 304:93–96 [DOI] [PubMed] [Google Scholar]
- 33. Lee K, Lee SE. 2007. Saccharomyces cerevisiae Sae2- and Tel1-dependent single-strand DNA formation at DNA break promotes microhomology-mediated end joining. Genetics 176:2003–2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee SE, Bressan DA, Petrini JH, Haber JE. 2002. Complementation between N-terminal Saccharomyces cerevisiae mre11 alleles in DNA repair and telomere length maintenance. DNA Repair (Amst.) 1:27–40 [DOI] [PubMed] [Google Scholar]
- 35. Lee SE, et al. 1998. Saccharomyces Ku70, mre11/rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell 94:399–409 [DOI] [PubMed] [Google Scholar]
- 36. Lee SE, Pellicioli A, Malkova A, Foiani M, Haber JE. 2001. The Saccharomyces recombination protein Tid1p is required for adaptation from G2/M arrest induced by a double-strand break. Curr. Biol. 11:1053–1057 [DOI] [PubMed] [Google Scholar]
- 37. Leroy C, et al. 2003. PP2C phosphatases Ptc2 and Ptc3 are required for DNA checkpoint inactivation after a double-strand break. Mol. Cell 11:827–835 [DOI] [PubMed] [Google Scholar]
- 38. Lydeard JR, Jain S, Yamaguchi M, Haber JE. 2007. Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature 448:820–823 [DOI] [PubMed] [Google Scholar]
- 39. Lydeard JR, Lipkin-Moore Z, Jain S, Eapen VV, Haber JE. 2010. Sgs1 and exo1 redundantly inhibit break-induced replication and de novo telomere addition at broken chromosome ends. PLoS Genet. 6:e1000973 doi:10.1371/journal.pgen.1000973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lydeard JR, et al. 2010. Break-induced replication requires all essential DNA replication factors except those specific for pre-RC assembly. Genes Dev. 24:1133–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ma JL, Kim EM, Haber JE, Lee SE. 2003. Yeast Mre11 and Rad1 proteins define a Ku-independent mechanism to repair double-strand breaks lacking overlapping end sequences. Mol. Cell. Biol. 23:8820–8828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Majka J, Burgers PM. 2007. Clamping the Mec1/ATR checkpoint kinase into action. Cell Cycle 6:1157–1160 [DOI] [PubMed] [Google Scholar]
- 43. Majka J, Burgers PM. 2003. Yeast Rad17/Mec3/Ddc1: a sliding clamp for the DNA damage checkpoint. Proc. Natl. Acad. Sci. U. S. A. 100:2249–2254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Makovets S, Blackburn EH. 2009. DNA damage signalling prevents deleterious telomere addition at DNA breaks. Nat. Cell Biol. 11:1383–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Matsuoka S, et al. 2007. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316:1160–1166 [DOI] [PubMed] [Google Scholar]
- 46. Mimitou EP, Symington LS. 2008. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 455:770–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mizuguchi G, et al. 2004. ATP-driven exchange of histone H2AZ variant catalyzed by SWR1 chromatin remodeling complex. Science 303:343–348 [DOI] [PubMed] [Google Scholar]
- 48. Moore JK, Haber JE. 1996. Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol. Cell. Biol. 16:2164–2173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Myung K, Pennaneach V, Kats ES, Kolodner RD. 2003. Saccharomyces cerevisiae chromatin-assembly factors that act during DNA replication function in the maintenance of genome stability. Proc. Natl. Acad. Sci. U. S. A. 100:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Neves-Costa A, Will WR, Vetter AT, Miller JR, Varga-Weisz P. 2009. The SNF2-family member Fun30 promotes gene silencing in heterochromatic loci. PLoS One 4:e8111 doi:10.1371/journal.pone.0008111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Oum JH, et al. 2011. RSC facilitates Rad59-dependent homologous recombination between sister chromatids by promoting cohesin loading at DNA double-strand breaks. Mol. Cell. Biol. 31:3924–3937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Papamichos-Chronakis M, Krebs JE, Peterson CL. 2006. Interplay between Ino80 and Swr1 chromatin remodeling enzymes regulates cell cycle checkpoint adaptation in response to DNA damage. Genes Dev. 20:2437–2449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pellicioli A, Lee SE, Lucca C, Foiani M, Haber JE. 2001. Regulation of Saccharomyces Rad53 checkpoint kinase during adaptation from DNA damage-induced G2/M arrest. Mol. Cell 7:293–300 [DOI] [PubMed] [Google Scholar]
- 54. Rowbotham SP, et al. 2011. Maintenance of silent chromatin through replication requires SWI/SNF-like chromatin remodeler SMARCAD1. Mol. Cell 42:285–296 [DOI] [PubMed] [Google Scholar]
- 55. Shim EY, Ma JL, Oum JH, Yanez Y, Lee SE. 2005. The yeast chromatin remodeler RSC complex facilitates end joining repair of DNA double-strand breaks. Mol. Cell. Biol. 25:3934–3944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shroff R, et al. 2004. Distribution and dynamics of chromatin modification induced by a defined DNA double-strand break. Curr. Biol. 14:1703–1711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Stralfors A, Walfridsson J, Bhuiyan H, Ekwall K. 2011. The FUN30 chromatin remodeler, Fft3, protects centromeric and subtelomeric domains from euchromatin formation. PLoS Genet. 7:e1001334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Strom L, Lindroos HB, Shirahige K, Sjogren C. 2004. Postreplicative recruitment of cohesin to double-strand breaks is required for DNA repair. Mol. Cell 16:1003–1015 [DOI] [PubMed] [Google Scholar]
- 59. Sugawara N, Wang X, Haber JE. 2003. In vivo roles of Rad52, Rad54, and Rad55 proteins in Rad51-mediated recombination. Mol. Cell 12:209–219 [DOI] [PubMed] [Google Scholar]
- 60. Thompson JS, Johnson LM, Grunstein M. 1994. Specific repression of the yeast silent mating locus HMR by an adjacent telomere. Mol. Cell. Biol. 14:446–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Toczyski DP, Galgoczy DJ, Hartwell LH. 1997. CDC5 and CKII control adaptation to the yeast DNA damage checkpoint. Cell 90:1097–1106 [DOI] [PubMed] [Google Scholar]
- 62. Unal E, et al. 2004. DNA damage response pathway uses histone modification to assemble a double-strand break-specific cohesin domain. Mol. Cell 16:991–1002 [DOI] [PubMed] [Google Scholar]
- 63. Usui T, Ogawa H, Petrini JH. 2001. A DNA damage response pathway controlled by Tel1 and the Mre11 complex. Mol. Cell 7:1255–1266 [DOI] [PubMed] [Google Scholar]
- 64. Vaze M, et al. 2002. Recovery from checkpoint-mediated arrest after repair of a double-strand break requires srs2 helicase. Mol. Cell 10:373. [DOI] [PubMed] [Google Scholar]
- 65. Wach A, Brachat A, Pohlmann R, Philippsen P. 1994. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast 10:1793–1808 [DOI] [PubMed] [Google Scholar]
- 66. Wu WH, et al. 2005. Swc2 is a widely conserved H2AZ-binding module essential for ATP-dependent histone exchange. Nat. Struct. Mol. Biol. 12:1064–1071 [DOI] [PubMed] [Google Scholar]
- 67. Yeung M, Durocher D. 2011. Srs2 enables checkpoint recovery by promoting disassembly of DNA damage foci from chromatin. DNA Repair 10:1213–1222 [DOI] [PubMed] [Google Scholar]
- 68. Yu Q, Zhang X, Bi X. 2011. Roles of chromatin remodeling factors in the formation and maintenance of heterochromatin structure. J. Biol. Chem. 286:14659–14669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yuan J, Adamski R, Chen J. 2010. Focus on histone variant H2AX: to be or not to be. FEBS Lett. 584:3717–3724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zhou J, Monson EK, Teng S, Schulz VP, Zakian VA. 2000. Pif1p helicase, a catalytic inhibitor of telomerase in yeast. Science 289:771–774 [DOI] [PubMed] [Google Scholar]
- 71. Zhu Z, Chung WH, Shim EY, Lee SE, Ira G. 2008. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 134:981–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.