

Abstract
Neutrophils are first responders rapidly mobilized to inflammatory sites by a tightly regulated, nonredundant hierarchy of chemoattractants. These chemoattractants engage neutrophil cell surface receptors triggering heterotrimeric G-protein Gαi subunits to exchange GDP for GTP. By limiting the duration that Gαi subunits remain GTP bound, RGS proteins modulate chemoattractant receptor signaling. Here, we show that neutrophils with a genomic knock in of a mutation that disables regulator of G-protein signaling (RGS)-Gαi2 interactions accumulate in the bone marrow and mobilize poorly to inflammatory sites. These defects are attributable to enhanced sensitivity to background signals, prolonged chemoattractant receptor signaling, and inappropriate CXCR2 downregulation. Intravital imaging revealed a failure of the mutant neutrophils to accumulate at and stabilize sites of sterile inflammation. Furthermore, these mice could not control a nonlethal Staphylococcus aureus infection. Neutrophil RGS proteins establish a threshold for Gαi activation, helping to coordinate desensitization mechanisms. Their loss renders neutrophils functionally incompetent.
INTRODUCTION
Neutrophil recruitment to a site of injury or infection helps organize a tissue response and depends upon negotiating multiple chemoattractant cues that include chemokines, peptides, and lipids (13, 24, 28). These chemoattractants engage G-protein-coupled receptors (GPCRs) that activate Gαi-containing heterotrimeric G proteins (12). The activated G proteins then trigger intracellular signaling cascades necessary for neutrophil migration and trafficking to inflammatory sites and for eliciting innate host defenses (8, 25, 42). Conversely, unrestrained neutrophil responses can injure healthy tissue by promoting vasculitis and impaired organ function (23, 25). Successful resolution of an inflammatory insult without excessive collateral damage requires careful regulation of neutrophil recruitment and function. Specific regulatory mechanisms that target chemoattractant receptors (G-protein receptor kinases and β-arrestins) exist, while others (regulators of G-protein signaling [RGS] proteins) affect the heterotrimeric G protein, the major signal transducer.
Most chemoattractant receptors undergo agonist-dependent phosphorylation that promotes the recruitment of nonvisual arrestins, β-arrestin-1 and β-arrestin-2 (9, 10). This uncouples the receptor from the heterotrimeric G proteins, targets the receptor for internalization, and can trigger G-protein-independent signaling. The internalized receptor has two fates: it can be recycled and returned to the cell surface as a resensitized receptor, or it can be sorted to lysosomes for degradation (5). Receptor degradation can serve to terminate signaling. Failure to appropriately downregulate chemoattractant receptor signaling is clinically relevant, as patients with warts, hypogammaglobulinemia, infections, and myelokathexis (WHIM) syndrome do not properly desensitize the chemoattractant receptor CXCR4 (22, 27). A feature of WHIM syndrome is myelokathexis, myeloid hyperplasia in the bone marrow with apoptosis, which results as a consequence of inappropriate retention of myeloid cells in the bone marrow.
Another mechanism that limits GPCR-mediated activation of signaling pathways is the targeting of activated Gαi and Gαq proteins by RGS proteins (12). More than 20 proteins with canonical RGS domains are encoded by the human genome. These proteins accelerate the intrinsic GTPase activity of Gαi and Gαq, thereby reducing the duration that the Gα subunit remains GTP bound. Despite the importance of Gαi-coupled receptors in neutrophil trafficking (32, 45), little is known about the expression and function of RGS proteins in neutrophils. One study provided evidence that RGS proteins might regulate myeloid cell trafficking, as the introduction of a mutation into the mouse Gnai2 locus (Gαi2 G184S), which abrogates RGS protein binding, led to splenomegaly and elevated blood neutrophil and monocyte counts (21). However, the focus of that study was on the description of a complex and pleomorphic mouse phenotype, which included reduced mouse viability, growth retardation, and cardiac hypertrophy.
Here, we have investigated the neutrophils from homozygous Gαi2G184S/G184S knock-in (KI) mice to assess the contribution of endogenous RGS proteins to neutrophil function with an emphasis on the neutrophil trafficking. We find that the loss of RGS protein-mediated attenuation of Gαi2 signaling severely affects neutrophil accumulation at inflammatory sites. These mice also exhibit myelokathexis similar to that observed in people suffering from WHIM syndrome.
MATERIALS AND METHODS
Mice.
A genomic knock-in of the G184S Gnai2 allele was backcrossed 15 times onto C57BL/6 mice. The mice used were 6 to 14 weeks of age and rederived from original mice (21). Mice were housed under specific-pathogen-free conditions. For adoptive transfer experiments, CD45.1 C57BL/6 mice were purchased from the Jackson Laboratory (Bar Harbor, ME). The LysM-enhanced green fluorescent protein (EGFP) mice were kindly provided by Ron Germain (NIAID) with permission from Thomas Graf (Center for Genomic Regulation, Barcelona, Spain). Mice were housed under specific-pathogen-free conditions and used in accordance with the guidelines of the Animal Care and Use Committee at the National Institutes of Health.
Cell isolation, calcium assay, flow cytometry, and RT-PCR.
To induce sterile peritonitis, mice were injected intraperitoneally with 3% (wt/vol) thioglycolate. Peritoneal lavage was performed to collect peritoneal cells. Bone marrow cells were isolated from the femur and tibia. For calcium assay, neutrophils were purified to a purity of ∼95% using anti-Ly-6G MicroBeads (Miltenyi Biotech) and a FLIPR calcium 3 assay kit (Molecular Devices Inc.). For flow cytometry, erythrocyte-depleted cells were preincubated with anti-CD16/CD32 monoclonal antibody to block FcγRII/III receptors and stained with various fluorochrome-conjugated antibodies on ice for 15 min. Anti-Gr-1 (clone RB6-8C5), anti-CD11b (clone M1/70), anti-CXCR2 (clone 242216), anti-CXCR4 (clone 2B11), anti-C5a receptor (anti-C5aR; clone 20/70), and isotype controls were purchased from BD Biosciences, eBiosciences Inc., R&D Systems, and Cedarlane Laboratories. Data were acquired using a FACSCanto II flow cytometer with FACSDiva (version 6.2) software (BD Biosciences) and analyzed using FlowJo software (Tree Star). For reverse transcription-PCR (RT-PCR) assays, bone marrow neutrophils were sorted positively with anti-Gr-1 and anti-CD11b antibodies and negatively against Ly6C, siglec-F, B220, and CD3e. Postsorting analysis showed a purity of 99.5%. RNA was isolated using an RNeasy kit (Qiagen), cDNAs were generated with an Advantage RT-for-PCR kit (Clontech), and PCR was performed using a Hot Start PCR kit (Qiagen). The PCR primers used were described previously (29, 38).
Migration assay and ELISA.
Migration assays were performed with freshly isolated bone marrow cells using Transwell plates with 5-μm inserts (Corning Incorporated). Cells were immunostained with anti-Gr-1 and anti-CD11b antibodies, and 2 × 105 cells in 200 μl were then added to the upper wells and various doses of chemoattractants were added to the lower wells. The plate was incubated at 37°C for 45 min and then at 4°C for 10 min, after adding a final concentration of 5 mM EDTA to the lower wells to detach bound cells. The Gr-1- and CD11b-positive cells that migrated to the lower wells were counted using a flow cytometer. Macrophage inflammatory protein 2 (MIP-2) was purchased from R&D Systems. Peritoneal keratinocyte-derived chemokine (KC) and MIP-2 were measured with commercial enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems) according to the manufacturer's protocol.
Cell staining and confocal microscopy.
Freshly isolated erythrocyte-depleted bone marrow cells were cytospun onto glass slides and stained with Hema 3 (Fisher Scientific) according to the manufacturer's instruction. For immunocytochemistry, the cells were plated onto glass coverslips and incubated at room temperature for 10 min to allow neutrophils to attach. After gently washing off unattached cells, the remaining cells were fixed in 4% paraformaldehyde–0.1% Triton X-100 for 10 min. Cells were then stained with anti-Gαi2 antibody (EMD Chemicals) and/or anti-GRK2 antibody (Santa Cruz Biotechnology). Images were acquired on a Leica TCS-SP5 confocal microscope (Leica Microsystems).
Adoptive transfer.
Isolated bone marrow cells were intravenously injected via the tail vein into 7- to 8-week-old irradiated (900 rads) CD45.1 C57BL/6 mice (3 × 106 cells per mouse). Establishment of injected CD45.2 cells in the host was checked at 4 weeks after the cell transfer by staining blood cells with anti-CD45.2 (clone 104) and anti-CD45.1 (clone A20) antibodies (eBiosciences Inc.). The chimeric mice were then analyzed 6 to 8 weeks after the adoptive transfer.
Intravital TP-LSM.
Two-photon laser scanning microscopy (TP-LSM) was performed with a Leica SP5 inverted 5-channel confocal microscope (Leica Microsystems) equipped with a 25× water-immersion objective (numerical aperture [NA], 0.7) as previously described (40). To image the ear skin, we followed a previously detailed procedure (24). Purified neutrophils were labeled with CellTracker green CMFDA or CellTracker red CMTPX (Invitrogen). The cells were transferred into the ear and imaged on the following day. Alternatively, we imaged neutrophils in the ears of LysM-EGFP, LysM-EGFP Gαi2G184S/WT, or Gαi2G184S/G184S LysM-EGFP mice following laser damage. In some instances, blood vessels were outlined by the intravenous injection of Qdot 655 (Invitrogen). Three-dimensional (x, y, and z) images of the ear skin were acquired (2-μm spacing in the z axis over a total distance of 10 to 25 μm) every 15 to 30 s for a total observation period of 1 to 3 h. Off-line cell tracking analyses and spectral unmixing were performed using Imaris software. For cell tracking, sequences of image stacks were transformed into volume-rendered four-dimensional movies using Imaris software, and spot analysis was used for semiautomated tracking of cell motility in three dimensions by using the following parameters: autoregressive motion algorithm, estimated diameter of 10 μm, background subtraction true, maximum distance of 20 μm, and maximum gap size of 3. The calculations of the cell velocity, straightness, track length, and displacement were performed using the Imaris software.
Adhesion assay.
Glass-bottom culture dishes (MatTek) were coated with intercellular adhesion molecule 1 (ICAM-1; 10 μg/ml) plus 100 ng/ml MIP-2 in phosphate-buffered saline (100 μl/well) at 4°C overnight. Alternatively, SVEC4-10 cells (CRL-2181; ATCC) were grown until confluent on the same culture dishes. ICAM-1 and vascular cell adhesion molecule 1 (VCAM-1) expression was induced by treating the monolayers with tumor necrosis factor alpha (TNF-α; 20 ng/ml, 24 h). Prior to neutrophil adhesion, the monolayer was immunostained with anti-VCAM-1 antibody conjugated to eFluor 450 (eBioscience). Gr-1-positive cells were purified from wild-type (WT) and KI mouse bone marrow as described above. Isolated cells were differentially labeled with CellTracker green CMFDA, CellTracker red CMTMR, or CellTracker blue CMAC (Invitrogen), and equal numbers of wild-type and KI cells (0.5 million of each per dish) were allowed to adhere to dishes at 37°C for 30 min. After washing three times with warm medium, time-lapse imaging of adherent cells was performed with a Leica SP5 confocal microscope (Leica Microsystems) equipped with a 25× water-immersion objective (NA, 0.7) and a CO2 incubation chamber. Images were acquired every 10 s for a period of 20 to 30 min, and the data were processed using Imaris software.
Statistical analysis.
A Student's unpaired two-tailed t test was used for all statistical analyses using Prism software (GraphPad Software), unless indicated otherwise. Differences with P values of 0.05 or less were considered significant.
RESULTS
RGS protein-mediated control of Gαi2 signaling is required for neutrophil recruitment to the inflamed peritoneum.
Gαi2 is critical for neutrophil recruitment to inflammatory sites (32, 45), and its interaction with RGS proteins appears to be important for neutrophil homeostasis (21). To further investigate the consequence of abolishing the Gαi2-RGS interaction in neutrophils, we used a sterile peritonitis model. We injected the KI mice with 3% thioglycolate intraperitoneally (i.p.) and counted infiltrating neutrophils as well as total peritoneal cells at 4 h postinjection (Fig. 1A and B). Strikingly, very few KI neutrophils trafficked to the inflamed peritoneum. The KI mean neutrophil count of 0.25 million was 22-fold lower than the WT mean count of 5.68 million. Surprisingly, this recruitment defect observed with KI mice was much more severe than the modest defect reported from a similar analysis of Gαi2-knockout mice (45). There was some compensation for the loss of neutrophil recruitment in the KI mice, as the mean count of total peritoneal cells was only approximately 3-fold less than that of wild-type mice.
Fig 1.

Neutrophil trafficking to inflamed peritoneum is impaired in KI mice. (A) Dot plots of WT and KI peritoneal cells at 4 h after intraperitoneal thioglycolate injection. SSC, side light scatter. (B) Total cell counts (filled symbols) of peritoneal exudates were obtained by flow cytometry. Neutrophil counts (open symbols) were calculated by multiplying the percentage of Gr-1+ CD11b+ cells by total cell counts. Data are means ± SEMs. (C) Surface expression of CXCR2, CXCR4, and C5aR on challenged peritoneal neutrophils. For intracellular CXCR2 (inCXCR2) expression, cells were fixed, permeabilized, and then stained for CXCR2. Since surface CXCR2 was not blocked prior to intracellular staining, intracellular CXCR2 represents a total CXCR2 level in the cell. (D) Surface CXCR2 expression on peritoneal KI monocytes was similar to that on WT cells. ISO, cells immunostained with isotype controls.
To examine the basis of this recruitment defect, we assessed surface receptor expression of chemoattractant receptors on peritoneal neutrophils as well as chemokine levels in the peritoneal cavity. CXCR2 binds KC, MIP-2, and other mouse glutamic acid-leucine-arginine motif (ELR)-positive CXC chemokines (6) and plays a major nonredundant role in neutrophil trafficking to the peritoneum (11). We found that the recruited KI neutrophils had a marked reduction in surface expression of CXCR2, while their total CXCR2 level was similar to that of WT cells, suggesting inappropriate internalization of the receptor in KI cells (Fig. 1C). In contrast, WT and KI peritoneal monocytes expressed similar levels of CXCR2 (Fig. 1D). We also examined the surface expression of CXCR4, another receptor critical for bone marrow retention and trafficking of neutrophils (15, 26), and C5aR as a nonchemokine receptor control (Fig. 1C). CXCR4 expression was not changed; however, the KI neutrophils expressed larger amounts of C5aR. We did not detect a significant difference in the amount of recruiting chemokines in the peritoneum, as an ELISA analysis showed similar levels of KC and MIP-2 in the peritoneum fluid from WT and KI mice (data not shown).
The loss of Gαi2-RGS interaction causes neutropenia and defective neutrophil mobilization from bone marrow.
Contrary to a previous study reporting higher neutrophil counts in the blood of naïve KI mice than wild-type controls (21), we observed that the KI mice are neutropenic (Fig. 2A). This discrepancy may be attributable to different microbial environments (15) or due to differences in genetic makeup. The KI mice used in this study were backcrossed 15 times onto a C57BL/6 background, while the previously reported study used mice backcrossed 5 to 6 times. Furthermore, we rederived the mice prior to establishing the colony in our animal facility. Besides the disturbed neutrophil counts, we also noted abnormal lymphocyte, monocyte, basophil, and eosinophil counts in the KI mouse blood. When challenged with thioglycolate, neutrophils from the KI mice poorly mobilized to the blood (Fig. 2B). The mean ± standard error of the mean (SEM) blood neutrophil counts were 4,500 ± 492 for WT mice and 1,567 ± 249 for the KI mice at 4 h postinjection. The lymphocyte counts were also lower in the challenged KI mice than the wild-type controls, with only 622 ± 210 lymphocytes (approximately 13% of the naïve count) and 2,140 ± 327 lymphocytes (approximately 31% of the naïve count) remaining, respectively (Fig. 2A and B). Naïve KI neutrophils had slightly less CXCR2 surface expression than naïve wild-type cells (Fig. 2C). As observed in the inflamed peritoneum, CXCR2 expression on the blood neutrophils from challenged KI mice was significantly lower than that on neutrophils from the challenged wild-type mice. There were no significant differences in surface expression of CXCR4, regardless of the strain or activation status of neutrophils, whereas C5aR expression was higher in the challenged KI neutrophils than the challenged wild-type neutrophils (Fig. 2C).
Fig 2.

KI mice show a modest neutropenia and inefficient neutrophil mobilization to blood upon challenge. (A) Complete blood cell counts of naïve mice. Data are means ± SEMs. WBC, white blood cells; Lym, lymphocytes; Neut, neutrophils; Mon, monocytes; Baso, basophils; Eos, eosinophils. (B) Blood leukocyte counts of challenged mice at 4 h after thioglycolate injection. Data are means ± SEMs. (C) Surface expression of CXCR2, CXCR4, and C5aR on naïve or challenged blood neutrophils. ISO, neutrophils immunostained with isotype controls.
To assess whether the inefficient mobilization into blood and poor recruitment to the peritoneum resulted from a production or a trafficking problem, we analyzed the bone marrow from WT and KI mice. On the basis of flow cytometric analysis of CD11b and Gr-1 expression, bone marrow neutrophils can be divided into three populations: promyelocytes/myelocytes, immature neutrophils, and mature neutrophils (18). Compared to WT bone marrow, we found a significant increase in the percentage and number of neutrophils in the KI bone marrow, with marked skewing toward a mature phenotype (Fig. 3A). Increased bone marrow neutrophil maturity is a key feature of myelokathexis, a disorder associated with poor neutrophil bone marrow egress (22). Cytospins of bone marrow cells from two independent experiments also confirmed the increased frequency of mature neutrophils (see Fig. S1A in the supplemental material). Even following thioglycolate challenge, most mature KI neutrophils remained in the bone marrow (Fig. 3B). CXCR2 expression on the KI bone marrow neutrophils declined dramatically following challenge, while naïve KI neutrophils had a slight decrease in CXCR2 expression (Fig. 3C). The mobilization of mature bone marrow neutrophils, which express high levels of CXCR2, into the blood and inflamed peritoneum explains the decreased CXCR2 on the remaining WT neutrophils (Fig. 3D). In contrast, the pronounced reduction in CXCR2 expression on the KI neutrophils that failed to mobilize indicates inappropriate receptor downregulation. WT and KI neutrophil CXCR4 expression was unaffected by thioglycolate challenge. C5aR expression showed a biphasic pattern on the naïve KI neutrophils and was upregulated following the challenge (see Fig. S1B in the supplemental material).
Fig 3.

RGS protein insensitivity of Gαi2 produces a myelokathexis-like phenotype. (A) Dot plots of erythrocyte-depleted live bone marrow cells from naïve mice (top). Percentages of Gr-1+ CD11b+ cells and total bone marrow cell counts from the right femur and tibia are shown on the right. Data are means ± SEMs. Paired t test. On the basis of the expression levels of CD11b and Gr-1, gates were drawn to distinguish three subpopulations (bottom): promyelocytes/myelocytes (P/M), immature neutrophils (ImN), and mature neutrophils (MaN) (18). Percentages of these three populations within the total Gr-1+ CD11b+ cells are shown on the right. Data are means ± SEMs. (B) Dot plots showing three populations of bone marrow Gr-1+ CD11b+ cells at 4 h after thioglycolate challenge. (C) Expression of CXCR2 on bone marrow neutrophils from naïve or challenged mice. ISO, neutrophils immunostained with an isotype control. (D) CXCR2 expression on naïve bone marrow and challenged bone marrow (4 h BM) neutrophils as well as on challenged peritoneal neutrophils (4 h PEC).
KI bone marrow neutrophils exhibit abnormal signaling and enhanced CXCR2 desensitization.
To gain insights into whether chemoattractant receptor signaling was perturbed in these mice, we tested the responses of bone marrow-derived neutrophils to MIP-2. Even prior to challenge, the KI neutrophils had an elevated intracellular calcium level compared to WT cells (Fig. 4A, left). When stimulated, intracellular calcium levels of KI neutrophils peaked in the same range as those of WT neutrophils but remained elevated considerably longer and returned to baseline more slowly (Fig. 4A, right, and B). Upon rechallenge, the WT neutrophils had a dampened secondary response consistent with partial receptor desensitization (35), while the KI neutrophils had almost no secondary response, indicating exaggerated receptor desensitization (Fig. 4B). Next, we examined the intracellular calcium responses of bone marrow-derived neutrophils to CXCL12. The exposure of WT and KI neutrophils to different concentrations of CXCL12 elicited similar peak intracellular calcium levels, much like exposure to MIP-2 had (Fig. 4C). Also, as did MIP-2, CXCL12 exposure triggered a more prolonged rise in intracellular calcium (Fig. 4D). However, in contrast to MIP-2, a secondary challenge with CXCL12 elicited an intracellular calcium signal of similar magnitude in the WT and KI cells (Fig. 4D). These data suggest that the loss of the Gαi2-RGS protein interactions had revealed an intrinsic difference between CXCR4 and CXCR2 desensitization.
Fig 4.

KI neutrophils exhibit both hypersensitivity and hyporesponsiveness. (A) WT and KI neutrophil basal fluorescence from 5 matched pairs of mice is shown (left; mean ± SE). Results were analyzed by paired t test. Peak fluorescence values minus basal fluorescence from WT and KI mice (right). Results are from duplicate determinations from one WT versus one KI neutrophil preparation and representative of four separate experiments. (B) The intracellular calcium response of naïve bone marrow neutrophils was monitored after stimulation with MIP-2 and then following restimulation 15 min later. Results are from duplicate determinations from one experiment and representative of three performed. (C) Peak intracellular calcium response minus the basal fluorescence of purified WT and KI bone marrow neutrophils in response to various amounts of CXCL12. Results are from duplicate determinations and representative of one of two experiments performed. (D) The intracellular calcium response of naïve bone marrow neutrophils was monitored after stimulation with CXCL12 and then following restimulation 15 min later. Results are from duplicate determinations and representative of one of two separate experiments. (E) Axial ratios of WT and KI neutrophils. The ratios were calculated by dividing the length of the long axis by that of the perpendicular axis passing through the cell nucleus of the immunostained cells. (F) Confocal images of Gαi2-immnostained bone marrow neutrophils. (Insets) Magnifications of ×2. Bar, 10 μm. (G) FSC plot of freshly isolated naïve WT and KI bone marrow neutrophils (top) and similar cells incubated in the presence of MIP-2 (20 ng/ml) for 15 min (bottom). The result is representative of five experiments. (H) Random migration of naïve bone marrow Gr-1+ CD11b+ cells in Transwell chambers. (I) In vitro migration assay with naïve bone marrow Gr-1+ CD11b+ cells. Various concentrations of MIP-2 (left) or CXCL12 (right) were used to induce cell migration. Three independent assays were performed in triplicate. Data are means ± SEMs. Paired t test.
KI bone marrow neutrophils exhibit signs of basal activation and a poor in vitro chemotactic response to a CXCR2 ligand.
Since we had noted an increase in basal intracellular calcium, we examined whether the KI neutrophils exhibited other signs of constitutive in vivo activation. A sensitive indicator of chemoattractant signaling is neutrophil polarization. While the majority of freshly isolated WT neutrophils were not polarized, many KI neutrophils had a polarized morphology. This was accompanied by an increase of Gαi2 at the leading edge of the polarized cells (Fig. 4E and F). Certain chemokines are known to trigger a robust concentration-dependent neutrophil shape change, which can be detected by an increase in forward light scatter (FSC) during flow cytometry. Pretreatment with a CXCR2 antagonist abolishes the shape change (26). Consistent with an in vivo activated phenotype, freshly isolated KI neutrophils had increased FSC compared to WT neutrophils (Fig. 4G, top), and while WT neutrophils underwent the usual MIP-2-induced shape change, the KI neutrophil had a much more dampened response (Fig. 4G, bottom). Finally, we noted an increased percentage of KI neutrophils that migrated spontaneously in the absence of chemokine (Fig. 4H). Despite or perhaps as a consequence of their preactivation, the KI neutrophils had a specific migratory defect which was more severe in response to MIP-2 than CXCL12. The higher concentrations of MIP-2 elicited a very poor KI neutrophil response; however, at lower concentrations, the KI cells migrated as well as or even better than WT cells. The KI neutrophil also had a subpar response at the higher CXCL12 concentrations (Fig. 4I, right).
GRK2 levels are elevated in neutrophils from KI thioglycolate-challenged mice.
GPCRs undergo receptor desensitization predominantly via a GPCR kinase (GRK)-dependent mechanism (33). In neutrophils, MIP-2 enhances the transcription of GRK2 and GRK5 in a phosphoinositide 3-kinase-γ-dependent manner (16). This promotes CXCR2 internalization, likely via β-arrestin recruitment. β-Arrestin-2-deficient neutrophils show decreased CXCR2 internalization and enhanced recruitment to inflammation sites (41). Stimulation of neutrophils with Toll-like receptor (TLR) ligands such as lipoteichoic acid and lipopolysaccharide is also known to downregulate CXCR2 via increased expression of GRK2 (2, 3). To determine whether enhanced GRK activity might contribute to the augmented CXCR2 downregulation in KI mice, we immunostained challenged bone marrow and peritoneal neutrophils for GRK2. We found high levels of GRK2 expression in ∼15% of the KI bone marrow neutrophils (likely the mature neutrophil population) and modestly increased GRK2 in many KI cells compared to WT cells (Fig. 5A and B). In the peritoneum, most KI neutrophils showed considerably higher GRK2 expression than WT cells (Fig. 5C and D). In contrast, prior to challenge we found no statistical difference in GRK2 expression between wild-type and KI bone marrow neutrophils (H. Cho, unpublished observation). Together, this suggests that during the thioglycolate challenge the increased sensitivity to CXCR2 ligands led to increased GRK2 expression.
Fig 5.

GRK2 expression is increased in thioglycolate-challenged KI neutrophils. Bone marrow (A) and peritoneal (C) cells were harvested from thioglycolate-challenged mice at 4 h postinjection. Neutrophils were fixed, permeabilized, and immunostained with rabbit anti-GRK2 and secondary Alexa 488-conjugated anti-rabbit antibodies. DNAs were stained with Hoechst 33324. (B and D) Mean fluorescence intensities of cells were obtained using Imaris software and plotted with Prism software. BM, bone marrow. (E) WT cells stained only with secondary antibody (2ndary only). Bars, 10 μm.
To assess which RGS proteins are likely to contribute to the observed phenotype, we examined the mRNA expression of RGS proteins in neutrophils and several other hematopoietic cell types. We queried the immunological genome microarray database (http://www.Immgen.org/databrowser/index.html) as well as performed RT-PCR on RNA extracted from bone marrow-derived neutrophils (see Fig. S2 in the supplemental material). We also examined the mRNA expression of several other neutrophil-relevant proteins. As expected, neutrophils highly expressed Gαi2, CXCR2, and β-arrestin-2. Of the 20 canonical RGS proteins, neutrophils expressed abundant mRNAs for RGS2, RGS14, RGS18, and RGS19 and smaller amounts for RGS1 and RGS10.
The defects observed with KI mice are neutrophil intrinsic.
To be certain that the phenotype that we had observed in KI mice was intrinsic to neutrophils and not due to defective stromal or endothelial cell function, chimeric mice were generated. Bone marrow cells isolated from either CD45.2 wild-type or CD45.2 KI mice were transferred into lethally irradiated wild-type CD45.1 mice. By 4 weeks after the adoptive transfer, greater than 99% of neutrophils were CD45.2 positive (data not shown). Thioglycolate challenge resulted in a similar impairment of neutrophil recruitment into the peritoneal cavity in mice into which KI cells had been transferred, as we had previously observed (Fig. 6A). Neutrophil mobilization into blood upon thioglycolate challenge was also defective in the mice into which KI cells had been transferred. Mice into which naïve KI cells had been transferred also showed higher percentages of total bone marrow neutrophils as well as mature neutrophils (Fig. 6B to D). Most of the mature KI neutrophils also remained in the bone marrow after the challenge. The surface expression of CXCR2, but not that of CXCR4, was also similarly reduced in the transferred KI neutrophils after the thioglycolate challenge (Fig. 6E). The shape change was also observed with adoptively transferred naïve KI neutrophils (data not shown). Together, these data indicate that the phenotype observed with the KI mice is neutrophil intrinsic.
Fig 6.

The phenotype associated with KI mice is neutrophil intrinsic. CD45.1 WT mice were irradiated and received WT CD45.2 or KI CD45.2 bone marrow cells. Data shown are from two independent adoptive transfers. (A) Total cell and neutrophil counts of peritoneal exudates from thioglycolate-challenged CD45.1/CD45.2 chimeras at 4 h postchallenge. Data are means ± SEMs. PECs, peritoneal cells. (B) Blood neutrophil counts of chimeric thioglycolate-challenged mice. Data are means ± SEMs. (C) Percentages of Gr-1+ CD11b+ bone marrow cells from naïve chimeric mice are shown. Data are means ± SEMs. (D) Dot plots and percentages of three populations, promyelocytes/myelocytes (P/M), immature neutrophils (ImN), and mature neutrophils (MaN), within the total Gr-1+ CD11b+ bone marrow cells from naïve and challenged chimeric mice. Data are means ± SEMs. (E) Expression of CXCR2 and CXCR4 on bone marrow and blood neutrophils from naïve and challenged mice to which cells were adoptively transferred. ISO, neutrophils immunostained with isotype controls.
Neutrophils from KI mice are poorly recruited and fail to stabilize sites of sterile inflammation.
To visualize the behavior of KI neutrophils in vivo, we used two approaches. First, we transferred differentially labeled wild-type and KI neutrophils into the ear of a wild-type mouse, induced laser damage, and examined the subsequent behavior of the neutrophils by intravital multiphoton imaging. Sterile inflammation triggers an initial scouting phase, whereby a few neutrophils investigate the area of damage (31). This is followed by amplification phase, where numerous neutrophils are recruited, and a stabilization phase, in which neutrophils persistently remain at the site of the inflammatory insult (31). Within 15 to 20 min of damage, occasional wild-type and KI neutrophils approached the site of damage (Fig. 7A; see Video S1 in the supplemental material). The KI neutrophils did not show any obvious defect in the initial scouting phase; however, during the amplification phase, fewer KI neutrophils accumulated at the site of photodamage. This was in part due to a failure of the KI neutrophils to remain localized at the site. KI neutrophils often wandered away from the site of injury, while the wild-type neutrophils remained focused. This can be readily appreciated at the last time point (Fig. 7A) and in Video S2 in the supplemental material. As a consequence, the KI neutrophils failed to fill and stabilize the injury site. Of note, since we did not use albino mice, melanocyte damage caused additional sites of inflammation, as noted in the second panel of Fig. 7A. As a second approach, we crossed the KI mice with LysM-EGFP mice (17) and obtained double heterozygotes for analysis. Gαi2G184S/WT mice possess many of the abnormalities observed with the homozygotic mice, although they are usually less severe (21). Following laser damage, we imaged neutrophils in the ears of LysM-EGFP and LysM-EGFP Gαi2G184S/WT mice (Fig. 7B; see Videos S3 and S4 in the supplemental material). The Gαi2G184S/WT mice also failed to fill and stabilize the injury site (Fig. 7B). With additional breeding, we obtained Gαi2G184S/G184S KI mice that also had a LysM-EGFP allele. These mice exhibited an even more severe phenotype than the heterozygote, with a dramatic paucity of neutrophils at the site of sterile inflammation (Fig. 7C; see Video S5 in the supplemental material). In addition, those neutrophils that approached the site of injury often wandered away again, failing to fill and stabilize the injury site. A comparison of the neutrophil motility of the wild-type and KI neutrophils in the ears of wild-type mice revealed that the KI neutrophils moved faster and with greater displacement than the WT neutrophils. This was the case whether we analyzed the behavior of the neutrophils close to (Fig. 7D) or remote from (data not shown) the site of photo damage. We also quantitated the number of neutrophils in a small volume surrounding sites of laser damage. As is apparent from the imaging, fewer KI cells accumulated around the sites of laser damage in the ear transfer model (Fig. 7E, top). Also, fewer neutrophils in the heterozygote and KI LysM-EGFP mice than neutrophils in the LysM-EGFP WT mice entered into the injury sites (Fig. 7E, bottom). Inspection of the imaging data revealed that fewer LysM-EGFP KI neutrophils than WT neutrophils accumulated in the adjacent blood vessels, a result consistent with a failure to mobilize cells from the bone marrow. In addition, many of the KI cells that adhered to the endothelium failed to transmigrate. Only 19% of the KI cells that remained adherent for 1 min crossed the blood vessel wall, while 46% of the WT cells did so (Fig. 7F). Next, we checked the adherence of WT and KI neutrophils to ICAM-1- and MIP-2-coated plates and to an SVEC4-10 endothelial cell monolayer that had been treated with tumor necrosis factor alpha. In both instances, similar numbers of WT and KI cells adhered (Fig. 7G). We also checked the motility of the WT and KI neutrophils on the ICAM-1- and MIP-2-coated plates. We found that the KI neutrophils moved faster than the WT neutrophils (see Fig. S3 in the supplemental material), a result similar to what we had observed with intravital imaging.
Fig 7.

Intravital imaging reveals aberrant trafficking of KI neutrophils around sites of sterile inflammation. (A) Images of WT and KI neutrophils in the ear of a WT mouse. Differentially labeled WT and KI bone marrow neutrophils were transferred into the ear of an anesthetized WT mouse. Following laser damage (white arrow), the cells were imaged by TP-LSM. Individual panels from the imaging are shown. Yellow arrows, a second inflammatory site that resulted from inadvertent laser damage. The left four panels show WT (green) and KI (red) cells immediately after injury and at 30, 45, and 75 min after injury. The right four panels show either WT or KI cells at the 45- and 75-min time points. Collagen is blue. (B) Images of neutrophils in the ears of LysM-EGFP WT and Gαi2G184S/WT mice. Individual images from TP-LSM of the indicated mice. Sites of laser damage are indicated with white arrows. Blood vessels (b.v.) are indicated, and collagen is blue. (C) Images of neutrophils in the ears of LysM-EGFP WT and KI mice. Individual images are shown. Sites of laser damage are indicated with white arrows. Blood vessels were outlined by fluorescent Qdots. Collagen is blue. (D) Analysis of WT and KI neutrophil migration in the ears of WT mice. Individual WT and KI neutrophils were tracked using the tracking function in Imaris software. (E) Failure of KI neutrophils to stabilize injury site. The accumulation of transferred WT and KI neutrophils (top) and endogenous WT, Gαi2G184S/WT (heterozygote [Het]), and KI neutrophils (bottom) to enter a space 150 mm by 150 mm by 10 mm centered on the site of laser damage. Each data point in the top panel is the mean ± SEM of triplicates. The accumulation of endogenous WT, heterozygous, and KI LysM-EGFP neutrophils are from one experiment of two performed. (F) Analysis of LysM-EGFP WT and KI transmigration. Shown are the percentages of WT and KI neutrophils that transmigrated among those cells that engaged the blood vessel wall (minimum, 1 min). The number of cells analyzed is indicated (two experiments). (G) Ratio of WT/KI neutrophils adherent in vitro. Equal numbers of WT and KI neutrophils were added to MIP-2- and ICAM-1-coated plates or an SVEC4-10 cell monolayer previously treated with TNF-α. Following washing, the ratio between WT and KI cells bound was determined by counting bound cells within a defined area. Each point is from an individual experiment.
KI mice exhibit increased susceptibility to Staphylococcus aureus infection.
Finally, to examine the role of RGS proteins in host response against infection, we infected 2- to 3-month-old mice with a sublethal dose of methicillin-resistant Staphylococcus aureus (MRSA) and monitored survival for 14 days. S. aureus is a major human pathogen that can cause life-threatening endocarditis and sepsis. Implying a crucial protective role of neutrophils, people with neutropenia or functionally defective neutrophils are more susceptible to S. aureus infection (19, 43). Among different inbred strains of mice tested, strain C57BL/6 is reported to be one of the most resistant to S. aureus infection. However, C57BL/6 mice become completely susceptible to S. aureus when neutrophils are depleted or neutrophil recruitment is blocked. Furthermore, the resistance is shown not to be dependent on T or B cells, indicating that neutrophils are essential (36, 44). Since the KI mice had lower body weights than wild-type mice of similar age, the sizes of the inocula were normalized on the basis of body weight (1.82 × 106 CFU/g of body weight). As expected, the WT mice survived, while 60% of the KI mice died within 2 to 3 days (Fig. 8). In contrast, the Gαi2G184S/WT mice survived similarly to WT mice (data not shown). These results suggest that RGS protein-mediated regulation of Gαi2 is an important factor contributing to the host response against MRSA infection. Interestingly, not neutrophil function but a delay in neutrophil recruitment to the site of infection appears to underlie the increased susceptibility to MRSA noted in certain inbred strains of mice (44). The increased mortality of the KI mice observed at 2 to 3 days after MRSA infection is probably caused by sepsis due to defective neutrophil recruitment. It has been previously shown that dramatic downregulation of CXCR2 expression via activation of TLR2 or TLR4 during severe sepsis accounts for impaired neutrophil migration and increased mortality (2, 3).
Fig 8.
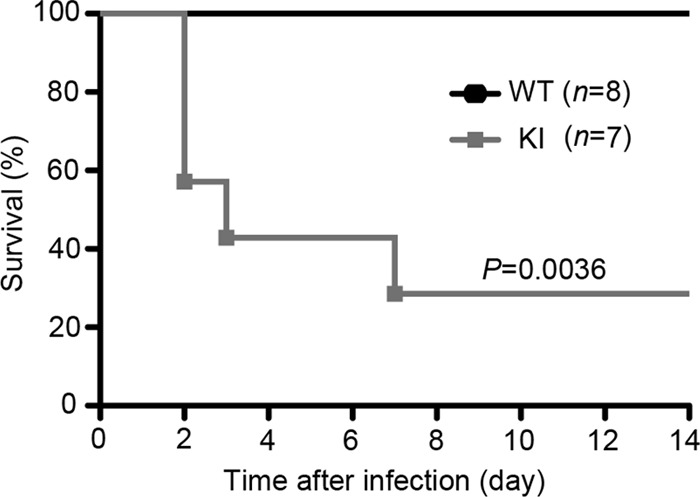
KI mice are more susceptible to Staphylococcus aureus infection. Two- to 3-month-old mice were intravenously infected with a clinical strain of methicillin-resistant Staphylococcus aureus (1.82 × 106 CFU/g). The underweight KI mice often received smaller inocula since the size of the inoculum was normalized on the basis of the body weight of each individual mouse. Survival rates were monitored for 14 days.
DISCUSSION
RGS domain-containing proteins appeared early in eukaryote evolution, often expanding in conjunction with lineage-specific expansion of Gα subunits, while the visual/β-arrestin subfamily branched more recently from the founding α-arrestins (4). In fungal cells, a GPCR-heterotrimeric GTPase-RGS switch mediates fungal mating interactions in the absence of β-arrestins (1, 14). The evolutionary expansion of RGS proteins has served as a primary mechanism to regulate the sensing of environmental signals by GPCRs (4), while the β-arrestin system evolved from the receptor trafficking system in the context of functional RGS proteins. Besides its role in GPCR desensitization, the β-arrestin system has conveyed upon GPCRs an alternative mechanism of activating downstream signaling (37). Our study shows that in neutrophils, the failure of RGS proteins to regulate Gαi2 GTPase activity can result in inappropriate engagement of receptor desensitization mechanisms with severe functional consequences.
Two antagonistic chemokine receptors, CXCR2 and CXCR4, both coupled to Gαi, regulate neutrophil release from the bone marrow (15). Cxcl12 expressed by bone marrow stromal cells provides a key retention signal via CXCR4, whereas CXCR2-binding chemokines promote neutrophil release. Patients with WHIM syndrome offer a compelling example of the consequences of disturbing this balance. The usual cause of the syndrome is a 10- to 19-amino-acid C-terminal truncation of CXCR4 (22). This impairs ligand-induced CXCR4 downregulation and receptor desensitization, thereby altering the equilibrium toward bone marrow neutrophil retention. When neutrophils lack both CXCR2 and CXCR4 (15), neutrophils mobilize constitutively from the bone marrow, suggesting that CXCR4 dominates over CXCR2 under basal conditions. As homeostatic KI neutrophils expressed normal levels of CXCR4 and CXCR2, overzealous CXCR4 signaling likely accounts for the constitutive bone marrow neutrophil retention observed in the KI mice. Normally during acute inflammation, CXCR2-binding ligands increase in the blood, helping to mobilize normal bone marrow neutrophils (34). Although they are neutropenic, WHIM patients readily mobilize their neutrophils following an inflammatory challenge. In contrast, even following a potent inflammatory challenge, the KI mice poorly mobilized their neutrophils. This suggested that the KI neutrophil phenotype would not be explained solely on the basis of increased CXCR4-mediated retention.
The bone marrow neutrophil retention in the KI mice was also surprising, as the opposite phenotype might have been expected because chemoattractant-triggered adhesion is quantitatively less sensitive to RGS proteins than chemotaxis (7). The striking reduction in CXCR2 membrane levels following an inflammatory challenge likely explains the phenotype that we observed. An obvious question is why the loss of RGS protein function so severely impacted CXCR2. Perhaps it is because CXCR2 functions as an intermediary chemoattractant receptor. Neutrophils must sort multiple competing cues to negotiate a complex environment such as an infected or inflamed tissue. To do so, they use a hierarchical response to chemoattractants, overriding initial recruitment signals such as those provided by CXCR2 to migrate toward end-target chemoattractants, such as formyl peptide (N-formyl-Met-Leu-Phe) and C5a (20). Helping the switch of allegiance, CXCR2 desensitizes, undergoing both heterologous desensitization to end-target recruitment signals and GRK-mediated homologous desensitization (30, 35). Our data indicate that the normal CXCR2 desensitization pathways have become hyperfunctional in the KI neutrophils, yet the biochemical mechanism that underlies the greater propensity of CXCR2 to desensitize remains unknown. Based on the rapid induction of GRK2 in the KI neutrophils, we are investigating whether GRK2 contributes. Further study of CXCR2 and CXCR4 phosphorylation, β-arrestin recruitment, and receptor recycling will be needed to understand how these receptors desensitize in neutrophils.
The loss of RGS protein function for Gαi2 in the KI neutrophils should lower the threshold of ligand needed to activate Gαi2-dependent signaling pathways (39). Inappropriate cellular responses to low levels of ligand that might otherwise be ignored likely occur. Our data indicate that this may be the case with the KI neutrophils. Compared to WT neutrophils, they had a higher basal intracellular calcium level, increased random migration, a tendency to spontaneously polarize, and constitutive shape changes normally associated with early responses to chemoattractants. However, we did not see a significant change in the sensitivity of the KI neutrophils in chemotaxis assays. Rather, we observed a flattening of the dose-response curve at higher ligand concentrations with a reduction in the magnitude of the peak response, which was much more striking in the response to MIP-2 than in that to CXCL12. This is not unique to neutrophils, as lymphocytes from the same KI mice exhibited similarly shaped dose-response curves in chemotaxis assays using other chemokines (I.-Y. Hwang et al., unpublished data), but this does contrast to what we have observed with lymphocytes and dendritic cells from mice lacking a single RGS protein, where we have found an enhanced migration at each tested chemokine concentration (12, 29). A likely explanation is that in the absence of all RGS protein function for Gαi2 in the KI leukocytes, there is premature and/or enhanced receptor desensitization during the course of the chemotaxis assay. This would result in a decrease in the responding cells. The more pronounced impairment of the MIP-2- versus CXCL12-mediated chemotaxis might also be explained by a greater propensity of CXCR2 to desensitize.
By directly injecting neutrophils into the ear, a side-by-side comparison of the WT and KI cells could be made without the necessity of bone marrow egress and transmigration. While the initial scouting of inflammatory sites depends upon Gαi signaling (31), we did not observe an obvious defect in this phase of the neutrophil response to a sterile inflammatory insult. However, the early KI neutrophils and many of those that arrived during the amplification stage failed to remain at the inflammatory site, often wandering away and sometimes returning only to leave again. As a consequence, the WT neutrophils greatly outnumbered the KI neutrophils at the later stages of the neutrophil response. This was also the case with the Gαi2G184S/WT and Gαi2G184S/G184S LysM-EGFP mice, as their neutrophils also exhibited poor accumulation at sites of laser damage. While lower blood neutrophil counts and poorer transmigration also contributed to the decreased neutrophil accumulation, the KI LysM-EGFP neutrophils exhibited a similar failure to focus at the site of inflammation. Thus, RGS protein regulation of Gαi2 not only is needed for the recruitment of neutrophils to inflammatory sites but also is needed for their proper retention. Since the adhesion of KI neutrophils to an endothelial monolayer or to a plate coated with ICAM-1 plus MIP-2 was not impaired, an inability to trigger integrins is unlikely to account for the failure of the cells to be retained at the site of injury.
The Gnai2−/− mice and the Gαi2G184S/G184S KI mice exhibit complex and partially overlapping phenotypes, illustrating the importance of not only the signal transducer but also its major regulator. Together our results indicate that RGS proteins in neutrophils establish a threshold for Gαi activation and assist in coordinating desensitization mechanisms for some chemoattractant receptors such as CXCR2. The loss of function of RGS18 and/or RGS19, the two most prominently expressed neutrophil RGS proteins, may account for the KI neutrophil phenotype, since Rgs2- and Rgs14-deficient neutrophils mobilized appropriately to inflamed peritoneum (H. Cho, unpublished data). RGS18 and RGS19 are also reasonable candidates to test in human myelokathexis syndromes of unknown etiology.
Supplementary Material
ACKNOWLEDGMENTS
We thank Mary Rust for her editorial assistance, Derek Gritz for help with some of the data analysis, L. Aravind (NCBI, NLM, NIH) for helpful discussion, and Anthony Fauci (NIAID, NIH) for his continued support.
This research was supported by the intramural research program of the National Institute of Allergy and Infectious Diseases.
Hyeseon Cho designed and performed the majority of the functional studies and helped write the manuscript, Olena Kamenyeva and Chung Park performed intravital microscopy, Olena Kamenyeva performed the adhesion assays, Sunny Yung performed the Staphylococcus infection studies, Ji-Liang Gao and Il-Young Hwang helped with the neutrophil functional studies, Philip M. Murphy provided advice for the study design, Richard R. Neubig provided the G184S KI mice and suggestions for the study, and John H. Kehrl oversaw all aspects of the study.
We have no conflicts of interest to declare.
Footnotes
Published ahead of print 10 September 2012
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1. Alvarez CE. 2008. On the origins of arrestin and rhodopsin. BMC Evol. Biol. 8:222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alves-Filho JC, et al. 2010. Interleukin-33 attenuates sepsis by enhancing neutrophil influx to the site of infection. Nat. Med. 16:708–712 [DOI] [PubMed] [Google Scholar]
- 3. Alves-Filho JC, et al. 2009. Regulation of chemokine receptor by Toll-like receptor 2 is critical to neutrophil migration and resistance to polymicrobial sepsis. Proc. Natl. Acad. Sci. U. S. A. 106:4018–4023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Anantharaman V, Abhiman S, de Souza RF, Aravind L. 2011. Comparative genomics uncovers novel structural and functional features of the heterotrimeric GTPase signaling system. Gene 475:63–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baugher PJ, Richmond A. 2008. The carboxyl-terminal PDZ ligand motif of chemokine receptor CXCR2 modulates post-endocytic sorting and cellular chemotaxis. J. Biol. Chem. 283:30868–30878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bizzarri C, et al. 2006. ELR+ CXC chemokines and their receptors (CXC chemokine receptor 1 and CXC chemokine receptor 2) as new therapeutic targets. Pharmacol. Ther. 112:139–149 [DOI] [PubMed] [Google Scholar]
- 7. Bowman EP, et al. 1998. Regulation of chemotactic and proadhesive responses to chemoattractant receptors by RGS (regulator of G-protein signaling) family members. J. Biol. Chem. 273:28040–28048 [DOI] [PubMed] [Google Scholar]
- 8. Brinkmann V, et al. 2004. Neutrophil extracellular traps kill bacteria. Science 303:1532–1535 [DOI] [PubMed] [Google Scholar]
- 9. Busillo JM, et al. 2010. Site-specific phosphorylation of CXCR4 is dynamically regulated by multiple kinases and results in differential modulation of CXCR4 signaling. J. Biol. Chem. 285:7805–7817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Byers MA, et al. 2008. Arrestin 3 mediates endocytosis of CCR7 following ligation of CCL19 but not CCL21. J. Immunol. 181:4723–4732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cacalano G, et al. 1994. Neutrophil and B cell expansion in mice that lack the murine IL-8 receptor homolog. Science 265:682–684 [DOI] [PubMed] [Google Scholar]
- 12. Cho H, Kehrl JH. 2009. Regulation of immune function by G protein-coupled receptors, trimeric G proteins, and RGS proteins. Prog. Mol. Biol. Transl. Sci. 86:249–298 [DOI] [PubMed] [Google Scholar]
- 13. Chou RC, et al. 2010. Lipid-cytokine-chemokine cascade drives neutrophil recruitment in a murine model of inflammatory arthritis. Immunity 33:266–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dohlman HG. 2009. RGS proteins the early days. Prog. Mol. Biol. Transl. Sci. 86:1–14 [DOI] [PubMed] [Google Scholar]
- 15. Eash KJ, Greenbaum AM, Gopalan PK, Link DC. 2010. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J. Clin. Invest. 120:2423–2431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fan J, Malik AB. 2003. Toll-like receptor-4 (TLR4) signaling augments chemokine-induced neutrophil migration by modulating cell surface expression of chemokine receptors. Nat. Med. 9:315–321 [DOI] [PubMed] [Google Scholar]
- 17. Faust N, Varas F, Kelly LM, Heck S, Graf T. 2000. Insertion of enhanced green fluorescent protein into the lysozyme gene creates mice with green fluorescent granulocytes and macrophages. Blood 96:719–726 [PubMed] [Google Scholar]
- 18. Fiedler K, et al. 2011. Neutrophil development and function critically depend on Bruton tyrosine kinase in a mouse model of X-linked agammaglobulinemia. Blood 117:1329–1339 [DOI] [PubMed] [Google Scholar]
- 19. Gresham HD, et al. 2000. Survival of Staphylococcus aureus inside neutrophils contributes to infection. J. Immunol. 164:3713–3722 [DOI] [PubMed] [Google Scholar]
- 20. Heit B, Tavener S, Raharjo E, Kubes P. 2002. An intracellular signaling hierarchy determines direction of migration in opposing chemotactic gradients. J. Cell Biol. 159:91–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huang X, et al. 2006. Pleiotropic phenotype of a genomic knock-in of an RGS-insensitive G184S Gnai2 allele. Mol. Cell. Biol. 26:6870–6879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kawai T, Malech HL. 2009. WHIM syndrome: congenital immune deficiency disease. Curr. Opin. Hematol. 16:20–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kessenbrock K, et al. 2009. Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 15:623–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lionakis MS, Lim JK, Lee CC, Murphy PM. 2011. Organ-specific innate immune responses in a mouse model of invasive candidiasis. J. Innate Immun. 3:180–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Marcos V, et al. 2010. CXCR2 mediates NADPH oxidase-independent neutrophil extracellular trap formation in cystic fibrosis airway inflammation. Nat. Med. 16:1018–1023 [DOI] [PubMed] [Google Scholar]
- 26. Martin C, et al. 2003. Chemokines acting via CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity 19:583–593 [DOI] [PubMed] [Google Scholar]
- 27. McCormick PJ, Segarra M, Gasperini P, Gulino AV, Tosato G. 2009. Impaired recruitment of Grk6 and beta-arrestin 2 causes delayed internalization and desensitization of a WHIM syndrome-associated CXCR4 mutant receptor. PLoS One 4:e8102 doi:10.1371/journal.pone.0008102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McDonald B, et al. 2010. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 330:362–366 [DOI] [PubMed] [Google Scholar]
- 29. Moratz C, Hayman JR, Gu H, Kehrl JH. 2004. Abnormal B-cell responses to chemokines, disturbed plasma cell localization, and distorted immune tissue architecture in Rgs1−/− mice. Mol. Cell. Biol. 24:5767–5775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nasser MW, Marjoram RJ, Brown SL, Richardson RM. 2005. Cross-desensitization among CXCR1, CXCR2, and CCR5: role of protein kinase C-epsilon. J. Immunol. 174:6927–6933 [DOI] [PubMed] [Google Scholar]
- 31. Ng LG, et al. 2011. Visualizing the neutrophil response to sterile tissue injury in mouse dermis reveals a three-phase cascade of events. J. Investig. Dermatol. 131:2058–2068 [DOI] [PubMed] [Google Scholar]
- 32. Pero RS, et al. 2007. Galphai2-mediated signaling events in the endothelium are involved in controlling leukocyte extravasation. Proc. Natl. Acad. Sci. U. S. A. 104:4371–4376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Premont RT, Gainetdinov RR. 2007. Physiological roles of G protein-coupled receptor kinases and arrestins. Annu. Rev. Physiol. 69:511–534 [DOI] [PubMed] [Google Scholar]
- 34. Rankin SM. 2010. The bone marrow: a site of neutrophil clearance. J. Leukoc. Biol. 88:241–251 [DOI] [PubMed] [Google Scholar]
- 35. Richardson RM, Pridgen BC, Haribabu B, Ali H, Snyderman R. 1998. Differential cross-regulation of the human chemokine receptors CXCR1 and CXCR2. Evidence for time-dependent signal generation. J. Biol. Chem. 273:23830–23836 [DOI] [PubMed] [Google Scholar]
- 36. Schmaler M, Jann NJ, Ferracin F, Landmann R. 2011. T and B cells are not required for clearing Staphylococcus aureus in systemic infection despite a strong TLR2-MyD88-dependent T cell activation. J. Immunol. 186:443–452 [DOI] [PubMed] [Google Scholar]
- 37. Shenoy SK, Lefkowitz RJ. 2011. beta-Arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol. Sci. 32:521–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shi GX, Harrison K, Han SB, Moratz C, Kehrl JH. 2004. Toll-like receptor signaling alters the expression of regulator of G protein signaling proteins in dendritic cells: implications for G protein-coupled receptor signaling. J. Immunol. 172:5175–5184 [DOI] [PubMed] [Google Scholar]
- 39. Signarvic RS, et al. 2010. RGS/Gi2alpha interactions modulate platelet accumulation and thrombus formation at sites of vascular injury. Blood 116:6092–6100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sinha RK, Park C, Hwang IY, Davis MD, Kehrl JH. 2009. B lymphocytes exit lymph nodes through cortical lymphatic sinusoids by a mechanism independent of sphingosine-1-phosphate-mediated chemotaxis. Immunity 30:434–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Su Y, et al. 2005. Altered CXCR2 signaling in beta-arrestin-2-deficient mouse models. J. Immunol. 175:5396–5402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Thelen M, Dewald B, Baggiolini M. 1993. Neutrophil signal transduction and activation of the respiratory burst. Physiol. Rev. 73:797–821 [DOI] [PubMed] [Google Scholar]
- 43. Verdrengh M, Tarkowski A. 1997. Role of neutrophils in experimental septicemia and septic arthritis induced by Staphylococcus aureus. Infect. Immun. 65:2517–2521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. von Kockritz-Blickwede M, et al. 2008. Immunological mechanisms underlying the genetic predisposition to severe Staphylococcus aureus infection in the mouse model. Am. J. Pathol. 173:1657–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zarbock A, Deem TL, Burcin TL, Ley K. 2007. Galphai2 is required for chemokine-induced neutrophil arrest. Blood 110:3773–3779 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.