

Abstract
Hepatitis C virus (HCV) infection remains a serious public health problem worldwide. Treatments are limited, and no preventive vaccine is available. Toward developing an HCV vaccine, we engineered two recombinant measles viruses (MVs) expressing structural proteins from the prototypic HCV subtype 1a strain H77. One virus directs the synthesis of the HCV capsid (C) protein and envelope glycoproteins (E1 and E2), which fold properly and form a heterodimer. The other virus expresses the E1 and E2 glycoproteins separately, with each one fused to the cytoplasmic tail of the MV fusion protein. Although these hybrid glycoproteins were transported to the plasma membrane, they were not incorporated into MV particles. Immunization of MV-susceptible, genetically modified mice with either vector induced neutralizing antibodies to MV and HCV. A boost with soluble E2 protein enhanced titers of neutralizing antibody against the homologous HCV envelope. In animals primed with MV expressing properly folded HCV C-E1-E2, boosting also induced cross-neutralizating antibodies against two heterologous HCV strains. These results show that recombinant MVs retain the ability to induce MV-specific humoral immunity while also eliciting HCV neutralizing antibodies, and that anti-HCV immunity can be boosted with a single dose of purified E2 protein. The use of MV vectors could have advantages for pediatric HCV vaccination.
INTRODUCTION
Hepatitis C virus (HCV) is the prototype member of the Hepacivirus genus within the family Flaviviridae. The virus is transmitted almost exclusively by the parenteral route, and acute infections, which are frequently subclinical, progress to chronicity in about 70% of cases. Persistent HCV carriers may develop liver cirrhosis, hepatocellular carcinoma, and end-stage liver disease. Despite an efficient preventive campaign based on the identification of HCV-infected blood donors, the prevalence of the virus among American young adults has not declined (35). Recent advances have improved treatment options for infections with certain viral genotypes, although limitations in efficacy remain and a preventive vaccine is not available.
HCV is an enveloped virus, with a positive-sense, single-stranded RNA genome. The genome is translated into a polyprotein which is proteolytically processed into 10 individual proteins (reviewed in reference 64). The structural proteins, core protein (C) and two envelope glycoproteins (E1 and E2), form the physical viral particle; C functions to encapsidate the RNA genome, while E1 and E2 mediate virus attachment and entry into host cells. E1 and E2 are highly glycosylated type I transmembrane proteins with an N-terminal ectodomain. Residues within the transmembrane domains are important for heterodimerization and dimer retention in the endoplasmic reticulum (ER) (22, 46). Replacement of the E1 or E2 transmembrane domains can direct transport of the corresponding chimeric proteins to the plasma membrane (1, 10). E2 also encompasses well-conserved antibody neutralization determinants, which are located near the binding sites for viral entry factors CD81 and scavenger receptor B1 (20, 29). There is also some evidence for the existence of neutralizing determinants in E1 (45). Patient neutralizing antibodies have been identified that target virus interactions with its coreceptors and block glycoprotein-mediated membrane fusion (31).
A major hurdle in the development of an effective HCV vaccine is the lack of an immunization strategy to elicit broadly protective antibodies and sustained cell-mediated immunity (32, 66). Studies with chimpanzees have shown the importance of total anti-E1/E2 antibody titers in conferring protection (15). Moreover, neutralizing immunity and functional CD4+ and CD8+ T-cell responses induced early in HCV infection correlate with clearance or viral control in patients (7, 37, 51).
In contrast, the measles virus (MV) vaccine has an outstanding record of efficacy and safety. The MV Moraten vaccine strain is credited with the temporary elimination of indigenous measles transmission on the American continent (14), and the World Health Organization is implementing a global measles eradication program (72, 73). After completion of the two-dose vaccination schedule, nearly 100% of recipients develop lasting neutralizing immunity that may be lifelong (2). In addition, the availability of established production methods makes MV an appealing platform for delivering foreign antigens (reviewed in reference 5). Several groups, including ours, have generated MV with additional vaccine specificities (17, 55). Surface proteins of hepatitis B (54, 55), severe acute respiratory syndrome coronavirus (41), HIV (17, 30, 42), simian immunodeficiency virus (74), West Nile virus (18), dengue virus (8, 9), mumps virus (69), and human papillomavirus (12) have been expressed and immunogenicity has been demonstrated in susceptible hosts. A recombinant MV-HBV divalent vaccine remains protective against measles in rhesus monkeys (55) and elicits protective anti-HBsAg antibody titers when combined with a single HBsAg protein boost (54).
In this study, we generated two recombinant MVs expressing HCV structural proteins, termed MV-CE1E2 and MV-E1/Ft-E2-Ft. Following characterization of protein expression in cultured cells, immunogenicity of the vectors was assessed in MV-susceptible mice. We show that both vectors elicited neutralizing antibodies against homologous envelope proteins and that the MV-CE1E2 vector also induced cross-neutralizing immune responses to heterologous virus envelopes.
MATERIALS AND METHODS
Cells and viruses. Vero/hSLAM cells (50) and the helper 293-3-46 cell line (52) were maintained as monolayers in Dulbecco's modified Eagle's medium (DMEM; Mediatech Inc., Herndon, VA) supplemented with 10% fetal bovine serum (FBS), 1% penicillin-streptomycin (Mediatech), and 0.5 and 1.2 mg/ml G418 (Mediatech), respectively. Huh-7.5 cells (6) were propagated in DMEM supplemented with 10% FBS and 0.1 mM nonessential amino acids (NEAA).
Recombinant MVs were generated as described by Radecke et al. (52). After detection of their cytopathic effect on Vero/hSLAM cells, single syncytia were picked and propagated on this cell line. To prepare virus stocks, Vero/hSLAM cells were infected at a multiplicity of infection (MOI) of 0.03 and incubated at 32°C. Cells were scraped in Opti-MEM (Gibco) and particles released by two freeze-thaw cycles.
Virus growth characteristics were determined by infecting Vero/hSLAM cell monolayers at an MOI of 0.03 and incubating them at 37°C. Infected cells and supernatants were collected and lysed by one cycle of freeze and thaw at different times postinfection, and the 50% tissue culture infectious dose (TCID50) was determined on Vero/hSLAM cells using the Spearman-Kärber method (34).
Plasmid construction.
For construction of the vectored MV, we started with pB(+)MVvac2(ATU)P (55). The MV genome in this plasmid has complete coding identity with the Moraten/Schwartz vaccine strain genomes and contains an additional transcription unit (ATU, a duplicated N/P intergenic region with a multicloning site) inserted downstream of the phosphoprotein gene. The HCV CE1E2 coding sequence of HCV subtype 1a, strain H77 (residues 1 to 747, numbering starting with the first amino acid of the C protein sequence), obtained from pBRTM/HCV1-809con (36) as an MluI-AatII restriction fragment, was then cloned into the corresponding sites of the ATU to obtain pB(+)MVvac2-HCV(CE1E2)P.
Plasmid pB(+)MVvac2-HCV(E1/Ft-E2/Ft)P was obtained after transferring an MluI-AatII restriction fragment containing the sequences encoding the ectodomains of E1 and E2 fused to the transmembrane and cytoplasmic region of MV F protein. For E1/Ft, the junction was His/Gly, where His is amino acid 352 of the HCV polyprotein (161 of the E1 protein ectodomain) and Gly is the first amino acid of the MV F protein transmembrane domain (56). For E2/Ft, the junction was Glu/Gly, where Glu is amino acid 717 of the HCV polyprotein (335 of the E2 protein ectodomain) and Gly the first amino acid of the transmembrane region of the MV F protein (56). Correct junctions were corroborated by sequencing. The corresponding recombinant MVs were then generated and named MV-CE1E2 and MV-E1/Ft-E2/Ft, respectively.
Analysis of HCV protein expression.
Vero/hSLAM cells were seeded in 35-mm-diameter 6-well plates and infected with MVvac2, MV-CE1E2, or MV-E1/Ft-E2/Ft at an MOI of 0.5. Twenty-four hours after infection, cells were collected and lysed with RSB–NP-40 buffer (10 mM NaCl, 10 mM MgCl2, 10 mM Tris-HCl [pH 7.5], 1% Nonidet P-40) plus protease inhibitors (protease inhibitor cocktail set I; Calbiochem, Darmstadt, Germany). Lysates were subjected to 4 to 15% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) and transferred to nitrocellulose using a semidry apparatus (Bio-Rad, Hercules, CA). Immunoblotting was performed using anti-C (MA1-080; Affinity BioReagents, Golden, CO) and anti-E1 (catalog no. 1879) and anti-E2 (catalog no. 1876) (both from ViroStat, Portland, MA) monoclonal antibodies (MAbs). As positive controls, rabbit anti-MV H cytoplasmic tail polyclonal serum (13) and a monoclonal antibody, antiactin (AC-15; Sigma, St. Louis, MO), were used. After incubation with the corresponding horseradish peroxidase (HRP)-conjugated secondary antibody, signals were detected with the SuperSignal West Femto maximum detection system (Pierce, Rockford, IL).
Immunoprecipitation.
For analysis of E1 and E2 protein conformation, Vero/hSLAM cells were seeded and infected as described above. Twenty hours after infection, the cells were methionine and cysteine starved for 15 min and labeled with 100 μCi/ml of Express protein labeling Mix 35S label (Perkin-Elmer, Boston, MA) for 4 h. After being washed with phosphate-buffered saline (PBS), cells were lysed by incubation with radioimmunoprecipitation assay buffer (50 mM Tris [pH 7.5], 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS) plus protease inhibitors. The antigenic material was immunoprecipitated with protein G-agarose (Roche, Mannheim, Germany) using 10 μl of each of the following monoclonal antibodies: anti-measles virus H clone 55 (27) and conformation-dependent anti-E2 antibodies H35, H53, and H60 (16), generously provided by J. Dubuisson; 10 μl of a goat polyclonal serum against E2 (AbD Serotec, Oxford, United Kingdom) was also used.
Glycosidase treatment and analysis of surface expression.
The HCV envelope proteins or MV H proteins were immunoprecipitated from nonlabeled cell lysates prepared as described above. Proteins were treated with endoglycosidase H (endo-H) and peptide N-glycosidase F (PNGase-F) (both from NEB, Ipswich, MA) as per the manufacturer's instructions for 1 h at 37°C. Protein material was analyzed by immunoblotting using anti-H- or anti-E1-specific antibodies. For analysis of surface expression, infected Vero/hSLAM cells were incubated with 0.2 mg/ml of Sulfo-NHS-LC biotin (Pierce, Rockford, IL) in PBS for 40 min at 4°C. After the cells were washed with PBS and nonincorporated biotin was quenched with DMEM, cells were lysed with RSB–NP-40 buffer. Biotinylated proteins were precipitated using streptavidin UltraLink beads (Pierce) overnight at 4°C. Labeled material was subjected to SDS-PAGE and protein immunoblotting using polyclonal antibodies against measles virus H and N proteins and monoclonal antibodies against HCV E1 and E2 proteins.
Preparation of viral particles.
Supernatants of 7 × 106 infected Vero/hSLAM cells were clarified by centrifugation at 8,000 rpm for 30 min in an SLA600TC rotor (Sorvall, Newtown, CT). Viral particles were then concentrated at the interphase of 20 and 60% sucrose layers in TNE buffer (1 mM Tris [pH 7.8], 100 mM NaCl, 10 mM EDTA) by ultracentrifugation at 28,000 rpm for 90 min in an SW41Ti rotor (Beckman, Palo Alto, CA). The interphase was brought to a 20% sucrose concentration with TNE buffer and pelleted by ultracentrifugation with the same conditions. Viral pellets resuspended in TNE buffer were subjected to immunoblotting.
Expression and purification of soluble homologous E2.
Purification of soluble H77 E2 ectodomain was performed as previously described (71). Briefly, HEK293T cells were transfected with an E2 expression plasmid under the control of a cytomegalovirus (CMV) promoter (pcDNA3.1). The construct included an amino-terminal prolactin signal sequence and a carboxyl-terminal Fc tag for affinity purification. A high-expression stable cell line was produced by hygromycin selection and expanded. Supernatant from the stable cell line was centrifuged to remove cellular debris, filtered through a 0.22-μm membrane, and applied to protein A-conjugated resin (GE Healthcare, Piscataway, NJ). Following extensive washing with HEPES-buffered saline, the resin was incubated with thrombin protease to remove the Fc tag. The final protein concentration was determined by Bradford protein assay (Bio-Rad, Hercules, CA).
Mouse inoculations.
All experimental procedures were performed according to a protocol previously approved by the Mayo Clinic Institutional Animal Care and Use Committee. MV-susceptible Ifnarko-CD46Ge mice (49) were used as hosts. Five to eight mice per group were inoculated twice by the intraperitoneal (i.p.) route with 105 TCID50s of the virus as indicated below. Mice were bled 28 days postinoculation, and serum was separated and stored at −20°C until use. For boosting 28 days after virus inoculation, 5 μg of HCV H77 E2 soluble protein in complete Freund's adjuvant (Sigma) was administered by the i.p. route. Blood was collected 28 days after protein boost, and serum was obtained and stored.
MV neutralization assay.
MV neutralizing antibody titers were determined in a plaque reduction assay by incubating heat-inactivated serum dilutions with 100 PFU of a green fluorescence protein-expressing virus [MVvac(GFP)N] (our unpublished data). Titers were expressed as 90% plaque reduction fluorescence-forming units at 72 h postinfection.
HCV ELISA.
Enzyme-linked immunosorbent assay (ELISA) using recombinant H77 E2 ectodomain measured specific anti-E2 reactivity. Purified protein was diluted in BBS (15 mM Na2B4O7, 120 mM NaCl [pH 8.5]) and 50 ng/well was adsorbed to Nunc MaxiSorp 96-well plates (VWR, West Chester, PA). Blocking was performed with phosphate-buffered saline containing 5% bovine serum albumin (BSA), 0.2% skim milk, and 0.1% Tween 20 for 2 h at room temperature (RT) and subsequently with antibody diluent (PBS with 5% BSA, 2% sheep serum, and 0.05% Tween 20) for 30 min at RT. Serum samples, diluted 1:400 in antibody diluent, and 0.5 μg/ml E2-specific monoclonal antibody (4F6/2; Austral Biologicals, San Ramon, CA) were incubated for 1 h at RT. The reaction was developed with the addition of a goat anti-mouse IgG-HRP antibody (Jackson ImmunoResearch, West Grove, PA) and TMB substrate (Sigma, St. Louis, MO) and quenched with 0.5 N sulfuric acid; absorbance was measured at 450 nm.
HCV neutralization assays.
HCV pseudoparticles (HCVpp) were generated by cotransfecting plasmids encoding an HIV provirus expressing secretable Gaussia luciferase (pNL4-3-.Gluc.R-E-) and the HCV envelope glycoproteins [pcDNA3.1-H77(CsigE1E2)] in 293T cells, as previously described (33). Cell culture-adapted HCV (HCVcc) was generated by electroporation of in vitro-transcribed RNA from full-length genomes (H77, Bi-Gluc-H77C/JFH [T2700C and A4080T]; J6, Bi-Gluc-J6/JFH; and Con1, Bi-Gluc-Con1/Jc1 [G2833C, T2910C, A4274G, A6558G, and A7136C]) into Huh-7.5 cells as previously described (40, 67). Supernatants were harvested, pooled, and filtered.
For neutralization assays, complement was inactivated, and the diluted sera were mixed with HCVpp or HCVcc in DMEM containing 2% FBS, 0.1 mM NEAA, 20 mM HEPES, and 4 μg/ml Polybrene at the dilution indicated below. Control antibodies against CD81 (2 μg/ml; JS81 clone; BD Pharmingen) and genotype 1a E2 (0.5 μg/ml; AR4A; generous gift from M. Law [26]) were utilized. The virus-serum mixture was incubated at 37°C for 1 h, transferred to Huh-7.5 cells seeded in 96-well plates (6.4 × 103 cells per well), and incubated at 37°C for 6 h. At this time, cells were washed three times and further incubated at 37°C in DMEM with 10% FBS and 0.1 mM NEAA. HCVpp and HCVcc infections were terminated after 72 h. Gaussia luciferase secreted within the supernatants was quantified using the Gaussia luciferase assay system (Promega, Madison, WI).
Neutralization of the mouse antisera using retroviral particles bearing no envelope or pseudotyped with the nonrelated feline immunodeficiency virus RD114 envelope were performed in parallel. Background levels of unspecific HCV neutralization were measured using sera from MVvac2-infected mice. In these mice, average luciferase relative light units (RLU) were approximately 20 to 25% below those of the untreated control (HCV-pseudotyped particles alone). Luciferase readings below this background were considered positive (with HCV neutralization potential). Sera neutralizing HCV 100 times more efficiently than the background level were considered 100% neutralizing. For HCVcc analysis, luciferase readings for preimmune and post-protein boost status were averaged for each HCV set. The averaged preimmune RLUs were used as baseline readings for comparison. Both CD81 and E2 were compared against the untreated control (HCVcc virus alone). All averages, both HCVpp and HCVcc, were calculated from 6 replicates.
RESULTS
Growth characteristics of vectored MV expressing HCV structural proteins.
We used the MV vaccine infectious cDNA, pB(+)MVvac2 (55), to create vectors encoding the HCV structural proteins (Fig. 1A). The MV-CE1E2 vector directs the expression of HCV C and the E1E2 envelope protein heterodimer. The MV-E1/Ft-E2/Ft vector expresses both HCV glycoprotein ectodomains fused to the transmembrane region and cytoplasmic tail of the MV F protein. The cytoplasmic tail of the MV F protein has been previously shown to enhance the incorporation of foreign glycoproteins into MV particles (60). To assess the replication efficiency of the MV vectors, multistep growth kinetics studies were performed. After inoculation with a starting MOI of 0.03, the unmodified MV vaccine strain (MVvac2) and MV-CE1E2 replicated with equivalent kinetics, reaching maximum titers of cell-associated virus of about 106.5 TCID50s/ml at 36 h postinfection (Fig. 1B). The growth kinetics of MV-E1/Ft-E2/Ft was slightly delayed, and maximum titers of 105.75 TCID50s/ml of cell-associated virus were reached at 48 h postinfection.
Fig 1.

Genome and growth characteristics of vectored MV expressing HCV proteins. (A) Diagram of the genomes of recombinant vectors. MV proteins are indicated by dark gray arrows, HCV proteins by inserts of light gray arrows. For details on construction, see Materials and Methods. The names of the recombinant MVs are indicated above the genome diagrams. (B) Time course of cell-associated (top) and cell-free virus production in Vero/hSLAM cells infected with MVvac2 (circles), MV-CE1E2 (squares), or MV-E1/Ft-E2/Ft (triangles). Viral titers are indicated on the vertical axes. These titers were measured at 12, 24, 36, 48, 72, and 96 h postinfection. For clarity, symbols were moved slightly sideways. Averages and standard deviations of three independent experiments are indicated.
HCV proteins expressed by vectored MV.
To assess the quality and quantity of HCV proteins expressed by the vectored MV, we first analyzed extracts of infected cells by Western blotting (Fig. 2A). Noninfected cells and cells infected with MVvac2 were used as controls. Only MV-CE1E2 expressed a protein of about 19 to 21 kDa, the expected size of the HCV C protein. Next, radiolabeled lysates from MVvac2-, MV-CE1E2-, or MV-E1/Ft-E2/Ft-infected cells were assayed by immunoprecipitation, using either rabbit anti-MV H (Fig. 2B, first three lanes) or goat anti-E2 antibodies (last three lanes). The anti-E2 serum immunoprecipitated a broad band of 60 to 70 kDa, as well as two distinct proteins with molecular masses of 32 and 29 kDa, from cells infected with MV-CE1E2. The 60- to 70-kDa band may correspond to E2 and the two smaller bands to glycosylated isoforms of E1. In contrast, a protein of approximately 72 kDa was the most prominent signal in lysates of MV-E1/Ft-E2/Ft-infected cells. As expected, no HCV proteins were detected in lysates of MVvac2-infected cells.
Fig 2.

Biochemical characterization of HCV proteins expressed by vectored MVs. (A) Expression of HCV core protein by vectored MV-CE1E2. Extracts from mock-, MVvac2-, MV-CE1E2-, or MV-E1/Ft-E2/Ft-infected cells were analyzed by immunoblotting. The reactivity of each antibody is indicated below each blot. (B) Immunoprecipitation of proteins produced by cells infected with MVvac2, MV-CE1E2, or MV-E1/Ft-E2/Ft at an MOI of 0.5. Proteins were labeled with [35S]methionine 24 h postinfection and precipitated with the antibody indicated below the lanes. The positions of molecular mass standards are indicated on the left. (C) Immunoprecipitation of HCV envelope proteins using conformation-dependent monoclonal antibodies H53 and H60, as indicated below the lanes. (D) Glycosidase treatment. MV H and HCV E1 complexes immunoprecipitated from cells infected with MV-CE1E2 or MV-E1/Ft-E2/Ft were treated with endoglycosidase H (E) or PNGase-F (F) or left untreated (U). MV H (top panel) and HCV E1 glycoproteins (botttom panel) were detected by immunoblotting using specific antibodies. The asterisk beside E1 indicates an endoglycosidase H-resistant form. (E) Surface biotinylation analysis. Cells were infected with MVvac2, MV-CE1E2, or MV-E1/Ft-E2/Ft at an MOI of 0.1. Forty-eight hours later, cells were labeled with biotin and lysed. Biotin-labeled proteins were pulled down with streptavidin-agarose and analyzed by immunoblotting. Antibodies used for specific detection are indicated at the bottom.
To further characterize the HCV envelope proteins expressed by the vectored MV, we performed immunoprecipitation using conformation-dependent anti-E2 monoclonal antibodies, H53 and H60 (16). Both antibodies precipitated the HCV envelope heterodimer from MV-CE1E2 infected cells (Fig. 2C, second and fifth lanes) with an efficiency similar to that of the goat polyclonal anti-E2 serum. In addition, a weak signal around 72 kDa was observed in the extracts of MV-E1/Ft-E2/Ft-infected cells (Fig. 2C, third and sixth lanes); this may correspond to the E2/Ft protein. Taken together, these results show that MV vaccine-based vectors in cell culture express the expected HCV proteins and that at least some of the expressed E2 protein is correctly folded.
Hybrid E2/Ft and E1/Ft proteins are transported to the cell surface.
HCV glycoproteins are retained in the ER during natural infection. By replacing the E1 and E2 transmembrane domains with the corresponding MV F protein sequence in the MV-E1/Ft-E2/Ft vector (Fig. 1A), we hoped to promote trafficking to the cell surface, where MV budding occurs. To characterize intracellular transport of the vector-expressed proteins, we analyzed the susceptibility of E1 and MV H proteins to digestion with endoglycosidase H (endo-H). This enzyme cleaves only high-mannose oligosaccharides; endo-H insensitivity would indicate that Golgi enzymes have modified the glycoprotein. As controls, we tested the susceptibility of immunoprecipitated samples to peptidyl-N glycosidase-F (PNGase-F), which cleaves all N-linked carbohydrates (i.e., high mannose, hybrid, and bi-, tri-, and tetra-antennary), or left the proteins undigested. As expected, MV H protein was partially resistant to endo-H, indicating transport through the Golgi (Fig. 2D, top). E1 expressed from MV-CE1E2 was endo-H sensitive, consistent with ER retention, while a fraction of E1 protein expressed from MV-E1/Ft-E2/Ft become endo-H resistant, indicating transport to the medial Golgi or a subsequent compartment (Fig. 2D, bottom).
To further characterize the localization of the E2/Ft and E1/Ft proteins, we labeled cell surface-exposed proteins with N-hydroxysulfosuccinimide (NHS)-biotin, followed by precipitation of cell extracts with beads coupled to streptavidin and immunoblotting with antibodies against MV H, MV nucleocapsid (N), or HCV E1 and E2 (Fig. 2E). As expected, MV H was detected on the surface of cells infected with each of the three viruses (Fig. 2E, top), whereas the cytoplasmic N protein was not detected (Fig. 2E, bottom). E2/Ft and E1/Ft proteins were also detected at the cell surface, whereas native E2 and E1 were not. These data indicate that the MV F transmembrane domain promotes trafficking of the hybrid E1 and E2 glycoproteins to the sites of MV assembly.
Hybrid E2/Ft and E1/Ft proteins are not incorporated into virions.
Since the E1/Ft and E2/Ft hybrid proteins express the MV F transmembrane domain and cytoplasmic tail and are expressed at the cell surface, they have the potential to be incorporated into chimeric MV virions. To investigate this, we gradient purified particles obtained from cells infected with vectored MV or the unmodified vaccine strain and analyzed them by nonreducing PAGE, followed by immunoblotting using a rabbit polyclonal serum specific for the cytoplasmic region of MV F. We observed mature MV F protein, but no hybrid proteins, in all the purified viral particles (Fig. 3). Parallel analysis of cell lysates (Fig. 3, right) showed that there was significant expression of the hybrid glycoproteins within the infected cells. These results indicate that no detectable E1/Ft and E2/Ft proteins are incorporated into MV particles.
Fig 3.

Hybrid HCV glycoproteins are not incorporated into measles virus virions. MV particles or infected-cell lysates, as indicated below each blot, were analyzed using a polyclonal serum specific for the F cytoplasmic tail. F0 and F1, precursor and mature forms of the F protein, respectively.
Vectored MV elicits neutralizing immune responses against HCV.
To test the immunogenic potential of the vectors, we inoculated each virus into MV-susceptible Ifnarko-CD46Ge mice. These mice express the MV vaccine strain receptor human CD46 with human-like tissue specificity in a type I interferon receptor knockout background (49). Groups of 7 mice were inoculated twice, at time zero and 4 weeks after the first infection, by the intraperitoneal route with MVvac2, MV-CE1E2, or MV-E1/Ft-E2/Ft. We assessed MV-neutralizing titers, E2-specific reactivity, and neutralization against HCV pseudotyped particles expressing a homologous luciferase at 4 and 6 weeks after the first inoculation.
Regardless of the inoculating virus, the MV average neutralization titers were in the range of 1:300 to 1:500 4 weeks after the first infection and between 1:800 and 1:1,500 2 weeks later (Fig. 4A). On the other hand, HCV E2 ELISA indicated reactivity only in mice inoculated with both vectored MVs: average optical densities at 450 nm (OD450) were about 1.6 and 1.0 6 weeks after infection with MV-CE1E2 and MV-E1/Ft-E2/Ft, respectively. In contrast, the average E2 ELISA OD450 in mice inoculated with MVvac2 was about 0.4 (Fig. 4B). The normalized HCV neutralization capacities were about 40% and 20% for mice inoculated with MV-CE1E2 and MV-E1/Ft-E2/Ft, respectively (Fig. 4C).
Fig 4.

Humoral immune response in genetically modified mice immunized with vectored MV expressing HCV structural proteins. Groups of 7 animals were immunized with two doses of MVvac2 (squares), CE1E2 (circles), or E1/Ft-E2/Ft (triangles). Each symbol represents one animal; a horizontal bar shows the average of the group. Sera were obtained either 4 weeks after the initial infection (4w) or 2 weeks after repeat infection (6w). All sera were assayed for MV neutralization (A; reciprocals of the titer), for reactivity against HCV E2 by ELISA (B; optical density at 450 nm), or for HCV neutralization using an H77-pseudotyped luciferase-expressing retrovirus (C; percent neutralization). In panel B, average readings for the background (−) and monoclonal antibody anti-E2 (+) are shown with gray and black bars, respectively. Even though most of the samples were anti-E2 seropositive as detected by ELISA after the second dose, one and three animals in the MV-CE1E2 and MV-E1/Ft-E2/Ft groups, respectively, had background level values. For panel C, two mice in each experimental group did not develop detectable levels of neutralizing antibodies.
E2 protein boosts the anti-HCV neutralizing immune response.
In an effort to enhance the neutralizing antibody response elicited by MV vectors, a prime-boost strategy was tested. In this approach, groups of eight animals received a single dose of a soluble HCV E2 protein (strain H77) 4 weeks after inoculation with MVvac2, MV-CE1E2, or MV-E1/Ft-E2/Ft. Neutralizing antibody responses against MV and HCV as well as HCV-E2 reactivity by ELISA were assessed over time.
As shown in Fig. 5A, average anti-MV neutralization titers of about 1:500 to 1:1,000 were documented 4 weeks after infection and increased 1.5-fold 4 weeks later. As in the previous experiment, 28 days after inoculation, all of the animals immunized with vectored MV were seropositive by ELISA (Fig. 5B, 4w). Importantly, the protein boost increased reactivity for all three experimental groups (Fig. 5B, 8w). Four weeks after inoculation of vectored MV, HCV neutralization was, on average, about 20% or 10% in MV-CE1E2- or MV-E1/Ft-E2/Ft-inoculated mice, respectively (Fig. 5C, 4w); however, two MV-CE1E2- and four MV-E1/Ft-E2/Ft-inoculated mice failed to develop HCV-neutralizing antibodies. Four weeks after protein boost (Fig. 5C, 8w), 90% or 70% of HCV-specific neutralization was observed in MV-CE1E2- or MV-E1/Ft-E2/Ft-inoculated mice, respectively. Indeed, highly diluted (up to 1:12,800) post-protein boost samples from animals primed with vectored MV still exhibited 45 to 50% neutralization (data not shown). Neutralization was not observed for control retroviral particles lacking envelope proteins or pseudotyped with the feline immunodeficiency virus RD114 envelope (data not shown).
Fig 5.

Humoral immune responses in genetically modified mice immunized with vectored MVs and boosted with a single dose of E2 protein. Groups of 5 to 8 animals were immunized with two doses of MVvac2 (squares), CE1E2 (circles), or E1/Ft-E2/Ft (triangles) and boosted with 5 μg of E2 protein treated in the presence of Freund's complete adjuvant. Sera obtained 4 weeks after viral inoculation (4w, solid symbols) or 4 weeks after subsequent homologous protein boost (8w, empty symbols) were assayed for MV neutralization (A; reciprocals of the titer), reactivity against HCV E2 by ELISA (B; optical density at 450 nm), or HCV neutralization using an H77-pseudotyped luciferase-expressing retrovirus (C; percentage of neutralization normalized by MVvac2 readings). In panel B, average readings for the background (−) and a monoclonal antibody, anti-E2 (+), are shown with gray and black bars, respectively. Each symbol represents one animal; a horizontal bar indicates the group average.
The E2 protein boost elicits cross-neutralizing HCV responses.
We measured the cross-neutralizing immune response in serum samples obtained from mice undergoing the prime-boost schedule. Neutralization was tested using a panel of infectious cell culture-adapted HCV intergenotypic chimeras, which consisted of genotype 2a strain JFH-1 genomes expressing the structural proteins of genotypes 1a (H77), 1b (Con1), or 2a (J6). Samples were analyzed 8 weeks after virus immunization, which was 4 weeks after the protein boost.
As expected, an antibody recognizing HCV entry factor CD81 neutralized chimeric virus infectivity to nearly 100% (Fig. 6, first group of three columns). An antibody targeting the genotype 1a glycoprotein (anti-E2) (26) neutralized about 61% of the infectivity of the homologous genotype H77, 30% of Con1 (genotype 1b), and 16% of J6 (genotype 2a) (Fig. 6, second group of three columns).
Fig 6.
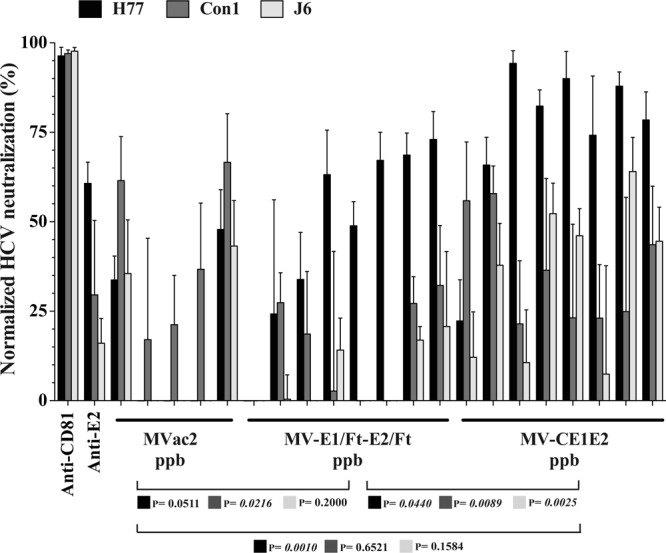
Cross-neutralizing immune responses against cell culture-replicating HCV. Huh-7.5 cells were infected with an MOI of 0.1. The first and second sets of 3 columns show neutralization with control antibodies anti-CD81 and anti-E2. Levels of neutralization of chimeric HCVcc containing structural proteins are indicated as follows: H77 (genotype 1a) with black columns, Con1 (genotype 1b) with dark gray columns, and J6 (genotype 2a) with light gray columns. Other sets of columns show neutralization levels observed in sera of 5 mice primed with control MVvac2, 8 mice primed with MV-E1/Ft-E2/Ft, or 8 mice primed with MV-CE1E2. All mouse serum samples were diluted 1:200. At the bottom is shown the statistical significance of the differences observed between the groups, using the same color-coding of the chimeric HCVcc genotypes as for the neutralization assay.
After control MVvac2 inoculation followed by H77 E2 protein boost, two out of five animals showed neutralization of at least 30% of the infectivity of 1a and 2a genotypes (Fig. 6, next five groups of columns); the heterologous Con1 chimeric virus was more sensitive to neutralization, averaging 40%, which was not statistically different from the result for mice primed with MV-CE1E2 (Fig. 6, bottom; P = 0.6521, two-tailed t test).
Sera from mice primed with MV-CE1E2, on the other hand, showed significantly higher homologous neutralization than sera from mice primed with MV-E1/Ft-E2/Ft (74% versus 47%; P = 0.044 [Fig. 6, bottom]). The cross-neutralizing profile for 1b and 2a chimeras had a similar trend: Con1 neutralization by serum from mice inoculated with MV-CE1E2 averaged 36% versus 13% for those inoculated with MV-E1/Ft-E2/Ft (P = 0.0089); J6 neutralization averaged 34% versus 4%, respectively (P = 0.0025). These results show that an MV vector expressing unmodified E1E2 glycoproteins, coupled with an E2 protein boost, can elicit cross-neutralizing HCV antibodies in vivo.
DISCUSSION
Public health policies alone are unlikely to control endemic HCV infections in countries with poor needle hygiene or nonstringent blood product usage (62). Moreover, the current therapeutic regimen is expensive and is curative in only about two-thirds of infected patients, depending on the viral genotype (43). Thus, it remains an urgent priority to develop a vaccine to protect against HCV infection, particularly in the developing world.
We produced two MV vectors, MV-CE1E2 and MV-E1/Ft-E2/Ft, which replicated in cultured cells and expressed high levels of HCV proteins. MV-E1/Ft-E2/Ft expresses both glycoprotein ectodomains fused to the transmembrane and cytoplasmic domain of the MV F protein. These modifications were expected to have two effects: to counteract retention of the HCV glycoproteins in the ER (16) and to enhance their incorporation in MV particles (60). While the hybrid proteins were indeed transported to the cell surface, their incorporation into MV particles was below the limit of detection. In contrast, analogous hybrid glycoproteins expressed from vesicular stomatitis virus (10, 63) and influenza virus (23) vectors are readily detected in viral particles. We do not know why this difference exists, but in our experimental system, based on an infectious MV cDNA that is vaccine equivalent, foreign glycoprotein incorporation may be more stringent. The fact that E1 and E2 glycoproteins are not detected in MV particles implies that the recombinant virus cannot use HCV receptors to enter cells. Thus, altered tropism of MV-E1/Ft-E2/Ft is unlikely.
For many viral infections, development of neutralizing antibodies is the clearest correlate of protection. In the case of HCV, however, clearance requires both humoral and cell-mediated immunity (reviewed in references 32 and 65). While viral clearance during acute infection is linked to the induction of cross-neutralizing antibodies, this correlation is not as evident in chronic HCV (4, 51). This is likely because the heterogeneity of E2 yields quasispecies that are able to escape neutralizing antibodies over long-term infection (68); association of virus particles with very-low-density lipoproteins that mediate their uptake through low-density lipoproteins or scavenger receptor class B type 1 (SR-BI) receptors (11), glycan interference, and interfering antibody epitopes also contribute to evasion (66). While the exact role of neutralizing immunity in the prophylaxis of HCV infection is debatable (21), several studies have demonstrated its importance. A recombinant form of the E1E2 (genotype 1a) heterodimer can induce protective immunity against homologous challenge in a chimpanzee model (15). More recently, samples from five of these animals were reexamined and neutralizing immune responses against genotypes 1a, 4a, 5a, and 6a were documented (44). A recent phase I clinical trial of a three-dose adjuvanted recombinant HCV E1E2 protein vaccine documented neutralization-of-binding responses in 11/14 subjects and HCV neutralization titers of ≥1:20 in 10/36 samples (24, 53). Similarly, Stamataki et al. showed that immunization of human volunteers with recombinant E1E2 (genotype 1a) glycoproteins induced cross-neutralization of heterologous strains derived from genotypes 1a, 1b, and 2a in vitro (61). These results provide encouragement that a recombinant protein vaccine approach is feasible for HCV.
Viral vectors have also previously been used to express HCV proteins and to elicit specific immunity. In particular, C, E1, E2, and NS3-specific immune responses were induced in four naïve chimpanzees using a DNA-prime, recombinant modified vaccinia virus Ankara (MVA) vector boost vaccine schedule (57). Despite control of acute-phase viremia after homologous challenge, three animals developed chronic, persistent infection. Interestingly, no neutralizing activity was detected against an HCV pseudotype recreating the envelope of the virus used for challenge, even when the amino acid sequences of the vaccine and challenge virus strains differed by approximately 5%. In another study which aimed to induce neutralizing antibodies against E1, inoculation of mice or macaques with retrovirus-derived virus-like particles pseudotyped with E1 and E2 efficiently primed a 3-dose boost (25). However, soluble E1 expressed alone from an MV vector failed to prime a cross-neutralizing response, indicating that either E2 or both glycoproteins in their native conformation are required. Finally, a rare-serotype adenoviral vector expressing genotype 1b nonstructural proteins was found to be highly immunogenic, with robust, broad T-cell-specific responses sustained for at least a year following heterologous boost (3).
While we have not yet characterized the immunogenicity of our vectors in primates, the studies presented here compare favorably with other viral vectors eliciting HCV neutralizing responses in mice. For example, three doses (4 × 106 infectious units) of a genotype 1a E1E2-vectored alphavirus elicited cross-neutralizing responses against a genotype 2a envelope in BALB/c mice. This response was higher when two 4-μg doses of adjuvanted recombinant E1E2 protein were given before a single dose of the alphavirus vector (39). Another study documented anti-HCV humoral immune responses after vaccination of mice with an MVA vector expressing hybrid or truncated E2 (1). However, neutralizing antibodies were not documented in that study, while in our studies homologous neutralizing antibodies were obtained in the presence or absence of a protein boost.
Despite eliciting similar E2 ELISA titers, our study indicates that MV-CE1E2 is a better vaccine candidate than MV-E1/Ft-E2/Ft. While the two vectors showed equivalent potentials for eliciting neutralizing antibodies against homologous pseudoparticles, neutralization of heterologous HCV was enhanced with MV-CE1E2. The C-terminal transmembrane domains of the glycoproteins have been shown to be necessary for efficient assembly of the E1E2 heterodimer (38); thus, the chimeric glycoproteins expressed from the MV-E1/Ft-E2/Ft virus may not contain conformationally relevant epitopes required for cross-neutralization. Furthermore, some neutralization of the heterologous Con1 chimeric virus could be generated with the recombinant E2 protein, suggesting that neutralization epitopes on Con1 E2 may be more accessible. We do not have an explanation for why this effect was more evident in animals primed with MVvac2. Priming with the MV-CE1E2 virus, however, was necessary to generate a broadly reactive humoral response. Indeed, we observed a variable capacity to neutralize HCV-pseudotyped retroviral particles, particularly in samples obtained 4 weeks postimmunization. For this time point, 80% of the animals responded to MV-CE1E2 immunization with an average neutralization of 20%, while for MV-E1/Ft-E2/Ft, 52% had a measurable neutralization response that averaged 10%. Finally, numerous groups have shown the importance of sustained T-cell responses targeting multiple regions of HCV for clearance; establishment of chronicity is marked by weak and narrowly targeted T-cell responses (7, 19, 28, 37, 58, 65, 70), and depletion of CD4+ and CD8+ cells after induction of protective T-cell responses within chimpanzees led to a reduction in control against repeated HCV challenge (28, 59). Therefore, additional studies are needed to determine whether broadly reactive cell-mediated immune responses can be generated with the MV-CE1E2 and recombinant E2 protein strategy.
There are strategies to further improve the HCV neutralization capacity of vectored MV. Higher expression of the HCV glycoprotein heterodimer may be obtained by moving the additional transcription unit toward the 3′ end of the MV genome; we have observed that higher antibody titers correlate with higher expression rates with another antigen (54). In addition, a broader cocktail of HCV glycoproteins could be incorporated in both MV vector priming and protein boost to optimize cross-reactive antibody responses. The current pediatric MV immunization schedule could be adapted to accommodate two doses of a divalent MV-HCV vaccine. For example, it could be given at 9 and 18 months of age, and the E2 protein dose could be administered along with the tetanus and diphtheria vaccine. Such enhanced vaccination strategies are particularly attractive in locations with high HCV infection rates, such as Egypt, or in selected populations. In particular, children with HCV-infected mothers might benefit from immunization, since an increased risk of infection not related to perinatal transmission has been reported for these children (47, 48).
ACKNOWLEDGMENTS
We thank Jean B. Dubuisson, University of Lille, France, for sharing anti-HCV E2 monoclonal antibodies H35, H53, and H60; Mansun Law, Scripps Research Institute, La Jolla, CA, for genotype 1a E2 antibody (clone AR4A); Mohsan Saeed for technical assistance; members of both the Cattaneo and Rice laboratories for helpful discussions and suggestions; Catherine Jones for editorial assistance; and Stephanie Ferguson for secretarial assistance.
This work was supported by the National Institutes of Health (AI 57761 to R.C., AI 080659 to J.M., and RR11069 and U19AI40034 to C.M.R.), the Mayo Foundation for Medical Education and Research, the Greenberg Medical Research Institute, the Starr Foundation, and the NJ Commission on Cancer Research.
Footnotes
Published ahead of print 15 August 2012
REFERENCES
- 1. Abraham JD, et al. 2004. Comparative immunogenicity analysis of modified vaccinia Ankara vectors expressing native or modified forms of hepatitis C virus E1 and E2 glycoproteins. Vaccine 22:3917–3928 [DOI] [PubMed] [Google Scholar]
- 2. Amanna IJ, Carlson NE, Slifka MK. 2007. Duration of humoral immunity to common viral and vaccine antigens. N. Engl. J. Med. 357:1903–1915 [DOI] [PubMed] [Google Scholar]
- 3. Barnes E, et al. 2012. Novel adenovirus-based vaccines induce broad and sustained T cell responses to HCV in man. Sci. Transl. Med. 4:115ra111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bartosch B, et al. 2003. In vitro assay for neutralizing antibody to hepatitis C virus: evidence for broadly conserved neutralization epitopes. Proc. Natl. Acad. Sci. U. S. A. 100:14199–14204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Billeter MA, Naim HY, Udem SA. 2009. Reverse genetics of measles virus and resulting multivalent recombinant vaccines: applications of recombinant measles viruses. Curr. Top. Microbiol. Immunol. 329:129–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Blight KJ, McKeating JA, Rice CM. 2002. Highly permissive cell lines for hepatitis C virus genomic and subgenomic RNA replication. J. Virol. 76:13001–13014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bowen DG, Walker CM. 2005. Adaptive immune responses in acute and chronic hepatitis C virus infection. Nature 436:946–952 [DOI] [PubMed] [Google Scholar]
- 8. Brandler S, et al. 2010. Pediatric measles vaccine expressing a dengue tetravalent antigen elicits neutralizing antibodies against all four dengue viruses. Vaccine 28:6730–6739 [DOI] [PubMed] [Google Scholar]
- 9. Brandler S, et al. 2007. Pediatric measles vaccine expressing a dengue antigen induces durable serotype-specific neutralizing antibodies to dengue virus. PLoS Negl. Trop. Dis. 1:e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Buonocore L, Blight KJ, Rice CM, Rose JK. 2002. Characterization of vesicular stomatitis virus recombinants that express and incorporate high levels of hepatitis C virus glycoproteins. J. Virol. 76:6865–6872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Burlone ME, Budkowska A. 2009. Hepatitis C virus cell entry: role of lipoproteins and cellular receptors. J. Gen. Virol. 90:1055–1070 [DOI] [PubMed] [Google Scholar]
- 12. Cantarella G, et al. 2009. Recombinant measles virus-HPV vaccine candidates for prevention of cervical carcinoma. Vaccine 27:3385–3390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cathomen T, Naim HY, Cattaneo R. 1998. Measles viruses with altered envelope protein cytoplasmic tails gain cell fusion competence. J. Virol. 72:1224–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Centers for Disease Control and Prevention 2008. Progress in global measles control and mortality reduction, 2000–2007. MMWR Morb. Mortal. Wkly. Rep. 57:1303–1306 [PubMed] [Google Scholar]
- 15. Choo Q, et al. 1994. Vaccination of chimpanzees against infection by the hepatitis C virus. Proc. Natl. Acad. Sci. U. S. A. 91:1294–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cocquerel L, Meunier JC, Pillez A, Wychowski C, Dubuisson J. 1998. A retention signal necessary and sufficient for endoplasmic reticulum localization maps to the transmembrane domain of hepatitis C virus glycoprotein E2. J. Virol. 72:2183–2191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Combredet C, et al. 2003. A molecularly cloned Schwarz strain of measles virus vaccine induces strong immune responses in macaques and transgenic mice. J. Virol. 77:11546–11554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Desprès P, et al. 2005. Live measles vaccine expressing the secreted form of the West Nile virus envelope glycoprotein protects against West Nile virus encephalitis. J. Infect. Dis. 191:207–214 [DOI] [PubMed] [Google Scholar]
- 19. Diepolder HM, et al. 1995. Possible mechanism involving T-lymphocyte response to non-structural protein 3 in viral clearance in acute hepatitis C virus infection. Lancet 346:1006–1007 [DOI] [PubMed] [Google Scholar]
- 20. Drummer HE, Boo I, Maerz AL, Poumbourios P. 2006. A conserved Gly436-Trp-Leu-Ala-Gly-Leu-Phe-Tyr motif in hepatitis C virus glycoprotein E2 is a determinant of CD81 binding and viral entry. J. Virol. 20:7844–7853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Edwards VC, Tarr AW, Urbanowicz RA, Ball JK. 2012. The role of neutralizing antibodies in hepatitis C virus infection. J. Gen. Virol. 93:1–19 [DOI] [PubMed] [Google Scholar]
- 22. Flint M, et al. 2000. Functional characterization of intracellular and secreted forms of a truncated hepatitis C virus E2 glycoprotein. J. Virol. 74:702–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Flint M, et al. 1999. Functional analysis of cell surface-expressed hepatitis C virus E2 glycoprotein. J. Virol. 73:6782–6790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Frey SE, et al. 2010. Safety and immunogenicity of HCV E1E2 vaccine adjuvanted with MF59 administered to healthy adults. Vaccine 28:6367–6373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Garrone P, et al. 2011. A prime-boost strategy using virus-like particles pseudotyped for HCV proteins triggers broadly neutralizing antibodies in macaques. Sci. Transl. Med. 3:94ra71. [DOI] [PubMed] [Google Scholar]
- 26. Giang E, et al. 2012. Human broadly neutralizing antibodies to the envelope glycoprotein complex of hepatitis C virus. Proc. Natl. Acad. Sci. U. S. A. 109:6205–6210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Giraudon P, Wild TF. 1985. Correlation between epitopes on hemagglutinin of measles virus and biological activities: passive protection by monoclonal antibodies is related to their hemagglutination inhibiting activity. Virology 144:46–58 [DOI] [PubMed] [Google Scholar]
- 28. Grakoui A, et al. 2003. HCV persistence and immune evasion in the absence of memory T cell help. Science 302:659–662 [DOI] [PubMed] [Google Scholar]
- 29. Grove J, et al. 2008. Identification of a residue in hepatitis C virus E2 glycoprotein that determines scavenger receptor BI and CD81 receptor dependency and sensitivity to neutralizing antibodies. J. Virol. 82:12020–12029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guerbois M, et al. 2009. Live attenuated measles vaccine expressing HIV-1 Gag virus like particles covered with gp160DeltaV1V2 is strongly immunogenic. Virology 388:191–203 [DOI] [PubMed] [Google Scholar]
- 31. Haberstroh A, et al. 2008. Neutralizing host responses in hepatitis C virus infection target viral entry at postbinding steps and membrane fusion. Gastroenterology 135:1719–1728.el [DOI] [PubMed] [Google Scholar]
- 32. Halliday J, Klenerman P, Barnes E. 2011. Vaccination for hepatitis C virus: closing in on an evasive target. Expert Rev. Vaccines 10:659–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hsu M, et al. 2003. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc. Natl. Acad. Sci. U. S. A. 100:7271–7276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kärber G. 1931. Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche. Arch. Exp. Pathol. Pharmakol. 162:480–483 [Google Scholar]
- 35. Klevens RM, et al. 2009. Population-based surveillance for hepatitis C virus, United States, 2006–2007. Emerg. Infect. Dis. 15:1499–1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kolykhalov AA, et al. 1997. Transmission of hepatitis C by intrahepatic inoculation with transcribed RNA. Science 277:570–574 [DOI] [PubMed] [Google Scholar]
- 37. Lauer GM, et al. 2004. High resolution analysis of cellular immune responses in resolved and persistent hepatitis C virus infection. Gastroenterology 127:924–936 [DOI] [PubMed] [Google Scholar]
- 38. Lavie M, Goffard A, Dubuisson J. 2007. Assembly of a functional HCV glycoprotein heterodimer. Curr. Issues Mol. Biol. 9:71–86 [PubMed] [Google Scholar]
- 39. Lin Y, et al. 2008. Induction of broad CD4+ and CD8+ T-cell responses and cross-neutralizing antibodies against hepatitis C virus by vaccination with Th1-adjuvanted polypeptides followed by defective alphaviral particles expressing envelope glycoproteins gpE1 and gpE2 and nonstructural proteins 3, 4, and 5. J. Virol. 82:7492–7503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lindenbach BD, et al. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626 [DOI] [PubMed] [Google Scholar]
- 41. Liniger M, et al. 2008. Induction of neutralising antibodies and cellular immune responses against SARS coronavirus by recombinant measles viruses. Vaccine 26:2164–2174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lorin C, et al. 2004. A single injection of recombinant measles virus vaccines expressing human immunodeficiency virus (HIV) type 1 clade B envelope glycoproteins induces neutralizing antibodies and cellular immune responses to HIV. J. Virol. 78:146–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Manns MP, Wedemeyer H, Cornberg M. 2006. Treating viral hepatitis C: efficacy, side effects, and complications. Gut 55:1350–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Meunier JC, et al. 2011. Vaccine-induced cross-genotype reactive neutralizing antibodies against hepatitis C virus. J. Infect. Dis. 204:1186–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Meunier JC, et al. 2008. Isolation and characterization of broadly neutralizing human monoclonal antibodies to the E1 glycoprotein of hepatitis C virus. J. Virol. 82:966–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Michalak JP, et al. 1997. Characterization of truncated forms of hepatitis C virus glycoproteins. J. Gen. Virol. 78(Part 9):2299–2306 [DOI] [PubMed] [Google Scholar]
- 47. Mohamed MK, et al. 2005. Intrafamilial transmission of hepatitis C in Egypt. Hepatology 42:683–687 [DOI] [PubMed] [Google Scholar]
- 48. Mohamed MK, et al. 2006. Transmission of hepatitis C virus between parents and children. Am. J. Trop. Med. Hyg. 75:16–20 [PubMed] [Google Scholar]
- 49. Mrkic B, et al. 1998. Measles virus spread and pathogenesis in genetically modified mice. J. Virol. 72:7420–7427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ono N, et al. 2001. Measles viruses on throat swabs from measles patients use signaling lymphocytic activation molecule (CDw150) but not CD46 as a cellular receptor. J. Virol. 75:4399–4401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pestka JM, et al. 2007. Rapid induction of virus-neutralizing antibodies and viral clearance in a single-source outbreak of hepatitis C. Proc. Natl. Acad. Sci. U. S. A. 104:6025–6030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Radecke F, et al. 1995. Rescue of measles viruses from cloned DNA. EMBO J. 14:5773–5784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ray R, et al. 2010. Characterization of antibodies induced by vaccination with hepatitis C virus envelope glycoproteins. J. Infect. Dis. 202:862–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Reyes-del Valle J, Hodge G, McChesney MB, Cattaneo R. 2009. Protective anti-hepatitis B virus responses in rhesus monkeys primed with a vectored measles virus and boosted with a single dose of hepatitis B surface antigen. J. Virol. 83:9013–9017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Reyes del Valle J, et al. 2007. A vectored measles virus induces hepatitis B surface antigen antibodies while protecting macaques against measles virus challenge. J. Virol. 81:10597–10605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Richardson C, et al. 1986. The nucleotide sequence of the mRNA encoding the fusion protein of measles virus (Edmonston strain): a comparison of fusion proteins from several different paramyxoviruses. Virology 155:508–523 [DOI] [PubMed] [Google Scholar]
- 57. Rollier CS, et al. 2007. Vaccine-induced early control of hepatitis C virus infection in chimpanzees fails to impact on hepatic PD-1 and chronicity. Hepatology 45:602–613 [DOI] [PubMed] [Google Scholar]
- 58. Schulze zur Wiesch J, et al. 2005. Broad repertoire of the CD4+ Th cell response in spontaneously controlled hepatitis C virus infection includes dominant and highly promiscuous epitopes. J. Immunol. 175:3603–3613 [DOI] [PubMed] [Google Scholar]
- 59. Shoukry NH, et al. 2003. Memory CD8+ T cells are required for protection from persistent hepatitis C virus infection. J. Exp. Med. 197:1645–1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Spielhofer P, et al. 1998. Chimeric measles viruses with a foreign envelope. J. Virol. 72:2150–2159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Stamataki Z, Coates S, Abrignani S, Houghton M, McKeating JA. 2011. Immunization of human volunteers with hepatitis C virus envelope glycoproteins elicits antibodies that cross-neutralize heterologous virus strains. J. Infect. Dis. 204:811–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Strickland GT, El-Kamary SS, Klenerman P, Nicosia A. 2008. Hepatitis C vaccine: supply and demand. Lancet Infect. Dis. 8:379–386 [DOI] [PubMed] [Google Scholar]
- 63. Tani H, et al. 2007. Replication-competent recombinant vesicular stomatitis virus encoding hepatitis C virus envelope proteins. J. Virol. 81:8601–8612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Tellinghuisen TL, Evans MJ, von Hahn T, You S, Rice CM. 2007. Studying hepatitis C virus: making the best of a bad virus. J. Virol. 81:8853–8867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Thimme R, et al. 2001. Determinants of viral clearance and persistence during acute hepatitis C virus infection. J. Exp. Med. 194:1395–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Torresi J, Johnson D, Wedemeyer H. 2011. Progress in the development of preventive and therapeutic vaccines for hepatitis C virus. J. Hepatol. 54:1273–1285 [DOI] [PubMed] [Google Scholar]
- 67. Tscherne DM, et al. 2006. Time- and temperature-dependent activation of hepatitis C virus for low-pH-triggered entry. J. Virol. 80:1734–1741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. von Hahn T, et al. 2007. Hepatitis C virus continuously escapes from neutralizing antibody and T-cell responses during chronic infection in vivo. Gastroenterology 132:667–678 [DOI] [PubMed] [Google Scholar]
- 69. Wang Z, et al. 2001. Recombinant measles viruses expressing heterologous antigens of mumps and simian immunodeficiency viruses. Vaccine 19:2329–2336 [DOI] [PubMed] [Google Scholar]
- 70. Wertheimer AM, et al. 2003. Novel CD4+ and CD8+ T-cell determinants within the NS3 protein in subjects with spontaneously resolved HCV infection. Hepatology 37:577–589 [DOI] [PubMed] [Google Scholar]
- 71. Whidby J, et al. 2009. Blocking hepatitis C virus infection with recombinant form of envelope protein 2 ectodomain. J. Virol. 83:11078–11089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. World Health Organization 2009. Global reductions in measles mortality 2000–2008 and the risk of measles resurgence. Wkly. Epidemiol. Rec. 84:505–516 [PubMed] [Google Scholar]
- 73. World Health Organization 2010. Monitoring progress towards measles elimination. Wkly. Epidemiol. Rec. 85:490–494 [PubMed] [Google Scholar]
- 74. Zuniga A, et al. 2007. Attenuated measles virus as a vaccine vector. Vaccine 25:2974–2983 [DOI] [PMC free article] [PubMed] [Google Scholar]