

Abstract
The henipaviruses, Hendra virus (HeV) and Nipah virus (NiV), are paramyxoviruses discovered in the mid- to late 1990s that possess a broad host tropism and are known to cause severe and often fatal disease in both humans and animals. HeV and NiV infect cells by a pH-independent membrane fusion mechanism facilitated by their attachment (G) and fusion (F) glycoproteins. Here, several soluble forms of henipavirus F (sF) were engineered and characterized. Recombinant sF was produced by deleting the transmembrane (TM) and cytoplasmic tail (CT) domains and appending a glycosylphosphatidylinositol (GPI) anchor signal sequence followed by GPI-phospholipase D digestion, appending a trimeric coiled-coil (GCNt) domain (sFGCNt), or deleting the TM, CT, and fusion peptide domain. These sF glycoproteins were produced as F0 precursors, and all were apparent stable trimers recognized by NiV-specific antisera. Surprisingly, however, only the GCNt-appended constructs (sFGCNt) could elicit cross-reactive henipavirus-neutralizing antibody in mice. In addition, sFGCNt constructs could be triggered in vitro by protease cleavage and heat to transition from an apparent prefusion to postfusion conformation, transitioning through an intermediate that could be captured by a peptide corresponding to the C-terminal heptad repeat domain of F. The pre- and postfusion structures of sFGCNt and non-GCNt-appended sF could be revealed by electron microscopy and were distinguishable by F-specific monoclonal antibodies. These data suggest that only certain sF constructs could serve as potential subunit vaccine immunogens against henipaviruses and also establish important tools for further structural, functional, and diagnostic studies on these important emerging viruses.
INTRODUCTION
Hendra virus (HeV) and Nipah virus (NiV) are closely related and recently emerged zoonotic pathogens that comprise the genus Henipavirus within the family Paramyxoviridae (28, 29). Both HeV and NiV are highly pathogenic, possessing an unusually broad species tropism and are classified as biosafety level 4 (BSL-4) select agents. Fruit bats, primarily of the genus Pteropus, which cover a broad geographic range over Southeast Asia and the western Pacific, are recognized as the principal host reservoir species of both NiV and HeV (21, 30, 32). Since the discovery of HeV in 1994 and later NiV in 1998 during outbreaks of infection and disease in humans associated with horses in Australia (54) and pigs in Malaysia, respectively (20), each has continued to cause spillover events resulting in morbidity and mortality in both humans and animals. To date, there have been 38 occurrences of HeV infection in horses in Australia involving 4 human fatalities among 7 cases of infection, along with the identification of a seropositive dog in 2011, and 24 of these episodes have occurred since June 2011 (reviewed in references 14 and 63; 3). Subsequent NiV outbreaks have been reported only in Bangladesh and India, with a total of 15 occurrences, with the most recent being in 2012 (4; reviewed in reference 58). Human cases of NiV infection in these areas have been associated with higher mortality rates of up to >75%, along with both food-borne and person-to-person transmission (16, 31, 36, 37, 46).
Serologic and nucleic acid evidence of henipavirus infection, along with a few cases of virus isolation, among various bat species has been reported across a wide geographic range from Oceania and Australia, China, and Southern Asia to Africa (5, 13, 26, 27, 30, 35, 36, 45, 61, 67). Although no connection to human disease occurrence has been reported, these observations suggest a potential threat of henipavirus spillovers in these regions. With no approved vaccines or therapeutics against NiV and HeV, they pose a continued emergent and biosecurity threat to both livestock and human populations.
HeV and NiV possess two membrane-anchored glycoproteins in the envelope of the viral particle. One glycoprotein is required for host cell receptor recognition and attachment and is a tetrameric dimer of homodimers designated the G glycoprotein which, unlike the majority of paramyxovirus attachment glycoproteins, has neither hemagglutination nor neuraminidase activities (29, 41). The other protein is the fusion (F) glycoprotein, which is a trimeric class I fusogenic envelope glycoprotein containing two heptad repeat (HR) regions and a hydrophobic fusion peptide (Fp) (reviewed in reference 62). The henipavirus F is synthesized as a precursor F0 that undergoes posttranslational cleavage by host cell cathepsin L within the endosomal compartment (25, 56, 57) during an endocytosis and recycling process (51), yielding the fusogenic F1 (a larger C-terminal fragment) and F2 (a smaller N-terminal fragment) subunits that are held together by a disulfide bond. In the mature form of F, the Fp is situated at the N terminus of F1, followed by the first HR (HRA) and the second HR (HRB) domains toward the C terminus of F1 and preceding its transmembrane (TM) domain (reviewed in reference 62).
Following henipavirus G-glycoprotein attachment to the host receptor protein ephrin-B2 or -B3 (reviewed in reference 69), virus infection occurs through a pH-independent membrane fusion process facilitated by F (reviewed in reference 64). The precise details of the receptor binding and fusion-triggering process remain poorly understood, but for the henipaviruses, it is believed to involve conformational changes in G upon receptor binding that lead to the activation and triggering of F (reviewed in reference 44). Upon triggering, F undergoes significant conformational rearrangements, folding from a metastable form to a lower energy state facilitating the exposure and insertion of the Fp into the target cell membrane and the refolding of the monomers within the F trimer. This refolding involves the reorganization of the HRA domains in the now Fp-anchored extended trimer core together with the refolding of the HRB helices within the F stalk that pack into the groves of the HRA trimer core in an antiparallel manner, forming the six-helix bundle structure concomitant with the merger of the virus and cell membranes (reviewed in reference 42). Multiple molecular details of the F-glycoprotein refolding process have been revealed in the structural solutions of both a post- and prefusion conformation of two paramyxovirus F glycoproteins, human parainfluenza virus type 3 (hPIV3) and parainfluenza virus type 5 (PIV5), respectively (72, 73).
The henipavirus F likely undergoes a similar refolding process from a prefusion metastable state to a postfusion configuration following its activation. To investigate this possibility, we explored a diverse set of strategies to engineer and produce soluble forms of henipavirus F (sF), including mutagenesis of hydrophobic residues, a glycosylphosphatidylinositol (GPI) anchoring technique (39), and appending of a trimeric coiled-coil domain (GCNt) (33). Using this battery of recombinant sF glycoprotein constructs, we detail several important biochemical, functional, structural, and immunogenic characteristics of the henipavirus F glycoprotein. The data indicate that sF is released from cells as an F0 precursor, all versions of sF could be purified as apparent trimers, and all were recognized by specific antiserum. Surprisingly, however, only a GCNt-appended sF trimer (sFGCNt) could elicit a virus-neutralizing antibody response in mice. In addition, purified sFGCNt trimer could be cleaved in vitro at the correct location into its disulfide-linked F1 plus F2 subunit form in a refolding process that could be captured by HRB peptide. These pre- and postfusion forms of sFGCNt trimer were also distinguishable by the binding of F-specific monoclonal antibodies (MAbs), and electron microscopy (EM) analysis of sFGCNt- and non-GCNt-appended sF trimers revealed distinct pre- and postfusion structures. Together, these findings indicate that recombinant henipavirus sFGCNt trimer retains important native structural and biochemical features, making it an ideal tool for future structural studies and diagnostics and vaccine development.
MATERIALS AND METHODS
Cells, viruses, antibodies, and peptides.
HeLa-USU cells have been described previously (8). 293T cells were provided by G. Quinnan (Uniformed Services University), and cells of the HeLa-PLD cell line stably expressing phospholipase D (PLD) were a gift from D. Sevlever (Mayo Clinic, Jacksonville, FL). All cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 2 mM l-glutamine and 10% cosmic calf serum (D-10). All medium reagents were obtained from Quality Biologicals, Gaithersburg, MD. G418 (Invitrogen Corp., Carlsbad, CA) was used at 400 μg/ml for culturing HeLa-PLD cells. Recombinant vaccinia viruses expressing full-length NiV F (vKB7) and HeV F (vKB1) have been previously described (11, 12). Polyclonal rabbit antisera against HeV F1 or F2 that are NiV cross-reactive have been described previously (11, 12). Rabbit anti-S-peptide-tag antibody, horseradish peroxidase (HRP) conjugated, was from Bethyl Laboratories, Inc., Montgomery, TX. Sera from nonimmune and gamma-irradiated NiV-infected African green monkeys (AGMs) were provided by T. Geisbert (University of Texas Medical Branch, Galveston, TX). The N-terminal biotinylated NiV-FC2 peptide corresponding to the predicted HRB region (residues 453 to 488) has been described previously (10). S peptide was synthesized by the Bioinstrumentation Center, Uniformed Services University.
Design of NiV and HeV sF constructs.
The predicted ectodomains of the NiV and HeV F sequences (10) were codon optimized and synthesized by (Geneart Inc., Germany). The NiV and HeV F sequences were synthesized on the basis of sequences cloned early (11, 12), which differed from sequences published later. These changes were N67D and N305D in NiV F and D255G and A263T in HeV F. The predicted TM anchor domain (residues 488 to 510) and the C-terminal cytoplasmic tail (CT) domain (residues 511 to 546) (10) of the NiV and HeV F-coding sequences were replaced by either the S-peptide tag (KETAAAKFERQHMDS) or the GCNt motif (MKQIEDKIEEILSKIYHIENEIARIKKLIGE) (33), followed by a factor Xa protease cleavage site (IEGR) and the S-peptide tag, generating NiV or HeV sF and NiV or HeV sFGCNt. In another construct, the TM and CT of NiV F were replaced by the S-peptide tag followed by the GPI anchor signal sequence (IDPNKGSGTTSGTTRLLSGHTCFTLTGLLGTLVTMGLLT) (39), generating NiV sFGPI. The predicted Fp domain (residues 110 to 122) (10) was deleted (dFp), generating HeV or NiV sFdFp and NiV sFGCNtdFp by site-directed mutagenesis using a QuikChange II site-directed mutagenesis kit (Stratagene, Cedar Creek, TX). Other mutants (NiV sF I114N, I120N, and GF330KY; HeV sF V114N and I120N; and NiV sFGCNt I120N and GF330KY) were prepared by a similar method. All the above-described mutants were derived in the context of the HeV and NiV sF or sFGCNt constructs containing a C-terminal S-peptide tag. All constructs were cloned into a promoter-modified pcDNA3.1 vector with a hygromycin selection marker (17).
Transient expression and generation of sF-expressing stable cell lines.
Human 293T cells were transfected with different sF plasmid constructs using Fugene (Roche, Indianapolis, IN). Cells were transfected with 2 μg DNA and 6 μl Fugene per well of a 60% confluent 6-well tissue culture plate. At 48 h posttransfection, the culture medium was either replaced with selection medium (D-10 supplemented with 150 μg/ml of hygromycin B [Invitrogen]) or harvested for S-protein agarose (EMD Biosciences Inc., Madison, WI) precipitation. Resistant cells were then subjected to two rounds of limiting-dilution cloning to obtain a stable sF-expressing clone (17). Expression of NiV sFGPI was performed by transfecting HeLa-PLD or HeLa-USU cells with the NiV sFGPI construct using 6 μl Lipofectamine (Invitrogen) with 3 μg DNA per well of a 60% confluent 6-well tissue culture plate following the manufacturer's instructions. At 48 h posttransfection, culture medium was replaced with DMEM without serum (D-0) with or without 0.1 unit of phospholipase C (PLC; Invitrogen). Cells and culture supernatant were harvested after 1 h of PLC treatment at 37°C. Culture supernatant of cells treated with D-0 without PLC was harvested after 2, 16, and 22 h. Cells were harvested at 22 h. The NiV sFGPI-expressing HeLa-PLD cells were prepared at 48 h posttransfection by replacing the culture medium with D-10 supplemented with 400 μg/ml of G418 and 300 μg/ml of hygromycin B for selection. Two rounds of limiting-dilution cloning were performed to select an expressing clone.
Large-scale expression and purification of sF.
Preparation of the various sF constructs from 293T stable cell lines or the NiV sFGPI construct from the HeLa-PLD cell line was carried out using serum-free conditions and employing a combination of affinity and size-exclusion chromatography steps (17). Briefly, a confluent 175-cm2 tissue culture flask of an sF-expressing culture maintained in D-10 containing 150 μg/ml hygromycin B was harvested and used to seed one 1,700-cm2 roller bottle in D-10. When cell cultures were ∼80 to 90% confluent, the cell monolayers were washed twice with phosphate-buffered saline (PBS), and the medium was replaced with Opti-MEM medium (Invitrogen). Culture medium was collected after 4 days and centrifuged at 4°C for 15 min at 5,000 × g, filtered through a 0.2-μm-pore-size low-protein-binding membrane (Corning, Inc., Lowell, MA), and passed through an S-protein agarose (EMD Biosciences) affinity column. The column was washed with PBS, 0.1% Triton X-100, 0.1 M arginine, and the bound sF was eluted with 0.2 M citrate, 0.2 M l-arginine, pH 2, followed by immediate pH neutralization of the eluent with 1 M HEPES buffer, pH 9.0. The eluate was concentrated (∼10-fold) to a volume of ∼2 to 3 ml and buffer exchanged into PBS, 0.01% Triton X-100 using Amicon Ultra centrifugal filter units (Millipore, Billerica, MA). The concentrated sF material was then purified by size-exclusion chromatography using a HiLoad 16/60 Superdex 200 preparative-grade gel filtration column (GE Healthcare, Piscataway, NJ), fractions were analyzed by 3 to 12% native PAGE (Invitrogen), and fractions containing sF trimer were pooled. In some experiments, the appended 15-amino-acid S-peptide tag within sFGCNt was removed using an immobilized factor Xa cleavage capture kit (EMD Biosciences).
Analytical size-exclusion chromatography.
To analyze the oligomeric profile of the various sF preparations, 0.3 to 1 mg of the S-agarose-purified sF material was applied to a Superdex 200 10/300 GL gel filtration column (GE Healthcare) calibrated with a series of standards. Fractions of 400 μl were collected, and 1 to 5 μl from each fraction encompassing the entire peak of separated material (monitored by absorbance at 280 nm) was analyzed on a 3 to 12% native polyacrylamide gel, followed by Western blotting and detection using a rabbit anti-S-peptide-tag antibody, HRP conjugated (Bethyl Laboratories). The molecular weights of the various sF construct preparations were calculated on the basis of the elution volume corresponding to the highest absorbance in the elution peak measured (17).
Sucrose gradient centrifugation analysis.
Soluble and full-length F proteins were analyzed by sucrose gradient centrifugation as described previously (9, 17). Briefly, 6 ml of 5% sucrose was underlaid with 6 ml of 20% sucrose in polyallomer 14- by 95-mm tubes. A linear sucrose gradient was generated using a Biocomp gradient master (Biocomp, Frederickton, NB, Canada) at an angle of 81.5° for 1 min 55 s at a speed of 15 rpm. Approximately 50 to 200 μg of sF was overlaid on top of the gradients. Analysis of full-length recombinant F was carried out by infection of 12 × 106 HeLa-USU cells using vKB1 or vKB7 with a multiplicity of infection (MOI) of 10 for 24 h. The cells were lysed in 0.5 ml of PBS, 5% n-dodecyl-β-d-maltoside (DDM) and clarified by centrifugation, and the lysate was overlaid onto the sucrose gradient. The gradients were centrifuged at 40,000 rpm for 20 h at 4°C using an SW40 rotor (Beckman Coulter, Inc.). A total of 14 fractions of ∼800 μl each were collected from the bottom to the top of the gradient using a Beckman fraction recovery system and automated fraction collector. To analyze the fractions, 5 μl and 15 μl of each collected fraction of the sF and the full-length F gradients were resolved on 3 to 12% blue native (BN) polyacrylamide gels (Invitrogen), followed by Western blotting and detection using a rabbit anti-HeV F1 for native F and rabbit anti-S-peptide tag for sF.
Sedimentation equilibrium analytical ultracentrifugation.
Analytical ultracentrifugation analysis was performed using a Beckman XL-A Optima analytical ultracentrifuge with an An-60 Ti rotor at 4°C. Samples of purified recombinant sF were dialyzed overnight into Tris-buffered saline (TBS; pH 8.0), applied at initial concentrations of 1, 2, and 4 mg/ml for NiV sFGCNt and 0.75, 1.5, and 3 mg/ml for HeV sFGCNt, and centrifuged at rotor speeds of 6,500 and 7,500 rpm. Data were acquired at two wavelengths per rotor speed, and data were fit, using the NONLIN analysis program, to a single-species model of the natural logarithm of the absorbance versus radial distance squared (38). Solvent density and protein partial specific volume parameters were calculated by taking into account the solvent and protein composition, respectively (43).
Specimen preparation and electron microscopic imaging of negative-stained sF samples.
Pre- and postfusion F glycoprotein was prepared for electron microscopy using the conventional negative-staining protocol (55). Briefly, 3 μl of sample was pipetted onto a glow-discharged carbon-coated grid and stained with 0.75% (wt/vol) uranyl formate. Protein samples were imaged at room temperature with a Tecnai T12 electron microscope operated at 120 kV using low-dose procedures. Images were recorded at a magnification of ×71,138 and a defocus value of ∼1.5 μm on a Gatan US4000 charge-coupled-device camera. All images were binned (2 by 2 pixels) to obtain a pixel size of 4.16 Å on the specimen level. A total of 4,778 and 4,024 pre- and postfusion F-glycoprotein particles were manually excised using the Boxer program (EMAN [version 1.9] software suite) (2). The two-dimensional reference free alignment and classification of the raw particles were performed using the EMAN (version 1.9) refine2d.py program (47). Pre- and postfusion F-glycoprotein particles were classified into 50 and 40 classes, respectively.
Deglycosylation and trypsin cleavage of sF.
Five micrograms of purified sF was digested with 2,500 units of peptide: N-glycosidase F (PNGase F) (New England BioLabs [NEB] Inc. Ipswich, MA) or 1,000 or 2,500 units of endoglycosidase H (EndoH) in a 30-μl reaction volume for 3 h at 37°C. To cleave purified F0 precursor to its F1 and F2 subunits, 1 μg of purified sF was incubated with 100, 50, or 10 ng of trypsin (NEB) in a 10-μl reaction volume of 1× buffer at 4°C for overnight. The reaction was stopped with 1 μl of 10× complete protease inhibitor cocktail (Roche). The digested product (1 μl) was analyzed by 4 to 12% Bis-Tris SDS-PAGE (Invitrogen) under reducing or nonreducing conditions (with or without 2% β-mercaptoethanol in NuPAGE sample buffer [SAB]), followed by Western blotting and detection using rabbit anti-S-peptide-tag antisera for deglycosylated F and rabbit anti-HeV F1 and F2 antisera for the cleaved F1 and F2 subunits.
N-terminal amino acid sequence analysis.
Purified NiV sFGCNt that was trypsin cleaved in vitro was separated by 4 to 12% BT SDS-PAGE and then transferred to polyvinylidene difluoride (PVDF) membranes, stained with 0.1% Coomassie brilliant blue R-250, and destained in 1% aldehyde-free acetic acid with 40% methanol, followed by a quick rinse with 100% methanol until protein bands were visible. The F1 and F2 protein bands were excised from the membrane and subjected to N-terminal amino acid sequence analysis. Analysis was performed on an automated protein/peptide sequencer (model 491A; Applied Biosystems, Foster City, CA) using the pulsed liquid module in conjunction with a phenylthiohydantoin amino acid separation system (model 140C microgradient delivery system; Applied Biosystems). Quantitative analysis of the sequence data was performed using a model 610A (version 2.1) data analysis program (Applied Biosystems).
Generation of F-specific monoclonal antibodies.
The sFGCNt with the S peptide removed by cleavage and the S-peptide-tagged sFdFp purified glycoproteins were used to immunize BALB/c mice. All animal studies were carried out under an approved protocol for animal experiments obtained from the Uniformed Services University Animal Care and Use Committee. All mice were immunized 4 times with various sF preparations, as indicated in Table 1, with 12 μg of protein in a Sigma adjuvant system (Sigma-Aldrich Co. LLC, St. Louis, MO) at intervals of 30 days by both intraperitoneal and subcutaneous inoculations. Serum samples were taken at 7 to 10 days following the third immunization. Mouse splenic lymphocytes were isolated 4 days following a final immunization without adjuvant and fused with SP2/0 cells using polyethylene glycol by standard methods. Hybridoma supernatants producing NiV F-specific antibodies were identified by enzyme-linked immunosorbent assay (ELISA) using purified S-peptide-cleaved NiV sFGCNt and confirmed by immunoprecipitation with native full-length F expressed in HeLa-USU cells. Positive hybridomas were subjected to two rounds of limiting-dilution cloning. The anti-F MAbs were prepared under serum-free conditions using HyClone SFM4MAb (Thermo Fisher Scientific Inc., Rockford, IL) and purified by protein G-Sepharose affinity chromatography.
Table 1.
Mouse henipavirus sF immunization scheme and endpoint ELISA titers of serum samples from mice immunized with different NiV and HeV sF glycoproteinsa
| Mouse no. | Immunogen |
ELISA antigen | Endpoint titer (103) |
||
|---|---|---|---|---|---|
| 1st, 2nd, and 3rd immunizations | Final immunization | 3rd bleed | Final bleed | ||
| 1 | NiV sFGCNt | NiV sFGCNt | NiV sFGCNt | 1,280 | 2,560 |
| 2 | NiV sFGCNt | NiV sFGCNt | NiV sFGCNt | 1,280 | 1,280 |
| 3 | HeV sFGCNt | HeV sFGCNt | HeV sFGCNt | 320 | 320 |
| 4 | HeV sFGCNt | HeV sFGCNt | HeV sFGCNt | 320 | 320 |
| 5 | NiV sFdFp | NiV sFGCNt | NiV sFdFp | 2,560 | 2,560 |
| 6 | NiV sFdFp | NiV sFGCNt | NiV sFdFp | 1,280 | 1,280 |
| 7 | HeV sFdFp | HeV sFGCNt | HeV sFdFp | 1,280 | 1,280 |
| 8 | HeV sFdFp | HeV sFGCNt | HeV sFdFp | 2,560 | 2,560 |
Each mouse was immunized a total of 4 times using the indicated sF glycoproteins. Serum samples were harvested following the 3rd immunization (3rd bleed). A final serum sample was also harvested following the 4th immunization (final bleed), prior to hybridoma development. An indirect ELISA was carried out using the indicated sF glycoprotein as the coat antigen and serial dilution of the serum samples. The S-peptide tag of all sFGCNt glycoproteins used in the ELISA was removed by factor Xa digestion and S agarose. Serum samples from mouse numbers 5 to 8 were preincubated with S peptide to quench any S-peptide-reactive antibodies in the samples. dFp, fusion peptide deleted.
ELISA.
MAbs and antibodies in serum samples specific for F were detected using 96-well ELISA plates coated with 30 ng of sF washed with PBS containing 0.05% Tween 20 (PBST) and blocked with PBST containing 5% bovine serum albumin (BSA)–PBST for 1 h at 37°C. Serum samples were diluted in 1% BSA–PBST in 2-fold series, incubated for 1 h at 37°C, and assayed in duplicate. When necessary, serum was preblocked in 0.5 mg/ml of S-peptide solution at 4°C for 1 h. For detection, goat anti-mouse HRP (1:10,000 in PBST; Thermo Fisher Scientific) was added; and the mixture was incubated for 1 h at 37°C, washed with PBST, and developed with ABTS [2,2′-azinobis(3-ethylbenzthiazolinesulfonic acid)] substrate (Roche). The absorbance of each well was measured at 405 nm, and the average value was calculated from duplicates.
HeV and NiV neutralization assays.
Infectious HeV and NiV assays were carried out at the BSL-4 laboratory of the CSIRO Livestock Industries, Australian Animal Health Laboratory, Geelong, Australia. Mouse serum samples or purified MAbs were serially diluted in duplicate or triplicate wells in a 96-well plate in Eagle's minimal essential medium (EMEM) and mixed with 200 50% tissue culture infective doses (TCID50) of either HeV or NiV for 30 min at 37°C. Dilutions of purified MAbs started at 200 μg/ml. A total of 2 × 104 Vero cells were added to each well, incubated for 3 days at 37°C, and observed for signs of viral cytopathic effect (CPE). The serum neutralization titer (SNT), or the lowest neutralizing concentration of MAb, was determined by the highest dilution in which a viral CPE was fully neutralized (absent) in at least one of the duplicate or triplicate wells.
Immunoprecipitation and NiV-FC2 peptide capture assay.
Precipitation of sF in culture supernatants and/or cell lysates using S agarose has been previously described (17). Briefly, sF-expressing cells were lysed in 500 μl of lysis buffer (1% Triton X-100, 0.1 M Tris, pH 8.0, 0.1 M NaCl) and clarified by centrifugation, or sF-expressing cell culture supernatants were used by adding 30 μl of 50% S-agarose slurry (EMD Biosciences), followed by 1 h incubation at room temperature. To immunoprecipitate sF, 1 or 0.5 μg of sF was mixed with either 1 μl NiV-immune AGM serum or 2 μg of MAb in 500 μl of lysis buffer containing 1× complete protease inhibitor, and the mixture was incubated at 4°C overnight. Complexes were precipitated with 50 μl of a 20% protein G-Sepharose slurry for 1 h at room temperature. Immunoprecipitation of native full-length F was performed by infection of 1 × 106 HeLa-USU cells using vKB1 or vKB7 with an MOI of 10 for 24 h, followed by cell lysis in 0.5 ml of lysis buffer supplemented with 1× complete protease inhibitor, and native full-length F was clarified by centrifugation. One microliter of mouse serum or 2 μg of purified MAb was added to the cell lysate, and the mixture was incubated at 4°C overnight. The sample was then precipitated with 50 μl of a 20% protein G-Sepharose slurry for 1 h at room temperature. For the FC2 peptide capture assay, 1 μg of purified sF was cleaved as described above, followed by addition of protease inhibitor. Biotinylated NiV-FC2 peptide (2 μg) was added to some samples. Samples were heated before or after trypsin cleavage or before or after NiV-FC2 peptide addition, as indicated. NiV sF-FC2 complex was precipitated for 1 h at 4°C using 30 μl of a 50% avidin agarose slurry (Thermo Fisher Scientific) in a 200-μl volume by adding 160 μl of lysis buffer with 1× complete protease inhibitor. In all cases, the precipitated beads with protein complex were washed 3 times with lysis buffer and boiled in 50 μl of SAB. To analyze the precipitated product, a 25-μl sample was applied to a 4 to 12% BT SDS-polyacrylamide gel (Invitrogen), followed by Western blotting and detection using rabbit anti-HeV F1 or rabbit anti-S-peptide-tag antibody.
RESULTS
Transient expression analysis of soluble F-glycoprotein constructs.
All sF constructs possessed a C-terminal S-peptide tag, and the expression of sF from plasmid-transfected cells in both cell lysates and culture supernatants was evaluated by S-protein agarose precipitation followed by SDS-PAGE analysis. Truncation of the wild-type NiV and HeV F-coding sequences to remove the TM and CT domains and appending of the S-peptide tag yielded constructs that expressed and that could be precipitated and detected from expressing-cell lysates, but only a smaller amount of HeV sF and no NiV sF was released from expressing cells (Fig. 1A). Codon optimization of both the NiV and HeV sF constructs resulted in only a minor increase in expression compared to that for each of the wild-type sequences, with little apparent enhancement of the release of sF into expressing-cell culture supernatants (Fig. 1A).
Fig 1.

Transient expression of a battery of sF glycoprotein constructs. A panel of NiV and HeV sF constructs and empty vector control (vector) were transfected into human 293T cells (A to D). At 48 h posttransfection, culture medium was harvested and cells were lysed and clarified by centrifugation. All constructs tested except wild type are codon optimized. (A) Expression of wild type and codon-optimized constructs without and with GCNt. (B) Expression of codon-optimized NiV and HeV sF and different mutants of NiV sF. (C) Expression of codon-optimized sF, sFGCNt, and different sF mutants of HeV. (D) Expression of codon-optimized NiV and HeV sFGCNt and different mutants of NiV sFGCNt. Bands migrating below F0 in panels C and D are most likely N-terminal degradation products (probing scheme using anti-S peptide). (E) Expression of GPI-anchored NiV F. The GPI-anchored NiV F construct was transfected into HeLa-USU (top) and HeLa-PLD (bottom) cells in a series of duplicate wells in 6-well tissue culture plates. At 48 h posttransfection, D-10 was replaced with either serum-free medium (D-0) or serum-free medium supplemented with 0.1 U of PLC (D-0+PLC). The culture supernatant containing PLC and the cells were harvested after 1 h; supernatants without PLC were harvested at 2, 16, and 22 h and cells were harvested at 22 h. A control D-10 supernatant was also harvested at 48 h. Cells were lysed and clarified by centrifugation. Cleared cell lysates and supernatants from panels A to E were precipitated with S-protein agarose. The precipitated proteins were resolved by 4 to 12% BT SDS-PAGE and detected by Western blotting with rabbit anti-S-peptide antibody. Sup, culture supernatant; Lys, cell lysate; dFp, fusion peptide deleted.
Addition of a GCN trimeric helix and mutation of select hydrophobic residues in soluble F glycoproteins.
It was previously demonstrated that appending a GCN trimeric helical domain (GCNt) (33) could stabilize trimers of the human immunodeficiency virus type 1 (HIV-1) truncated envelope glycoprotein known as gp140 (70, 71), and more recently, the GCNt domain was used to maintain metastable prefusion conformations of soluble F glycoproteins of several paramyxoviruses (65, 68, 73). In the attempt to enhance the release of sF from expressing cells, we examined this technique in conjunction with altering select hydrophobic residues within the predicted hydrophobic Fp domain or deleting the Fp domain entirely. In addition, a GPI anchoring strategy was also tested (39).
Using the codon-optimized sequences and appending the NiV and HeV sF glycoproteins with the GCNt-coding sequence downstream of their HRB domains resulted in comparable expression in cell lysates but also significantly enhanced the amount of sF released into the culture supernatants of construct-transfected cells in comparison to that for the wild-type and codon-optimized-expression constructs that were assayed in parallel (Fig. 1A). Presumably, on the basis of the previous report by Yin et al. (73), which used a PIV5 sF construct appended with a GCNt domain, the HeV and NiV sFGCNt glycoproteins produced here are likely folded into a prefusion conformation. Thus, for the purposes of comparing pre- and postfusion forms of henipavirus sF in subsequent experiments, we also constructed non-GCNt-appended versions of sF using several strategies aimed at enhancing the release of protein from expressing cells, which would presumably be in a postfusion conformation. A further consideration was that the non-GCNt-appended versions of sF produced within expressing cells were poorly released into culture supernatants because of protein aggregation due to the hydrophobic features of the glycoprotein. In an attempt to ameliorate this possibility, the Fp domain of F was mutated in the non-GCNt-appended sF constructs, and isoleucine and valine residues were replaced by asparagine within the Fp domain, a strategy previously shown to reduce aggregation in the human respiratory syncytial virus (RSV) F glycoprotein (48). Using the codon-optimized coding sequences of NiV and HeV sF glycoproteins, replacing the hydrophobic residues I120N and I114N and the hydrophobic residues I120N and V114N, respectively, or deleting the entire Fp domain, the release of the non-GCNt-appended sF constructs could be significantly enhanced for both NiV (Fig. 1B) and HeV (Fig. 1C).
It was also consistently observed that some of the unaltered wild-type HeV sF construct was released from expressing cells more efficiently than NiV sF (Fig. 1A and B). A comparison of the hydrophobicity plots by graphic display of Kyte and Doolittle analysis (40) of NiV and HeV sF highlighted two amino acids (at positions 329 and 330) in NiV F notably more hydrophobic than those in HeV F: glycine and phenylalanine versus lysine and tyrosine at positions 329 and 330, respectively. We hypothesized that mutation of these two residues within the NiV sF-coding sequence to the corresponding residues in HeV sF (mutation GF330KY) could result in a more comparable production and release of NiV sF. However, the expression and release of this mutant (NiV sFGF330KY) showed little enhancement (Fig. 1B). Importantly, the combination of either the Fp or GF330KY mutation in the NiV sFGCNt construct had no deleterious effect on the already enhanced release of NiV sFGCNt from expressing cells (Fig. 1D).
Phospholipase D and C digestion of GPI-anchored NiV F ectodomain.
Because of the poor release of the unaltered wild-type NiV sF construct in comparison to wild-type HeV sF from expressing cells, we examined the possibility of incorporating a GPI anchor signal sequence for producing non-GCNt-appended NiV sF. This GPI-anchored NiV F construct was generated by replacing the F-glycoprotein TM and CT domains with the S-peptide tag as before and then appending a GPI signal sequence downstream. The GPI-anchored NiV F could then be released from the surface of expressing cells by PLD- or PLC-mediated cleavage. The plasmid expression construct was transfected into cells of the HeLa-USU or HeLa-PLD cell line, the latter of which is a HeLa cell line stably expressing the bovine GPI-specific PLD. As shown in Fig. 1E (top), the GPI-anchored NiV sF could be cleaved off expressing cells by the addition of exogenous PLC and precipitated from the cell culture supernatant using S-protein agarose. In addition, when the construct was transfected and expressed in HeLa-PLD cells, the GPI-anchored NiV sF was spontaneously released from cells without a need for any phospholipase supplement and could be precipitated from the cell culture medium using S-protein agarose (Fig. 1E, bottom). This GPI-anchored NiV sF construct was subsequently used to produce a stably expressing cell line using the HeLa-PLD cells. The remaining henipavirus sF constructs within this panel, with the exception of the wild-type NiV sF, were used to generate stably expressing cell lines using human 293T cells. On the basis of the levels of sF glycoprotein released into culture supernatants, the HeV and NiV sFGCNt and sFdFp, NiV FGPI, NiV sFGCNtdFp, and wild-type HeV sF were selected for purification and further analyses.
Size-exclusion chromatography analysis of the sF glycoproteins.
Affinity chromatography purification of the various sF preparations using S agarose revealed that the GCNt-appended sF constructs produced the greatest yield of protein, followed by dFp. The lowest yields of purified sF glycoproteins were obtained from the wild-type HeV sF and the NiV sFGPI. The sF glycoprotein yields ranged from 4.3 μg/106 cells to 0.43 μg/106 cells. Using these purified sF preparations, we determined their apparent molecular masses by size-exclusion chromatography with a calibrated Superdex 200 10/300 analytical-grade column, and the elution profile of each sF preparation is shown in Fig. 2. The apparent molecular mass of each sF preparation was calculated and is shown above the principal elution peak observed for each sF construct. When samples of each fraction obtained across the eluted peak were analyzed by native PAGE, the sF glycoproteins migrated between the 242- and 480-kDa markers, as shown below each sF elution profile in Fig. 2, which was consistent with their calculated molecular masses based on size-exclusion chromatography analysis. Both the NiV sFGCNt and NiV sFGPI glycoprotein preparations contained a significant amount of material that separated as large aggregates which were eluted near the void volume (Vo). These aggregated sF materials also separated as high-molecular-mass material by native PAGE, and notably, the NiV sFdFp construct yielded a distinct-molecular-mass species of sF that migrated above the 480-kDa marker and that was also observed in NiV sFGCNtdFp. By comparison, all the HeV sF preparations contained smaller amounts of aggregated material, and the major peak of eluted sF glycoprotein migrated between the 242- and 480-kDa markers when samples of the fractions across the eluted peak were analyzed by native PAGE (Fig. 2). Overall, the analysis of the panel of sF glycoproteins by gel filtration revealed that the major species of glycoprotein possessed comparable apparent molecular masses ranging from 263 to 357 kDa, which suggests a trimeric oligomeric form of the sF glycoprotein.
Fig 2.

Size-exclusion chromatography analysis of sF glycoproteins. Various S-protein agarose-purified sF glycoprotein preparations (0.3 to 1 mg each) were fractionated on a calibrated Superdex 200 gel filtration column. Each fraction of the separation from the void volume (Vo) to the end of the major protein peak was analyzed by 3 to 12% native PAGE (Invitrogen), followed by Western blotting with rabbit anti-S-peptide antibody to detect sF. Each sF glycoprotein construct is indicated at the top of each elution profile, and the apparent molecular mass of each sF major peak was calculated and is indicated.
Sucrose gradient centrifugation analysis of sF glycoproteins.
To further characterize the oligomeric nature of the sF glycoprotein preparations and to compare them to the native full-length F glycoproteins, we subjected samples of purified sF and cell lysates containing full-length F expressed in HeLa-USU cells to sucrose gradient centrifugation, followed by analysis of gradient fractions by native PAGE. Consistent with the gel filtration chromatography data, all the sF glycoprotein preparations contained some aggregated material which sedimented toward the bottom of the gradient along with a major species that separated toward the middle of the gradient (Fig. 3). The apparent molecular masses of the major sF species ranged from 242 to 480 kDa, which was consistent with the separation pattern of full-length F analyzed in parallel (Fig. 3). The full-length wild-type F glycoprotein did fractionate more toward the top of the gradient in comparison to sF, and this could be due to the presence of detergent or cellular material in the gradient or because wild-type F also retains the TM and CT domains. However, inclusion of the same concentration of n-dodecyl-β-maltoside (DDM) detergent in purified sF did not appear to alter its separation profile (Fig. 3). Taken together, these analyses indicate that sF and full-length F have a similar apparent trimeric oligomeric configuration.
Fig 3.

Sucrose gradient ultracentrifugation analysis of sF glycoproteins and full-length native F glycoprotein. Various S-protein agarose-purified sF glycoprotein preparations (50 to 200 μg each) or a lysate of HeLa-USU cells expressing full-length native F, as indicated, was layered onto individual continuous (5 to 20%) sucrose gradients and fractionated by centrifugation. Each fraction was analyzed on a 3 to 12% native polyacrylamide gel (Invitrogen), followed by Western blotting with rabbit anti-S-peptide antibody to detect sF or rabbit anti-HeV F1 to detect full-length F. Cells expressing full-length F glycoprotein were prepared in buffer containing 5% DDM, and a sample of sFdFp was also prepared in DDM for comparison. The bottom and top of each gradient is indicated. The trimer oligomer species (T) and apparent high-molecular-mass aggregates (A) in each type of sF glycoprotein preparation and the full-length F glycoprotein are indicated.
Sedimentation equilibrium analytical ultracentrifugation and cross-linking analysis of sF glycoproteins.
To confirm that the major species of recombinant-expressed sF is trimeric, we prepared samples of both the NiV and HeV sFGCNt glycoproteins by purification using S-agarose affinity chromatography, followed by preparative size-exclusion chromatography, and subjected these purified sF glycoprotein preparations to analytical ultracentrifugation and cross-linking analyses. A nonglycosylated sF monomer is ∼55 kDa, thus accounting for the 3 to 4 predicted N-linked glycosylation modifications (2, 15, 53); a trimer would be expected to be ∼210 to 225 kDa. Sedimentation equilibrium measurements of NiV and HeV sFGCNt revealed glycoproteins with an apparent molecular mass of ∼221 ± 26 kDa for NiV and 215 ± 18 kDa for HeV, both consistent with a trimeric configuration (Fig. 4A). There was no systematic dependence of apparent molecular mass on protein concentration over a 4-fold range of protein concentrations studied. Nonetheless, analysis of residual differences from the trimeric model revealed a systematic error, suggesting that the sFGCNt indicates a tendency to aggregate, which was also consistent with the gel filtration and sucrose gradient results obtained earlier. However, incubation with bis(sulfosuccinimidy) suberate (BS3) cross-linker indicated that the NiV and HeV sFGCNt purified trimer migrated with an apparent molecular mass that was 3 times that of the non-cross-linked glycoprotein when analyzed by SDS-PAGE (Fig. 4B), indicating that purified sF glycoproteins exist as stable trimers in solution.
Fig 4.

Equilibrium sedimentation analysis of purified NiV and HeV sFGCNt. (A) Analytical ultracentrifugation of NiV and HeV sFGCNt. Representative equilibrium sedimentation data (6,000 rpm) for NiV sFGCNt (2 mg/ml) and HeV sFGCNt (1.5 mg/ml) in TBS (pH 8.0) buffer at 4°C. Data are plotted as absorbance versus the radius from the axis of rotation. The data fit closely to a trimeric complex. The deviation in the data from the linear fit for a trimeric model is plotted (top). (B) Cross-linking of each sFGCNt preparation with BS3. NiV or HeV purified sFGCNt (1 μg per sample) was incubated with different concentrations of BS3 cross-linker, as indicated, for 30 min at room temperature. The reactions were then quenched with 50 mM Tris, and 10 ng of the cross-linked sF samples was analyzed on a 4 to 12% BT SDS-polyacrylamide gel and detected by Western blotting with rabbit anti-S-peptide antibody.
Trypsin cleavage of sF glycoprotein.
We next investigated whether the recombinant sF could be cleaved into its F1 and F2 subunits in vitro. It has been shown that cathepsin L is required to process HeV and NiV F0 to the mature F1 and F2 forms (56, 57); however, our attempts to carry out in vitro cleavage of sF by cathepsin L were unsuccessful. Experiments with a variety of incubation periods, concentrations, and temperatures were carried out, and although immunopurified native HeV F0 has been demonstrated to be cleaved into the F1 and F2 subunits (57), cleavage of purified sF by cathepsin L resulted only in degradation of the protein preparations (data not shown). However, when recombinant sF was treated with trypsin with a concentration ratio of 1:0.01 to 1:0.05 (substrate to enzyme), a specific band with a molecular mass consistent with that of the F1 subunit was observed when the digested sF material was analyzed by SDS-PAGE followed by Western blotting and detection using rabbit anti-HeV F1-specific antisera (Fig. 5A). When the digested sF material was analyzed under nonreducing conditions, the cleaved sF migrated with an apparent molecular mass that was comparable to that of the sF0 precursor (Fig. 5B), indicating that the cleaved product also remained linked by a disulfide bond, as predicted. The apparent molecular mass difference between uncleaved sF and the nonreduced cleaved soluble F1 plus F2 is most likely due to the trypsin cleavage of the factor Xa site and removal of the S-peptide-tag sequence. The trypsin-cleaved sF was also not detectable by anti-S-peptide antibody (data not shown). In addition, when the cleaved sF material was probed using rabbit anti-HeV F2-specific antisera, the F1 band was not detected but the F2 subunit was present (Fig. 5C), and when analyzed under nonreducing conditions in parallel, a band corresponding to the soluble F1 plus F2 glycoprotein that migrated with a molecular mass similar to that of the uncleaved sF was observed, confirming that the cleaved F2 subunit is disulfide bond linked to F1. Taken together, these data indicate that the recombinant sF trimer is unprocessed but can be cleaved by in vitro trypsin treatment to a soluble F1 and F2 disulfide-linked complex.
Fig 5.

Deglycosylation and trypsin cleavage analysis of sF glycoproteins. (A) Digestion of 1 μg NiV sFGCNt with different amounts of trypsin, as indicated, at 4°C overnight. (B, C) Digestion of 1 μg NiV or HeV sFGCNt with 50 ng of trypsin at 4°C overnight. + and −, samples with or without trypsin treatment, respectively, or in reducing or nonreducing β-mercaptoethanol (BME) sample buffer, respectively. (D) Digestion of 5 μg of NiV or HeV sFGCNt with 2,500 units of PNGase F and EndoH for 3 h at 37°C. + and −, with and without PNGase F, respectively; numbers above the lanes for EndoH indicate the amount (103) of EndoH enzyme units. (E) Digestion of 1 μg NiV and HeV sFGCNt with 2,500 units of PNGase F for 3 h at 37°C, followed by 50 ng of trypsin at 4°C overnight. For all experimental samples, 0.1 μg of the undigested and digested products was analyzed by 4 to 12% BT SDS-PAGE and detected by Western blotting with rabbit anti-HeV F1 antibody (A and B), rabbit anti-HeV F2 antibody to detect F2 (C and E), and rabbit anti-S-peptide-tag antibody (D).
Identification of trypsin cleavage site in soluble F0.
To determine whether the in vitro trypsin cleavage described here produced soluble F1 and F2 subunits that were processed at the predicted authentic cleavage site in F0, the trypsin-processed NiV soluble F1 plus F2 glycoprotein was subjected to N-terminal sequencing analysis. N-terminal amino acid sequencing of the F2 subunit generated a 13-residue sequence (ILHYEKLSKIGLV) that was identical to the NiV F amino acid sequence after removal of the predicted leader sequence and consistent with the sequence data derived from infectious HeV (52). N-terminal amino acid sequencing of the soluble F1 subunit yielded a 13-residue sequence of LAGVIMAGVAIGI identical to residues 110 to 122 of HeV and NiV F0, which also confirmed that the cleavage site in soluble F0 by trypsin in vitro was the correct lysine residue (residue 109 of HeV and NiV F0). This result was also consistent with the data obtained from native F glycoprotein from infectious HeV (52).
N-glycosylation analysis of sF glycoproteins.
The ectodomain of both NiV and HeV F is predicted to contain six N-linked glycosylation sites, three on each of F1 and F2 (34). On the basis of mutagenesis experiments, only four sites (two on each of F1 and F2) of NiV F have been shown to be glycosylated when expressed in human 293T cells (2) or Madin-Darby canine kidney (MDCK) cells (53). The same four sites have also been observed to be N glycosylated in HeV F expressed in Vero cells (15). As noted in Materials and Methods, the NiV F-coding sequence used here, derived from a gene cloned early, has an alteration (N67D), in comparison to other published sequences, which is also one of the utilized N-glycosylation sites in F2. Indeed, it was consistently noted that HeV sF migrated at a slightly higher apparent molecular mass in comparison to that for NiV sF when analyzed by SDS-PAGE (Fig. 1 and 5). To determine if this apparent molecular mass difference observed by SDS-PAGE was due to altered N glycosylation, the NiV and HeV sFGCNt glycoproteins were digested with PNGase F, which removes all N-glycosylation moieties, and EndoH, which removes only high mannose and some hybrid types of N-linked carbohydrates. As shown in Fig. 5D, PNGase F digestion of both the NiV and HeV sFGCNt glycoproteins eliminated the difference in migration pattern by SDS-PAGE, suggesting that the two glycoproteins are glycosylated differently but that the difference is apparently not due to high mannose and hybrid types of carbohydrates, as shown by Endo H digestion. These results, together with those data derived from the trypsin cleavage studies in conjunction with the sequence data, indicate that the difference in N glycosylation appears to be in the F2 subunit, also because the cleaved soluble F1 subunit of both NiV and HeV migrates with a similar apparent molecular mass by SDS-PAGE (Fig. 5B). Further, the cleaved HeV and NiV F2 subunits also migrated differently by SDS-PAGE, with the F2 subunit of HeV possessing a higher apparent molecular mass (Fig. 5C). To confirm that this difference in F2 migration was attributable to a different N-glycosylation pattern, the HeV and NiV sFGCNt glycoproteins were treated with PNGase F, followed by trypsin treatment, and the digestion products were then compared to those of trypsin-treated sFGCNt glycoproteins by SDS-PAGE. As predicted, PNGase F treatment of the trypsin-cleaved sF glycoprotein eliminated the apparent molecular mass difference between the NiV and HeV F2 subunits (Fig. 5E), confirming that amino acid position N67 of NiV and HeV F is N glycosylated in this construct.
Immunoreactivity of recombinant sF.
To evaluate if the sF trimers produced here were immunologically relevant, we tested whether serum derived from an AGM experimentally infected with NiV could immunoprecipitate the sF glycoproteins. All the sF glycoproteins tested could react with the immune AGM serum but not with serum derived from a noninfected control animal (Fig. 6A). A control S-peptide-tagged protein of 45 kDa was included to confirm that the reactivity of the AGM serum with all sF glycoprotein preparations was specific (Fig. 6B). In addition, preparations of recombinant NiV and HeV soluble G (sG) were employed as a positive control for the immunoprecipitation assay and also confirmed that the immune AGM serum was polyreactive (Fig. 6B).
Fig 6.

Immunoreactivity of sF glycoprotein constructs with NiV-immune African green monkey serum and analysis of sF glycoprotein-immunized mouse serum. Serum harvested from an experimentally NiV-infected African green monkey was used to immunoprecipitate different sF glycoprotein preparations. Infected or immune (i) and noninfected or control (c) monkey serum (1 μl each) was added to a 0.5-μg sample of the indicated sF glycoprotein (A) or to S-tagged NiV sG or HeV sG and an S-tagged control protein (soluble ephrin-B2) (B). The antibody-protein complexes were precipitated with protein G-Sepharose beads, washed three times, and boiled in sample buffer. The precipitated proteins were then resolved by 4 to 12% BT SDS-PAGE, followed by Western blotting and detection with HRP-conjugated rabbit anti-S-peptide-tag antibody. (C) Serum samples harvested following the third immunization of mice with sF glycoprotein. Numbers above the lanes represent mouse numbers 1 and 2 (NiV sFGCNt), 3 and 4 (HeV sFGCNt), 5 and 6 (NiV sFdFp), and 7 and 8 (HeV sFdFp). Pooled prebleed (pb) serum was used as a negative control, serum was used to immunoprecipitate full-length wild-type NiV F (top) and HeV F (bottom) expressed in HeLa-USU cells, and the antibody-protein complexes were processed as described in Materials and Methods and analyzed by 4 to 12% BT SDS-PAGE, followed by Western blotting and detection with rabbit anti-HeV F1.
Immunization of mice with sFGCNt elicits a more potent neutralizing antibody response than immunization with non-GCNt-appended sF.
To explore the hypothesis that the predicted prefusion conformational form of sF (sFGCNt) could elicit a virus-neutralizing antibody response stronger than that elicited by the predicted postfusion conformational form of sF (wild-type sF), an immunization protocol was carried out in mice by comparing GCNt-appended and non-GCNt-appended sF glycoproteins (Table 1). The sFdFp was chosen as the non-GCNt-appended sF glycoprotein so that both the HeV and NiV sF glycoproteins could be compared. All mice generated a high titer of immunoreactivity against the respective immunogen employed, as measured by ELISA (Table 1). Importantly, the immune serum from all animals was also able to react with full-length wild-type HeV and NiV F in an immunoprecipitation analysis (Fig. 6C). The serum obtained following the third immunization of all mice was able to precipitate full-length F (Fig. 6C), with serum samples from mice immunized with the non-GCNt-appended HeV sFdFp (mouse numbers 7 and 8; Fig. 6C) having a slightly reduced profile of precipitated full-length F, suggesting that the sFGCNt could be more antigenically similar to the full-length F than a non-GCNt-appended sF. To further examine this possibility, these serum samples were also tested in an SNT against infectious HeV and NiV (Table 2). Surprisingly, it was immediately apparent that those sera from mice immunized with the GCNt-appended sF glycoproteins could more effectively neutralize the respective homologous infectious virus (mouse numbers 1 to 4), as well as had some lower cross-neutralizing activity, whereas serum from the non-GCNt-appended sF glycoprotein-immunized mice had either very low SNT titers (mouse numbers 5 and 6) or no detectable neutralizing titers (mouse numbers 7 and 8). These data indicate that GCNt-appended sF glycoprotein-immunized mice were able to generate a higher neutralizing antibody titer than those mice immunized with non-GCNt-appended sF glycoprotein. As a result of this initial observation, the non-GCNt-appended sF-immunized mice were boosted a fourth time using their respective homologous GCNt-appended sF (Table 1). Following this immunization, serum samples were examined by SNT, and the neutralizing serum titers of antibodies against NiV did increase (mouse numbers 5 and 6, samples 5F and 6F), while no increase in SNT titers of antibodies against HeV was observed (mouse numbers 7 and 8, samples 7F and 8F) (Table 2). Although the number of animals used here was small, the data show that both the HeV and NiV sFGCNt can elicit a strong homologous virus-neutralizing antibody response along with a lower level of cross-neutralizing activity and suggest that the GCNt-appended sF glycoprotein immunogens are superior to the non-GCNt-appended sF glycoproteins, perhaps because they are presented as the prefusion conformational form of F glycoprotein.
Table 2.
Serum neutralization titer of mice immunized with different sF glycoproteinsa
| Mouse no. or sample | Serum neutralization titer |
|
|---|---|---|
| NiV | HeV | |
| 1 | 1:640 | 1:320 |
| 2 | 1:320 | 1:80 |
| 3 | 1:20 | 1:320 |
| 4 | 1:40 | 1:80 |
| 5 | 1:5 | 1:10 |
| 6 | 1:40 | 1:20 |
| 7 | − | − |
| 8 | − | − |
| 5F | 1:80 | 1:10 |
| 6F | 1:640 | 1:20 |
| 7F | − | − |
| 8F | − | − |
| Pooled prebleed | − | − |
Serum samples were harvested following the third immunization of mice with sF glycoprotein. Serum samples from mouse numbers 1 and 2 (NiV sFGCNt), 3 and 4 (HeV sFGCNt), 5 and 6 (NiV sFdFp), and 7 and 8 (HeV sFdFp), along with serum samples collected following the final and fourth immunization from mouse numbers 5 to 8 (5F to 8F), were assayed by SNT using 200 TCID50 HeV or NiV in Vero cell culture and scored by CPE as described in the Materials and Methods. The SNT analysis was carried out twice, and a representative result is shown. SNT was determined as the highest dilution in which a viral CPE was still fully neutralized (absent) in at least one well. −, negative or neutralization was not observed in any of the dilutions tested.
Activation and refolding of sFGCNt from a pre- to postfusion conformation.
It was shown by Yin et al. (72, 73) that appending the GCNt helix to the HRB of a truncated PIV5 sF construct yielded a prefusion structure in comparison to the structure for the non-GCNt-appended hPIV3 sF glycoprotein, which spontaneously folds to a postfusion configuration. In addition, it was demonstrated that the prefusion PIV5 sFGCNt could be activated and refolded to a postfusion conformation in vitro by heat and trypsin treatment (23). These earlier findings, together with the data described above, suggested that the HeV and NiV sFGCNt glycoproteins are also likely folded into a prefusion conformation, whereas the non-GCNt-appended forms are not and likely have a configuration similar to a postfusion configuration. To examine this possibility, the purified sFGCNt, presumably in a prefusion conformation, was activated and triggered using techniques similar to those reported by Connolly et al. (23). Because sFGCNt could be properly cleaved in vitro by trypsin cleavage, shown earlier, we could examine whether the HeV and NiV sFGCNt could be cleaved and triggered to fold into a postfusion configuration by applying heat (50°C, 15 min) and trypsin. We previously demonstrated that short peptides derived from the HRB sequence of HeV and NiV F (FC2 peptides) were potent inhibitors of virus-mediated cell-cell fusion as well as live virus infection (10, 12). Here we used a biotinylated NiV F-derived FC2 peptide and included it during a series of heat and trypsin treatments of sFGCNt carried out in several combinations in an attempt to trap and capture an intermediate form during pre- to postfusion conversion of the sF glycoprotein by complex formation with the HRA domain during sF triggering. This would essentially prevent the final six-helix bundle formation and form an sF-FC2 complex that could be precipitated by streptavidin.
The following proteins were examined in this experiment: NiV and HeV sFGCNt, NiV and HeV sFdFp, NiV sFGCNtdFp, NiV sFGPI, and HeV sF. We also added heat, trypsin, and FC2 peptide to the different sF glycoproteins in several different combinations and orders. Of the entire panel of sF glycoprotein tested, we found that only the NiV and HeV sFGCNt could be precipitated by streptavidin beads and could be precipitated only when a specific sequence of trypsin digestion followed by biotin-FC2 peptide addition and, subsequently, heating was carried out. These results are shown in Fig. 7A. These data indicate that a mature form of soluble F1 plus F2 derived from the NiV or HeV sFGCNt can convert from a prefusion conformation by heat application into a postfusion conformation which can be captured by the biotinylated FC2 peptide only if it is present during the reaction, whereas the addition of biotinylated FC2 peptide after triggering (heat) did not allow sF-FC2 complex formation (Fig. 7A). To probe for conformational changes that should occur during the in vitro triggering process, we also analyzed the treated glycoproteins by native PAGE. As shown in Fig. 7B, applying heat only to NiV sFGCNt resulted in protein aggregation with the appearance of high-molecular-mass species, and this aggregation was eliminated by trypsin treatment. In contrast, NiV and HeV sFdFp glycoproteins, which, based on the data detailed above, were predicted to be in a postfusion conformation, did not reveal any changes, as assessed by native PAGE analysis, as a result of any of the treatments applied. No aggregation was observed for NiV sFGCNtdFp across all treatments, whereas higher aggregations were observed in all trypsin treatments for NiV sFGPI, HeV sF, and HeV sFGCNt. Taken together, these observations reveal that the various sF glycoproteins examined here display different conformations that can be altered by heat and/or trypsin treatments. Further, the data suggest that the GCNt helices stabilize sF in a prefusion configuration which can be triggered to fold into a presumed postfusion conformation that can be captured by the presence of an HRB peptide. In contrast, the non-GCNt-appended sF glycoproteins could not be triggered and detected by this approach, strongly suggesting that these constructs likely form a postfusion configuration that is prone to aggregation.
Fig 7.

In vitro processing of purified sFGCNt from a pre- to postfusion conformation and analysis of conformational changes. (A) Capture of a conformational intermediate during in vitro processing of sF by biotinylated FC2 heptad peptide. The NiV (top) and HeV (bottom) sFGCNt were untreated (−) or treated (+) with combinations of heat (50°C, 15 min), trypsin, and FC2 heptad peptide in a sequence indicated by the numbers (1 followed by 2 followed by 3). The sF-FC2 complexes were precipitated with avidin agarose and boiled in sample buffer. The precipitated proteins were resolved by 4 to 12% BT SDS-PAGE, followed by Western blotting. Blots were probed with rabbit anti-HeV F1 to probe for F. (B) Mobility shift in native PAGE of the different sF glycoprotein samples that were alternatively treated as indicated: untreated (−) or treated using heat only (H), trypsin only (T), heat followed by trypsin (HT), or trypsin followed by heat (TH). The treated proteins were resolved by 3 to 12% native PAGE, followed by Western blotting using anti-S-peptide-tag antibody to detect sF.
We next sought to examine whether pre- and postfusion forms of sF glycoprotein could be directly visualized as distinct conformational forms. Here, we applied single-particle electron microscopic analysis of NiV sF glycoproteins embedded in negative stain (Fig. 8). The NiV sFGCNt glycoprotein was examined as the prefusion conformation glycoprotein, and the non-GCNt-appended NiV sFdFp was examined as the representative postfusion conformation. Figure 8A shows examples of both raw images and representative class averages of the pre- and postfusion sF particles. The prefusion sFGCNt particles adopted primarily two preferred orientations on the carbon support of the EM grid, displaying a top view of the trimer head region and a side view of the spherical head region with a thin stalk domain similar to the lollipop structure previously observed for the prefusion F of PIV5 (23), Newcastle disease virus (NDV) (65), and human metapneumovirus (hMPV) (68). The particles with the postfusion conformation of the non-GCNt-appended NiV sFdFp adopted a single preferred orientation revealing a side view of the trimer head region and stalk domain, with the head region being distinctly less round, more box shaped, and similar to the golf tee shape of postfusion PIV5, NDV, and hMPV (23, 65, 68).
Fig 8.
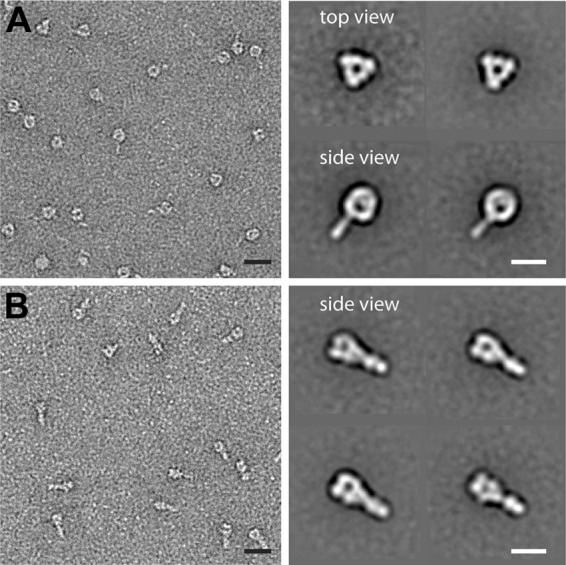
Single-particle electron microscopy analysis of NiV sF glycoproteins. Raw images (left) and representative class averages (right) of prefusion sF (A) and postfusion sF (B) embedded in negative stain. The prefusion sF assumes primarily two preferred orientations on the carbon support, displaying a top view of the trimer head region and a side view of the trimer head region with a stalk. The postfusion conformation of the non-GCNt-appended NiV sFdFp adopted a single preferred orientation, revealing a side view of the trimer head region and stalk domain. Bars, 20 nm (left) and 10 nm (right).
Characterization of pre- and postfusion sF by MAb binding.
As an additional way of characterizing the pre- and postfusion configurations of the sF glycoproteins, we next examined the binding of a series of MAbs that were derived from mice that had been immunized with the non-GCNt-appended NiV sFdFp followed by NiV sFGCNt (Table 1) and that we hypothesized would potentially elicit antibodies specific for either the pre- or postfusion conformational forms of the F glycoprotein. Of a panel of F-specific MAb-secreting hybridomas generated, three that displayed distinctly different profiles of binding to the different conformational forms of sF were identified. MAb 5B3 was determined to recognize a conformation-dependent epitope (no binding was observed by Western blotting) and could also completely neutralize infectious NiV and HeV at concentrations of 1.5 and 12.5 μg/ml, respectively, as detailed in Materials and Methods. Using an immunoprecipitation followed by Western blot analysis, MAb 5B3 was determined to bind only to NiV and HeV sFGCNt and the full-length wild-type NiV and HeV F-glycoprotein forms of both sF and native F that evidently exist in the prefusion conformational state. In stark contrast, MAb 5B3 was unable to recognize the non-GCNt-appended sF and the sF glycoprotein with the Fp deletion that the previous analyses strongly suggested were in a postfusion conformational form (Fig. 9A). However, MAb 5B3 lost all binding reactivity to the prefusion conformational form of NiV sFGCNt when the glycoprotein was properly triggered by heat (50°C, 15 min) and trypsin cleavage (Fig. 9B), while importantly, it retained binding activity to the NiV sFGCNt when it was exposed only to either heat or trypsin alone. By comparison, another MAb, 7C5, which is a nonneutralizing conformation-independent antibody, recognized all forms of sF glycoprotein (Fig. 9A) and also demonstrated a strong ability to bind to the properly triggered (heat- and trypsin-treated) sFGCNt, the only form of the protein that could also be recognized in the biotinylated HRB peptide capture assay described earlier. MAb 7C5 also retained binding activity to heat-treated-only NiV sFGCNt but not trypsin-cleaved-only sFGCNt (Fig. 9B).
Fig 9.

Characterization of pre- and postfusion sF glycoproteins by monoclonal antibody binding. (A) Binding of MAbs to sF and full-length F glycoproteins. Various sF glycoproteins or lysates of HeLa-USU cells expressing full-length F glycoprotein, as indicated, were immunoprecipitated by MAb 5B3, 7C5, or 1E11. (B) Binding of MAbs to in vitro-processed NiV sFGCNt. Samples of NiV sFGCNt that were untreated (−) or treated with heat (50°C, 15 min) only (H), trypsin only (T), or trypsin followed by heat (TH) were precipitated with the indicated MAb. In all cases, 0.5 μg of purified sF glycoprotein was added to 2 μg of MAb, and the MAb-protein complexes were precipitated with protein G-Sepharose, washed three times, and boiled in sample buffer. The precipitated proteins were resolved by 4 to 12% BT SDS-PAGE, followed by Western blotting using rabbit anti-HeV F1 to probe for F.
In addition, another conformation-dependent MAb, 1E11, was able to bind all forms of sF glycoprotein, regardless of treatment, with the single exception of NiV sFGPI (Fig. 9). The binding of MAb 1E11 supports the conclusion that the in vitro-triggered sFGCNt, which can be captured by HRB peptide, is refolded into a relevant postfusion conformation and not simply denatured by the treatment conditions. Together, these MAb binding analyses further demonstrate, by epitope exposure modulation, that distinct conformational forms of sF have been developed where the GCNt-appended and non-GCNt-appended forms of sF adopt the pre- and postfusion configurations, respectively. Further, the prefusion sFGCNt glycoprotein can be triggered in vitro into an apparent and relevant postfusion conformation.
DISCUSSION
Although all paramyxovirus F glycoproteins are apparent class I membrane fusion proteins, which undergo a significant refolding transition during the virus entry process, most function in concert with a partner attachment glycoprotein, either a hemagglutinin-neuraminidase (HN), hemagglutinin (H), or G, that binds the virion to specific receptors on host cells, and F activation and membrane fusion occur only upon proper receptor engagement (reviewed in references 44, 59, and 62). The majority of well-studied paramyxoviruses attach to sialic acid receptors mediated by an HN, whereas others bind to host cell protein receptors, such as the henipaviruses, which bind to ephrin-B2 and -B3 (reviewed in reference 69). A complete understanding of the F triggering process remains to be elucidated and may be different depending on whether the virus employs an HN versus a G or H attachment glycoprotein (reviewed in reference 64).
There are two principal models for the role of the attachment glycoprotein in the paramyxovirus fusion process. The first example is described as a clamp or dissociation model, which adheres to the premise that the attachment glycoprotein restrains F in a nonfusogenic or metastable conformation and that upon receptor engagement the attachment protein dissociates from F, initiating the conformational changes in F leading to its postfusion configuration and in so doing facilitates the membrane merger process. The clamp or dissociation model is largely supported with data from an extensive analysis of measles virus (MeV) (24, 60) and also the henipaviruses (1, 7), whereby membrane fusion activity is inversely correlated with the F to H or G avidity. In contrast, a second alternative association, or provocateur, model is suggested, which implies that the F association with its attachment glycoprotein partner, as in the case of HN, forms a complex at the cell surface only in the presence of receptor, and this scenario is supported by data showing that mutations which alter receptor binding decrease both membrane fusion activity and the F-HN interaction. This model is supported by studies on PIV5 by Connolly et al. (22) demonstrating that the enhancement of a strong interaction of the HN and F oligomers at the cell surface promotes membrane fusion activity, suggesting that receptor engagement of an attachment glycoprotein promotes the conformational transition and fusogenic activity of F by inducing its destabilization through an association of F and HN. In addition, findings on the interactions between the NDV F and HN in relation to membrane fusion activity have demonstrated that the fusion activity of NDV F and its HN partner was directly proportional to the extent of the HN-F interaction at the cell surface (49, 50). Together, these observations have led to the suggestion that paramyxoviruses that use protein entry receptors (MeV and henipaviruses) employ a mechanism more in line with a clamp or dissociation model, while those making use of sialic acid moieties as receptors (NDV, hPIV3, and PIV5) employ the association or provocateur model (reviewed in references 62 and 64).
Several soluble forms of paramyxovirus F glycoproteins have been reported, including those from NDV (18, 65), RSV (48), hMPV (66), hPIV3 (72), and PIV5 (73). The characterization of some of these sF glycoproteins facilitated the solution structures of both pre- and postfusion F trimers of sialic acid receptor-binding paramyxoviruses, providing significant detail to the molecular changes in F that occur during its triggering processes (19, 65, 72, 73). Here, we set out to characterize the structural, functional, and immunological properties of recombinant-expressed soluble forms of the henipavirus F (sF), which could provide insights into how protein receptor-using paramyxoviruses might differ mechanistically from those that employ sialic acid receptors.
While the recombinant hPIV3 and PIV5 and the postfusion NDV sF glycoproteins were produced in insect cells (19, 65, 72, 73), the RSV (48) and hMPV (66) sF glycoproteins and another version of NDV sF (18) were produced in mammalian cell culture systems. Initial attempts to produce henipavirus sF by recombinant gene expression in insect cells proved unsuccessful, and we converted to mammalian cell culture systems for making sF. Our studies revealed that a simple truncation of the wild-type or mammalian codon-optimized F-coding sequences to remove the TM and CT domains did not afford adequate protein production and release of sF from expressing cells. In addition, there was also an apparent and significant level of aggregation of the sF glycoprotein which presumably resulted from misfolding and retention of the glycoprotein within the expressing cell, with low levels of subsequent release into cell culture supernatants. Although TM and CT deletion did allow some HeV sF to be recovered from culture supernatants, only a GPI anchor and cleavage method with the HeLa-PLD cell line was useful, and the method was successfully used to produce soluble rubella virus E1 (6) and influenza virus hemagglutinin (39). However, only low yields of sF could be achieved with this method, and the sF may have an altered structure, as observed for GPI-H (39). Also, the inefficient recognition of NiV sFGPI by the conformation-dependent MAb 1E11 suggested that misfolding of this sF may be occurring.
Paramyxovirus F glycoproteins demonstrate a propensity to aggregate in solution or when extracted out of membranes, which may be the result of an exposed Fp (23, 48). The alteration of hydrophobic residues within the Fp of RSV F yielded an sF construct with a reduced tendency to aggregate (48). Similarly, our studies here revealed that reducing the hydrophobicity of the NiV and HeV sF by deletion or mutation of the Fp domain not only enhanced the secretion of the mutant sF but also reduced the amounts of aggregated sF observed during size-exclusion chromatography analysis. An optimal method for both recombinant expression and release of sF from cells was achieved by deletion of the TM and CT domains and appending of the helical GCNt trimerization motif. In addition, expression could be further enhanced by combining the Fp deletion strategy with appending of the GCNt motif, which was noted with the NiV sFdFpGCNt. The GCNt appeared to aid the stabilization of the sF trimers, as was observed for soluble and secreted envelope glycoprotein from HIV-1 (gp140) (70, 71), and/or the maintenance of a prefusion metastable form of the glycoprotein (68, 73). While the use of GCNt to stabilize NDV sF produced a heterogeneous population of pre- and postfusion F (65), our current MAb binding data and EM analysis indicate that GCNt provided complete stabilization of NiV sF with homogeneous prefusion lollipop structures.
Of particular importance, the entire panel of recombinant sF configurations engineered and analyzed in the present study shared the common feature of harboring a distinct trimeric glycoprotein species, with the single exception of the NiV sFGPI, which was produced as primarily high-molecular-mass aggregates. The apparent size of the various sF trimers ranged from 263 to 357 kDa, as measured by size-exclusion chromatography and sucrose gradient centrifugation analyses. In addition, the analytical ultracentrifugation and cross-linking analysis confirmed this trimer species, with apparent molecular masses of 221 ± 26 kDa for NiV sFGCNt and 215 ± 18 kDa for HeV sFGCNt. All of the sF glycoprotein configurations detailed here shared a similar separation profile by sucrose gradient centrifugation, consisting of various amounts of high-molecular-mass aggregates along with a principal trimeric species, which was found to be consistent with the analysis of full-length native F glycoprotein conducted in parallel.
Previous observations reported that the henipavirus F0 precursor is cleaved by host cell cathepsin L protease (56, 57) during an endocytosis and recycling trafficking of the glycoprotein (51). Indeed, the henipavirus sF constructs engineered here were expressed and released from cells as an F0 precursor without alterations to the predicted cleavage site described by others (18, 65, 72), in accord with an absence of any cellular membrane recycling process of the sF glycoprotein. However, we were able to carry out an in vitro process of protease cleavage of henipavirus sF (F0 precursor) into its disulfide-linked F1 and F2 subunits using a specific amount of trypsin in place of cathepsin L. The in vitro trypsin cleavage of sF at the native cleavage site was confirmed by N-terminal sequencing of the F1 cleavage product. A similar trypsin treatment was employed to produce cleaved sF from RSV (48), PIV5 (23), and NDV (18, 65) F glycoproteins. Although the alternate NiV sF produced in the present study lacked a single N glycosylation in F2, all sF glycoproteins, including those from HeV, could be precipitated by NiV-specific monkey serum, indicating the retention of native immunologically relevant epitopes.
We also demonstrated here that using the NiV and HeV sFGCNt as an immunogen was able to elicit cross-reactive virus-neutralizing responses in mice. Of particular importance, the observation that only the sFGCNt glycoproteins were able to induce a virus-neutralizing antibody response indicated that there are important structural and antigenic differences between the GCNt-appended and non-GCNt-appended forms of recombinant sF. These differences, likely in glycoprotein conformation, were further revealed using an NiV-FC2 capture and precipitation assay. Here, trypsin-treated and -cleaved NiV and HeV sFGCNt could be triggered by heat, resulting in sF refolding by a conformational transition, whereby an intermediate sF structure could be captured by a biotinylated NiV-FC2 peptide. The natural trigger for paramyxovirus F activation is the receptor binding event by its partner attachment glycoprotein, such as a G, H, or HN molecule. In our heptad peptide NiV-FC2 binding assay, heat was apparently able to substitute for the role of the attachment G glycoprotein that is required for F triggering, in addition to the requirement of F to be in its mature cleaved F1 plus F2 subunit form. Similar observations were earlier made in the GCNt-appended F of PIV5 (23), and here, distinct structural rearrangements of pre- and postfusion purified sF were revealed by single-particle EM analysis. In addition, however, the sFGCNt prefusion glycoprotein form could be captured and precipitated by a NiV-FC2 heptad peptide and only by a specific treatment sequence of trypsin digestion and NiV-FC2 addition followed by heating, suggesting that both the NiV and HeV sFGCNt glycoproteins were produced in a prefusion metastable conformation. The inability of the non-GCNt-appended sF to be captured in the NiV-FC2 heptad peptide assay also suggested that this protein was likely folded in a postfusion conformation, similar to soluble paramyxovirus F glycoproteins reported by others (19, 48, 72). The present studies revealed conformational changes as mobility shifts when the sF glycoproteins were analyzed by native PAGE, whereby heat treating NiV sFGCNt caused aggregation and a shift to an oligomeric species with a higher apparent molecular mass, while trypsin cleavage could eliminate this aggregation. On the other hand, either trypsin treatment only or trypsin with heat treatment caused HeV sFGCNt aggregation, as observed by native PAGE, and this was also consistent with the EM observations made by Connolly et al. (23), where protease cleavage exposed the Fp and heat facilitated folding to the postfusion conformation, which also aggregated the sF glycoprotein into rosette structures. The sF-triggering aggregation that we observed by native PAGE also occurred in both HeV sF and NiV sFGPI glycoproteins and likely occurs through Fp exposure, as none of the constructs of sF with Fp deletions could be triggered to form such aggregates. This conclusion is further supported by prior observations suggesting that paramyxovirus F aggregation occurs through Fp exposure (19, 48, 72) and that mutation or deletion of the Fp could ameliorate the aggregation event (48). It will be particularly interesting to further examine how the trypsin treatment could prevent NiV sFGCNt from aggregating, perhaps by similar EM studies done by others.
The results obtained in the NiV-FC2 heptad peptide capture assay and our EM study, together with the mobility shift assay observations made by native PAGE, suggest that sFGCNt is in a prefusion conformation and the non-GCNt-appended versions of sF are in a postfusion conformational state. This conclusion is further supported by the results obtained from the MAb binding analyses, where the conformation-dependent, cross-reactive, murine MAb 5B3 produced here was able to neutralize infectious NiV and HeV and was able to recognize only the NiV and HeV sFGCNt glycoproteins and none of the other postfusion sF glycoprotein forms described here. Of further importance, the MAb 5B3 also lost the ability to immunoprecipitate the triggered sFGCNt (postfusion conformation), while in contrast, MAb 7C5, which is a nonneutralizing antibody, could recognize all forms of sF, including the triggered sFGCNt. Altogether, it appeared that both the GCNt helices and Fp may be necessary for sF to maintain a metastable prefusion conformation. A further important observation made in the present study is that the prefusion-specific F-reactive MAb 5B3 was able to precipitate full-length F expressed in the absence of G, indicating that the stable prefusion conformation of F is apparently not dependent on an interaction with its partner G glycoprotein. These observations suggest that the F-G interaction may be important for F triggering upon receptor binding of G (the provocateur fusion model) rather than solely maintaining F in its prefusion metastable form (the clamp fusion model). However, we cannot exclude the possibility that the F and G glycoproteins simply have a specific affinity for each other, which has been demonstrated by coprecipitation studies.
Our findings reported here demonstrate that biologically relevant versions of a soluble henipavirus F glycoprotein (sF) have been successfully engineered. These materials will be important for further studies on the henipavirus entry mechanism, for it will be of considerable interest in future studies to explore whether such sF glycoproteins might serve as biochemical probes to explore the native F and G interaction and how receptor engagement modulates their interactions during virus infection. In addition, these soluble trimeric forms of henipavirus F glycoprotein retain epitopes that can be recognized by antibodies from an experimentally infected nonhuman primate and, when used as an immunogen in mice, were able to elicit a response by antibodies that could bind to full-length native F glycoprotein. Of particular interest, only the sFGCNt glycoprotein was able to stimulate the production of virus-neutralizing antibodies, which further suggests a prefusion conformation of this sF glycoprotein, a conclusion supported by both the NiV-FC2 heptad peptide capture assay and specific MAb binding results.
Together, these findings suggest that sFGCNt could be an effective subunit vaccine against henipaviruses or a diagnostic reagent for the detection of F-specific antibodies in animals or humans. The sF glycoproteins produced here also revealed measureable conformational differences, representing pre- versus postfusion conformational states which, together with the distinct anti-F MAbs described here, could facilitate further structural and functional studies on the henipavirus F-mediated fusion and virus entry process.
ACKNOWLEDGMENTS
We thank D. Sevlever (Mayo Clinic, Jacksonville, FL) for providing the HeLa-PLD cells.
This work was supported by NIH grants AI054715 and AI077995 to C.C.B.
Footnotes
Published ahead of print 22 August 2012
REFERENCES
- 1. Aguilar HC, et al. 2007. Polybasic KKR motif in the cytoplasmic tail of Nipah virus fusion protein modulates membrane fusion by inside-out signaling. J. Virol. 81:4520–4532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aguilar HC, et al. 2006. N-Glycans on Nipah virus fusion protein protect against neutralization but reduce membrane fusion and viral entry. J. Virol. 80:4878–4889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Anonymous 2012. New Hendra virus case in Cairns area. Department of Agriculture, Fisheries and Forestry, Brisbane, Queensland, Australia: www.daff.qld.gov.au/30_21974.htm [Google Scholar]
- 4. Anonymous 2012. Nipah encephalitis, human—Bangladesh: Jipurhat. Pro-MED-mailArchive no. 20120212.1040138. February 7. International Society for Infectious Diseases, Brookline, MA: www.promedmail.org [Google Scholar]
- 5. Arankalle VA, et al. 2011. Genomic characterization of Nipah virus, West Bengal, India. Emerg. Infect. Dis. 17:907–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bernasconi E, Fasel N, Wittek R. 1996. Cell surface expression of a functional rubella virus E1 glycoprotein by addition of a GPI anchor. J. Cell Sci. 109(Pt 6):1195–1201 [DOI] [PubMed] [Google Scholar]
- 7. Bishop KA, et al. 2007. Identification of Hendra virus G glycoprotein residues that are critical for receptor binding. J. Virol. 81:5893–5901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bonaparte MI, et al. 2005. Ephrin-B2 ligand is a functional receptor for Hendra virus and Nipah virus. Proc. Natl. Acad. Sci. U. S. A. 102:10652–10657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bossart KN, et al. 2005. Receptor binding, fusion inhibition, and induction of cross-reactive neutralizing antibodies by a soluble G glycoprotein of Hendra virus. J. Virol. 79:6690–6702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bossart KN, et al. 2005. Inhibition of henipavirus fusion and infection by heptad-derived peptides of the Nipah virus fusion glycoprotein. Virol. J. 2:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bossart KN, Wang LF, Eaton BT, Broder CC. 2001. Functional expression and membrane fusion tropism of the envelope glycoproteins of Hendra virus. Virology 290:121–135 [DOI] [PubMed] [Google Scholar]
- 12. Bossart KN, et al. 2002. Membrane fusion tropism and heterotypic functional activities of the Nipah virus and Hendra virus envelope glycoproteins. J. Virol. 76:11186–11198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Breed AC, et al. 2010. Prevalence of henipavirus and rubulavirus antibodies in pteropid bats, Papua New Guinea. Emerg. Infect. Dis. 16:1997–1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Broder CC. 2012. Henipavirus outbreaks to antivirals: the current status of potential therapeutics. Curr. Opin. Virol. 2:176–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Carter JR, Pager CT, Fowler SD, Dutch RE. 2005. Role of N-linked glycosylation of the Hendra virus fusion protein. J. Virol. 79:7922–7925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chadha MS, et al. 2006. Nipah virus-associated encephalitis outbreak, Siliguri, India. Emerg. Infect. Dis. 12:235–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chan YP, Yan L, Feng YR, Broder CC. 2009. Preparation of recombinant viral glycoproteins for novel and therapeutic antibody discovery. Methods Mol. Biol. 525:31–58, xiii [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen L, et al. 2001. Cloning, expression, and crystallization of the fusion protein of Newcastle disease virus. Virology 290:290–299 [DOI] [PubMed] [Google Scholar]
- 19. Chen L, et al. 2001. The structure of the fusion glycoprotein of Newcastle disease virus suggests a novel paradigm for the molecular mechanism of membrane fusion. Structure (Camb.) 9:255–266 [DOI] [PubMed] [Google Scholar]
- 20. Chua KB, et al. 2000. Nipah virus: a recently emergent deadly paramyxovirus. Science 288:1432–1435 [DOI] [PubMed] [Google Scholar]
- 21. Chua KB, et al. 2002. Isolation of Nipah virus from Malaysian Island flying-foxes. Microbes Infect. 4:145–151 [DOI] [PubMed] [Google Scholar]
- 22. Connolly SA, Leser GP, Jardetzky TS, Lamb RA. 2009. Bimolecular complementation of paramyxovirus fusion and hemagglutinin-neuraminidase proteins enhances fusion: implications for the mechanism of fusion triggering. J. Virol. 83:10857–10868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Connolly SA, Leser GP, Yin HS, Jardetzky TS, Lamb RA. 2006. Refolding of a paramyxovirus F protein from prefusion to postfusion conformations observed by liposome binding and electron microscopy. Proc. Natl. Acad. Sci. U. S. A. 103:17903–17908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Corey EA, Iorio RM. 2009. Measles virus attachment proteins with impaired ability to bind CD46 interact more efficiently with the homologous fusion protein. Virology 383:1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Diederich S, Moll M, Klenk HD, Maisner A. 2005. The Nipah virus fusion protein is cleaved within the endosomal compartment. J. Biol. Chem. 280:29899–29903 [DOI] [PubMed] [Google Scholar]
- 26. Drexler JF, et al. 2009. Henipavirus RNA in African bats. PLoS One 4:e6367 doi:10.1371/journal.pone.0006367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Drexler JF, et al. 2012. Bats host major mammalian paramyxoviruses. Nat. Commun. 3:796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Eaton BT, Broder CC, Middleton D, Wang LF. 2006. Hendra and Nipah viruses: different and dangerous. Nat. Rev. Microbiol. 4:23–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Eaton BT, Mackenzie JS, Wang L-F. 2007. Henipaviruses, p 1587–1600 In Knipe DM, et al. (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 30. Field HE, Mackenzie JS, Daszak P. 2007. Henipaviruses: emerging paramyxoviruses associated with fruit bats. Curr. Top. Microbiol. Immunol. 315:133–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gurley ES, et al. 2007. Person-to-person transmission of Nipah virus in a Bangladeshi community. Emerg. Infect. Dis. 13:1031–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Halpin K, et al. 2011. Pteropid bats are confirmed as the reservoir hosts of henipaviruses: a comprehensive experimental study of virus transmission. Am. J. Trop. Med. Hyg. 85:946–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Harbury PB, Kim PS, Alber T. 1994. Crystal structure of an isoleucine-zipper trimer. Nature 371:80–83 [DOI] [PubMed] [Google Scholar]
- 34. Harcourt BH, et al. 2000. Molecular characterization of Nipah virus, a newly emergent paramyxovirus. Virology 271:334–349 [DOI] [PubMed] [Google Scholar]
- 35. Hayman DT, et al. 2008. Evidence of henipavirus infection in West African fruit bats. PLoS One 3:e2739 doi:10.1371/journal.pone.0002739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Homaira N, et al. 2010. Cluster of Nipah virus infection, Kushtia District, Bangladesh, 2007. PLoS One 5:e13570 doi:10.1371/journal.pone.0013570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hsu VP, et al. 2004. Nipah virus encephalitis reemergence, Bangladesh. Emerg. Infect. Dis. 10:2082–2087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Johnson ML, Correia JJ, Yphantis DA, Halvorson HR. 1981. Analysis of data from the analytical ultracentrifuge by nonlinear least-squares techniques. Biophys. J. 36:575–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kemble GW, Henis YI, White JM. 1993. GPI- and transmembrane-anchored influenza hemagglutinin differ in structure and receptor binding activity. J. Cell Biol. 122:1253–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kyte J, Doolittle RF. 1982. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157:105–132 [DOI] [PubMed] [Google Scholar]
- 41. Lamb RA, Parks GD. 2007. Paramyxoviridae: the viruses and their replication, p 1449–1496 In Knipe DM, et al. (ed), Fields virology, 5 ed, vol 1 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 42. Lamb RA, Paterson RG, Jardetzky TS. 2006. Paramyxovirus membrane fusion: lessons from the F and HN atomic structures. Virology 344:30–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Laue TM, Shah BD, Ridgeway TM, Pelletier SL. 1992. Computer-aided interpretation of analytical sedimentation data for proteins, p 90–125 In Harding SE, Rowe AJ, Horton JC. (ed), Analytical ultracentrifugation in biochemistry and polymer science. Royal Society of Chemistry, Cambridge, United Kingdom [Google Scholar]
- 44. Lee B, Ataman ZA. 2011. Modes of paramyxovirus fusion: a henipavirus perspective. Trends Microbiol. 19:389–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li Y, et al. 2008. Antibodies to Nipah or Nipah-like viruses in bats, China. Emerg. Infect. Dis. 14:1974–1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Luby SP, et al. 2009. Recurrent zoonotic transmission of Nipah virus into humans, Bangladesh, 2001-2007. Emerg. Infect. Dis. 15:1229–1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ludtke SJ, Baldwin PR, Chiu W. 1999. EMAN: semiautomated software for high-resolution single-particle reconstructions. J. Struct. Biol. 128:82–97 [DOI] [PubMed] [Google Scholar]
- 48. Martin D, Calder LJ, Garcia-Barreno B, Skehel JJ, Melero JA. 2006. Sequence elements of the fusion peptide of human respiratory syncytial virus fusion protein required for activity. J. Gen. Virol. 87:1649–1658 [DOI] [PubMed] [Google Scholar]
- 49. Melanson VR, Iorio RM. 2006. Addition of N-glycans in the stalk of the Newcastle disease virus HN protein blocks its interaction with the F protein and prevents fusion. J. Virol. 80:623–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Melanson VR, Iorio RM. 2004. Amino acid substitutions in the F-specific domain in the stalk of the Newcastle disease virus HN protein modulate fusion and interfere with its interaction with the F protein. J. Virol. 78:13053–13061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Meulendyke KA, Wurth MA, McCann RO, Dutch RE. 2005. Endocytosis plays a critical role in proteolytic processing of the Hendra virus fusion protein. J. Virol. 79:12643–12649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Michalski WP, Crameri G, Wang L, Shiell BJ, Eaton B. 2000. The cleavage activation and sites of glycosylation in the fusion protein of Hendra virus. Virus Res. 69:83–93 [DOI] [PubMed] [Google Scholar]
- 53. Moll M, Kaufmann A, Maisner A. 2004. Influence of N-glycans on processing and biological activity of the Nipah virus fusion protein. J. Virol. 78:7274–7278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Murray K, et al. 1995. A morbillivirus that caused fatal disease in horses and humans. Science 268:94–97 [DOI] [PubMed] [Google Scholar]
- 55. Ohi M, Li Y, Cheng Y, Walz T. 2004. Negative staining and image classification—powerful tools in modern electron microscopy. Biol. Proced. Online 6:23–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pager CT, Craft WW, Jr, Patch J, Dutch RE. 2006. A mature and fusogenic form of the Nipah virus fusion protein requires proteolytic processing by cathepsin L. Virology 346:251–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pager CT, Dutch RE. 2005. Cathepsin L is involved in proteolytic processing of the Hendra virus fusion protein. J. Virol. 79:12714–12720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pallister J, Middleton D, Broder CC, Wang LF. 2011. Henipavirus vaccine development. J. Bioterr. Biodef. S1:005. doi:10.4172/2157-2526.S1-005 [Google Scholar]
- 59. Plemper RK, Brindley MA, Iorio RM. 2011. Structural and mechanistic studies of measles virus illuminate paramyxovirus entry. PLoS Pathog. 7:e1002058 doi:10.1371/journal.ppat.1002058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Plemper RK, Hammond AL, Gerlier D, Fielding AK, Cattaneo R. 2002. Strength of envelope protein interaction modulates cytopathicity of measles virus. J. Virol. 76:5051–5061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Reynes JM, et al. 2005. Nipah virus in Lyle's flying foxes, Cambodia. Emerg. Infect. Dis. 11:1042–1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Smith EC, Popa A, Chang A, Masante C, Dutch RE. 2009. Viral entry mechanisms: the increasing diversity of paramyxovirus entry. FEBS J. 276:7217–7227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Smith I, et al. 2011. Identifying Hendra virus diversity in pteropid bats. PLoS One 6:e25275 doi:10.1371/journal.pone.0025275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Steffen DL, Xu K, Nikolov DB, Broder CC. 2012. Henipavirus mediated membrane fusion, virus entry and targeted therapeutics. Viruses 4:280–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Swanson K, et al. 2010. Structure of the Newcastle disease virus F protein in the post-fusion conformation. Virology 402:372–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ulbrandt ND, et al. 2006. Isolation and characterization of monoclonal antibodies which neutralize human metapneumovirus in vitro and in vivo. J. Virol. 80:7799–7806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wacharapluesadee S, et al. 2010. A longitudinal study of the prevalence of Nipah virus in Pteropus lylei bats in Thailand: evidence for seasonal preference in disease transmission. Vector Borne Zoonotic Dis. 10:183–190 [DOI] [PubMed] [Google Scholar]
- 68. Wen X, et al. 2012. Structure of the human metapneumovirus fusion protein with neutralizing antibody identifies a pneumovirus antigenic site. Nat. Struct. Mol. Biol. 19:461–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Xu K, Broder CC, Nikolov DB. 2012. Ephrin-B2 and ephrin-B3 as functional henipavirus receptors. Semin. Cell Dev. Biol. 23:116–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yang X, Farzan M, Wyatt R, Sodroski J. 2000. Characterization of stable, soluble trimers containing complete ectodomains of human immunodeficiency virus type 1 envelope glycoproteins. J. Virol. 74:5716–5725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Yang X, et al. 2000. Modifications that stabilize human immunodeficiency virus envelope glycoprotein trimers in solution. J. Virol. 74:4746–4754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yin HS, Paterson RG, Wen X, Lamb RA, Jardetzky TS. 2005. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc. Natl. Acad. Sci. U. S. A. 102:9288–9293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yin HS, Wen X, Paterson RG, Lamb RA, Jardetzky TS. 2006. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 439:38–44 [DOI] [PMC free article] [PubMed] [Google Scholar]