

Abstract
DNAJC14 is an Hsp40 family member that broadly modulates flavivirus replication. The mechanism by which DNAJC14 stoichiometrically participates in flavivirus replication complex (RC) formation is unknown; both reduced and elevated levels result in replication inhibition. Using yellow fever virus (YFV), we demonstrate that DNAJC14 redistributes and clusters with YFV nonstructural proteins via a transmembrane domain and a newly identified membrane-binding domain (MBD), which both mediate targeting to detergent-resistant membranes. Furthermore, the RC and DNAJC14 reside as part of a protein interaction network that remains after 1% Triton solubilization. Mutagenesis studies demonstrate that entry into this protein interaction network requires the DNAJC14 C-terminal self-interaction domain. Fusion of the DNAJC14 MBD and self-interaction domain with another Hsp40 family protein is sufficient to confer YFV-inhibitory activity. Our findings support a novel model of DNAJC14 action that includes specific membrane targeting of both DNAJC14 and YFV replication proteins, the formation of protein interactions, and a microdomain-specific chaperone event leading to RC formation. This process alters the properties of the RC membrane and results in the formation of a protein scaffold that maintains the RC.
INTRODUCTION
The family Flaviviridae, consisting of the Flavivirus, Pestivirus, and Hepacivirus genera, causes a significant worldwide disease burden (30). Within the Flavivirus genus are numerous arthropod-borne human pathogens (reviewed in references 16 and 17), including yellow fever virus (YFV), dengue virus (DENV), West Nile virus (WNV), Japanese encephalitis virus, and tick-borne encephalitis viruses, each associated with clinically important diseases. Viruses in the Flavivirus genus have similar genome organization and replication strategies (30). Translation of the viral positive-sense genomic RNA produces a polyprotein that is co- and posttranslationally cleaved by host and viral proteases. Structural proteins C, prM, and E are located in the polyprotein amino terminus, followed by nonstructural (NS) proteins NS1, NS2A, NS2B, NS3, NS4A, 2K, NS4B, and NS5. Similar to the expression of other positive-sense RNA viruses, the expression of NS proteins induces intracellular membrane modification, resulting in a virus-specific membrane structure housing the viral replication complex (RC) (9, 35, 54). Electron microscope tomography studies have revealed that the flavivirus RC results from invagination of the endoplasmic reticulum (ER) membrane with the RC interior connected to the cytoplasm by a pore (15, 54). Virus RC formation is probably driven by viral NS protein self-interactions (34, 41, 45) and also needs concerted viral protein and viral protein-host factor interactions (10, 11, 37). In addition, many positive-strand viruses exploit host lipids to facilitate RC assembly and the specific lipids potentially provide a scaffold or modify membrane curvature to help generate the RC (20, 21, 33, 43, 48).
DNAJC14 (also designated DRIP78, Jiv, and LIP6) was identified as interacting with the dopamine D1 receptor and modulates receptor transport from the ER to the plasma membrane (2). It interacts with the lysosomal trafficking regulator protein that is implicated in SNARE complex-mediated transport (50). It is also involved in the life cycles of several Flaviviridae family members. The bovine homolog of this factor, Jiv, is essential for polyprotein cleavage and replication of the pestivirus bovine viral diarrhea virus (BVDV) and acts as a cofactor for NS2-3 cleavage by the NS2 autoprotease (27, 38, 44). Like other Hsp40 cochaperones, DNAJC14 contains a conserved 70-amino-acid J domain that interacts with Hsp70 family members to stimulate ATP hydrolysis during the chaperone process (24, 52). In the case of BVDV, bovine DNAJC14 or a 90-amino-acid domain downstream of the J domain, designated Jiv90 and containing putative zinc fingers, can function to facilitate NS2-3 cleavage (44). We identified DNAJC14 as a broadly acting flavivirus replication modulator that also affects members of the Hepacivirus and Flavivirus genera when overexpressed. Using YFV as a model flavivirus, our initial mutagenesis studies implicated the J domain and a C-terminal domain capable of self-interaction, but not the zinc fingers, as important for YFV inhibition upon DNAJC14 overexpression. The inhibition occurred after viral genome translation and prior to genome amplification, suggesting inhibition at the step of RC formation. DNAJC14 is likely involved in YFV RC assembly in a stoichiometric manner, since both overexpression and knockdown of DNAJC14 reduce YFV replication (56). We hypothesized that endogenous DNAJC14, which colocalized with double-stranded RNA (dsRNA) in YFV-infected cells, was likely involved in the regulation of RC assembly (56).
Despite extensive progress in the characterization of flaviviral RCs, the mechanism by which viral proteins interact with host membranes and host factors to assemble the RC is largely unknown. In this study, we used DNAJC14's dual role in RC formation, namely, its requirement for viral replication and its interference with the same process when overexpressed, to probe the events that lead to RC formation. Through mapping of the domains of the DNAJC14 protein that are functional in YFV replication modulation, we elucidate a model of DNAJC14 action and propose an RC assembly model involving protein targeting to detergent-resistant membrane (DRM) subdomains within the ER, followed by DNAJC14-facilitated protein-protein interactions and RC formation.
MATERIALS AND METHODS
Plasmids.
Retroviral vectors pV1, pTrip-EGFP, and pTrip-TagRFP and plasmids pV1-hDNAJC14-FL, pV1-hDNAJC14-CT1, pV1-hDNAJC14-NT5, and pV1-hDNAJC14-NT5CT1 have been described previously (6, 13, 22, 56, 57). Derivatives were generated by standard methods; all PCR-generated sequences were verified by sequencing. The sequences of the primers used are listed in Table 1.
Table 1.
Sequences of primers used in this study
| Primer | Sequence (5′→3′) |
|---|---|
| RU-O-13506 | TGGCATATTCCAGTCAACCTT |
| RU-O-13507 | GAAGCCCAAGATGGAATCAACT |
| RU-O-14383 | AAATGTACAAGGCCCAGAAGCACCCCGGAG |
| RU-O-14384 | TTTCTCGAGTCAACGTTGGAAGGGCCTCCTC |
| RU-O-15362 | AAATGTACAAGGCAGGCTTTTGGTGGCTGATTG |
| RU-O-15363 | TTTCTCGAGTCAGTCACCTAGTCCCACCAGAAACCG |
| RU-O-16562 | AAATGTACAAGGGAGTGTGGACAGGGCGGT |
| RU-O-16563 | AAATGTACAAGTTTACTCGTTTTCTTAAGCTGCTGGGTGCT |
| RU-O-15162 | AAATGTACAAGCTGGTGGGACTAGGTGACCGG |
| RU-O-16543 | AAATGTACAAGTTTTTGGGCTTTCTACAGTTGGGATGG |
| RU-O-15159 | AAATGTACAAGATGGCTGGGGTTCCTGAGG |
| RU-O-15971 | ATGGACGAGCTGTACAAGATCTATGCCTGCAGGCAA |
| RU-O-15972 | TTGCCTGCAGGCATAGATCTTGTACAGCTCGTCCAT |
| RU-O-15977 | TTTCGAGTTTGGATGGGACTGTTTACTCGTTTTCTT |
| RU-O-15978 | AAGAAAACGAGTAAACAGTCCCATCCAAACTCGAAA |
| RU-O-15973 | ATGGACGAGCTGTACAAGCTGTTTACTCGTTTTCTT |
| RU-O-15974 | AAGAAAACGAGTAAACAGCTTGTACAGCTCGTCCAT |
| RU-O-16162 | GTACAAGCGTTTTCTTAAGCTGCTGGGTGCTTTGCTGCTCCTGGCTCTGGCCCTCTTTTTGGGCTTTCTACAGTTGGGATGGCGGTTTTGAC |
| RU-O-16163 | TCGAGTCAAAACCGCCATCCCAACTGTAGAAAGCCCAAAAAGAGGGCCAGAGCCAGGAGCAGCAAAGCACCCAGCAGCTTAAGAAAACGCTT |
| RU-O-16191 | GTACAAGCGTTTTTTGCTGCTCCTGGCTCTGGCCCTCTTTTTGGGCTTTCTACAGTTGGGATGGCGGTTTTGAC |
| RU-O-16192 | TCGAGTCAAAACCGCCATCCCAACTGTAGAAAGCCCAAAAAGAGGGCCAGAGCCAGGAGCAGCAAAAAACGCTT |
| RU-O-16189 | GTACAAGCGTTTTGGTGCTTTGCTGCTCCTGGCTCTGGCCCTCTTTTTGGGCTTTCTACAGTTGGGATGGCGGTTTTGAC |
| RU-O-16190 | TCGAGTCAAAACCGCCATCCCAACTGTAGAAAGCCCAAAAAGAGGGCCAGAGCCAGGAGCAGCAAAGCACCAAAACGCTT |
| RU-O-17194 | AAAGGCCATTACGGCCATGGGTAAAGACTACTACCAGACGTTGG |
| RU-O-17195 | AAAGGCCGAGGCGGCCTCACAGATCCTCTTCTGAGATGAGTTTTTGTTCTATTGGAAGAACCTGCTCAAGTACG |
| RU-O-16849 | AAAGGCCATTACGGCCATGGGCTGGAGGGATAAGGCTACC |
| RU-O-16957 | TAGTCTTTACCCATTAGCTCATCCTCAGGAACCCCAG |
| RU-O-16958 | AGGATGAGCTAATGGGTAAAGACTACTACCAGACGTTGG |
| RU-O-17195 | AAAGGCCGAGGCGGCCTCACAGATCCTCTTCTGAGATGAGTTTTTGTTCTATTGGAAGAACCTGCTCAAGTACG |
| RU-O-15154 | GGTGCCTGGTATTGGAAGAACCTGCTCAAGTACG |
| RU-O-15155 | GCTAAAAGCCCAGGCACCAGAGGGCG |
| RU-O-15153 | AAAGGCCGAGGCGGCCTC |
| RU-O-13945 | AAAGGCCGAGGCGGCCTCACAGATCCTCTTCTGAGATGAGTTTTTGTTCACGTTGGAAGGGCCTCCTCACTTTC |
| RU-O-16850 | AAAGGCCATTACGGCCATGTCTCCAGCCTTGCAGCGTTG |
| RU-O-16851 | AAAGGCCATTACGGCCATGGATAGCAGGCCATGGCAGC |
| RU-O-16852 | AAAGGCCATTACGGCCATGTGGGGCTGGCTGGAGTTG |
| RU-O-16853 | AAAGGCCATTACGGCCATGCCTGAAGAGGAAGTGGCTCGAC |
| RU-O-16893 | AAACTCGAGATGCTGGTGGGACTAGGTGACCGGT |
| RU-O-16897 | TTTGGATCCCGTAGCTCATCCTCAGGAACCCCAG |
Plasmids pV1-hDNAJC14-NT5.1 (primers, RU-O-16849/RU-O-13945), pV1-hDNAJC14-NT5.2 (primers, RU-O-16850/RU-O-13945), pV1-hDNAJC14-NT5.3 (primers, RU-O-16851/RU-O-13945), pV1-hDNAJC14-NT5.4 (primers, RU-O-16852/RU-O-13945), and pV1-hDNAJC14-NT5.5 (primers, RU-O-16853/RU-O-13945) were generated by PCR amplification using PfuTurbo DNA polymerase (Stratagene) from the hDNAJC14 cDNA plasmid (Open Biosystems), followed by SfiI digestion and ligation into similarly digested pV1.
Plasmid pTrip-EGFP-hDNAJC14-FL was generated by PCR amplification from the hDNAJC14 cDNA plasmid by using primers RU-O-14383 and RU-O-14384. After BsrG1 and XhoI digestion, the PCR product was ligated into similarly digested pTrip-EGFP. Plasmids pTrip-EGFP-hDNAJC14-pTM and pTrip-RFP-hDNAJC14-pTM were generated by PCR amplification with primers RU-O-15362 and RU-O-15363. After BsrG1 and XhoI digestion, the PCR products were ligated into similarly digested pTrip-EGFP or pTrip-TagRFP. These plasmids express hDNAJC14 (or the pTM region of DNAJC14) fused in frame to the C terminus of enhanced green fluorescent protein (EGFP) or red fluorescent protein (RFP).
Plasmids pTrip-EGFP-hDNAJC14-NT3 (primers, RU-O-16562/RU-O-14384), pTrip-EGFP-hDNAJC14-NT4 (primers, RU-O-16563/RU-O-14384), pTrip-EGFP-hDNAJC14-NT5 (primers, RU-O-15162/RU-O-14384), pTrip-EGFP-hDNAJC14-NT4.1 (primers, RU-O-16543/RU-O-14384), and pTrip-EGFP-hDNAJC14-NT6 (primers, RU-O-15159/RU-O-14384) were generated by PCR amplification, BsrG1 and XhoI digestion, and ligation into similarly digested pTrip-EGFP.
Plasmids pTrip-EGFP-hDNAJC14-ΔTM1, pTrip-EGFP-hDNAJC14-ΔTM2, and pTrip-EGFP-hDNAJC14-ΔTM1+2 were generated by site-directed mutagenesis with primer pairs RU-O-15971/RU-O-15972, RU-O-15977/RU-O-15978, and RU-O-15973/RU-O-15974, respectively. Plasmid pTrip-EGFP-hDNAJC14-TM3 was generated by directly ligating synthesized oligonucleotides RU-O-16162/RU-O-16163 into BsrG1- and XhoI-digested pTrip-EGFP. Plasmids pTrip-EGFP-hDNAJC14-TM17 (primers, RU-O-16191/RU-O-16192) and pTrip-EGFP-hDNAJC14-TM19 (primers, RU-O-16189/RU-O-16190) were generated by ligating the oligonucleotide pairs indicated into BsrG1- and XhoI-digested pTrip-EGFP.
Plasmid pV1-DNAJB1 (primers, RU-O-17194/RU-O-17195) was generated by PCR amplification from the hDNAJB1 cDNA plasmid (Open Biosystems), followed by SfiI digestion and ligation into similarly digested pV1. To generate DNAJC14-DNAJB1 hybrids, first fragment I (DNAJC14 membrane-binding domain [MBD]) was generated by PCR amplification from the hDNAJC14 cDNA plasmid by using primers RU-O-16849 and RU-O-16957. Fragment II (DNAJB1 with a C-terminal myc tag) was generated by amplification from hDNAJB1 cDNA using primers RU-O-16958 and RU-O-17195. Fragment III (DNAJB1) was generated by amplification from hDNAJB1 cDNA by using primers RU-O-17194 and RU-O-15154. Fragment IV (DNAJC14 C-terminal self-interaction domain with a myc tag) was generated by amplification from plasmid pV1-hDNAJC14-FL using primers RU-O-15155 and RU-O-15153. Purified fragments I and II were fused by PCR amplification with primer RU-O-16849 and primer RU-O-17195 to generate pV1-MDNAJB1. Fragment III and fragment IV were fused by PCR amplification with primer RU-O-17194 and primer RU-O-15153 to generate pV1-DNAJB1C. Fragment V (DNAJC14 MBD fused to DNAJB1) was amplified with primers RU-O-16849 and RU-O-15154 using pV1-MDNAJB1 as the template. Fragments V and IV were fused by PCR with primers RU-O-16849 and RU-O-15153 to generate pV1-MDNAJB1C.
Plasmid pEGFP-N1-MBD was generated by PCR amplification from the hDNAJC14 cDNA plasmid with primers RU-O-16893 and RU-O-16897, and after XhoI and BamHI digestion, the PCR product was ligated into similarly digested pEGFP-N1 (Clontech).
Antibodies.
YFV NS3/4A, NS4B, and NS5 rabbit polyclonal antisera were previously described (5). NS3/4A and NS4B antisera were used at a 1:10,000 dilution for Western analyses and at 1:1,000 for immunofluorescence analyses. NS5 antiserum was used at 1:1,000 for Western blot analysis. Rabbit polyclonal anti-GFP antiserum was generated as described previously (7) and used at a 1:20,000 dilution in Western analyses. Mouse monoclonal anti-calnexin antibody (610523; BD Biosciences) was used in Western and immunofluorescence analyses at 1:500 and 1:50 dilutions, respectively. Mouse monoclonal anti-actin (A5441; Sigma) antibodies were used in Western analyses at a 1:5,000 dilution. Rabbit polyclonal anti-DNAJC14 antibody (HPA017653; Sigma) was used in Western and immunofluorescence analyses at 1:2,000 and 1:200 dilutions, respectively. Mouse monoclonal anti-dsRNA J2 antibody (English & Scientific Consulting, Bt., Szirak, Hungary) was reconstituted and diluted with an equal volume of glycerol and used at a 1:1,000 dilution in immunofluorescence analyses. Guinea pig polyclonal anti-adipophilin (ADRP) antibody (20R-AP002; Fitzgerald) was used at a 1:500 dilution in immunofluorescence analyses. Rabbit polyclonal anti-calnexin antibody (sc-11397; Santa Cruz Biotechnology, Inc.) was used at a 1:1,000 dilution in Western blot analyses. Mouse monoclonal anti-flotillin-1 antibody (610820; BD Biosciences) was used at 1:2,000 in Western blot analyses. Mouse monoclonal anti-Hsp70 antibody (SPA-810; Stressgen) was used at a 1:2,000 dilution in Western blot analyses. Mouse monoclonal anti-myc antibody (sc-40; Santa Cruz Biotechnology, Inc.) was used at 1:50 in coimmunoprecipitation analyses. Mouse monoclonal anti-GFP antibody (B-2; Santa Cruz Biotechnology, Inc.) was used at 1:50 in coimmunoprecipitation analyses. Alexa Fluor 488 donkey anti-mouse IgG (A-212020; Invitrogen), donkey anti-guinea pig IgG (706-546-148; Jackson ImmunoResearch), and Alexa Fluor 594 goat anti-rabbit IgG (A-11012; Invitrogen) were used in immunofluorescence analyses at a 1:1,000 dilution. Horseradish peroxidase (HRP)-conjugated secondary anti-mouse (115-035-146; Jackson ImmunoResearch) and anti-rabbit (31462; Pierce) IgG antibodies were used at a 1:20,000 dilution. Normal mouse IgG used in immunoprecipitations was from Santa Cruz Biotechnology, Inc. (sc-2025).
Cell lines, viruses, and virus titration.
SW13 (human adrenal carcinoma), Huh-7.5 hepatoma (3), and HEK293T cells and the BHK-J line of BHK-21 cells (29) were maintained as described previously (56). SW13 and Huh-7.5 cells were used interchangeably, and assays with the two cell lines gave similar results. YFV (strain 17D) stocks were generated by electroporation with in vitro-transcribed RNA (26) from plasmid pACNR-YF17D (4), and titers were determined as described previously (56). Lentiviral stock generation and transduction of cells were performed as described previously (56).
Cell fractionation.
SW13 cells were seeded into six-well plates and, after transduction, scraped into 400 μl hypotonic buffer (5 mM Tris-Cl [pH 7.5], 15 mM KCl, 2.5 mM MgCl2). After 15 min swelling on ice, the cells were passed 20 times through a 27-gauge needle and then centrifuged (900 × g for 5 min) to remove nuclei. One-tenth volume of 5 M NaCl was added to the postnuclear supernatants, and after incubation on ice for 20 min, the membranes were collected by centrifugation at 15,000 × g for 20 min. Pellets (membrane fraction, P) were resuspended in 50 μl sodium dodecyl sulfate (SDS) loading buffer. Proteins in the supernatant (cytosol fraction, S) were concentrated by adding 4 volumes of methanol, centrifuged (10 min, 12,000 × g), and resuspended in 50 μl SDS loading buffer.
In situ cell permeabilization and solubilization.
SW13 cells were washed once with cold buffer C (20 mM HEPES-KOH [pH 7.7], 110 mM potassium acetate, 2 mM magnesium acetate, 1 mM EDTA) and permeabilized by incubation in room temperature buffer C containing 50 μg/ml digitonin for 5 min or solubilized by incubation in room temperature buffer C containing 1% Triton X-100. Supernatants were collected if necessary. The reaction was stopped by three washes with cold buffer C. The cells were fixed for immunofluorescence analyses, scraped into cold buffer C for proteinase K digestion, or lysed in 2× SDS loading buffer (100 mM Tris-Cl [pH 6.8], 20% glycerol, 4% SDS, 3% β-mercaptoethanol, 0.02% bromophenol blue).
Immunoprecipitation.
To demonstrate the GFP-pTM and NT5CT1 interaction with YFV NS3, SW13 cells were transduced with pTrip-GFP-DNAJC14-pTM or pV1-DNAJC14-NT5CT1 and 2 days later infected with YFV (multiplicity of infection [MOI], 5). After 1.5 days, the cells were scraped into ice-cold phosphate-buffered saline (PBS), solubilized with 300 μl buffer A (10 mM HEPES [pH 7.5], 150 mM KCl, 3 mM MgCl2, 0.5% NP-40, 1× Roche proteinase inhibitor cocktail), and disrupted by being passed through a 27-gauge needle 10 times. After clarification by centrifugation twice at 1,000 × g for 5 min at 4°C, the soluble fraction was incubated overnight at 4°C with anti-myc antibody at a dilution of 1:50. Prewashed protein G-Dynabeads (100.03D; Invitrogen) were then added, and after 2 h of incubation, protein-antibody complexes were captured by a magnet and washed four times with 600 μl buffer B (10 mM HEPES [pH 7.5], 150 mM KCl, 3 mM MgCl2, 0.5% NP-40). The bound proteins were eluted by boiling in SDS sample buffer and subjected to Western blot analysis.
To demonstrate the DNAJC14-NS3 interaction after in situ solubilization, SW13 cells were transduced with pV1-DNAJC14-FL and 1.5 days later infected with YFV (MOI, 10). After 4 days, cells were solubilized in situ with buffer C (see above) supplemented with 1% Triton X-100. After being washed with buffer C, the remaining material was scraped into buffer B (10 mM HEPES [pH 7.5], 150 mM KCl, 3 mM MgCl2, 0.5% NP-40) and disrupted by being passed through a 27-gauge needle 10 times. After clarification by centrifugation at 15,000 × g for 10 min at 4°C, the soluble fraction was incubated overnight at 4°C with anti-myc antibody at a dilution of 1:50. Prewashed protein G-Dynabeads (100.03D; Invitrogen) were then added, and after 2 h of incubation, protein-antibody complexes were captured by a magnet and washed four times with 600 μl buffer B. The bound proteins were eluted by boiling in SDS sample buffer and subjected to Western blot analysis.
Proteinase K digestion.
After in situ cell permeabilization or solubilization, and being scraped into cold buffer C, the samples were incubated with or without 10 μg/ml proteinase K at 37°C for 5 min; the reaction was terminated by the addition of phenylmethylsulfonyl fluoride to 1 mM. Proteins were concentrated by trichloroacetic acid (TCA) precipitation by adding an equal volume of 40% TCA. After the precipitates were washed with acetone and solubilized in SDS loading buffer, the samples were boiled for 10 min and used in Western blot analyses.
Membrane flotation.
Membrane flotation assays were conducted essentially as described previously (47, 51). Cells grown in 60-mm dishes were washed, scraped into PBS, and collected by centrifugation (500 × g, 5 min). After resuspension in 200 μl hypotonic buffer (10 mM Tris-HCl [pH 7.8], 10 mM NaCl) and swelling (15 min on ice), the cells were disrupted by being passed 25 times through a 27-gauge needle. After centrifugation (900 × g for 5 min), postnuclear factions were treated with Triton X-100 (final concentration, 1%) on ice for 30 min. The entire 0.2-ml fraction was mixed with 1.6 ml 72% (wt/wt) sucrose in low-salt buffer (LSB; 50 mM Tris-HCl [pH 7.5], 25 mM KCl, 5 mM MgCl2) overlaid with 2 ml of 55% (wt/wt) sucrose in LSB, followed by 0.6 ml of 10% (wt/wt) sucrose in LSB. For flotation after in situ solubilization, cells from 100-mm dishes were solubilized with 2.5 ml buffer C containing 1% Triton X-100. Supernatants (S1) were collected. After washing, cells were scraped into 600 μl buffer C containing 1% Triton X-100, disrupted by needle passage, and clarified by centrifugation at 15,000 × g for 5 min. Supernatants (S2) were collected. A 0.2-ml fraction of S1 or S2 was mixed with 1.6 ml 72% (wt/wt) sucrose in low-salt buffer and overlaid with 55% and 10% (wt/wt) sucrose in LSB as described above. The sucrose gradient was centrifuged at 46,000 rpm in a Beckman Coulter MLS-50 Optimax ultracentrifuge rotor for 16 h at 4°C. Fractions (400 μl) were taken from the top of the gradient, and proteins were precipitated from 300 μl of each fraction by the addition of 4 volumes of methanol and centrifugation at 12,000 × g for 10 min. RNAs were extracted from 100 μl of each fraction by dilution with 100 μl H2O, addition of 700 μl RLT (Qiagen) buffer, and purification using the RNeasy minikit (Qiagen).
Western blot analysis.
Cells were directly lysed with 2× SDS loading buffer and boiled for 5 min. Proteins were separated by SDS-polyacrylamide gel electrophoresis and transferred to a Hybond ECL nitrocellulose membrane (GE Healthcare Life Sciences). The membrane was incubated in blocking buffer (PBS, 0.05% Tween 20, 5% dried milk) for 2 h and then incubated with primary antibody diluted in blocking buffer at 4°C overnight. The membrane was washed three times in PBS supplemented with 0.05% Tween 20 and incubated for 2 h at room temperature with HRP-conjugated secondary antibody. After three washes, the membrane was visualized by ECL Supersignal West Pico or Femto chemiluminescent substrate (Thermo Scientific). Protein bands were quantified by densitometry with ImageJ if necessary.
Immunofluorescence and microscopy.
Cells were fixed with 4% paraformaldehyde in PBS and permeabilized with 0.2% Triton X-100 or 0.5% Triton X-100 (for dsRNA) in PBS for 5 min at room temperature. Permeabilization was omitted for in situ-solubilized cells. After being washed with PBS, samples were blocked and incubated overnight with primary antibody in PBS containing 3% bovine serum albumin at 4°C. After three washes with PBS, samples were incubated at room temperature for 2 h with an Alex488- or Alex594-conjugated secondary antibody. For confocal microscopy, coverslips were mounted with Mowiol mounting medium and observed with a Zeiss LSM510 confocal microscope equipped with a 100× 1.3 numerical aperture oil immersion objective. Images were captured by using the LSM software and processed by ImageJ. Photoshop was used to adjust the contrast and brightness; all changes for a given channel were applied to the entire image. In some images, Pearson (rp) and Spearman (rs) correlation coefficients were used to calculate colocalization as described previously (14). For high-resolution microscopy, coverslips were mounted with ProLong Gold antifade reagent (Invitrogen). Samples were initially observed using a Nikon Eclipse TE300 inverted fluorescence microscope. Images were acquired with a Nikon Plan Fluor 20× 0.45 numerical aperture objective, a Digital Sight DS2MBW camera, and Nikon Elements software. For high-resolution images, the samples were observed with an OMX Blaze 3D-SIM superresolution microscope (Applied Precision) equipped with a 100× 1.40 numerical aperture UPLSAPO oil immersion objective. Images were captured and deconvoluted by using SoftWoRx software and processed by ImageJ. All of the data shown are representative results obtained with cells in at least two independent experiments.
Reverse transcription-quantitative PCR (RT-qPCR).
RNAs from each sucrose fraction were purified by using the RNeasy minikit (Qiagen) and eluted in 40 μl H2O. After heating at 85°C for 10 min, 8 μl RNA sample was used for RT with random primers and the Superscript III first-strand synthesis kit (Invitrogen). Each sample was reverse transcribed in triplicate. Four microliters of cDNA was used for qPCR with the QuantiTect SYBR green PCR kit (Qiagen) and a LightCycler 480 (Roche) for detection as previously described (56). The primers used for YFV genome amplification (oligonucleotides RU-O-13506 and RU-O-13507, Table 1) were as described previously (28).
Bioinformatics.
The transmembrane (TM) prediction program SOSUI (http://bp.nuap.nagoya-u.ac.jp/sosui) was used for membrane helix prediction. The program for secondary structure prediction was PSIPRED (http://bioinf.cs.ucl.ac.uk/psipred/).
RESULTS
DNAJC14 is a polytopic membrane protein with both N and C termini in the cytoplasm.
In order to gain insight into its function in YFV RC assembly, we first undertook a topology study of DNAJC14. DNAJC14 contains three potential TM domains (referred to as pTM1, pTM2, and pTM3, with the region designated the pTM region), although it has been hypothesized to have both its N and C termini in the cytoplasm (2). We used selective permeabilization and protection from protease digestion to determine the orientation of DNAJC14 with respect to the ER membrane. We generated constructs expressing GFP fused to the amino termini of DNAJC14 N-terminal truncation constructs containing successively fewer of the putative TMs (Fig. 1A). SW13 cells expressing these mutant constructs were treated with digitonin to permeabilize the plasma membrane and left without further treatment or treated with proteinase K. Proteinase K digestion efficiency and ER membrane integrity were monitored with an antibody that recognizes the calnexin N terminus. Upon proteinase K digestion, full-length calnexin was cleaved and the N terminus within the ER lumen was protected (Fig. 1B, bottom panels), indicating that proteinase K digestion was essentially complete and that the ER membrane remained intact under our experimental conditions. We monitored the location of the C termini by using an antibody that recognizes the DNAJC14 C terminus. Proteinase K digested all of the C termini (Fig. 1B, middle panels), indicating that the DNAJC14 C terminus resides in the cytoplasm. In the absence of proteinase K digestion, faster-migrating bands were apparent for the most highly expressed mutant constructs (GFP-FL, GFP-NT3, and GFP-NT4), most likely because of nonspecific degradation. A very small fraction of GFP-NT3 and GFP-NT4 remained resistant to proteinase K digestion. We monitored the location of the N termini with an antibody to GFP. GFP is known to be resistant to protease digestion (reviewed in references 8 and 53). In cells expressing GFP, which is known to be present in the cytosol (see Fig. 5E), exposure to proteinase K had no discernible effect on the size or levels of GFP, confirming the resistance of the GFP domain to proteinase K digestion (Fig. 1C, left panels). We thus expected that cytosolic exposure of the GFP-DNAJC14 fusion proteins to proteinase K would result in release of the protease-resistant GFP domain, while protection within the ER lumen would result in protected fragments larger than GFP but smaller than the intact fusion protein. A protease-resistant GFP domain remaining after digestion was detected for constructs GFP-FL, GFP-NT3, GFP-NT5, and GFP-NT6, suggesting that the GFP domain was present in the cytoplasm and susceptible to proteinase K digestion (Fig. 1B, top panels). GFP-containing larger protected fragments were observed for GFP-NT3, GFP-NT4, and GFP-NT4.1. This indicates that the amino-terminal GFP tag for each of these constructs was located in the ER lumen, even though no signal sequence was present in the amino termini of the constructs. The ER localization was likely mediated by pTM3, since fusion of pTM3 (data not shown) or 19 residues from this region of DNAJC14 to the C terminus of GFP (GFP-pTM3-19; see schematic in Fig. 3A) results in the translocation of GFP into the ER lumen and protection from proteinase K digestion (Fig. 1C, right panels). The apparent molecular masses of the protected GFP fragments of GFP-NT3, GFP-NT4, and GFP-NT4.1 (∼37, 34, and 32 kDa, respectively) are slightly larger than the predicted molecular masses that would occur upon the digestion of each protein C terminal to pTM3 (∼33, 30, and 28 kDa, respectively). This could be due to features inherent to this protein (e.g., charge) or due to the protection of a portion of a membrane-binding domain (MBD; see below) localized downstream of pTM3. GFP-NT3 was an interesting case, in that evidence for both cytosolic localization (GFP-sized band) and luminal location (larger band) was found. This suggests that in the absence of the N terminus and pTM1, pTM2 may adopt an alternative TM topology that places the GFP moiety in the cytosol. Whether pTM2 inserts itself into the membrane bilayer or whether the pTM region can take on alternate conformations in the context of full-length DNAJC14 (DNAJC14-FL) is an interesting area for future study. Despite this uncertainty, the model of DNAJC14 topology shown in Fig. 1D is the most consistent with the results; pTM1 and pTM3 serve as TM domains, and both the N and C termini reside on the cytoplasmic side of the ER membrane.
Fig 1.

Studies of DNAJC14 topology. (A) Schematic of DNAJC14-FL and pTrip-GFP mutant constructs fused in frame to the C terminus of GFP. The three individual putative TMs (pTMs) are indicated. (B) SW13 cells transduced with the indicated DNAJC14 mutant constructs were permeabilized with 50 μg/ml digitonin, scraped, and resuspended in buffer for treatment with 10 μg/ml proteinase K (pk) or no treatment. Proteins were TCA precipitated and analyzed by Western blotting with the antibodies indicated (IB). Top panels assessed N-terminal protection (N-), while the middle panels assessed C-terminal protection (-C). Arrowheads in the top panel indicate protected N-terminal GFP-containing proteins. Asterisks indicate the undigested DNAJC14 mutant constructs. The diamond indicates the protease-resistant GFP domain released after exposure to proteinase K. Calnexin (bottom panels) serves as a control for proteinase K digestion. (C) SW13 cells expressing GFP or GFP with 19 amino acids from pTM3 fused to the C terminus were treated and analyzed as in panel B. The protease-resistant GFP domain released by proteinase K is indicated by the diamond, and the protected larger species containing the TM sequence is indicated by the arrowhead. (D) Summary of proteinase K digestion results. The N-terminal GFP tag location is represented by a circle; green indicates that the GFP was not protected, while red indicates protection from proteinase K digestion. The alternative topology of pTM2 is indicated (dashed line). (E) Subcellular fractionation of DNAJC14 mutant constructs. Cells were disrupted by hypotonic buffer, and postnuclear lysates were separated by centrifugation into a crude membrane fraction (P) and a cytosolic fraction (S), which were analyzed by Western blotting.
Fig 5.

Identification a new MBD in DNAJC14. (A) Schematic of NT5 and truncation mutant constructs. All constructs expressed the MBD region (or truncation constructs thereof) at the amino terminus. MBD truncation mutant constructs contained a myc epitope tag at the carboxyl terminus. (B, C) SW13 cells transduced with V1-NT5 or the indicated truncation mutant constructs were challenged with YFV (MOI, 5). (B) Cells were harvested at 1 day postinfection and analyzed by Western immunoblotting (IB) and (C) virus in the medium was enumerated by plaque assay. Data are the mean titer ± the standard error of the mean; n = 3. Abbreviations: pfu, plaque-forming units; V, V1 vector control. (D) The intracellular distribution of mutant constructs was examined by immunostaining with anti-myc antibody (green) and confocal microscopy 1.5 days after transduction. (E) Localization of the DNAJC14 MBD fused to GFP. SW13 cells were transfected to express GFP or MBD-GFP and mock infected or infected with YFV (MOI, 5) 1 day later. After 1 day, the cells were fixed for immunostaining and observed by confocal microscopy. Green (GFP), red (NS4B), and merged channels of a representative image are shown. Bars, 20 μm. (F) The DNAJC14 MBD confers detergent resistance and membrane flotation on GFP. SW13 cells were cotransfected to express both GFP and MBD-GFP. Postnuclear lysates were treated with 1% Triton X-100 and subjected to membrane flotation; fractions were taken from the top to the bottom. Proteins from each fraction were methanol precipitated and analyzed by Western blotting with the antibodies indicated. The values to the left of panels B and F are molecular sizes in kilodaltons.
Fig 3.

DNAJC14 pTM3 mediates recruitment to YFV NS protein clustering sites. (A) Schematic of DNAJC14 pTM mutant constructs. Various combinations of the individual TMs (gray) of the pTM region were fused in frame to the C terminus of GFP in the pTrip-GFP plasmid as indicated. (B, C) Huh-7.5 cells transduced to express the indicated DNAJC14 pTM mutant constructs were infected with YFV (MOI, 5) or not infected and fixed 1 day later. Cells were stained with antibodies to NS3 or calnexin as indicated and examined by confocal microscopy for DNAJC14 mutant construct or calnexin (green) and NS3 (red) localization. Representative images are shown. Bars, 10 μm.
To confirm the membrane association of these mutant constructs, cells were fractionated by differential centrifugation into membrane (pellet, P) and cytosolic (soluble, S) fractions. Calnexin served as a membrane protein marker, and Hsp70 served as a cytosolic protein marker. Mutant constructs containing pTM3 (GFP-NT3, GFP-NT4) or even a portion of pTM3 (GFP-NT4.1) were almost totally membrane associated (Fig. 1E). Similar results were observed with equivalent C-terminally myc-tagged proteins (data not shown). These data confirm that pTM3 is indeed a TM helix. Although a portion of GFP-NT5 resided in the cytosolic fraction, a similar amount resided in the membrane fraction, while GFP-NT6 resided totally in the cytosolic fraction (Fig. 1E). Therefore, a region in NT5 not found in NT6 is able to mediate membrane association by a pTM3-independent mechanism.
DNAJC14 is redistributed and recruited to YFV NS protein clustering sites.
Overexpressed DNAJC14 localized both with the ER and in cytoplasmic structures that we identified as probable lipid droplets (LD, Fig. 2B). DNAJC14-FL and the CT1 mutant construct lacking the C-terminal self-interaction domain (schematic mutant construct representations are shown in Fig. 2A) colocalized with the LD marker adipocyte differentiation-related protein (ADRP, Fig. 2B, left panel, top and middle). The pTM region likely mediated DNAJC14 colocalization with the LD marker, since it conferred ADRP colocalization on RFP (RFP-TM, Fig. 2B, left panel, bottom), a heterologous protein that is normally distributed diffusely in the cytoplasm and nucleus (data not shown). Since endogenous DNAJC14 was not found to localize with the LD marker at steady state (see below), similar to caveolin-1 (40), detection of the LD localization is likely due to the high level of protein expressed during transduction and might not reflect DNAJC14's normal localization. We then examined the localization of DNAJC14 in YFV-infected cells (Fig. 2B, right panels) by using the noninhibitory CT1 mutant construct. Electron microscopy of infected cells suggested a large increase in RC formation between 18 and 24 h (data not shown), so we examined the cells 1 day after infection, when RC formation is likely ongoing. We found DNAJC14-CT1 and NS3 colocalized in aggregates that did not appear to be vesicular in nature. As had been previously observed in DENV-infected cells (19), we also noticed reduced LD staining in YFV-infected cells, with the LD marker ADRP present in smaller punctate structures (compare Fig. 2B, middle panels with and without YFV). Unlike in uninfected cells, where ADRP and DNAJC14-CT1 strongly colocalized (Fig. 2B, left middle panels), in YFV-infected cells, much of the DNAJC14-CT1 was found unassociated with the LD marker (Fig. 2B, right middle panels), presumably because of the recruitment of DNAJC14-CT1 to sites of YFV NS proteins. Consistent with this, YFV NS3 also did not colocalize with ADRP (Fig. 2B, right bottom panels). Determination that the NS3-DNAJC14-CT1 colocalization was reflecting the biological properties of DNAJC14-FL is complicated by the inhibitory activity exerted on YFV by increased levels of DNAJC14. We previously found that although DNAJC14 overexpression inhibited YFV RNA replication, at late times after infection, the virus was able to overcome the inhibition (56). Huh-7.5 cells expressing DNAJC14-FL were therefore infected with YFV and fixed late (4 days) after infection. Transduced DNAJC14-FL colocalized exclusively with NS3 and NS4B, which displayed a punctate staining pattern (Fig. 2C). Taken together, overexpressed DNAJC14 resides in the ER and LD, but during YFV infection, DNAJC14 has reduced association with the LD marker ADRP because of DNAJC14 recruitment to NS protein clustering sites.
Fig 2.

DNAJC14 is redistributed and recruited to YFV NS protein clustering sites. (A) Schematic of DNAJC14 mutant constructs. DNAJC14-FL and the NT5CT1 and CT1 mutant constructs are in plasmid pV1 and contain a C-terminal myc epitope tag. Putative TM helices (pTM, black), the J domain (red) with a conserved HPD motif, a zinc finger motif (blue) in the JIV90 domain, and the self-interaction domain (gray) are shown. The pTM region was expressed fused to the C terminus of GFP or RFP in a pTrip-GFP (or -RFP) plasmid. The predicted hydrophobic helices are in gray. (B) Huh-7.5 cells were transduced with lentiviruses expressing FL-myc, CT1-myc, RFP-pTM (left panels), or CT1-myc (right panels). Cells were left uninfected or infected with YFV (MOI, 5) and fixed 1 day later and stained with antibodies against myc, the LD marker ADRP, or YFV NS3 as indicated. (C) Huh-7.5 cells expressing FL-myc were infected with YFV (MOI, 10) and fixed 4 days later. Cells were stained with antibody against myc and YFV NS3 or NS4B and examined by confocal microscopy. (D) SW13 cells expressing myc-tagged NT5CT1 or GFP-pTM were left uninfected or infected with YFV (MOI, 5) and fixed 1 day later. The cells were examined by confocal microscopy after staining for myc or NS proteins as indicated. Representative images are shown; similar results were obtained in independent experiments. Bars, 10 μm.
We then mapped the minimal elements that mediate DNAJC14 recruitment to YFV NS protein clustering sites. We found that mutant construct NT5CT1, lacking both the pTM region and the C-terminal interaction domain, and GFP fused to the pTM region (Fig. 2A) could colocalize with NS3; moreover, GFP-pTM colocalization with NS4B was also confirmed (Fig. 2D). This indicated that while the pTM region can confer localization with NS proteins, another region, present in NT5CT1, is also capable of this activity. We focused on the pTM element and further deleted the individual TMs. GFP-ΔpTM1, GFP-ΔpTM2, and GFP-ΔpTM1+2, each containing pTM3, all colocalized with NS3 (Fig. 3A and B), suggesting that pTM3 mediates NS3 colocalization. For unknown reasons, we were unable to stably express GFP-ΔpTM3 lacking pTM3. We next focused on pTM3 to define the minimal sequence necessary for targeting to NS protein clustering sites. We fused pTM3 to the C terminus of GFP and found that it redistributed and colocalized with NS3 (Fig. 3C). Mutant constructs with reduced TM3 lengths as small as 19 or 17 amino acids (aa) were able to confer NS3 colocalization on GFP (Fig. 3C). In contrast, the ER protein calnexin only partially colocalized with NS3 (Fig. 3C). Given the inability to express GFP-pTM with the pTM3 sequences deleted, it remains possible that other pTM sequences can also mediate localization to NS protein clustering sites. However, DNAJC14 recruitment to and colocalization with YFV NS protein clustering sites can be mediated by its third putative TM with a length as short as 17 aa. We noted that in the absence of infection, DNAJC14 mutant constructs lacking pTM1 or pTM2 but not both (ΔpTM1+2, Fig. 3B; GFP-pTM3, Fig. 3C) were associated with vesicular structures suggestive of LDs (inserts in Fig. 3B, left panels). Clarification of whether pTM1 or pTM2 is sufficient for LD recruitment requires further investigation.
DNAJC14 and YFV NS proteins are targeted to DRMs.
Since DNAJC14 subdomains mediate recruitment to YFV NS protein clustering sites and NS proteins of hepatitis C virus (HCV) and DENV reside on DRMs (39, 47), we wondered if DNAJC14 and YFV NS proteins were targeted to DRMs. We conducted membrane flotation assays with 1% Triton X-100-treated cell lysates to evaluate the flotation behavior of GFP fused to the DNAJC14 pTM region (GFP-pTM) and the noninhibitory mutant construct lacking TMs (NT5CT1) in the absence or presence of YFV infection. NS3 and NS4B floated to lighter membrane fractions (Fig. 4A and B). GFP-pTM, in contrast to GFP alone, floated as efficiently as NS4B in the presence or even the absence of YFV infection (Fig. 4A). Furthermore, GFP fused to the third putative TM (GFP-pTM3) floated as efficiently as GFP-pTM (data not shown). NT5CT1 (Fig. 4B) also floated, albeit less efficiently. Surprisingly, neither endogenous nor overexpressed DNAJC14 floated to lighter fractions in the presence or absence of YFV infection (Fig. 4A). We believe that this is due to the presence of the C-terminal self-interaction domain, which mediates interaction, allowing chaperone activity and the formation of protein interactions that preclude flotation (see below and Discussion). Taken together, the results show that two subregions of DNAJC14, pTM3 and NT5CT1 lacking both pTM and the carboxyl-terminal self-interaction domain (Fig. 2A), colocalized and cofractionated with YFV NS proteins on DRMs. We next tested if GFP-pTM and the NT5CT1 mutant construct physically associate with YFV proteins in detergent-containing lysis buffer. SW13 cells expressing GFP-pTM or NT5CT1 were infected with YFV, and cell lysates were prepared for GFP-pTM or NT5CT1 immunoprecipitation. In both situations, NS3 was detected in the precipitated complex, while the ER protein calnexin was not (Fig. 4C). We were unable to assess the coprecipitation of NS4B because of the presence of a nonspecific band of the same size. These results indicate that the pTM region of DNAJC14 and the NT5CT1 DNAJC14 mutant construct each physically associate with YFV NS proteins in a membrane microdomain.
Fig 4.

DNAJC14 mutant constructs are associated with YFV NS proteins on DRMs. SW13 cells expressing GFP, GFP-pTM, DNAJC14-FL (A), or NT5CT1 (B) were infected with YFV (MOI, 5) or not infected. Cells were harvested 1.5 days postinfection, and 1% Triton X-100-treated lysates were subjected to membrane flotation assay. Fractions were taken from the top to the bottom, and proteins from each fraction were methanol precipitated and analyzed by Western blotting with the antibodies indicated. The identity of the endogenous DNAJC14 protein band was previously verified by small interfering RNA-mediated silencing (56). (C, D) Coimmunoprecipitation of GFP-DNAJC14-pTM (GFP-pTM) and NT5CT1-myc with YFV NS3. SW13 cells left untransduced or transduced to express GFP-pTM (C) or NT5CT1-myc (D) as indicated were challenged with YFV (MOI, 5). Cells were harvested 1.5 days later and immunoprecipitated with anti-GFP or anti-myc antibodies, respectively. Precipitated proteins were subjected to Western blot analysis with antibodies to NS3, calnexin, GFP, or myc, and relevant proteins are indicated. Similar results were obtained in several independent experiments. The values to the left are molecular sizes in kilodaltons.
A newly identified MBD is essential for DNAJC14 function.
As DNAJC14 NT5 is still membrane associated while NT6 is not (Fig. 1E), we proposed that there is another MBD in DNAJC14 following the pTM3 and before the J domain (Fig. 5A). To map the MBD and test whether it is important for antiviral activity, we first generated truncated forms of the putative MBD (Fig. 5A) and examined the resultant anti-YFV activity and intracellular distribution. There are three predicted α-helices within the putative MBD. On the basis of these, we generated constructs with truncations in this region (Fig. 5A) and examined their relative expression levels (Fig. 5B) and anti-YFV activities (Fig. 5C). The expression levels of the mutant proteins were similar, although some were expressed more robustly than others. This is unlikely to contribute substantially to differences in antiviral efficacy, since our previous work demonstrated that mutant constructs with very low expression levels could demonstrate potent antiviral activity (56). NT5.1 and NT5.2, each containing all three putative α-helices, had anti-YFV activities comparable to that of NT5, causing a 2-log reduction in YFV titers (Fig. 5C). NT5.3 lacking the first putative α-helix (H1) had partially impaired anti-YFV activity and reduced virus titers by about 1 log. Truncation mutant constructs NT5.4 and NT5.5, each lacking both the first (H1) and second (H2) putative α-helices, were no longer antiviral (Fig. 5A and C). These two mutant constructs also failed to localize in the cytosol and resided primarily in the nucleus (Fig. 5D), which may explain their failure to inhibit YFV replication. These results indicate that the first and second putative α-helices in the MBD are required for DNAJC14's maximal antiviral function. Next, we fused the putative MBD to GFP to examine if the MBD could mediate membrane targeting of GFP and recruitment to YFV NS protein clustering sites. Similar to NT5 truncation constructs NT5.1, NT5.2, and NT5.3, MBD-GFP displayed a cytosolic pattern and also colocalized with YFV NS4B in YFV-infected cells (Fig. 5E). These data demonstrate that the MBD of DNAJC14 is required for anti-YFV activity and mediates recruitment to YFV NS protein clustering sites.
We also verified that the MBD, like NT5CT1, could be targeted to DRMs. GFP was used as an internal nonfloating control, and cells were cotransfected with plasmids expressing GFP and MBD-GFP. Triton-treated cell lysates were subjected to membrane flotation assays, and fractions were examined by Western blotting. As shown in Fig. 5F, flotillin-1, a protein known to float, was found in the floating fractions, while GFP and calnexin, as expected, did not float. MBD-GFP displayed an obvious floating pattern, although it did not float as efficiently as flotillin-1. Thus, either the DNAJC14 pTM region or MBD can mediate localization to DRMs, where YFV NS proteins also reside.
The YFV RC resides on a detergent-insoluble fraction.
Since DNAJC14 subdomains colocalized and cofractionated with YFV NS proteins on DRMs, we wondered if DRMs might be the site of YFV RC assembly. We carried out in situ permeabilization with the mild detergent digitonin, which retains ER membrane integrity, or in situ solubilization with 1% Triton X-100, which presumably disrupts the ER membrane (Fig. 6A). We then examined if viral proteins remained after permeabilization or solubilization and the remaining viral proteins' sensitivity to proteinase K digestion, since viral proteins within the RC interior are presumably protected from proteinase K digestion (36, 42, 51). In addition, we tracked the RC by staining for dsRNA (15, 25, 54) in the cell fractions remaining after permeabilization or solubilization. Upon digitonin permeabilization (Fig. 6B, left panels), the majority of NS3, NS4B, calnexin, and DNAJC14 remained with the insoluble fraction. The majority of the insoluble NS3 was resistant to protease digestion, whereas only a small part of NS4B was resistant and almost all of DNAJC14 was sensitive to proteinase K digestion, which indicates that DNAJC14 is unlikely to be incorporated into the RC interior. Proteinase K digestion was efficient, as monitored by the complete loss of full-length calnexin in the protease-treated samples. Upon in situ solubilization with 1% Triton, cell morphology was lost while the nuclear shape was maintained (see Fig. 8B). The ER integral protein calnexin was completely solubilized and was detected only in the supernatant (S) fractions (Fig. 6B, right panels). Although some NS3 was solubilized and detected in the supernatant, substantial NS3 remained insoluble (IS). Even after Triton treatment, a small portion of NS3 remained proteinase K resistant. Compared to NS3, NS4B was solubilized more efficiently, with a greater portion detected in the supernatants (S). The insoluble NS4B was largely protease sensitive. Substantial endogenous DNAJC14 remained in the IS fraction and was protease sensitive.
Fig 6.

The YFV RC is on a detergent-insoluble fraction. (A) Schematic of experimental procedures for panels B and C. WB, Western blotting; IFA, immunofluorescent staining. (B) Proteinase K digestion of YFV-infected cells. SW13 cells were infected with YFV (MOI, 5) and permeabilized in situ 1 day later with 50 μg/ml digitonin or solubilized with 1% Triton X-100. The supernatant (S) was collected for Western blotting. The remaining detergent-insoluble cell material (IS) was then washed, scraped, and treated with 10 μg/ml proteinase K or not treated. Proteins were TCA precipitated and analyzed by Western blotting with the antibodies indicated. The values to the left are molecular sizes in kilodaltons. (C) After in situ permeabilization and washing, cells were fixed and permeabilized with 0.5% Triton for immunofluorescence analyses and examination by confocal microscopy. The results shown are representative of multiple similar experiments. Bar, 10 μm.
Fig 8.

DNAJC14 is associated with the YFV RC. (A) Schematic of the experimental procedures for panels B and C. coIP, coimmunoprecipitation; IFA, immunofluorescent staining; RT, room temperature. (B, C) SW13 cells were infected with YFV (MOI, 5), and 1 day later they were in situ solubilized with 1% Triton X-100, fixed, and then subjected to immunostaining. In panel B, samples were examined using an inverted fluorescence microscope (TE300; Nikon). In panel C, samples were observed by high-resolution microscopy. Uninfected cells stained with anti-DNAJC14 antibody are shown on the left. YFV-infected cells were stained with antibodies to dsRNA and DNAJC14, NS3, or NS4B, as indicated. Enlarged images of the boxed areas are shown below. Scale bars are as indicated. (D, E) SW13 cells were transduced with DNAJC14-FL and infected with YFV (MOI, 10). After 4 days, the cells were in situ solubilized with 1% Triton. In panel D, the cells were fixed and stained with the antibodies indicated and examined by confocal microscopy. Bar, 10 μm. (E) After in situ solubilization, the remaining insoluble cell fractions (IS) were scraped into lysis buffer and disrupted by needle passage. Clarified cell lysates were immunoprecipitated with anti-myc antibody or an IgG control, and immunoprecipitates were analyzed by Western blotting with the antibodies indicated. Asterisks indicate IgG heavy chain. Input, 6.7%. Similar results were obtained in multiple independent experiments. The values to the left are molecular sizes in kilodaltons.
When what remained of the cells after digitonin permeabilization or Triton solubilization was examined by confocal microscopy (Fig. 6C), dsRNA foci were detected near YFV NS proteins and DNAJC14 but were not completely colocalized. Although good colocalization of dsRNA and NS protein has been demonstrated with DENV (54), in some viral studies, such as those with severe acute respiratory syndrome virus and HCV (25, 49), only partial colocalization was noted. Failure to completely colocalize may be explained by differences in the relative antigen abundance of viral proteins and dsRNA within the RCs and/or by the participation of only a small part of the viral proteins in RC function (42, 51). Upon Triton solubilization, calnexin was totally removed. Although NS protein signals were reduced, in contrast, the dsRNA signal was enhanced, indicating that dsRNA is tightly associated with the NS proteins and likely protected within a Triton-sensitive membrane structure as described previously (36, 42, 51). After in situ Triton treatment, the dsRNA remains associated with the NS protein and DNAJC14 network but is more accessible because of the Triton-mediated membrane disruption. Although Triton treatment may remove some factors important for RC function (55), these data indicate that the YFV RC, along with DNAJC14, resides on a Triton-insoluble fraction.
The YFV RC is maintained by a protein-protein interaction network.
To further address the relationship between YFV RCs and DRMs and examine if the Triton-insoluble fraction containing the YFV RC is a DRM, we used progressive solubilization steps (Fig. 7A). We first solubilized YFV-infected cells in situ with 1% Triton as in Fig. 6 and collected the solubilized supernatant fraction (S1). The remaining insoluble fraction (IS1), which contains YFV RCs, was scraped off and solubilized again in Triton buffer by being passed through a needle. This physically disrupted sample was fractionated into solubilized supernatant (S2) and the insoluble nuclear pellet (IS2). As shown in Fig. 7B, and consistent with results in Fig. 6B, a greater proportion of NS4B than of NS3 was solubilized with Triton (S1). Substantial amounts of the NS3 and NS4B proteins (Fig. 7B) and the YFV genome (Fig. 7C) remained associated with the insoluble nuclear pellet (IS2) and failed to be solubilized in the S2 fraction. We subjected S1 and S2 (derived from the YFV RC-containing insoluble fraction, IS1) to the membrane flotation assay and examined the distribution of NS3, NS4B, and the viral genome. As shown in Fig. 7D, analysis of the S1 fraction revealed that NS4B floated very efficiently, while NS3 also partially floated, but not as efficiently. In contrast, in the S2 fraction, the flotation of NS4B was reduced compared to that of NS4B in the S1 fraction. NS3 present in S2 was found primarily in the nonfloating fraction. The YFV genome in both the S1 and S2 fractions failed to float (Fig. 7E). This, together with the results in Fig. 6, demonstrates that the YFV RC components could survive after Triton solubilization and fail to float. Thus, although the YFV NS proteins are targeted to DRM domains within the ER and can float in the membrane flotation assay, the RC components (dsRNA and NS proteins) are not present in the floating fractions but instead reside in a Triton-resistant network.
Fig 7.

The YFV RC is maintained by a protein-protein interaction network. (A) Schematic of the experimental procedures for panels B to E. RT, room temperature. (B) Proteins from the supernatants (S1 and S2) were precipitated with methanol, and the pellets from each of the three fractions (S1, S2, and IS2) were solubilized in equal volumes of SDS loading buffer and analyzed by Western blotting with anti-NS3 and -NS4B antisera. Bands were quantified by densitometry, and the percentage of the total NS protein found in each fraction is indicated at the bottom. Note that only one-fourth of the S1 fraction was run on the gel. (C) Distribution of RNA genomes in the fractions. YFV RNA levels were quantified by RT-qPCR and normalized to the level found in S1. (D) YFV-infected SW13 cells were solubilized and fractionated as outlined in panel A. Fractions S1 and S2 were subjected to a membrane flotation assay. Proteins from each fraction were analyzed by Western blotting with the antibodies indicated. (E) YFV RNA levels from each fraction of S1 and S2 after membrane flotation were analyzed by RT-qPCR, and the mean values from triplicate cDNA syntheses are plotted. Error bars indicate standard deviations. (F) SW13 cells expressing the indicated DNAJC14 mutant constructs were infected or not infected with YFV (MOI, 5), and then cells were in situ permeabilized with 1% Triton as described in the legend to Fig. 6A. After supernatant (S) collection, the remaining Triton-insoluble material was washed and harvested (IS). Proteins in the supernatants were precipitated with methanol. Proteins from S and IS were solubilized in an equal amount of SDS loading buffer and analyzed by Western blotting with the antibodies indicated. Arrowheads indicated nonspecific bands. (G) SW13 cells expressing DNAJC14 NT5CT1 and NT5 mutant constructs were treated with Triton, and lysates were subjected to a sucrose gradient membrane flotation assay. Proteins from each fraction were analyzed by Western blotting with anti-myc antibodies (NT5 and NT5CT1) or the antibodies indicated. For panels D and E, similar results were obtained in independent experiments. The values to the left of panels B, D, F, and G are molecular sizes in kilodaltons.
Interestingly, endogenous DNAJC14, like YFV RCs, resides on a Triton-insoluble but nonfloating fraction in the membrane flotation assay (Fig. 4A). This is in contrast to the behavior of the DNAJC14 pTM or NT5CT1 subdomain, each of which can reside on floating fractions (Fig. 4). To address this apparent contradiction and further define the relationship between Triton solubility and flotation ability, we analyzed the in situ Triton solubility of DNAJC14 mutant constructs with different flotation behaviors (Fig. 7F). We analyzed GFP-pTM3, NT5CT1, and CT1, each of which could be recruited to and colocalized with YFV NS protein clustering sites (Fig. 3C and 2D and B, respectively). Of these, DNAJC14-pTM3 (data not shown) and NT5CT1 (Fig. 4) were able to float. We also included DNAJC14-FL and mutant construct NT5 lacking the pTM region, both of which are capable of YFV inhibition. Cells were transduced to express the proteins, infected with YFV or left uninfected, and then in situ solubilized with 1% Triton. Solubilized supernatants (S) and the remaining insoluble fractions (IS) were collected and analyzed by Western blotting. In the presence or absence of YFV infection, GFP-pTM3, NT5CT1, and CT1, each lacking the C-terminal self-interaction domain, were efficiently solubilized and detected only in the supernatant fraction. In contrast, NT5 and FL (each containing the C-terminal self-interaction domain) were partially resistant to Triton solubilization (Fig. 7F). Note that NS4B was not detected in the NT5 sample because of very efficient transduction levels and the strong inhibitory effect of NT5 on YFV replication. These data suggest that self-interaction of DNAJC14 is required for resistance to in situ Triton solubilization and suggest that the ability to partition into a Triton-insoluble fraction correlates with DNAJC14-mediated YFV inhibition. We examined whether the Triton-insoluble NT5 mutant construct, in contrast to NT5CT1, resides predominantly in a nonfloating fraction, similar to endogenous DNAJC14 (Fig. 4). Membrane flotation studies (Fig. 7G) demonstrated that NT5CT1 was able to float while NT5 floated much less efficiently. Taken together, the YFV RC component resides on a nonfloating structure or complex that is likely mediated by protein-protein interactions and this structure is resistant to in situ Triton solubilization. Only a portion of the YFV NS proteins enters into the RC. Self-interaction of DNAJC14, which is essential for its anti-YFV activity, also is required for entering into this nonfloating structure.
DNAJC14 associates with the YFV RC via direct protein-protein interaction.
Since both the YFV RC and DNAJC14 reside in a nonfloating, Triton-insoluble fraction, we wondered if the YFV RC and DNAJC14 both reside within a protein network and whether DNAJC14 physically associates with YFV RC components. We first examined the viral RC structure and endogenous DNAJC14 by high-resolution microscopy. After in situ Triton solubilization (Fig. 8A and B), dsRNAs and NS3 and NS4B appeared as a clustered network and dsRNAs were attached to the NS3 and NS4B network (Fig. 8C). In YFV-infected cells, DNAJC14 was redistributed and recruited to a dsRNA-containing network. We next examined if transduced DNAJC14, like endogenous DNAJC14, associates with the YFV RC-containing protein network after in situ Triton solubilization. Since overexpressed DNAJC14 inhibits YFV replication, we took advantage of the fact that, at late time points after infection (Fig. 2C), YFV can overcome DNAJC14-mediated inhibition in DNAJC14-FL-transduced cells (56). Colocalization of DNAJC14-FL with NS3, NS4B, and dsRNA after Triton solubilization was analyzed by confocal microscopy (Fig. 8D). DNAJC14-FL colocalized well with NS3 (rp, 0.67; rs, 0.75) and NS4B (rp, 0.76; rs, 0.81). As demonstrated above for endogenous DNAJC14 (Fig. 8C and 6C), dsRNA foci were also found in association with overexpressed DNAJC14 (rp, 0.51; rs, 0.54).
To test for a physical interaction of RC components with DNAJC14, we performed coimmunoprecipitation using the RC-containing insoluble fraction. Using in situ solubilization, as outlined in Fig. 8A, of YFV-infected cells expressing DNAJC14-FL-myc, an S2 fraction was prepared and DNAJC14 was immunoprecipitated using anti-myc antibody. YFV NS3 and NS5, but not NS4B, were coprecipitated with DNAJC14-FL-myc, while flotillin-1 and β-actin were not (Fig. 8E), indicating that DNAJC14-FL associates with NS3 and NS5 either directly or indirectly within a protein-protein interaction network. These results imply a protein scaffold within the YFV RC and support the presence of a protein-protein interaction between DNAJC14 and components of the YFV RC.
The DNAJC14 MBD and C-terminal self-interaction domain can confer activity to a heterologous Hsp40 family member.
On the basis of our findings described above, we hypothesized that DNAJC14 first is targeted via its TM or MBD to a unique DRM microdomain within the ER where YFV NS proteins also reside. Within this DRM, we hypothesized that DNAJC14 then interacts with a substrate viral protein(s) and itself via protein-protein interactions and serves to chaperone conformational changes of viral proteins to allow formation of the RC. In this model, overexpression of DNAJC14 would alter the stoichiometry of this process and disrupt RC formation. Overexpression of DNAJC14 mutant constructs unable to enter into this protein-protein interaction network would fail to inhibit YFV replication. This model predicts that the MBD (or TM) and the C-terminal interaction domain are key elements of DNAJC14 that play important roles in its function. To test this hypothesis, we asked whether the MBD and the C-terminal self-interaction domain could confer antiviral activity on an unrelated noninhibitory Hsp40 protein. We selected DNAJB1, another Hsp40 family member that has a similar domain arrangement but lacks an MBD or a membrane-targeting motif (23). We fused the DNAJC14 MBD or the C-terminal self-interaction domain individually or in combination to DNAJB1 (Fig. 9A) and then examined each hybrid protein's localization and anti-YFV activity. DNAJB1 was distributed throughout the cell in both the nucleus and the cytoplasm, and fusion of the DNAJC14 self-interaction domain (DNAJB1C) did not change the localization of DNAJB1 (Fig. 9B). DNAJB1 with an N-terminal fusion of the DNAJC14 MBD (MDNAJB1), like DNAJC14-NT5, displayed a cytosolic distribution, although it resided more peripherally than DNAJC14-NT5. Further fusion of the DNAJC14 self-interaction domain to MDNAJB1 (MDNAJB1C) changed its distribution and resulted in a cytoplasmic localization more similar to that of DNAJC14-NT5 (Fig. 9B). When tested for the ability to inhibit YFV replication, DNAJB1 did not exhibit significant anti-YFV activity, lowering titers less than 1 log (Fig. 9D). Fusion of either the DNAJC14 self-interaction domain (DNAJB1C) or the MBD (MDNAJB1) to DNAJB1 did not confer anti-YFV activity. However, fusion of both the MBD and the self-interaction domain of DNAJC14 (MDNAJB1C) conferred significant (P < 0.05) anti-YFV activity on DNAJB1 (Fig. 9D), reducing viral titers by greater than 1 log compared to those obtained with the vector alone. These data indicate that DNAJC14 MBD-mediated membrane targeting and self-interaction are each essential but not sufficient for YFV-inhibitory activity.
Fig 9.

The DNAJC14 MBD and self-interaction domain together confer anti-YFV activity on a noninhibitory Hsp40 family member. (A) Schematic of DNAJC14-NT5 and DNAJB1 mutant constructs. The DNAJC14 MBD (green) or self-interaction domain (red) was fused to the amino or carboxyl terminus, respectively, of DNAJB1, either alone or in combination. The chimeric proteins were cloned into the V1 plasmid and contained a C-terminal myc epitope tag. (B) Intracellular distribution of DNAJB1 and mutant constructs. SW13 cells were transduced to express DNAJB1 or the mutant constructs, and after 1.5 days, they were fixed for immunostaining with anti-myc antibody and analysis by confocal microscopy. Bar, 10 μm. (C) Protein expression of DNAJB1 and mutant constructs. Cells transduced to express the indicated proteins were analyzed by Western blotting with anti-myc antibody. (D) Anti-YFV activity of DNAJB1 and mutant constructs. SW13 cells transduced to express the V1 vector alone (V) or the proteins indicated were infected with YFV (MOI, 5). After 1 day, the virus in the medium was quantified by plaque assay. Data represent the mean titer ± the standard error of the mean; n = 3. Asterisks indicate statistically significant differences from titers obtained in cells transduced with the vector alone or between the indicated pairs (*, P < 0.05; **, P < 0.01; ns, not significant [unpaired t test]). pfu, plaque-forming units.
DISCUSSION
We recently identified DNAJC14 as a broadly active modulator of flavivirus replication. It plays a role after viral genome translation, likely in YFV RC function (56). In the present study, we extended our investigation to further characterize the individual domains of DNAJC14 and their roles in YFV RC assembly. We found that DNAJC14, like the YFV NS proteins, could be targeted to DRMs within the ER via its TM or MBD (Fig. 4). DRMs or lipid rafts, which are found mainly in the plasma membrane, are minor fractions of cell membranes enriched in sphingolipid and cholesterol that are preserved after mild detergent (e.g., 1% Triton X-100) extraction (31, 46). DRMs or lipid rafts provide the platform for lipid and protein interaction and function in membrane signaling and trafficking (31). Flavivirus infection is known to alter cellular lipid homeostasis. Infection alters cellular cholesterol levels, with cholesterol enrichment at viral replication sites (33), and fatty acid synthase activity is modulated (18), which likely facilitates RC assembly. Flavivirus NS proteins and likely RCs have been reported to localize on DRMs (1, 39, 51).
Here we found that YFV NS3 and NS4B, together with subdomains of DNAJC14, including the TM (GFP-pTM) and NT5CT1 lacking the C-terminal interaction domain, target DRMs (Fig. 4). In addition, GFP-pTM and NT5CT1 both are redistributed and recruited to YFV NS protein clustering sites, as judged by immunostaining (Fig. 2). Coimmunoprecipitation of either GFP-pTM or NT5CT1 with YFV NS proteins in detergent-containing buffer verified association of GFP-pTM and NT5CT1 with YFV NS proteins within an ER membrane microdomain (Fig. 4C and D). However, infection with YFV is not required for GFP-pTM and NT5CT1 distribution to these membrane microdomains, since in flotation assays both float efficiently in YFV-infected or mock-infected cells (Fig. 4A and B). Thus, YFV infection does not change the biochemical behavior of GFP-pTM and NT5CT1, even though their subcellular localization is altered upon infection (Fig. 2). This could be explained by infection resulting in the clustering of these subdomains, which might be driven by protein-protein interactions between viral proteins (see model in Fig. 10).
Fig 10.
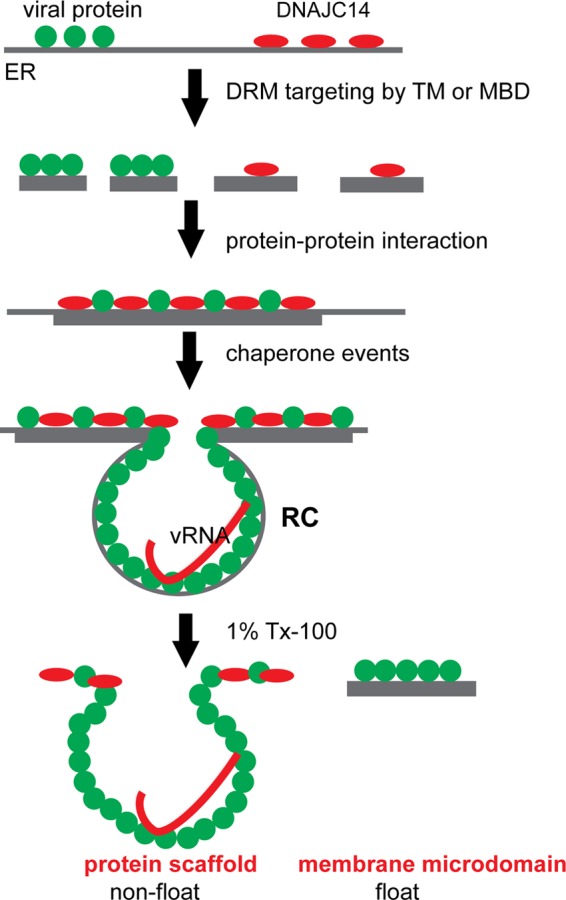
Proposed model of DNAJC14 action and RC formation. Viral proteins (green) and DNAJC14 (red) are targeted to DRMs within the ER membrane. Membrane targeting of DNAJC14 can be mediated by its TM or MBD. Viral infection prompts the clustering of these DRMs, which in turn facilitates protein-protein interactions between the viral proteins and between a viral protein(s) and DNAJC14, allowing chaperone events that facilitate RC formation to occur. The RC, once formed, is maintained by a protein scaffold consisting of viral proteins and possibly host proteins and also contains the viral dsRNA replication intermediate. The RC protein scaffold remains intact after Triton solubilization and fails to float in membrane flotation assays, while viral NS proteins that failed to undergo the chaperone process remain in the DRMs after Triton solubilization and float in membrane flotation assays.
Although the YFV NS proteins and DNAJC14 and DNAJC14 subdomains target DRMs, we demonstrate that the YFV RC is probably formed and maintained by protein-protein interactions or a protein scaffold that is distinct from the DRM. First, after in situ Triton solubilization, substantial amounts of YFV NS3, NS4B, and DNAJC14 are retained (Fig. 6B) and found attached to the remainder of the cells that failed to be solubilized (Fig. 6C). In contrast, despite efficient targeting to DRMs and flotation by GFP-pTM (Fig. 4A), upon similar treatment, this protein is solubilized in the supernatant (Fig. 7F), which indicates efficient removal of lipid components by in situ Triton solubilization. In addition, dsRNA staining, used as an RC marker, is not reduced but, in contrast, is enhanced upon in situ Triton solubilization (Fig. 6C). The dsRNA colocalized or associated with YFV NS protein- and DNAJC14-containing clustering networks (Fig. 8). These findings indicate that there is a protein network containing substantial viral NS protein and dsRNA, which survives after in situ solubilization with a nondenaturing detergent, such as Triton X-100. Further, after in situ Triton solubilization, the insoluble fraction, which contains the YFV RC, fails to float when applied to a membrane flotation assay (Fig. 7D), verifying that it is a protein complex that is not part of the floatable DRM. Last, in addition to the MBD or the TM domain, DNAJC14 mutant constructs must contain the self-interaction domain in order enter into the protein network (Fig. 7F), which indicates that the protein network is mediated by self-interaction of DNAJC14 and probably DNAJC14-viral protein interaction (Fig. 10).
Studies of other RNA viruses also support the requirement for a protein interaction network for viral RC function. Self-interaction of the NS4B integral membrane protein of HCV is required for viral RC assembly (41). Bromovirus RC formation requires the precise action of domains of its viral replication protein 1a, with individual domains capable of forming multimers and lattices (10). Poliovirus polymerase also could form lattices in vitro and polymerase arrays in infected cells (32). The ER membrane-shaping reticulon proteins are incorporated into the Bromovirus RC via protein interaction with viral proteins and probably are crucial for maintenance of the RC (11).
In this study, we have capitalized on the ability of overexpression of the host protein DNAJC14 to inhibit YFV RC formation to gain insight into the normal function of DNAJC14 in the virus replication cycle. Taking a mutagenesis approach, we used biochemical, virological, and cell biological assays to characterize DNAJC14 action with respect to virus inhibition and viral RC formation. On the basis of our findings, we propose a model wherein DNAJC14 functions to chaperone the assembly of the YFV RC (Fig. 10). DRM targeting of both YFV NS proteins and DNAJC14 facilitates the clustering of viral proteins and DNAJC14, allowing subsequent protein-protein interactions to occur to form a protein network, within which DNAJC14 likely chaperones a conformation change in its viral substrate protein, resulting in RC assembly. A portion of the NS proteins remains associated with DRMs and floats in flotation assays. In contrast, the RC-associated NS proteins no longer reside in floating DRMs; instead, they reside in a nonfloating protein scaffold formed by interactions between viral proteins and probably additional host factors and the viral dsRNA. This interaction network survives after in situ Triton solubilization and fails to float in flotation assays (Fig. 10). This mechanism may be analogous to the T cell receptor complex-containing microdomain that is created by protein-protein networks (12).
It is interesting that DNAJC14 has two potential mechanisms for membrane targeting, the TM region and the MBD. While each domain can mediate membrane association, the two may have distinct roles in DNAJC14's normal cellular roles and its role in Flavivirus replication modulation and RC formation. It is possible that the MBD may bind different lipid components at the membrane surface to regulate protein conformation or membrane curvature. Whether DNAJC14 plays a role in the lipid alterations present at sites of Flavivirus replication would be interesting to study further. It is also remarkable that the closest DNAJC14 homologue in mosquitoes has no predicted TMs. It would be interesting to determine why DNAJC14 evolved in mammals to contain TM helices and to identify lipid components targeted by the DNAJC14 MBD.
ACKNOWLEDGMENTS
We are grateful to Alison North and the Rockefeller University Bio-Imaging Resource Center staff for their assistance in this project.
This work was supported in part by NIH grants AI089062 (M.R.M.) and CA057973 (C.M.R.), the Greenberg Medical Research Institute, the Starr Foundation, and the Irma T. Hirschl/Monique Weill-Caulier Trust (M.R.M.). This project was also supported by award S10RR031855 from the National Center For Research Resources.
The content of this report is solely our responsibility and does not necessarily represent the official views of any funding sources. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Published ahead of print 22 August 2012
REFERENCES
- 1. Aizaki H, Lee KJ, Sung VM, Ishiko H, Lai MM. 2004. Characterization of the hepatitis C virus RNA replication complex associated with lipid rafts. Virology 324:450–461 [DOI] [PubMed] [Google Scholar]
- 2. Bermak JC, Li M, Bullock C, Zhou QY. 2001. Regulation of transport of the dopamine D1 receptor by a new membrane-associated ER protein. Nat. Cell Biol. 3:492–498 [DOI] [PubMed] [Google Scholar]
- 3. Blight KJ, McKeating JA, Rice CM. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 76:13001–13014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bredenbeek PJ, et al. 2003. A stable full-length yellow fever virus cDNA clone and the role of conserved RNA elements in flavivirus replication. J. Gen. Virol. 84:1261–1268 [DOI] [PubMed] [Google Scholar]
- 5. Chambers TJ, McCourt DW, Rice CM. 1990. Production of yellow fever virus proteins in infected cells: identification of discrete polyprotein species and analysis of cleavage kinetics using region-specific polyclonal antisera. Virology 177:159–174 [DOI] [PubMed] [Google Scholar]
- 6. Cowan S, et al. 2002. Cellular inhibitors with Fv1-like activity restrict human and simian immunodeficiency virus tropism. Proc. Natl. Acad. Sci. U. S. A. 99:11914–11919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cristea IM, Williams R, Chait BT, Rout MP. 2005. Fluorescent proteins as proteomic probes. Mol. Cell. Proteomics 4:1933–1941 [DOI] [PubMed] [Google Scholar]
- 8. Cubitt AB, et al. 1995. Understanding, improving and using green fluorescent proteins. Trends Biochem. Sci. 20:448–455 [DOI] [PubMed] [Google Scholar]
- 9. den Boon JA, Ahlquist P. 2010. Organelle-like membrane compartmentalization of positive-strand RNA virus replication factories. Annu. Rev. Microbiol. 64:241–256 [DOI] [PubMed] [Google Scholar]
- 10. Diaz A, Gallei A, Ahlquist P. 2012. Bromovirus RNA replication compartment formation requires concerted action of 1a's self-interacting RNA capping and helicase domains. J. Virol. 86:821–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Diaz A, Wang X, Ahlquist P. 2010. Membrane-shaping host reticulon proteins play crucial roles in viral RNA replication compartment formation and function. Proc. Natl. Acad. Sci. U. S. A. 107:16291–16296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Douglass AD, Vale RD. 2005. Single-molecule microscopy reveals plasma membrane microdomains created by protein-protein networks that exclude or trap signaling molecules in T cells. Cell 121:937–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Evans MJ, et al. 2007. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446:801–805 [DOI] [PubMed] [Google Scholar]
- 14. French AP, Mills S, Swarup R, Bennett MJ, Pridmore TP. 2008. Colocalization of fluorescent markers in confocal microscope images of plant cells. Nat. Protoc. 3:619–628 [DOI] [PubMed] [Google Scholar]
- 15. Gillespie LK, Hoenen A, Morgan G, Mackenzie JM. 2010. The endoplasmic reticulum provides the membrane platform for biogenesis of the flavivirus replication complex. J. Virol. 84:10438–10447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gould EA, Solomon T. 2008. Pathogenic flaviviruses. Lancet 371:500–509 [DOI] [PubMed] [Google Scholar]
- 17. Gubler DJ, Kuno G, Markoff L. 2007. Flaviviruses, p 1153–1252 In Knipe DM, Howley PM. (ed), Fields virology, fifth ed, vol 1 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 18. Heaton NS, et al. 2010. Dengue virus nonstructural protein 3 redistributes fatty acid synthase to sites of viral replication and increases cellular fatty acid synthesis. Proc. Natl. Acad. Sci. U. S. A. 107:17345–17350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Heaton NS, Randall G. 2010. Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microbe 8:422–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Heaton NS, Randall G. 2011. Multifaceted roles for lipids in viral infection. Trends Microbiol. 19:368–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hsu NY, et al. 2010. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 141:799–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jones CT, et al. 2010. Real-time imaging of hepatitis C virus infection using a fluorescent cell-based reporter system. Nat. Biotechnol. 28:167–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kampinga HH, Craig EA. 2010. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 11:579–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kelley WL. 1998. The J-domain family and the recruitment of chaperone power. Trends Biochem. Sci. 23:222–227 [DOI] [PubMed] [Google Scholar]
- 25. Knoops K, et al. 2008. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 6:e226 doi:10.1371/journal.pbio.0060226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kümmerer BM, Rice CM. 2002. Mutations in the yellow fever virus nonstructural protein NS2A selectively block production of infectious particles. J. Virol. 76:4773–4784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lackner T, Thiel HJ, Tautz N. 2006. Dissection of a viral autoprotease elucidates a function of a cellular chaperone in proteolysis. Proc. Natl. Acad. Sci. U. S. A. 103:1510–1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Leyssen P, Balzarini J, De Clercq E, Neyts J. 2005. The predominant mechanism by which ribavirin exerts its antiviral activity in vitro against flaviviruses and paramyxoviruses is mediated by inhibition of IMP dehydrogenase. J. Virol. 79:1943–1947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lindenbach BD, Rice CM. 1997. trans-complementation of yellow fever virus NS1 reveals a role in early RNA replication. J. Virol. 71:9608–9617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lindenbach BD, Thiel H-J, Rice CM. 2007. Flaviviridae: the viruses and their replication, p 1101–1152 In Knipe DM, Howley PM. (ed), Fields virology, fifth ed, vol 1 Lippincott, Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 31. Lingwood D, Simons K. 2010. Lipid rafts as a membrane-organizing principle. Science 327:46–50 [DOI] [PubMed] [Google Scholar]
- 32. Lyle JM, Bullitt E, Bienz K, Kirkegaard K. 2002. Visualization and functional analysis of RNA-dependent RNA polymerase lattices. Science 296:2218–2222 [DOI] [PubMed] [Google Scholar]
- 33. Mackenzie JM, Khromykh AA, Parton RG. 2007. Cholesterol manipulation by West Nile virus perturbs the cellular immune response. Cell Host Microbe 2:229–239 [DOI] [PubMed] [Google Scholar]
- 34. Miller S, Kastner S, Krijnse-Locker J, Buhler S, Bartenschlager R. 2007. The non-structural protein 4A of dengue virus is an integral membrane protein inducing membrane alterations in a 2K-regulated manner. J. Biol. Chem. 282:8873–8882 [DOI] [PubMed] [Google Scholar]
- 35. Miller S, Krijnse-Locker J. 2008. Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 6:363–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Miyanari Y, et al. 2003. Hepatitis C virus non-structural proteins in the probable membranous compartment function in viral genome replication. J. Biol. Chem. 278:50301–50308 [DOI] [PubMed] [Google Scholar]
- 37. Nagy PD, Pogany J. 2012. The dependence of viral RNA replication on co-opted host factors. Nat. Rev. Microbiol. 10:137–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Neill JD, Ridpath JF. 2001. Recombination with a cellular mRNA encoding a novel DnaJ protein results in biotype conversion in genotype 2 bovine viral diarrhea viruses. Virus Res. 79:59–69 [DOI] [PubMed] [Google Scholar]
- 39. Noisakran S, et al. 2008. Association of dengue virus NS1 protein with lipid rafts. J. Gen. Virol. 89:2492–2500 [DOI] [PubMed] [Google Scholar]
- 40. Ostermeyer AG, et al. 2001. Accumulation of caveolin in the endoplasmic reticulum redirects the protein to lipid storage droplets. J. Cell Biol. 152:1071–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Paul D, et al. 2011. NS4B self-interaction through conserved C-terminal elements is required for the establishment of functional hepatitis C virus replication complexes. J. Virol. 85:6963–6976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Quinkert D, Bartenschlager R, Lohmann V. 2005. Quantitative analysis of the hepatitis C virus replication complex. J. Virol. 79:13594–13605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reiss S, et al. 2011. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 9:32–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rinck G, et al. 2001. A cellular J-domain protein modulates polyprotein processing and cytopathogenicity of a pestivirus. J. Virol. 75:9470–9482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Roosendaal J, Westaway EG, Khromykh A, Mackenzie JM. 2006. Regulated cleavages at the West Nile virus NS4A-2K-NS4B junctions play a major role in rearranging cytoplasmic membranes and Golgi trafficking of the NS4A protein. J. Virol. 80:4623–4632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schuck S, Honsho M, Ekroos K, Shevchenko A, Simons K. 2003. Resistance of cell membranes to different detergents. Proc. Natl. Acad. Sci. U. S. A. 100:5795–5800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shi ST, Lee KJ, Aizaki H, Hwang SB, Lai MM. 2003. Hepatitis C virus RNA replication occurs on a detergent-resistant membrane that cofractionates with caveolin-2. J. Virol. 77:4160–4168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stapleford KA, Miller DJ. 2010. Role of cellular lipids in positive-sense RNA virus replication complex assembly and function. Viruses 2:1055–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Targett-Adams P, Boulant S, McLauchlan J. 2008. Visualization of double-stranded RNA in cells supporting hepatitis C virus RNA replication. J. Virol. 82:2182–2195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tchernev VT, et al. 2002. The Chediak-Higashi protein interacts with SNARE complex and signal transduction proteins. Mol. Med. 8:56–64 [PMC free article] [PubMed] [Google Scholar]
- 51. Uchil PD, Satchidanandam V. 2003. Architecture of the flaviviral replication complex. Protease, nuclease, and detergents reveal encasement within double-layered membrane compartments. J. Biol. Chem. 278:24388–24398 [DOI] [PubMed] [Google Scholar]
- 52. Vos MJ, Hageman J, Carra S, Kampinga HH. 2008. Structural and functional diversities between members of the human HSPB, HSPH, HSPA, and DNAJ chaperone families. Biochemistry 47:7001–7011 [DOI] [PubMed] [Google Scholar]
- 53. Ward WW. 2006. Biochemical and physical properties of green fluorescent protein, p 39–65 In Chalfie M, Kain SR. (ed), Green fluorescent protein: properties, applications, and protocols, vol 47, 2nd ed John Wiley & Sons, Inc., Hoboken, NJ: [PubMed] [Google Scholar]
- 54. Welsch S, et al. 2009. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 5:365–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yang G, et al. 2004. Newly synthesized hepatitis C virus replicon RNA is protected from nuclease activity by a protease-sensitive factor(s). J. Virol. 78:10202–10205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yi Z, et al. 2011. Identification and characterization of the host protein DNAJC14 as a broadly active flavivirus replication modulator. PLoS Pathog. 7:e1001255 doi:10.1371/journal.ppat.1001255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zennou V, et al. 2000. HIV-1 genome nuclear import is mediated by a central DNA flap. Cell 101:173–185 [DOI] [PubMed] [Google Scholar]