

Abstract
It is well known that proteins in the tegument (located between the viral capsid and envelope proteins) play critical roles in the assembly and budding of herpesviruses. Tegument proteins UL16 and UL11 of herpes simplex virus (HSV) are conserved among all the Herpesviridae. Although these proteins directly interact in vitro, UL16 was found to colocalize poorly with UL11 in cotransfected cells. To explain this discrepancy, we hypothesized that UL16 is initially made in an inactive form and is artificially transformed to the binding-competent state when cells are disrupted. Consistent with a regulated interaction, UL16 was able to fully colocalize with UL11 when a large C-terminal segment of UL16 was removed, creating mutant UL16(1-155). Moreover, membrane flotation assays revealed a massive movement of this mutant to the top of sucrose gradients in the presence of UL11, whereas both the full-length UL16 and the C-terminal fragment (residues 156 to 373) remained at the bottom. Further evidence for the presence of a C-terminal regulatory domain was provided by single-amino-acid substitutions at conserved cysteines (C269S, C271S, and C357S), which enabled the efficient interaction of full-length UL16 with UL11. Lastly, the binding site for UL11 was further mapped to residues 81 to 155, and to our surprise, the 5 Cys residues within UL16(1-155) are not required, even though the modification of free cysteines in UL16 with N-ethylmaleimide does in fact prevent binding. Collectively, these results reveal a regulatory function within the C-terminal region of UL16 that controls an N-terminal UL11-binding activity.
INTRODUCTION
All herpesviruses have electron-dense material located between the nucleocapsid and the viral membrane, and this layer is known as the tegument. In the case of herpes simplex virus (HSV), there are more than 20 different protein species in this compartment (54), and their structural organization is almost entirely unknown. However, it has become quite clear that the tegument is more than a simple layer of proteins but in fact contains machinery that rearranges when the glycoproteins on the outside of the virus engage receptors on the cell surface (52).
Tegument assembly begins in the nucleus, where some proteins are added to the DNA-containing capsid (8, 32, 53, 54). Additional proteins are added as the capsid travels to the cytoplasmic site of budding on Golgi-derived membranes, which are studded with various viral membrane proteins whose cytoplasmic tails are bound with specific tegument proteins. At this step, multiple and complex interactions between the capsid-bound tegument proteins and the membrane protein complexes provide linkages that drive the budding process as the mature virion emerges (9, 23, 25, 44, 53, 54, 57, 74, 75). Two of the bridging interactions that are thought to be important for envelopment are those between capsid-bound UL16 and the membrane-bound proteins UL11 and glycoprotein E (gE) (44, 74, 75). The focus of studies reported here is the UL11-UL16 interaction.
UL11 is a small (96-amino-acid [aa]) protein, conserved among all herpesviruses, that has long been known to be important for budding (4, 5, 7, 11, 18, 19, 38–40, 46, 47, 63, 66, 68). In particular, UL11-null mutants accumulate capsids in the cytoplasm and exhibit a greater-than-100-fold reduction in the release of virus particles (4, 5, 39, 47, 68). UL11 is peripherally bound to membranes via a myristate (on its N-terminal glycine) and a palmitate (on one or more cysteines at residues 11 to 13) (43, 46). It accumulates on Golgi-derived membranes (43) and has been found to traffic through lipid rafts (6), the significance of which remains unknown. Like its UL16 binding partner (44), UL11 also interacts directly with the cytoplasmic tail of gE (a virus-encoded glycoprotein suggested to have a role in cell-to-cell spread of herpesviruses) (14–16, 25, 74).
UL16 is a 373-aa tegument protein that is also conserved among all herpesviruses (24, 34, 48, 55, 58, 73). It contains 20 cysteines (51, 75), 8 of which are conserved. UL16 is found in both the nucleus and cytoplasm of infected cells (51, 55, 58), but a stable association with capsids has been found only in the cytoplasm (51). UL16-null mutants have a 10-fold reduction in virus titers (3, 35), and for various other herpesviruses, such mutants have defects in cytoplasmic budding, based on the accumulation of capsids in the cytoplasm (24, 48, 61). Curiously, the UL16-capsid interaction is weakened within extracellular virions (51), indicating that tegument assembly is a dynamic process. Moreover, UL16 appears to be completely released from the capsid when the virus binds to attachment receptors on the host cell (52), providing evidence for a complicated molecular machine within the tegument—one that is intimately linked to the viral membrane proteins.
The interaction of UL16 with UL11 is highly specific. Most importantly, UL16 recognizes an acidic-cluster (AC) motif, which is located in the first half of UL11 (44, 75). When this motif is deleted, UL11 accumulates to higher levels in lipid rafts (6), and in the context of a virus infection, such mutants fail to package UL16 into virions (25). Substitution mutants that have foreign acidic clusters restore the membrane-trafficking properties of UL11, but not UL16 binding (43–45). This observation suggests that host factors involved in the trafficking of UL11 are less discriminatory in their recognition of acidic clusters than UL16, which has a strong preference for the native motif. In spite of this specificity and the robust binding properties observed for UL11 and UL16 in vitro (44, 75), these two proteins do not recognize each other very well when they are expressed alone in transfection experiments (44). Most of the UL16 molecules remain distributed throughout the cytoplasm and nucleus with only a small population relocalized to the juxtanuclear position where UL11 accumulates. Nevertheless, this inefficient colocalization is lost when the acidic cluster is removed from UL11 (44), again emphasizing the specificity of the interaction.
While the critical residues of UL11 are quite clear, the location of the UL11-binding site within UL16 has been difficult to identify. Studies of a large collection of deletion mutants failed to reveal any fragments that could bind in vitro to UL11 (75) or to UL21, another conserved tegument protein (2, 13, 50) and a binding partner of UL16 (26, 35). Only the first 40, nonconserved residues were found to be dispensable (75). On the other hand, treatment of UL16 with N-ethylmaleimide (NEM) (a thiol blocker) was found to efficiently block binding to UL11 (but not UL21) (26, 75), suggesting that the interaction may require one or more free cysteines within UL16. In contrast, none of the four cysteines in UL11 are required (75). Unfortunately, cysteines are located throughout UL16, and approximately 10 of them were estimated to be modified by NEM (51). Hence, the NEM experiments are noninformative with regard to mapping the UL11-binding site.
A powerful insight regarding the binding mechanism of UL16 was recently obtained in studies of its interaction with the cytoplasmic tail of gE (74). In brief, these two proteins interact well in vitro but colocalize poorly in transfection experiments, as is the case for the interaction of UL16 and UL11. Surprisingly, a spontaneous deletion mutant that retains only the first 155 residues of UL16 was found to efficiently colocalize with gE (74). On the basis of this observation, we proposed that a structure in the C-terminal portion of UL16 negatively regulates an N-terminal site that binds to the tail of gE. According to this model, the binding site is artificially activated in vitro when UL16 is extracted from cells or when the regulatory domain is deleted. Moreover, we presume that binding is normally activated in infected cells by the interaction of UL16 with other viral proteins (UL21, for example).
In experiments described below, we addressed the hypothesis that the UL11 binding in UL16 also occurs within the first 155 residues of UL16. This was found to be the case, and the binding site was mapped to an even smaller region of this fragment. Surprisingly, the interaction does not require cysteines at all, suggesting that previously reported modifications with NEM serve to distort the actual binding site. Further evidence for a regulatory domain was also revealed by single-amino-acid substitutions in the C-terminal region that activate the ability of full-length UL16 molecules to efficiently colocalize with UL11 in cotransfected cells. These findings suggest that the assembly of the tegument is a highly dynamic and regulated process.
MATERIALS AND METHODS
Cells and antibodies.
Vero cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum (FBS), penicillin (65 μg/ml), and streptomycin (131 μg/ml). Rabbit anti-green fluorescent protein (GFP) serum (Cocalico Biologicals, Inc.) was raised against His6-GFP and recognizes both GFP and the His6 tag (6). Anti-UL16 serum was raised against glutathione S-transferase (GST)-UL16 in rabbits (51, 75). Antibodies used in immunoprecipitation or GST pulldown assays specifically recognize an N-terminal peptide (21 to 32 aa) in UL16 and were produced in rabbits (74). Rabbit anti-UL11 serum was raised against GST-UL11 (44). Mouse monoclonal antibodies against the hemagglutinin (HA) epitope (Sigma) were used for immunofluorescence assays at 1:3,000.
Construction of His-UL16 and UL16-GFP derivatives.
The plasmid expressing His6-tagged UL16 in Escherichia coli has been described previously (75). The plasmid pHis6-UL16(1-155) was generated by inserting a stop codon after that for the 155th amino acid of His6-UL16 (74). Plasmid pCMV.UL16-GFP expresses GFP-tagged UL16 from the cytomegalovirus (CMV) promoter (44). Construct pCMV.UL16(1-155)-GFP (74) was used to create the following cysteine-to-serine replacement mutants: C69S, C78S, C93S, C125S, C142S, C78S/C93S, C125S/C142S, C69S/C78S/C125S, C78S/C93S/C125S, C69S/C78S/C93S/C125S, and C69S/C78S/C93S/C125S/C142S. The mutant that lacks all five cysteines in UL16(1-155)-GFP is referred to as (5C−)-GFP. The UL16-GFP mutants bearing single cysteine-to-serine substitutions, C221S, C244S, C247S, C269S, C271S, and C357S, were described previously (75). The remaining 14 cysteines in UL16-GFP were individually replaced with serine by using site-directed QuikChange mutagenesis. Three cysteines at positions 269, 271, and 357 in UL16-GFP were replaced with alanines to create cysteine-to-alanine substitution mutants.
The plasmid pUL16(156-373)-GFP expressing the C-terminal 156 to 373 aa of UL16 was cloned into the pEGFP-N2 vector (Clontech) with an In-Fusion Advantage PCR cloning kit (Clontech) using forward (5′-GGACTCAGATCTCGAGGCCACCATGGAGGAAACCCCCGACCCAACC-3′) and reverse (5′-GTCGACTGCAGAATTCTTCGGGATCGCTTGAGGAGGCCCG-3′) primers. Cysteines at positions 269, 271, and 357 in UL16(156-373)-GFP were also replaced individually with either serine or alanine.
UL11 constructs used in the study.
Plasmids coding for GST, GST-UL11, and GST-UL11(AC−) were described previously (44). The C-terminal, HA-tagged UL11 and UL11(AC−) constructs were derived from the UL11-GFP plasmid (43) and have been described previously (45). The Src-UL11 constructs sUL11-HA and sUL11(AC-)-HA have the membrane-binding peptide (10 aa) of the Src oncoprotein fused to the N termini of the UL11 proteins (43, 45).
Preparation of GST- or His-tagged fusion proteins.
E. coli BL21-codon plus cells transformed with GST constructs or pHis6-UL16 and its derivatives were grown overnight at 37°C. The cultures were diluted at 1:100 in 2× YT (yeast extract-tryptone) medium and grown until the optical density at 600 nm reached 0.6 at 37°C, and then protein expression was induced by adding 0.1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) to the cultures for 3 h. For pHis6-UL16 constructs, the cultures were moved to room temperature after addition of IPTG. The cultures were pelleted at 8,000 rpm for 5 min and resuspended in PBS supplemented with protease inhibitors (Sigma), sonicated, and lysed by incubation with 0.1% Triton X-100 on ice for 30 min. The samples were then spun at 14,000 × g for 20 min to remove the cell debris and insoluble material. The lysates for GST constructs were incubated with glutathione-Sepharose 4B beads (GE Healthcare) for 1 h at room temperature (RT), pelleted, and washed three times with PBS for 10 min each time. The beads were resuspended in 300 μl of PBS and stored at 4°C. Cleared lysates for His-UL16 proteins were used directly in GST pulldown assays.
GST pulldown assays.
To analyze the interaction of UL16-GFP mutants with GST-UL11, pCMV.UL16-GFP or its cysteine mutants and pCMV.UL16(1-155)-GFP or its corresponding cysteine mutants were transfected individually into Vero cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. Briefly, 10 μg of plasmid DNA was mixed with 20 μl of Lipofectamine, diluted with 1.0 ml of Opti-MEM (Invitrogen), and incubated at RT for 20 min, and the mixtures were added on top of Vero cells grown to 70% confluence in 100-mm cell culture dishes. At 16 to 20 h posttransfection, the cells were harvested, washed twice with PBS, lysed in NP-40 lysis buffer (0.5% NP-40, 150 mM NaCl, 50 mM Tris-HCl [pH 8.0]) containing protease inhibitors (P8340; Sigma), precleared with glutathione-Sepharose 4B beads for 2 h at room temperature, and incubated with 2 μl of bead-bound GST-UL11 or its derivatives for 5 h at RT or overnight at 4°C. The beads were pelleted, washed three times with 0.5% NP-40 lysis buffer for 10 min each time, resuspended in 1× sample buffer, boiled for 5 min, separated by SDS-PAGE, and transferred to nitrocellulose membranes. The blots were probed with appropriate antibodies and developed with enhanced chemiluminescence (ECL) reagents (Pierce). The GST pulldown assays in the absence of detergent were performed for cells transfected with pUL16(1-155)-GFP or (5C−)-GFP by using the same protocol described above, except that the cells were resuspended in hypotonic buffer (10 mM Tris-HCl, pH 7.4, 0.2 mM MgCl2), incubated on ice for 20 min, lysed by 35 strokes with a Dounce homogenizer, and spun to remove the nuclei and unbroken cells. The supernatants obtained were then subjected to pulldown assays. The in vitro binding assays for interaction between the E. coli-expressed His-tagged N-terminal domain of UL16 [His-UL16(1-155)] and GST-UL11 were performed in essentially the same manner as described above for the GFP-tagged version of the protein.
NEM treatment.
To determine the effect of NEM on the UL16-UL11 interaction, the Vero cells expressing UL16-GFP, UL16(1-155)-GFP, or their cysteine mutant derivatives were treated with 10 mM NEM to covalently modify free cysteines in UL16 before lysing the cells with NP-40, essentially as described previously (75). For in vitro binding assays involving His6-UL16 or its mutant proteins, the bacterial lysates were treated with 10 mM NEM on ice for 30 min prior to use in GST pulldown assays.
Immunofluorescence and confocal microscopy.
To determine the localization of UL16-GFP or its deletion or point mutants in the presence or absence of UL11, confocal immunofluorescence microscopy was performed. Vero cells were grown to 60 to 70% confluence on coverslips in six-well dishes and transfected with plasmids encoding UL16-GFP, its N- or C-terminal deletion mutants, or its cysteine substitution mutants using Lipofectamine 2000 (Invitrogen). For cotransfections of UL16 or its mutant constructs with UL11-HA or sUL11-HA derivatives, either 1:1 (UL16/sUL11) or 1:2 (UL16/UL11) DNA ratios were used to equalize the expression levels of both proteins. At 16 to 20 h posttransfection, cells were washed three times with PBS and fixed with 3.7% paraformaldehyde in PBS for 10 min. The cells were washed three times with PBS for 5 min each time and permeabilized for 15 min with 0.1% Triton X-100 in blocking buffer (2% bovine serum albumin in PBS). The cells were then incubated in blocking buffer for 30 min, followed by staining with monoclonal antibody specific for the HA tag (1:3,000) for 1 hour. After three washes with PBS, the cells were incubated with goat anti-mouse secondary antibody conjugated with Alexa Fluor 568 (Invitrogen) for 1 h and washed 3 times with PBS. Cells were imaged using the GFP and Alexa Fluor 568 channels and a 60× oil immersion objective of a Leica TCS SP2 AOBS confocal microscope (Penn State University Core Facility).
Membrane flotation assay.
Vero cells grown to 65 to 70% confluence in 100-mm dishes were transfected or cotransfected with constructs that express UL16-GFP or its derivatives and/or plasmids encoding UL11-HA or sUL11-HA, as described above. The expression levels of cysteine substitution mutants were adjusted empirically by increasing the amounts of DNA until levels comparable to those of wild-type UL16-GFP were achieved. The total-membrane samples were prepared as described previously (6) Briefly, at 16 to 20 h posttransfection, cells were harvested, washed twice with cold NTE buffer (10 mM Tris-HCl, pH 7.4, 100 mM NaCl, 1 mM EDTA), and then resuspended in 300 μl of hypotonic lysis buffer (10 mM Tris-HCl, pH 7.4, 0.2 mM MgCl2) on ice for 20 min. Swollen cells were lysed on ice by 35 strokes with a Dounce homogenizer and then centrifuged at low speed to remove unbroken cells and nuclei. Postnuclear supernatants were mixed with 1.7 ml of 65% (wt/wt) sucrose, placed at the bottom of a Beckman SW55Ti tube, and sequentially overlaid with 2.5 ml of 45% and 0.5 ml of 2.5% sucrose. The sucrose solutions used were made in NTE buffer. The samples were spun for 20 h at 200,000 × g and 4°C in a Beckman ultracentrifuge, and six equal-volume fractions (∼833 μl) were collected from the top. The collected fractions were solubilized by adding 5× RIPA buffer (50 mM Tris-HCl, pH 8.0, with 150 mM sodium chloride, 1.0% Triton X-100, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate) and immunoprecipitated with either anti-GFP serum to precipitate UL16 proteins or anti-UL11 serum to immunoprecipitate UL11 proteins. Immunocomplexes were captured using protein A agarose beads (Roche), washed three times with 1× RIPA buffer, and lysed in Laemmli sample buffer. Samples were separated on 12% SDS-PAGE, followed by Western blot analysis using the respective antibodies.
RESULTS
A fundamental limitation in understanding how tegument protein UL16 interacts with its known binding partners—UL11, UL21, and gE—is an absence of information regarding its functional domains. Recently, we fortuitously discovered a mutant of UL16 that retained only the first 155 residues but gained the ability to efficiently bind to gE (74). Those results suggested the possibility that a C-terminal domain (located within residues 156 to 373) negatively regulates the gE-binding activity within the N-terminal 155 residues. This in turn raises the possibility that other binding activities of UL16 might reside within the N-terminal segment, including that for UL11. However, it seemed more likely that the site that binds UL11 would be located within the C-terminal portion of UL16 because it contains all the conserved cysteines, some of which have been implicated in the UL11-UL16 interaction (75). The following experiments tested these hypotheses.
Interaction between UL16(1-155) and UL11 in GST pulldown assays.
To determine whether UL16(1-155) interacts with UL11, we first used a GST pulldown assay. His6-tagged versions of full-length UL16 or the UL16(1-155) mutant (Fig. 1A) were expressed in E. coli, and cell lysates were mixed with equal amounts of glutathione beads bound with GST only or GST-UL11 (Fig. 1B). Following incubation, the beads were washed and analyzed for UL16 by immunoblotting with antibodies specific for the His6 tag. As expected (44, 75), GST-UL11 pulled down full-length UL16 while the GST-only construct did not (Fig. 1C). In support of the binding site being located within the N-terminal fragment, UL16(1-155) was also readily and efficiently pulled down by GST-UL11 (Fig. 1C). As a positive control, a GST fusion with the cytoplasmic tail of gE was used, and it also pulled down the N-terminal fragment (not shown), as previously reported (74). To ascertain whether binding of UL16(1-155) is specific, a GST-UL11 mutant lacking the acidic cluster was used (44). This small motif is essential for UL16 recognition and for the ability of UL11 to be efficiently endocytosed and to exit lipid rafts (6, 43). As expected GST-UL11(AC−) did not pull down full-length UL16, and it also failed to interact with the truncated UL16 protein (data not shown). Thus, UL16(1-155) behaves identically to the full-length protein in this in vitro binding assay.
Fig 1.

Interaction of UL16(1-155) with UL11 in vitro. (A) The His-tagged UL16 constructs used in the GST pulldown assay. They were expressed in E. coli, and cell lysates were either treated with NEM or left untreated. (B) As demonstrated by Ponceau S staining, equal amounts of GST and GST-UL11 beads were added to the lysates. (C) After incubating and washing the beads, they were resuspended in sample buffer prior to the analysis of bound proteins via Western blotting with UL16-specific antibodies. (D) Direct loading and Western blotting of input lysate samples show the amounts of wild-type and mutant UL16 that were initially present and the shift in migration that results from NEM modification.
Modification of UL16 with NEM, a small membrane-permeable thiol blocker that modifies free cysteines has been shown to block the interaction with UL11 (75). Inspection of the first 155 amino acids of UL16 revealed the presence of five cysteines. If UL16(1-155) retains the binding properties of the full-length molecule, then its binding to UL11 should be sensitive to NEM, too. To test this, E. coli lysates containing either His6-UL16 or His6-UL16(1-155) were treated with NEM prior to use in binding assays. Shifts in their molecular masses confirmed that both proteins were modified (Fig. 1D). In the pulldown assay, both proteins were unable to bind to GST-UL11 (Fig. 1C). This finding reduced the number of cysteines that could be involved in the UL11 interaction from 20 to only 5. We later narrowed this number further (see below).
Colocalization of UL16(155) with UL11 in cotransfected cells.
To test whether the interaction between UL16(1-155) and UL11 can occur within mammalian cells (and not just in vitro), cotransfection experiments were performed. Previous studies have shown that some full-length UL16 molecules colocalize with UL11 in transfected cells and in a manner that is dependent upon the acidic cluster. However, the interaction is not very efficient, and most of the molecules remain in the nucleus or dispersed throughout the cytoplasm (44). If a negative regulatory domain resides in the C-terminal residues of UL16, then UL16(1-155) might exhibit enhanced colocalization with UL11, as was found for the UL16-gE interaction (74).
When expressed individually, UL11-HA accumulated in the cytoplasm at a juxtanuclear location, while full-length UL16-GFP was found to be distributed throughout the cell, with a greater accumulation within the nucleus (Fig. 2A). In contrast, UL16(1-155)-GFP was found throughout the cell, including the nucleus in ∼80% of the cells, but in ∼20% of the cells, it also accumulated in punctate structures in the cytoplasm (Fig. 2A). The exact nature of these puncta is unknown, but some were found to colocalize with Hsp70 (data not shown), a marker for aggresomes (37). In any case, when UL16(1-155)-GFP and UL11-HA were expressed together, they colocalized at a juxtanuclear location, and the diffuse nuclear and cytoplasmic patterns characteristic of UL16 were absent (Fig. 2B). Colocalization was specific, as it was dependent on the presence of the acidic-cluster motif in UL11 (Fig. 2B). Interestingly, in ∼40% of the cells, either UL11 and UL16(1-155) were dramatically relocalized to speckles throughout the nucleus, along with cytoplasmic colocalization (Fig. 2B, third row from top), or, in some cases, the proteins were localized only in the nuclear speckles with no signal in the perinuclear region in the cytoplasm (data not shown). These findings were surprising, because UL11 is never found in the nucleus when expressed alone (43, 44) (Fig. 2A), presumably because it is tightly associated with membranes via its modification with both myristate and palmitate (6, 43). On the other hand, a population of UL11 molecules has been observed in the nuclei of HSV-infected cells (1), but whether it serves a function there remains unknown. To be relocalized to the nucleus, loss of the palmitate moiety would be required. Depalmitoylation enzymes are abundant in cells (12, 64, 69), and perhaps binding of UL16 captures some UL11 molecules for a role in the nucleus. In any case, the nuclear speckles observed with these two proteins in cotransfections offer further support for their ability to interact within mammalian cells.
Fig 2.

Coexpression of UL16(1-155)-GFP with UL11 in mammalian cells. (A) Cells were singly transfected with vectors that express UL16-GFP, UL16(1-155)-GFP, or UL11-HA to show the sites where these proteins accumulate on their own. (B) UL16-GFP and UL16(1-155)-GFP were coexpressed with UL11-HA to look for changes in subcellular localization. In all cases, the cells were fixed at 16 to 20 h posttransfection and stained with DAPI (4′,6-diamidino-2-phenylindole) to reveal nuclei (blue). UL16 and UL16(1-155) were revealed by the fluorescence of their GFP tags (green). The position of UL11 was revealed by a monoclonal antibody specific for the HA peptide (red).
Evidence for the interaction obtained from membrane flotation assays.
To examine the interaction of UL16(1-155) and UL11 in a different and unbiased assay, membrane flotation experiments were performed (Fig. 3). For this, transfected cells were osmotically lysed, cytoplasmic membranes were placed at the bottom of the sucrose step gradient, and the samples were centrifuged. Membranes and their associated proteins rise to the upper fractions of the gradient and can be detected by immunoblotting. In contrast to membrane proteins UL11-HA and UL11(AC−)-HA, neither UL16-GFP nor UL16(1-155)-GFP was capable of floating when expressed alone. A small increase in flotation was observed when full-length UL16 was coexpressed with UL11, which is consistent with previous studies (44) showing limited colocalization of the two proteins (Fig. 2B). This increase does not take into account the large population of UL16 that remains within nuclei, which are discarded when preparing the membrane samples. Nevertheless, the amount of UL16-GFP floating was reduced when UL11(AC−)-HA was used. The properties of UL16(1-155)-GFP were strikingly different. This derivative was able to float substantially (∼80%) to the top when coexpressed with UL11-HA (Fig. 3). As expected, this activity was lost when the acidic-cluster motif was absent from UL11.
Fig 3.

Membrane flotation analyses of UL16 derivatives in the presence or absence of UL11. (A) Vero cells were transfected with the indicated UL16 constructs, either alone or with UL11-HA or its acidic-cluster mutant (AC−). At 16 to 20 h posttransfection, the cells were osmotically disrupted, and the ability of each protein to float to the upper fractions of sucrose step gradients was examined. Six equal fractions were collected, and detergent was added to solubilize the membranes. Proteins were concentrated by immunoprecipitation with antibodies specific for either GFP or UL11 and analyzed by Western blotting with the indicated antibodies. The tops and bottoms (Bot.) of the gradients are indicated. (B) Densitometry was used to quantitate the immunoblots from three independent experiments. The results are shown as the percentage of floating protein (top three fractions) relative to the total protein (all fractions). The error bars indicate standard deviations.
Relocating the subcellular site of interaction.
Although the colocalization experiments provide strong evidence for an interaction between UL16(1-155) and UL11, the results were somewhat “tarnished” by the ability of the N-terminal fragment to aggregate by itself in a location that resembles that of UL11 (described above). To address this, we attempted to move the site of the interaction by utilizing sUL11-HA, a construct that has its first 10 amino acids derived from the Src oncoprotein (44). It has long been known that the Src membrane-binding motif can direct a variety of proteins to the plasma membrane (60, 67, 71, 72), and confocal microscopy showed that sUL11 has a dramatically different distribution from that of wild-type UL11 (compare Fig. 4A and Fig. 2A). When UL16(1-155)-GFP was coexpressed with sUL11-HA, it also exhibited a massive change in its localization relative to what was observed with wild-type UL11-HA (compare Fig. 4B with Fig. 2B). Relocalization was specific, depending on the acidic-cluster motif of UL11. Speckles were not observed in the nucleus, which is not surprising, because the Src membrane-binding motif does not rely on palmitate (60, 67), and hence, sUL11 is unlikely to be released from the membrane to be available for nuclear entry. In contrast to these observations, full-length UL16-GFP showed only partial relocalization with sUL11, and most of the molecules remained in the nucleus or were found dispersed throughout the cytoplasm. In further support of these findings, UL16(1-155)-GFP was able to rise to the top fractions of sucrose gradients in membrane flotation assays, but full-length UL16-GFP was not (Fig. 4C).
Fig 4.
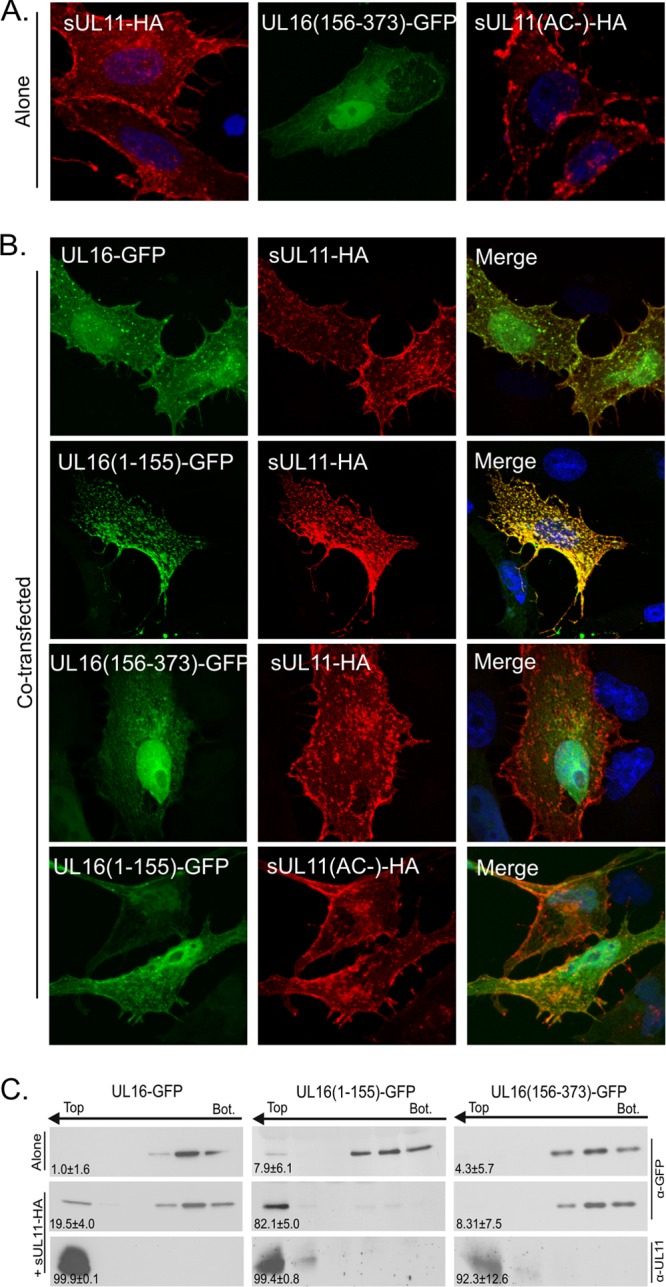
Relocalization of UL16(1-155)-GFP by sUL11. (A) Vero cells were singly transfected with expression vectors to show where the indicated proteins accumulate when expressed alone. (B) The indicated derivatives of UL16 were coexpressed with either sUL11-HA or sUL11(AC−)-HA to see which could be relocated. UL11 constructs were detected with a monoclonal antibody specific for the HA peptide (red), while the UL16 constructs were detected by the fluorescence of their GFP tags (green). DAPI was used to stain nuclei (blue). The images were captured with a confocal microscope. (C) Sucrose step gradients were used to examine the ability of the UL16 derivatives to associate with coexpressed, membrane-bound sUL11 (as described in the legend to Fig. 3). Representative immunoblots are shown. The number at the bottom left of each panel is the percentage of floating protein (top three fractions) relative to the total protein (all fractions) from three independent experiments.
Absence of a UL11-binding site in the C-terminal segment of UL16.
While it appears that a negative regulatory domain resides within residues 156 to 373 of UL16, it was possible that this C-terminal fragment might have a separate UL11-binding region. To address this, a mutant was constructed that expresses only the C-terminal portion of UL16 (construct CT-GFP). When expressed alone, this fragment was found in the nucleus and throughout the cytoplasm of transfected cells (Fig. 4A). CT-GFP did not respond at all to the presence of UL11-HA in the cells (Fig. 4B), suggesting the absence of a binding site. Moreover, it was unable to respond to UL11-HA in the membrane flotation assay (Fig. 4C). Thus, it appears that the only binding site for UL11 is located somewhere within the first 155 amino acids of UL16.
Single-amino-acid substitutions in UL16 that activate binding to UL11.
If a C-terminal negative regulatory domain is present in UL16, then it might be possible to activate binding to UL11 by making single-amino-acid substitutions that disrupt the mechanism. The most striking sequence feature in this part of UL16 is eight conserved cysteines (Fig. 5). Previously, we made serine substitutions at six of these positions (C221 to C275), but they were only tested in pulldown experiments with GST-UL11 (75). Four of the Cys-Ser mutants (C247S to C275S) lost the ability to interact with UL11, but if the binding site actually resides in the first 155 residues of UL16, then these mutants were likely misfolded in the in vitro assay. Therefore, we decided to use the in vivo colocalization assay to test the complete set of mutants in which all 20 cysteines were individually changed within the context of full-length UL16. All of the mutants produced proteins of the expected size, but two (C93S and C357S) exhibited somewhat reduced levels of expression (data not shown). When transfected alone, 17 of the mutants (not shown) showed a distribution similar to that of wild-type UL16 (Fig. 6A), with accumulation in the nucleus and a diffuse pattern throughout the cytoplasm. The three exceptional mutants had changes at conserved cysteines C269, C271, and C357, all present within the C-terminal portion of UL16. These mutants showed a heterogeneous phenotype in terms of their subcellular localization (Fig. 6A). In particular, they each had three distinct distribution patterns. (i) Some cells showed the same distribution as the wild type, with diffuse signals in the nucleus and cytoplasm. (ii) In other cells, the mutants were either localized to puncta at a juxtanuclear position or occasionally found in puncta distributed throughout the cytoplasm. (iii) In the most striking phenotype, these mutants were exclusively localized to the cytoplasm and absent from the nucleus.
Fig 5.

Positions of cysteines and putative domain organization of UL16. The 20 cysteines contained within the 373 amino acids of UL16 are indicated, along with their residue numbers. Five are found in the N-terminal fragment (NTD) (residues 1 to 155), which binds to UL11 and gE. The eight conserved cysteines (boldface) are located in the putative C-terminal regulatory domain (CTD). Amino acid substitutions at 3 of the 20 cysteines (indicated with asterisks) enabled the full-length protein to bind to UL11 in vivo. The position of the CXXC-X21-CXC motif that resembles the unusual zinc finger of E. coli chaperone Hsp33 is shown (see Discussion).
Fig 6.

Substitutions in the putative C-terminal regulatory domain activate sUL11 binding in cells. (A) Cells were singly transfected to express either UL16-GFP or the three cysteine substitution mutants that enable binding to UL11. Confocal microscopy revealed heterogeneous phenotypes for the mutants, examples of which are shown. (B) Cells were cotransfected with the indicated UL16-GFP derivatives and sUL11-HA. After 16 to 20 h of transfection, the cells were fixed, stained for UL11 with monoclonal anti-HA antibodies, and visualized by confocal microscopy.
To see whether any of the 20 individual cysteine substitutions activate the ability of full-length UL16 to find UL11, cotransfections were performed. Because some of the mutants exhibited juxtanuclear accumulation when expressed alone, much like UL16(1-155), we used the sUL11-HA chimera for these experiments. Of the 20 mutants tested, three exhibited a massive relocalization to sUL11-HA, namely, C269S, C271S, and C357S (Fig. 6B), which are the mutants that also exhibited the heterogeneous phenotype when expressed alone (Fig. 6A). The other 17 mutants all behaved like wild-type UL16, which barely moved in response to sUL11 (Fig. 6B). Identical results were obtained when each of the three cysteines was replaced with alanine (data not show), which has hydrophobic properties similar to those of cysteine, whereas serine shares the hydrogen-bonding properties of cysteine. Independent evidence that these changes activate full-length UL16 was provided by the flotation assay, which revealed that all three mutants were able to efficiently rise to the upper fractions of the gradient when coexpressed with sUL11-HA, but none were able to float substantially when expressed alone (Fig. 7). One of the nonactivating mutants (C68S) was also tested, and as expected, it behaved like wild-type UL16 and remained in the lower fractions (data not shown).
Fig 7.

Membrane flotation analyses of cysteine mutants in the presence or absence of sUL11. Cells were transfected to express either UL16-GFP, the full-length cysteine substitution mutants, or a C-terminal fragment (CT-GFP.C269S) by themselves or with sUL11. The abilities of the UL16 derivatives to float to the top fractions in sucrose step gradients were measured as described in the legend to Fig. 3. (A) Representative immunoblots. (B) Data from three independent experiments are shown as the percentage of protein in the top three fractions relative to the total protein. The error bars indicate standard deviations.
To test the possibility that the activating substitutions work by creating or opening up another UL11-binding site within the C-terminal domain, these alterations were examined in the context of CT-GFP, which lacks the first 155 residues of UL16. As was the case with wild-type CT-GFP (Fig. 4), none of the three constructs was able to relocalize to sUL11 (data not shown), and none was able to float in its presence (shown for CT-GFP.C269S in Fig. 7). Hence, it appears that changes in the C-terminal portion of UL16 activate the UL11-binding site located somewhere in the N-terminal portion.
Because UL16(1-155) has been shown to be capable of interacting with the tail of gE (74), the three substitution mutants that activate binding to UL11 were tested for the ability to interact with this viral glycoprotein. Confocal microscopy experiments revealed that all three were incapable of relocalizing to gE (data not shown). This may not be surprising, because there are distinct binding sites for UL11 and gE within UL16 (74, 75). Hence, these results suggest that the two sites are regulated in different ways and perhaps in an ordered manner.
Cysteine-independent interaction of UL16(1-155) with UL11.
Having found that NEM blocks the ability of the N-terminal portion of UL16 to bind UL11 (see above), the five cysteines in this region (Fig. 5) were examined within the context of UL16(1-155)-GFP to see which ones are required. The mutants were coexpressed with sUL11-HA in mammalian cells, and confocal microscopy revealed that all five were efficiently relocalized (data not shown) in a manner similar to that of the wild-type fragment (Fig. 4B). Additional constructs were made that have double, triple, and quadruple cysteine substitutions. A mutant having no cysteines (named 5C−) was also constructed. To our great surprise, all of these derivatives of UL16(1-155) retained the ability to massively relocalize with sUL11-HA (shown for the full substitution mutant in Fig. 8A). Moreover, mutant 5C− exhibited a robust ability to float to the upper fractions of sucrose gradients when coexpressed with sUL11-HA, but not when the acidic-cluster motif was absent (Fig. 8B and C). Thus, in the complete absence of cysteines, the N-terminal fragment of UL16 retains the ability to specifically recognize UL11.
Fig 8.

Cysteines are not required for interaction with UL11. Mutant UL16(1-155)-GFP or its derivative that lacks all five cysteines, (5C−)-GFP, was either expressed alone or coexpressed with sUL11-HA or its acidic-cluster derivative (AC−), as indicated. (A) Cells were fixed and stained for sUL11 with anti-HA antibodies (red) and viewed by confocal microscopy. (B) The ability of the cysteine mutant to float when coexpressed with sUL11-HA, but not with the acidic cluster mutant, is shown in immunoblots from a membrane flotation assay. (C) The combined results from three independent flotation experiments are shown. The error bars indicate standard deviations.
Mapping of the UL11-binding site within UL16(1-155).
To more precisely define the location of the UL11-binding site, the first 155 residues of UL16 were divided into approximately equal halves, and each was expressed as a GFP chimera (Fig. 9A). By themselves, both mutants exhibited the heterogeneous localization pattern seen for the parental construct, UL16(1-155). That is, some cells had a diffuse pattern of fluorescence throughout the nucleus and cytoplasm, and some also produced puncta at a juxtanuclear location (Fig. 9B). Because of this heterogeneity, the relocalization assay with sUL11-HA was used (Fig. 9C). Fragment 1 to 80 was unable to be relocalized and remained distributed throughout the cell. In contrast, fragment 81 to 155 was dramatically relocalized by sUL11-HA, with no GFP signal found in the nucleus, similar to the UL16(1-155) control. Importantly, redistribution was dependent upon the presence of the acidic cluster in UL11, indicating that the interaction was specific (data not shown). Fragment 81 to 155 was also able to rise to the upper fractions of sucrose gradients in the flotation assay (Fig. 9D), providing further proof that the binding site is contained within this segment of UL16. The flotation profile of fragment 1 to 80 remained unaltered in the presence or absence of UL11 (Fig. 9D).
Fig 9.

Mapping of the UL11-binding region within the N-terminal fragment. (A) Diagrams of UL16(1-155) mutants used. WT, wild type. (B and C) Cells were transfected with the indicated GFP-tagged plasmids, either alone (B) or cotransfected with sUL11-HA (C), and examined by confocal microscopy. (D) The interactions of these deletion mutants with sUL11-HA were analyzed by membrane flotation assays; representative immunoblots are shown.
DISCUSSION
Tegument proteins UL11 and UL16 are conserved among all the herpesviruses, and two fundamental observations have been made regarding their mechanism of interaction. First, the UL11-binding site was found to be contained within residues 81 to 155 of UL16 and to have no requirement for cysteines, even though it is normally sensitive to NEM. Second, the interaction was found to be controlled by a regulatory domain contained within the second half of UL16. The significance of these findings is discussed below.
The UL11-binding domain in UL16.
The experiments described here have clearly shown that UL11 binds within the N-terminal 155 aa of UL16. The same fragment was recently found to interact with the cytoplasmic tail of gE, too (74); however, it is quite clear that two distinct sites for binding are present. For example, treatment of UL16 or UL16(1-155) with NEM blocks the interaction with UL11 but has no effect on the interaction with gE (74, 75). Prior to this study, the only part of UL16 found to be dispensable for interaction with UL11 was its first 40 residues, which are the least conserved and absent from most homologs (75). All other deletion mutants failed to bind to GST-UL11 in pulldown assays, including one that retained residues 41 to 155 (and the UL11-binding site). It now seems clear that this in vitro binding assay, which was used for the discovery and initial characterization of the UL11-UL16 interaction (44, 75), simply does not work reliably for N-terminal fragments of UL16. Based on the tendency of UL16(1-155)-GFP to aggregate in mammalian cells when expressed by itself, it seems likely that N-terminal fragments of UL16 are sensitive to misfolding in vitro. The cysteines in this region of UL16 may compound the problem by enabling the formation of inappropriate disulfide bonds when the protein is isolated under oxidizing and detergent-containing conditions. Consistent with misfolding, His-tagged UL16(1-155) was able to bind to GST-UL11 only when the E. coli cells producing it were grown at 30°C. In contrast, the in vivo relocalization assay was quite robust for UL16(1-155)-GFP but did not work well for the full-length molecule (Fig. 2). The simplest interpretation of these dichotomous observations is that the full-length and truncated forms of UL16 respond differently to NP-40 lysis buffer and/or oxygen. In particular, it seems clear that full-length UL16 exists in a “closed” state (unable to bind to UL11) within the cytoplasm, and we presume that cell lysis induces an “open” state by enabling the formation of specific disulfide bonds in the C-terminal regulatory domain, an activating mechanism that has been shown to occur in other proteins (see the discussion of Hsp33 below). In contrast, the N-terminal fragment of UL16 exists in the open state (capable of binding to UL11) within the cytoplasm, and we presume that cell lysis adversely affects the conformation of the binding site when the C-terminal domain is absent.
The N-terminal position of the UL11-binding site (within residues 81 to 155) and the lack of a requirement for cysteines were unexpected. Previous studies of UL16 had shown that treatment with NEM or substitutions of several conserved cysteines in the C-terminal half of the molecule blocked the interaction in pulldown assays with GST-UL11 (75). Accordingly, we had anticipated that the binding site would be C-terminally located. Instead, those Cys substitutions that block the pulldown assay had the effect of activating the ability of UL16 to colocalize with UL11 within cells. These results raise two questions. First, if wild-type UL16 is induced to bind GST-UL11 upon exposure to NP-40 lysis buffer and/or oxygen, then why is it unable to bind in this assay when “activating” cysteine substitutions are introduced? We speculate that these particular cysteines work with others to form zinc fingers or disulfide bonds, and changing one residue leaves another unpaired. Thus, when cells are disrupted, the unpaired cysteine forms an inappropriate disulfide bond that disrupts the conformation of the binding site. Second, if cysteines are not important for the UL11-binding site, then why does NEM block the interaction? The most likely explanation is that the addition of hydrophobic NEM moieties distorts the UL11-binding site, similar to its affect on the conformation of other proteins, such as Hsp70 chaperone Ssa1p from Saccharomyces cerevisiae (10, 27, 41). However, NEM does not globally disrupt the conformation of UL16, because the modified protein is still capable of binding UL21 (26) and gE (74). In any case, it is clear that caution is needed in interpreting experiments in which NEM is used. Further support for our finding is provided by the UL16 homolog of pseudorabies virus (PRV) (35, 36). This protein has only 11 cysteines but is capable of binding to UL11 (44). The three nonconserved cysteines are scattered among the other eight, with none being located within the N-terminal portion of this homolog, where the binding site is most likely to be located.
Regulation of the UL16-UL11 interaction.
We recently proposed that residues 156 to 373 serve to negatively regulate the binding of UL16 to the tail of gE (74). That is, removal of this segment enabled UL16(1-155) to bind to gE in cotransfected cells, whereas the full-length molecule could not. As shown here, the same is true for the UL16-UL11 interaction. Specifically, full-length UL16 mostly remains distributed throughout the cell—including the nucleus—when it is coexpressed with UL11 (44), but removal of the C-terminal fragment enables highly efficient colocalization at the trans-Golgi network (TGN) (Fig. 2). Further support for a regulatory domain comes from single-amino-acid substitutions at three conserved cysteines (C269, C271, and C357), which enabled the full-length molecule to bind to UL11, presumably by inducing a conformational change that is similar to what occurs upon exposure to NP-40 lysis buffer and/or oxygen.
While the mutational analyses are insightful, they do not explain how UL16 is normally activated during the course of an infection and how its regulatory domain works. Because UL16 has other cytoplasmic binding partners (e.g., UL21), we presume that one or more of these may induce the active conformation. However, the data presented here, combined with a particular sequence motif located among the conserved cysteines, suggest that a second mechanism for activating UL16 may be possible.
Sequence alignments of all UL16 homologs reveal eight conserved cysteines, and it has been suggested that some of these might chelate zinc (73). Unfortunately, computer analyses do not reveal any matches with known eukaryotic zinc fingers. On the other hand, we have noticed that UL16 does have a motif (CXXC-X21-CXC) that is similar to an unusual zinc-binding motif (CXC-X27-CXXC) found in the E. coli chaperone protein Hsp33 (29). The “ends” of the motif are swapped, but that would not be expected to preclude the ability of the UL16 peptide to chelate zinc. Hsp33 also has a two-domain structure. Its N-terminal domain has the chaperone activity, and its C-terminal domain regulates that activity using the cysteine motif as a switch (28–30). In brief, Hsp33 is normally turned “off” unless the bacterium encounters oxidative stress. When that happens, the zinc finger motif is oxidized, creating two disulfide bonds, and the molecule dimerizes (22). These events turn “on” the chaperone activity of the N-terminal domain (i.e., it binds to the exposed hydrophobic regions of misfolded bacterial proteins to help rescue them). When the stress is gone and the reducing environment of the cytoplasm is regenerated, the disulfide bonds are converted back to a zinc finger, and the N-terminal domain is once again turned off. We hypothesize that the C-terminal regulatory domain of UL16 may also contain a switch that is activated in response to oxidation. In fact, it has been shown that HSV-infected cells undergo oxidative stress (17, 21, 33, 49, 56, 59, 65, 70). Hence, it is possible that UL16 utilizes this change to modulate its function. That is, a zinc finger formed by some of the conserved cysteines might undergo oxidation to trigger an “open” conformation that allows the UL11-binding site to be exposed. Indeed, it is possible that UL16 is a virus-specific chaperone that is recruited by the acidic cluster of UL11 to help with the assembly of various viral components. Unfortunately, it remains to be seen whether UL16 actually chelates zinc. If it does, this might only occur in the reducing environment of the cytoplasm (i.e., zinc may be lost when disulfide bonds are formed upon cell lysis). In any case, this model predicts that at least some of the conserved cysteines will be found in disulfide bonds when UL16 is in the active state. Also, if UL16 is regulated like Hsp33, then it would be reasonable to think that four, rather than three, activating cysteine substitutions would be found within the regulatory domain. However, it is well known that some zinc fingers include histidine (20), and two such residues are conserved in UL16. The complexity of the activation mechanism is further emphasized by the observation that the three Cys substitutions that enable binding to UL11 do not activate binding to gE (data not shown). Given the number of conserved residues and our paucity of structural information for UL16 or any of its homologs, it is clear that a great deal more investigation will be needed to understand the fundamental mechanism(s) by which this protein is regulated.
Relevance to the nuclear trafficking of UL16.
In HSV-infected cells (and in transfections), UL16 is found mostly in the nucleus once its expression begins, but at later times, it becomes cytoplasmic (51, 55, 58). The function of UL16 within the nucleus is unknown, as are the mechanisms for its movement between the nucleus and the cytoplasm. We hypothesize that it shuttles between these compartments, but the protein does not have obvious nuclear import and export signals. Mutants of UL16 that gained the ability to colocalize with UL11 were strikingly absent from the nuclei of some of the cotransfected cells (Fig. 6). Binding to membrane-bound UL11 may simply prevent UL16 from reentering the nucleus by either overriding or hiding its import signals. None of these mutants are defective for nuclear import, although they do exhibit heterogeneous patterns of subcellular localization. Thus, in some cells, the distribution had the appearance of the wild type (i.e., mostly in the nucleus), but an exclusively cytoplasmic distribution was found in adjacent cells. It is difficult to explain this strange phenotype, but perhaps the activated mutants have a metastable conformation in which they are poised to bind to UL11, but in its absence, they may simply aggregate via their artificially exposed binding sites. Alternatively, it may be that activated UL16 is poised to multimerize (much like Hsp33 dimerizes) (22), and once a few molecules do so, others are triggered to aggregate in the cytoplasm, thereby preventing nuclear accumulation. Whatever accounts for the heterogeneous phenotype, it is not observed when UL11 is present. Alterations that activate UL16 enable the highly efficient colocalization of the two molecules.
Relevance to other herpesviruses.
It remains to be seen to what extent the mechanism of regulation will be found in other UL16 homologs; however, there are reasons to believe that the details will be different for the beta- and gammaherpesviruses. For example, the homologs of these viruses are different in being mostly capsid associated (31, 73, 76), even in the absence of NEM, unlike UL16 in HSV-1 or PRV (51). More pertinent to this study, it has been shown that the UL16 homolog of human cytomegalovirus (HCMV) (UL94) can relocalize to the UL11 homolog (UL99) in an unregulated manner (42, 62). However, it is difficult to predict whether the UL99-binding site is N terminally located in UL94 because of a lack of obviously conserved residues. Additionally, a recent report on murine cytomegalovirus appears to be in line with our findings because an insertion mutant in the N terminus of M94, which is the homolog of UL16, was shown to disrupt its interaction with M99, the UL11 homolog (48), indicating that the nonconserved N terminus might have a function. Without a doubt, the most important insights regarding the functions and mechanisms of UL16 and its homologs remain to be discovered.
ACKNOWLEDGMENTS
We thank our colleagues Carol B. Wilson and Jacob A. Marsh for their helpful discussions and encouragement. We also thank the Penn State Hershey College of Medicine Imaging Core for use of the confocal microscope. Special thanks are due to Rebecca Craven for critical review of the manuscript.
This work was supported by an NIH grant to J.W.W. (RO1 AI071286). J.L.S. was supported in part by a training grant from the NIH (T32 CA60395).
Footnotes
Published ahead of print 22 August 2012
REFERENCES
- 1. Baines JD, Jacob RJ, Simmerman L, Roizman B. 1995. The herpes simplex virus 1 UL11 proteins are associated with cytoplasmic and nuclear membranes and with nuclear bodies of infected cells. J. Virol. 69:825–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baines JD, Koyama AH, Huang T, Roizman B. 1994. The UL21 gene products of herpes simplex virus 1 are dispensable for growth in cultured cells. J. Virol. 68:2929–2936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baines JD, Roizman B. 1991. The open reading frames UL3, UL4, UL10, and UL16 are dispensable for the replication of herpes simplex virus 1 in cell culture. J. Virol. 65:938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Baines JD, Roizman B. 1992. The UL11 gene of herpes simplex virus 1 encodes a function that facilitates nucleocapsid envelopment and egress from cells. J. Virol. 66:5168–5174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baird NL, Starkey JL, Hughes DJ, Wills JW. 2010. Myristylation and palmitylation of HSV-1 UL11 are not essential for its function. Virology 397:80–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Baird NL, Yeh PC, Courtney RJ, Wills JW. 2008. Sequences in the UL11 tegument protein of herpes simplex virus that control association with detergent-resistant membranes. Virology 374:315–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Britt WJ, Jarvis M, Seo JY, Drummond D, Nelson J. 2004. Rapid genetic engineering of human cytomegalovirus by using a lambda phage linear recombination system: demonstration that pp28 (UL99) is essential for production of infectious virus. J. Virol. 78:539–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bucks MA, O'Regan KJ, Murphy MA, Wills JW, Courtney RJ. 2007. Herpes simplex virus type 1 tegument proteins VP1/2 and UL37 are associated with intranuclear capsids. Virology 361:316–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chi JH, Harley CA, Mukhopadhyay A, Wilson DW. 2005. The cytoplasmic tail of herpes simplex virus envelope glycoprotein D binds to the tegument protein VP22 and to capsids. J. Gen. Virol. 86:253–261 [DOI] [PubMed] [Google Scholar]
- 10. Chirico WJ, Markey ML, Fink AL. 1998. Conformational changes of an Hsp70 molecular chaperone induced by nucleotides, polypeptides, and N-ethylmaleimide. Biochemistry 37:13862–13870 [DOI] [PubMed] [Google Scholar]
- 11. Chiu YF, et al. 2012. Characterization and intracellular trafficking of Epstein-Barr virus BBLF1, a protein involved in virion maturation. J. Virol. 86:9647–9655 doi:10.1128/JVI.01126-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Conibear E, Davis NG. 2010. Palmitoylation and depalmitoylation dynamics at a glance. J. Cell Sci. 123:4007–4010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. de Wind N, Wagenaar F, Pol J, Kimman T, Berns A. 1992. The pseudorabies virus homology of the herpes simplex virus UL21 gene product is a capsid protein which is involved in capsid maturation. J. Virol. 66:7096–7103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dingwell KS, et al. 1994. Herpes simplex virus glycoproteins E and I facilitate cell-to-cell spread in vivo and across junctions of cultured cells. J. Virol. 68:834–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dingwell KS, Doering LC, Johnson DC. 1995. Glycoproteins E and I facilitate neuron-to-neuron spread of herpes simplex virus. J. Virol. 69:7087–7098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dingwell KS, Johnson DC. 1998. The herpes simplex virus gE-gI complex facilitates cell-to-cell spread and binds to components of cell junctions. J. Virol. 72:8933–8942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fraternale A, et al. 2009. GSH and analogs in antiviral therapy. Mol. Aspects Med. 30:99–110 [DOI] [PubMed] [Google Scholar]
- 18. Fuchs W, Granzow H, Veits J, Mettenleiter TC. 2012. Identification and functional analysis of the small membrane-associated protein pUL11 of avian infectious laryngotracheitis virus. Virus Res. 163:599–608 [DOI] [PubMed] [Google Scholar]
- 19. Fulmer PA, Melancon JM, Baines JD, Kousoulas KG. 2007. UL20 protein functions precede and are required for the UL11 functions of herpes simplex virus type 1 cytoplasmic virion envelopment. J. Virol. 81:3097–3108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gamsjaeger R, Liew CK, Loughlin FE, Crossley M, Mackay JP. 2007. Sticky fingers: zinc-fingers as protein-recognition motifs. Trends Biochem. Sci. 32:63–70 [DOI] [PubMed] [Google Scholar]
- 21. Gonzalez-Dosal R, et al. 2011. HSV infection induces production of ROS, which potentiate signaling from pattern recognition receptors: role for S-glutathionylation of TRAF3 and 6. PLoS Pathog. 7:e1002250 doi:10.1371/journal.ppat.1002250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Graumann J, et al. 2001. Activation of the redox-regulated molecular chaperone Hsp33—a two-step mechanism. Structure 9:377–387 [DOI] [PubMed] [Google Scholar]
- 23. Gross ST, Harley CA, Wilson DW. 2003. The cytoplasmic tail of Herpes simplex virus glycoprotein H binds to the tegument protein VP16 in vitro and in vivo. Virology 317:1–12 [DOI] [PubMed] [Google Scholar]
- 24. Guo H, Wang L, Peng L, Zhou ZH, Deng H. 2009. ORF33 of a gammaherpesvirus encodes a tegument protein essential for virion morphogenesis and egress. J. Virol. 83:10582–10595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Han J, Chadha P, Meckes DG, Jr, Baird NL, Wills JW. 2011. Interaction and interdependent packaging of tegument protein UL11 and glycoprotein e of herpes simplex virus. J. Virol. 85:9437–9446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Harper AL, et al. 2010. Interaction domains of the UL16 and UL21 tegument proteins of herpes simplex virus. J. Virol. 84:2963–2971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hermawan A, Chirico WJ. 1999. N-Ethylmaleimide-modified Hsp70 inhibits protein folding. Arch. Biochem. Biophys. 369:157–162 [DOI] [PubMed] [Google Scholar]
- 28. Ilbert M, et al. 2007. The redox-switch domain of Hsp33 functions as dual stress sensor. Nat. Struct. Mol. Biol. 14:556–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jakob U, Eser M, Bardwell JC. 2000. Redox switch of hsp33 has a novel zinc-binding motif. J. Biol. Chem. 275:38302–38310 [DOI] [PubMed] [Google Scholar]
- 30. Jakob U, Muse W, Eser M, Bardwell JC. 1999. Chaperone activity with a redox switch. Cell 96:341–352 [DOI] [PubMed] [Google Scholar]
- 31. Johannsen E, et al. 2004. Proteins of purified Epstein-Barr virus. Proc. Natl. Acad. Sci. U. S. A. 101:16286–16291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Microbiol. 9:382–394 [DOI] [PubMed] [Google Scholar]
- 33. Kavouras JH, et al. 2007. Herpes simplex virus type 1 infection induces oxidative stress and the release of bioactive lipid peroxidation by-products in mouse P19N neural cell cultures. J. Neurovirol. 13:416–425 [DOI] [PubMed] [Google Scholar]
- 34. Kelly BJ, Fraefel C, Cunningham AL, Diefenbach RJ. 2009. Functional roles of the tegument proteins of herpes simplex virus type 1. Virus Res. 145:173–186 [DOI] [PubMed] [Google Scholar]
- 35. Klupp BG, Bottcher S, Granzow H, Kopp M, Mettenleiter TC. 2005. Complex formation between the UL16 and UL21 tegument proteins of pseudorabies virus. J. Virol. 79:1510–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Klupp BG, Hengartner CJ, Mettenleiter TC, Enquist LW. 2004. Complete, annotated sequence of the pseudorabies virus genome. J. Virol. 78:424–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kopito RR. 2000. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 10:524–530 [DOI] [PubMed] [Google Scholar]
- 38. Kopp M, Granzow H, Fuchs W, Klupp B, Mettenleiter TC. 2004. Simultaneous deletion of pseudorabies virus tegument protein UL11 and glycoprotein M severely impairs secondary envelopment. J. Virol. 78:3024–3034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kopp M, et al. 2003. The pseudorabies virus UL11 protein is a virion component involved in secondary envelopment in the cytoplasm. J. Virol. 77:5339–5351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Leege T, et al. 2009. Effects of simultaneous deletion of pUL11 and glycoprotein M on virion maturation of herpes simplex virus type 1. J. Virol. 83:896–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu Q, Levy EJ, Chirico WJ. 1996. N-Ethylmaleimide inactivates a nucleotide-free Hsp70 molecular chaperone. J. Biol. Chem. 271:29937–29944 [DOI] [PubMed] [Google Scholar]
- 42. Liu Y, et al. 2009. The tegument protein UL94 of human cytomegalovirus as a binding partner for tegument protein pp28 identified by intracellular imaging. Virology 388:68–77 [DOI] [PubMed] [Google Scholar]
- 43. Loomis JS, Bowzard JB, Courtney RJ, Wills JW. 2001. Intracellular trafficking of the UL11 tegument protein of herpes simplex virus type 1. J. Virol. 75:12209–12219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Loomis JS, Courtney RJ, Wills JW. 2003. Binding partners for the UL11 tegument protein of herpes simplex virus type 1. J. Virol. 77:11417–11424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Loomis JS, Courtney RJ, Wills JW. 2006. Packaging determinants in the UL11 tegument protein of herpes simplex virus type 1. J. Virol. 80:10534–10541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. MacLean CA, Clark B, McGeoch DJ. 1989. Gene UL11 of herpes simplex virus type 1 encodes a virion protein which is myristylated. J. Gen. Virol. 70:3147–3157 [DOI] [PubMed] [Google Scholar]
- 47. MacLean CA, Dolan A, Jamieson FE, McGeoch DJ. 1992. The myristylated virion proteins of herpes simplex virus type 1: investigation of their role in the virus life cycle. J. Gen. Virol. 73:539–547 [DOI] [PubMed] [Google Scholar]
- 48. Maninger S, et al. 2011. M94 is essential for the secondary envelopment of murine cytomegalovirus. J. Virol. 85:9254–9267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mathew SS, Bryant PW, Burch AD. 2010. Accumulation of oxidized proteins in Herpesvirus infected cells. Free Radic. Biol. Med. 49:383–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mbong EF, Woodley L, Frost E, Baines JD, Duffy C. 2012. Deletion of UL21 causes a delay in the early stages of the herpes simplex virus 1 replication cycle. J. Virol. 86:7003–7007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Meckes DG, Jr, Wills JW. 2007. Dynamic interactions of the UL16 tegument protein with the capsid of herpes simplex virus. J. Virol. 81:13028–13036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Meckes DG, Jr, Wills JW. 2008. Structural rearrangement within an enveloped virus upon binding to the host cell. J. Virol. 82:10429–10435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mettenleiter TC. 2002. Herpesvirus assembly and egress. J. Virol. 76:1537–1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mettenleiter TC. 2004. Budding events in herpesvirus morphogenesis. Virus Res. 106:167–180 [DOI] [PubMed] [Google Scholar]
- 55. Nalwanga D, Rempel S, Roizman B, Baines JD. 1996. The UL16 gene product of herpes simplex virus 1 is a virion protein that colocalizes with intranuclear capsid proteins. Virology 226:236–242 [DOI] [PubMed] [Google Scholar]
- 56. Nucci C, et al. 2000. Imbalance in corneal redox state during herpes simplex virus 1-induced keratitis in rabbits. Effectiveness of exogenous glutathione supply. Exp. Eye Res. 70:215–220 [DOI] [PubMed] [Google Scholar]
- 57. O'Regan KJ, Bucks MA, Murphy MA, Wills JW, Courtney RJ. 2007. A conserved region of the herpes simplex virus type 1 tegument protein VP22 facilitates interaction with the cytoplasmic tail of glycoprotein E (gE). Virology 358:192–200 [DOI] [PubMed] [Google Scholar]
- 58. Oshima S, et al. 1998. Characterization of the UL16 gene product of herpes simplex virus type 2. Arch. Virol. 143:863–880 [DOI] [PubMed] [Google Scholar]
- 59. Palamara AT, et al. 1995. Evidence for antiviral activity of glutathione: in vitro inhibition of herpes simplex virus type 1 replication. Antiviral Res. 27:237–253 [DOI] [PubMed] [Google Scholar]
- 60. Pellman D, Garber EA, Cross FR, Hanafusa H. 1985. An N-terminal peptide from p60src can direct myristylation and plasma membrane localization when fused to heterologous proteins. Nature 314:374–377 [DOI] [PubMed] [Google Scholar]
- 61. Phillips SL, Bresnahan WA. 2012. The human cytomegalovirus (HCMV) tegument protein UL94 is essential for secondary envelopment of HCMV virions. J. Virol. 86:2523–2532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Phillips SL, Cygnar D, Thomas A, Bresnahan WA. 2012. Interaction between the human cytomegalovirus tegument proteins UL94 and UL99 is essential for virus replication. 86:9995–10005 J. Virol. doi:10.1128/JVI.01078-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sadaoka T, Yoshii H, Imazawa T, Yamanishi K, Mori Y. 2007. Deletion in open reading frame 49 of varicella-zoster virus reduces virus growth in human malignant melanoma cells but not in human embryonic fibroblasts. J. Virol. 81:12654–12665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Salaun C, Greaves J, Chamberlain LH. 2010. The intracellular dynamic of protein palmitoylation. J. Cell Biol. 191:1229–1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Schachtele SJ, Hu S, Little MR, Lokensgard JR. 2010. Herpes simplex virus induces neural oxidative damage via microglial cell Toll-like receptor-2. J. Neuroinflammation 7:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Schimmer C, Neubauer A. 2003. The equine herpesvirus 1 UL11 gene product localizes to the trans-Golgi network and is involved in cell-to-cell spread. Virology 308:23–36 [DOI] [PubMed] [Google Scholar]
- 67. Sigal CT, Zhou W, Buser CA, McLaughlin S, Resh MD. 1994. Amino-terminal basic residues of Src mediate membrane binding through electrostatic interaction with acidic phospholipids. Proc. Natl. Acad. Sci. U. S. A. 91:12253–12257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Silva MC, Yu QC, Enquist L, Shenk T. 2003. Human cytomegalovirus UL99-encoded pp28 is required for the cytoplasmic envelopment of tegument-associated capsids. J. Virol. 77:10594–10605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Triola G, Waldmann H, Hedberg C. 2012. Chemical biology of lipidated proteins. ACS Chem. Biol. 7:87–99 [DOI] [PubMed] [Google Scholar]
- 70. Vogel JU, et al. 2005. Effects of S-acetylglutathione in cell and animal model of herpes simplex virus type 1 infection. Med. Microbiol. Immunol. 194:55–59 [DOI] [PubMed] [Google Scholar]
- 71. Wills JW, Craven RC, Achacoso JA. 1989. Creation and expression of myristylated forms of Rous sarcoma virus gag protein in mammalian cells. J. Virol. 63:4331–4343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wills JW, Craven RC, Weldon R, Jr, Nelle TD, Erdie CR. 1991. Suppression of retroviral MA deletions by the amino-terminal membrane-binding domain of p60src. J. Virol. 65:3804–3812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wing BA, Lee GC, Huang ES. 1996. The human cytomegalovirus UL94 open reading frame encodes a conserved herpesvirus capsid/tegument-associated virion protein that is expressed with true late kinetics. J. Virol. 70:3339–3345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yeh PC, et al. 2011. Direct and specific binding of the UL16 tegument protein of herpes simplex virus to the cytoplasmic tail of glycoprotein E. J. Virol. 85:9425–9436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Yeh PC, Meckes DG, Jr, Wills JW. 2008. Analysis of the interaction between the UL11 and UL16 tegument proteins of herpes simplex virus. J. Virol. 82:10693–10700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zhu FX, Chong JM, Wu L, Yuan Y. 2005. Virion proteins of Kaposi's sarcoma-associated herpesvirus. J. Virol. 79:800–811 [DOI] [PMC free article] [PubMed] [Google Scholar]