

Abstract
Phylogenetic analysis has demonstrated that some positive-sense RNA viruses can be classified into the picornavirus-like supercluster, which includes picornaviruses, caliciviruses, and coronaviruses. These viruses possess 3C or 3C-like proteases (3Cpro or 3CLpro, respectively), which contain a typical chymotrypsin-like fold and a catalytic triad (or dyad) with a Cys residue as a nucleophile. The conserved key sites of 3Cpro or 3CLpro may serve as attractive targets for the design of broad-spectrum antivirals for multiple viruses in the supercluster. We previously reported the structure-based design and synthesis of potent protease inhibitors of Norwalk virus (NV), a member of the Caliciviridae family. We report herein the broad-spectrum antiviral activities of three compounds possessing a common dipeptidyl residue with different warheads, i.e., an aldehyde (GC373), a bisulfite adduct (GC376), and an α-ketoamide (GC375), against viruses that belong to the supercluster. All compounds were highly effective against the majority of tested viruses, with half-maximal inhibitory concentrations in the high nanomolar or low micromolar range in enzyme- and/or cell-based assays and with high therapeutic indices. We also report the high-resolution X-ray cocrystal structures of NV 3CLpro-, poliovirus 3Cpro-, and transmissible gastroenteritis virus 3CLpro- GC376 inhibitor complexes, which show the compound covalently bound to a nucleophilic Cys residue in the catalytic site of the corresponding protease. We conclude that these compounds have the potential to be developed as antiviral therapeutics aimed at a single virus or multiple viruses in the picornavirus-like supercluster by targeting 3Cpro or 3CLpro.
INTRODUCTION
Positive-sense RNA viruses possess RNA that is translated directly into one or more polyproteins which are subsequently cleaved by virus proteases into mature or intermediate viral proteins. Genetic analysis of RNA-dependent RNA polymerase has demonstrated that some positive-sense RNA viruses can be classified further into the picornavirus-like supercluster, which includes viruses belonging to the Picornaviridae, Caliciviridae, and Coronaviridae families (16). Viruses in the picornavirus-like supercluster include important classical and emerging human and animal pathogens. They include noroviruses (Norwalk virus [NV] and MD145 virus) and feline calicivirus (FCV), in the Caliciviridae family; human rhinovirus (HRV), enterovirus 71 (EV71), poliovirus (PV), foot-and-mouth disease virus (FMDV), hepatitis A virus (HAV), and porcine teschovirus (PTV), in the Picornaviridae family; and human coronavirus 229E, transmissible gastroenteritis virus (TGEV), murine hepatitis virus (MHV), bovine coronavirus (BCV), feline infectious peritonitis virus (FIPV), and severe acute respiratory syndrome coronavirus (SARS-CoV), in the Coronaviridae family. Therefore, great efforts have been made to discover effective preventive and therapeutic measures, including vaccines and antiviral agents, against these viruses. The development and implementation of the PV vaccine in children for the prevention of poliomyelitis represent an example of successful control of viral diseases. Still, there are many challenges on the way toward the development of effective vaccines for some of these viruses, such as diverse serotypes, short-term immunity, or an inability to grow the virus in cell culture.
At present, no antiviral drug specific for viruses belonging to the picornavirus-like supercluster has been approved by the FDA. The importance of these RNA viruses as human and animal pathogens and the lack of antiviral drugs make it imperative to develop therapeutics against these viruses. Traditionally, antiviral drug development has focused on virus-specific approaches due to the widely diverse replication strategies and antigenicity of viruses and the limited knowledge of a common therapeutic target. Exceptions are interferons, which are a part of innate immunity that act as natural antivirals to counteract various viral pathogens. Recently, synthetic compounds and several protease inhibitors were shown to be effective against multiple viruses in single (6, 12, 44) or multiple (18, 25, 26, 38, 43) virus families. For example, the protease inhibitor rupintrivir, originally developed for HRV, and/or its derivatives also showed broad-spectrum antiviral activity against picornaviruses and coronaviruses in cell culture (6, 12, 44), underscoring the potential development of broad-spectrum antivirals.
A common feature of the viruses in the picornavirus-like supercluster is that they possess a viral 3C or 3C-like protease (3Cpro or 3CLpro, respectively) which is responsible for the majority of cleavages of the corresponding viral polyprotein into mature or intermediate virus proteins (4, 41). The 3Cpro and 3CLpro share several common characteristics, including a typical chymotrypsin-like fold; a Cys residue as an active site nucleophile in the catalytic triad (or dyad), composed of Cys, His, and Glu (or Asp) residues; and a preference for a Glu or Gln residue at the P1 position on the substrate (in the nomenclature of Schechter and Berger [36]). The structural conservation of the active sites containing the catalytic triad or dyad and of substrate binding pockets has been shown by high-resolution three-dimensional (3D) structures of the 3Cpro and 3CLpro of these viruses (2, 19, 29, 30, 45, 46). Since virus proteases are essential for virus replication, the conserved key sites of 3Cpro or 3CLpro may serve as attractive targets for the design of antiviral drugs.
We previously identified a series of protease inhibitors (28, 40), including a dipeptidyl aldehyde (GC373) and an α-ketoamide (GC375), and evaluated their efficacy against NV in enzyme- and cell-based assays. In this study, we synthesized a dipeptidyl bisulfite adduct salt (GC376) from GC373 and evaluated the activities of GC373, GC375, and GC376 against various viruses in the picornavirus-like supercluster in enzyme- and/or cell-based assays. The viruses and virus proteases tested in this study included caliciviruses (NV, MD145, murine norovirus [MNV], and FCV), coronaviruses (229E, TGEV, MHV, BCV, FIPV, and SARS-CoV), and picornaviruses (HAV, PV, FMDV, EV71, HRV, and PTV). All compounds were highly effective against most picornaviruses, caliciviruses, and coronaviruses tested, with half-maximal inhibitory concentrations (IC50s) in the high nanomolar or low micromolar range in cell-based or protease assays and with high therapeutic indices. The crystal structures of NV 3CLpro-, PV 3Cpro-, and TGEV 3CLpro-GC376 complexes, determined at 1.65, 1.60, and 2.25 Å, respectively, revealed that GC376 covalently bound to Cys 139, Cys 147, and Cys 144 of NV 3CLpro, PV 3Cpro, and TGEV 3CLpro, respectively. Based on our results, these compounds have broad-spectrum antiviral activity against multiple viruses in the picornavirus-like supercluster, including important classical and emerging animal and human pathogens. We conclude that members of these compound series may have the potential to be developed as antiviral therapeutics targeting a specific virus or, more importantly, as broad-spectrum antivirals targeting multiple viruses.
MATERIALS AND METHODS
Compounds.
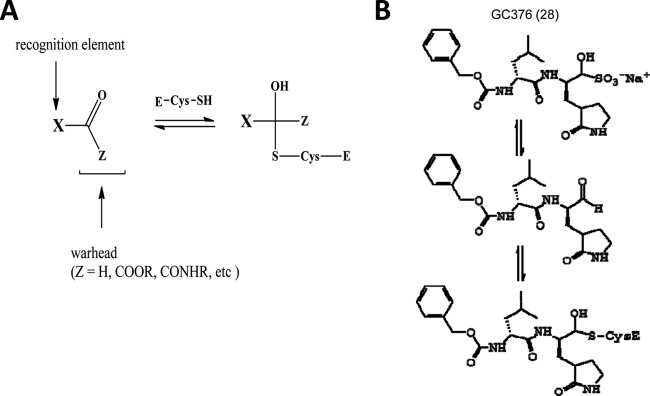
GC373 and GC375 were synthesized as reported elsewhere (28, 40). The aldehyde bisulfite adduct salt GC376 was synthesized as described in the supplemental material. The structures of the dipeptide inhibitors are shown in Fig. 1. Rupintrivir, a protease inhibitor designed against HRV 3Cpro, was purchased from Axon Medchem (Groningen, Netherlands) and used as a control.
Fig 1.

Structures (A) and summary of synthesis (B) of dipeptide inhibitors. GC373, GC375, and GC376 are shown, with aldehyde, α-ketoamide, and aldehyde bisulfite adduct salt, respectively, as warheads.
Cells, viruses, and reagents.
Various cell lines, including HG23 (Huh-7 cells containing an NV replicon) (8), CRFK, RAW267.4, ST, CCL-9.1, MRC-5, FRhK-4, HeLa, and Vero cells, were maintained in Dulbecco's minimal essential medium (DMEM) or MEM containing 5% fetal bovine serum and antibiotics (chlortetracycline [25 μg/ml], penicillin [250 U/ml], and streptomycin [250 μg/ml]). All cells except for HG23 cells were obtained from ATCC (Manassas, VA). Viruses used in this study were FCV (strain Urbana), MNV-1, TGEV (strain Miller), BCV (a field isolate from the Kansas State University [KSU] diagnostic lab), FIPV (strain 1146), human coronavirus 229E, MHV (strain A59), HAV (strain HM175), PTV (a field isolate from the KSU diagnostic lab), enterovirus 71 (strain H), and HRV (strains 18, 51, and 68). FCV and MNV-1 were obtained from K. Green at the NIH and from H. Virgin at Washington University (St. Louis, MO), respectively. BCV and PTV were obtained from the KSU diagnostic lab. All other viruses were obtained from ATCC.
Expression and purification of 3Cpro and 3CLpro.
The cDNAs encoding full-length viral 3Cpro or 3CLpro of TGEV and HAV were amplified by reverse transcription-PCR (RT-PCR) as previously described (2, 37). Primers contained the nucleotide sequences of each corresponding protease, for cloning, as well as the nucleotides for 6 His residues (in the forward primers). The codon-optimized cDNAs for 3Cpro or 3CLpro of NV, MD145, SARS-CoV (3), PV (31), and FMDV (7) were synthesized fused with 6 His at the N terminus (Genscript, Piscataway, NJ). Each synthesized gene or amplified product was subcloned into the pET-28a(+) vector. The expression and purification of each protease were performed by a standard method described previously by our lab (39). Recombinant HRV 3Cpro was purchased from EMD Chemicals, Inc. (Gibbstown, NJ).
FRET protease assays.
Fluorogenic substrates with Edans and Dabcyl as a donor and quencher pair were purchased from Bachem (coronavirus substrate) or synthesized by GenScript. The viral proteases and the corresponding fluorogenic substrates are listed in Table 1, along with their sources. The designation of substrate residues for P1 and P1′ starts at the scissile bond and counts toward the N and C termini, respectively (36). The fluorescence resonance energy transfer (FRET) protease assay was performed as follows. Stock solutions (10 mM) of the substrates and the compounds were prepared in dimethyl sulfoxide (DMSO) and diluted in assay buffer. The assay buffer comprised 20 mM HEPES buffer containing NaCl (0 mM for HAV 3Cpro and 200 mM for all other proteases), 0.4 mM EDTA, glycerol (60% for NV and MD145 3CLpro and 30% for TGEV and 229E 3CLpro), and 6 mM (NV and MD145 3CLpro and HAV 3Cpro) or 4 mM (all other proteases) dithiothreitol (DTT) at pH 6 (coronavirus 3CLpro) or 8 (all other proteases) (9). Each protease was mixed with serial dilutions of each compound or with DMSO in 25 μl of assay buffer and incubated at 37°C for 30 min, followed by the addition of 25 μl of assay buffer containing substrate. Fluorescence readings were obtained using an excitation wavelength of 360 nm and an emission wavelength of 460 nm on a fluorescence microplate reader (FLx800; Biotek, Winooski, VT) 1 h following the addition of substrate. Relative fluorescence units (RFU) were determined by subtracting background values (substrate-containing well without protease) from the raw fluorescence values as described previously (9, 22). The dose-dependent FRET inhibition curves were fitted with a variable slope by using GraphPad Prism software (GraphPad, La Jolla, CA) in order to determine the IC50s of compounds.
Table 1.
Virus proteases and fluorogenic substrates used for FRET protease assaysa
| Virus family and virus | Fluorogenic substrate | Source and/or reference for fluorogenic substrate | Buffer conditionsb |
|||
|---|---|---|---|---|---|---|
| pH | Glycerol (%) | DTT (mM) | NaCl (mM) | |||
| Caliciviridae | ||||||
| NV | Edans-DFHLQ/GP-Dabcyl (truncated) | 46 | 8 | 60 | 6 | 120 |
| MD145 | Edans-DFHLQ/GP-Dabcyl | 8 | 60 | 6 | 120 | |
| Coronaviridae | ||||||
| TGEV | Dabcyl-KTSAVLQ/SGFRKME-Edans | Bachem; 24 | 6 | 30 | 4 | 120 |
| SARS-CoV | Dabcyl-KTSAVLQ/SGFRKME-Edans | 6 | 30 | 4 | 120 | |
| Picornaviridae | ||||||
| PV | Dabcyl-KTSAVLQ/SGFRKME-Edans | 8 | 20 | 4 | 120 | |
| HRV | Edans-DFHLQ/GP-Dabcyl | 7 | 20 | 4 | 120 | |
| HAV | Dabcyl-GLRTQ/SFS-Edans | Bachem | 7 | 20 | 4 | 120 |
| FMDV | Edans-APAKQ/LLN-Dabcyl | 22 | 8 | 50 | 4 | 120 |
NV, norovirus strain Norwalk; MD145, norovirus strain MD145; TGEV, transmissible gastroenteritis virus; SARS-CoV, severe acute respiratory syndrome coronavirus; PV, poliomyelitis virus; HRV, human rhinovirus; HAV, human hepatitis A virus; FMDV, foot-and-mouth disease virus.
The buffer contained 20 mM HEPES and 0.4 mM EDTA.
Cell-based inhibition assays.
The effects of each compound on viral replication were examined in cell culture systems. Virus-infected cells were incubated at 37°C, except for HRV-infected HeLa cells, which were maintained at 33°C. The viruses and corresponding cell lines are listed in Table 2. Briefly, confluent or semiconfluent cells were inoculated with virus at a multiplicity of infection of 0.05 for 1 h, and the inoculum was replaced with medium containing DMSO (<0.1%) or each compound (up to 100 μM). The virus-infected cells were further incubated for up to 168 h, and the replication of virus was measured by the 50% tissue culture infective dose (TCID50) method (35) and/or real-time quantitative RT-PCR (qRT-PCR). The TCID50 method was used for titration of viruses showing apparent cell cytopathic effects, which included FCV, MNV-1, TGEV, FIPV, MHV, BCV, HAV, 229E, EV71, and PTV. Real-time qRT-PCR was performed for titration of NV (replicon-harboring cells) and HRV. For HAV and 229E, real-time qRT-PCR was also used to confirm the TCID50 results. For real-time qRT-PCR, RNA was extracted from each sample (cell lysates for HG23 cells and viral suspensions for HAV, HRV, and 229E) by use of an RNeasy kit (Qiagen, Valencia, CA), followed by amplification in a Cepheid SmartCycler with the following parameters: 45°C for 30 min and 95°C for 10 min followed by 40 cycles of denaturation at 95°C for 30 s, annealing at 50°C for 1 min, and elongation at 72°C for 30 s. The primers and probes used for real-time qRT-PCR included the following: for NV, 5′-CGYTGGATGCGNTTYCATGA-3′, 5′-CTTAGACGCCATCATCATTYAC-3′, and 6-carboxyfluorescein (FAM)–5′-AGATYGCGATCYCCTGTCCA-3′–6-carboxytetramethylrhodamine (TAMRA); for HAV, 5′-ACTGCAGTGACTGGTGCTTC-3′, 5′-CCGGGTTTATCAACAGAGGT-3′, and FAM-5′-CCTGGTGTGATCCAACCTCAGCTG-3′-IABkFQ; for HRV, 5′-TGTTCYAGCCTGCGTGGC-3′, 5′-GAAACACGGACACCCAAAGTA-3′, and FAM-5′-TCCTCCGGCCCCTGAATGYGGC-3′-IABkFQ; and for 229E, 5′-TTCCGACGTGCTCGAACTTT-3′, 5′-CCAACACGGTTGTGACAGTGA-3′, and FAM-5′-TCCTGAGGTCAATGCA-3′-IABkFQ. The IC50s were determined by GraphPad Prism software.
Table 2.
Viruses and corresponding cell lines used in cell culture
| Virus family | Virusa | Cell line |
|---|---|---|
| Caliciviridae | NV | HG23 |
| FCV | CRFK | |
| MNV-1 | RAW267.4 | |
| Coronaviridae | TGEV | ST |
| FIPV | CRFK | |
| 229E | MRC-5 | |
| MHV | CCL-9.1 | |
| BCV | HRT-18 | |
| Picornaviridae | HAV | FRhK-4 |
| EV71 | Vero | |
| HRV 18, 51, and 68 | HeLa | |
| PTV | ST |
MNV-1, murine norovirus 1; FIPV, feline infectious peritonitis virus; 229E, human coronavirus 229E; MHV, mouse hepatitis virus; BCV, bovine coronavirus; EV71, enterovirus 71; HRV 18, 51, and 68, human rhinovirus strains 18, 51, and 68; PTV, porcine teschovirus.
Nonspecific cytotoxic effects.
The toxic dose for 50% cell death (TD50) for each compound was determined for the various cells used in this study. Confluent cells grown in 96-well plates were treated with various concentrations (1 to 500 μM) of each compound for 72 h. Cell cytotoxicity was measured by a CytoTox 96 nonradioactive cytotoxicity assay kit (Promega, Madison, WI) and crystal violet staining. The in vitro therapeutic index was calculated by dividing the TD50 by the IC50.
X-ray crystallography: crystallization and data collection.
Details regarding crystallization, data collection, structure solution, and refinement are provided in Table S1 in the supplemental material. Briefly, all crystallization trials were conducted in high-throughput Compact Jr. (Emerald Biosystems) sitting-drop vapor diffusion plates at 20°C, using equal volumes of protein and crystallization solution equilibrated against 75 μl of the latter. Crystals of apo-NV 3CLpro or an NV 3CLpro-, PV 3Cpro-, or TGEV 3CLpro-GC376 complex were obtained within 48 h (see Fig. S1), and X-ray diffraction data were collected at Advanced Photon Source beamline 17-ID, using a Dectris Pilatus 6 M pixel array detector. Structure solution was conducted by molecular replacement with Molrep (42), and refinement and model building were carried out with Phenix (1) and Coot (15), respectively.
Protein structure accession numbers.
Coordinates and structure factors for apo-NV 3CLpro and the NV 3CLpro-, PV 3Cpro-, and TGEV 3CLpro-GC376 complexes have been deposited in the Protein Data Bank (PDB) under accession numbers 3UR6, 3UR9, 4DCD, and 4F49, respectively.
RESULTS
Effects of compounds against 3Cpro or 3CLpro of viruses in the picornavirus-like supercluster.
Viral 3Cpro or 3CLpro, except for HRV 3Cpro, which was purchased from a commercial source, were expressed and optimized for protease assays with the fluorogenic substrates presented in Table 1. The effects of the compounds as well as rupintrivir in optimized FRET protease assays are summarized in Table 3. GC373 (dipeptidyl aldehyde) (Fig. 1), previously shown to be effective against NV 3CLpro (40), inhibited the activities of all viral proteases except for HAV 3Cpro; it was effective against the proteases of caliciviruses (NV and MD145 virus), coronaviruses (TGEV and SARS-CoV), and picornaviruses (PV, FMDV, and HRV), with IC50s ranging from 0.61 to 3.48 μM under our assay conditions (Table 3). The effects and range of inhibition of GC376 were comparable to those of GC373 against various 3Cpro and 3CLpro. The inhibitory effects of GC375 (a dipeptidyl α-ketoamide) were moderate (2.87 to 4.02 μM) against the proteases of caliciviruses (Table 3). However, the inhibitory activities of GC375 against the proteases of picornaviruses and coronaviruses were comparable to those of GC373 and GC376 (Table 3). In addition, GC375 showed weak but appreciable inhibitory effects on HAV 3Cpro compared to GC373 and GC376. Rupintrivir, used as a control, showed similar activities against calicivirus and picornavirus proteases compared to GC373 and GC376 but was substantially less effective against the coronavirus proteases (Table 3).
Table 3.
Effects of GC373, GC375, and GC376 on 3Cpro or 3CLpro of viruses in the FRET protease assay
| Drug | Inhibition (IC50 [μM]) of recombinant 3Cpro or 3CLproa |
|||||||
|---|---|---|---|---|---|---|---|---|
| Caliciviruses |
Coronaviruses |
Picornaviruses |
||||||
| NV | MD145 | TGEV | SARS-CoV | HAV | HRV | PV | FMDV | |
| GC373 | 0.64 ± 0.27 | 1.18 ± 0.52 | 0.99 ± 0.12 | 3.48 ± 1.59 | >50 | 0.65 ± 0.36 | 2.02 ± 1.56 | 0.61 ± 0.32 |
| GC375 | 2.87 ± 0.39 | 4.02 ± 2.19 | 1.55 ± 0.10 | 4.66 ± 0.19 | 42.59 ± 2.61 | 0.55 ± 0.39 | 2.84 ± 0.25 | 0.55 ± 0.32 |
| GC376 | 0.49 ± 0.05 | 0.96 ± 0.65 | 0.82 ± 0.47 | 4.35 ± 0.47 | >50 | 0.20 ± 0.14 | 1.77 ± 0.31 | 1.16 ± 0.75 |
| Rupintrivir | 0.83 ± 0.19 | 0.70 ± 0.19 | >50 | >50 | >50 | 0.34 ± 0.05 | 1.83 ± 0.38 | 4.21 ± 1.97 |
The IC50s are means ± standard errors of the means for two or three independent tests with each optimized protease assay.
Effects of compounds on the replication of viruses in the picornavirus-like supercluster.
The overall effects of the compounds on the replication of various viruses in the cell-based assays were in line with those in the protease assay (Tables 3, 4, and 5). Both GC373 and GC376 were significantly effective against caliciviruses (NV and MNV-1), coronaviruses (TGEV, FIPV, MHV, 229E, and BCV), and picornaviruses (HRVs 18, 51, and 68, EV71, and PTV), with nanomolar or low micromolar IC50s, except for FCV and HAV (Tables 4 and 5). Interestingly, FCV was less sensitive to GC373 and GC376, with IC50s of 65 and 35 μM, respectively (Table 4). As shown with the protease assay, GC373 and GC376 showed no or weak effectiveness against the replication of HAV in cells. GC375 showed significant antiviral effects against NV and all coronaviruses and picornaviruses, including HAV, but no effects against FCV and MNV-1 at concentrations up to 50 μM (Tables 4 and 5). Rupintrivir inhibited the replication of NV (in HG23 cells), TGEV, FIPV, 229E, BCV, HRV strains 18, 51, and 68, EV71, and PTV, with various potencies (Tables 4 and 5). Note that rupintrivir did not inhibit the replication of MNV-1, FCV, and MHV at concentrations up to 50 μM or 100 μM (Tables 4 and 5). All compounds, including rupintrivir, did not show any nonspecific cytotoxicity at concentrations up to 500 μM in the cells used for virus replication in this study.
Table 4.
Effects of GC373, GC375, and GC376 on replication of viruses in cell culture as determined by the TCID50 method
| Drug | Inhibition (IC50 [μM]) of virus (mean ± SEM)a |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Caliciviruses |
Coronaviruses |
Picornaviruses |
||||||||
| FCV | MNV-1 | TGEV | FIPV | MHV | 229E | BCV | HAV | EV71 | PTV | |
| GC373 | 65.1 ± 2.1 | 6.5 ± 1.2 | 0.3 ± 0.2 | 0.3 ± 0.1 | 2 ± 0.2 | 0.2 ± 0.1 | 0.7 ± 0.2 | >100 | 11.1 ± 1.2 | 0.15 ± 0.1 |
| GC375 | >50 | >50 | 0.2 ± 0.1 | 1.5 ± 0.3 | 4.5 ± 0.4 | 0.25 ± 0.1 | 0.8 ± 0.2 | 19.5 ± 2.1 | 15.2 ± 1.3 | 0.2 ± 0.1 |
| GC376 | 35.2 ± 3.6 | 5.3 ± 1.6 | 0.15 ± 0.1 | 0.2 ± 0.1 | 1.1 ± 0.3 | 0.15 ± 0.1 | 0.6 ± 0.2 | 50.2 ± 5.6 | 10.3 ± 2.4 | 0.15 ± 0.1 |
| Rupintrivir | >50 | >50 | 2.5 ± 0.7 | 10.3 ± 1.3 | >100 | 0.3 ± 0.1 | 15.3 ± 2.5 | >100 | 0.5 ± 0.2 | 0.15 ± 0.1 |
The IC50s are averages for 3 independent tests and were determined by the TCID50 method. SEM, standard error of the mean.
Table 5.
Effects of GC373, GC375, and GC376 on replication of viruses in cell culture as determined by real-time qRT-PCR
| Drug | Inhibition (IC50 [μM]) of virus (mean ± SEM)a |
|||
|---|---|---|---|---|
| Caliciviruses |
Picornaviruses |
|||
| HG23 | HRV18 | HRV51 | HRV68 | |
| GC373 | 0.2 ± 0.1 | 1.1 ± 0.3 | 1.8 ± 0.3 | 1.6 ± 0.2 |
| GC375 | 2.8 ± 0.3 | 0.09 ± 0.03 | 0.08 ± 0.02 | 0.07 ± 0.02 |
| GC376 | 0.3 ± 0.1 | 1.2 ± 0.2 | 1.5 ± 0.2 | 0.8 ± 0.2 |
| Rupintrivir | 2.3 ± 0.5 | 0.03 ± 0.01 | 0.02 ± 0.01 | 0.04 ± 0.02 |
The IC50s are averages for 3 independent tests and were determined by real-time qRT-PCR. SEM, standard error of the mean.
Crystal structure of NV 3CLpro refined to 1.50-Å resolution.
The apo-NV 3CLpro structure, which represents a new crystal form of NV 3CLpro (Fig. 2A), was similar to a previously reported crystal structure of NV 3CLpro (PDB accession no. 2FYQ) (46). Superposition of residues Ala 1 to Ala 173 of our structure with those of structure 2FYQ by using Superpose (23) via the CCP4 (10) interface yielded root mean square deviations (RMSD) of 0.84 Å and 0.77 Å between the Cα atoms for chains A and B, respectively. The largest differences were observed in certain loop regions of the protease, including Met 101 to Arg 112, Lys 146 to Val 152, and Thr 161 to Thr 166, and are not likely due to crystal contacts. These loop regions are known to be flexible and involved in the substrate recognition and interaction for the protease activity (19, 30, 46).
Fig 2.
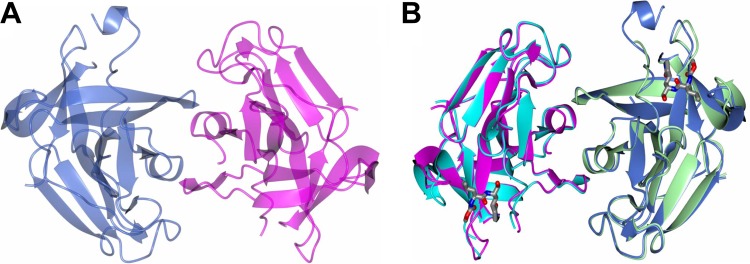
Crystal structures of apo-NV 3CLpro and NV 3CLpro in complex with GC376. (A) Noncrystallographic symmetry (NCS) dimer of NV 3CLpro showing chain A (blue) and chain B (magenta). (B) NCS dimer of apo-NV 3CLpro showing chain A (magenta) and chain B (blue) superimposed with the GC376-bound form showing chain A (cyan) and chain B (green).
Crystal structure of NV 3CLpro in complex with GC376 refined to 1.65-Å resolution.
NV 3CLpro existed as a noncrystallographic dimer that was nearly identical to that observed for the apo crystal form (Fig. 2B). Examination of the active site revealed a prominent difference in electron density (Fo − Fc) of >3σ in each subunit, consistent with GC376. However, the bisulfite group appeared to have been removed, and the compound was converted to the aldehyde form, which created a covalent bond with Cys 139 (Fig. 3A). The 6-membered aromatic ring of the compound is not included in the modeling due to disorder, which may have resulted from the possibility that the aromatic ring does not bind to the S3 site but faces outwards toward the solvent (Fig. 3A). The glutamine surrogate ring and the Leu of GC376 fit into the S1 and S2 sites, respectively, as expected (Fig. 3B). The hydrogen bond interactions between amino acid residues His 30, Gln 110, Thr 134, His 157, Ala 158, and Ala 160 in NV 3CLpro and GC376 are shown in Fig. 3C. Superposition of residues Ala 1 to Ala 173 of apo-NV 3CLpro with those of the NV 3CLpro-GC376 complex yielded RMSD of 0.80 Å and 0.68 Å between the Cα atoms for chains A and B, respectively. The largest differences were observed in the loop containing Gln 110, which undergoes a conformational change to accommodate hydrogen bonding to GC376 (Fig. 4A; see Movie S1 in the supplemental material). Gln 100 and Ala 160 are involved in tight binding to GC376, with large conformational changes in the loops containing those amino acids. All other residues that form hydrogen bonds to the compound are in similar positions in both the apo and ligand-bound forms. Comparison of the binding interactions between 3CLpro of norovirus strain Southampton and a Michael acceptor inhibitor, acetyl-Glu-Phe-Gln-Leu-Gln-CH CHCOO− (19), and between NV 3CLpro and dipeptidyl GC376 demonstrated that the same amino acids were involved in the interactions with the inhibitors.
CHCOO− (19), and between NV 3CLpro and dipeptidyl GC376 demonstrated that the same amino acids were involved in the interactions with the inhibitors.
Fig 3.

Cocrystal structures of NV 3CLpro, PV 3Cpro, and TGEV 3CLpro in complex with GC376. (A to C) NV 3CLpro-GC376 complex. (A) Fo − Fc omit map contoured at 3σ for GC376. The aromatic ring of the inhibitor (green) was not included in the model due to disorder. (B) GC376 (red) in the S1 and S2 positions of the active site of NV 3CLpro (gray). (C) Hydrogen bonding (dashed lines) interactions between NV 3CLpro (cyan) and GC376 (gray). (D to F) PV 3Cpro-GC376 complex. (D) Fo − Fc omit map contoured at 3σ for GC376. (E) GC376 (red) in the S1 and S2 positions of the active site of PV 3Cpro (gray). (F) Hydrogen bonding (dashed lines) interactions between PV 3Cpro (magenta) and GC376 (gray). (G to I) TGEV 3CLpro-GC376 complex. (G) Fo − Fc omit map contoured at 3σ for GC376. (H) GC376 (red) in the S1 and S2 positions of the active site of TGEV 3CLpro (gray). (I) Hydrogen bonding (dashed lines) interactions between PV 3Cpro (magenta) and GC376 (blue). The red spheres in panels F and I are water molecules.
Fig 4.
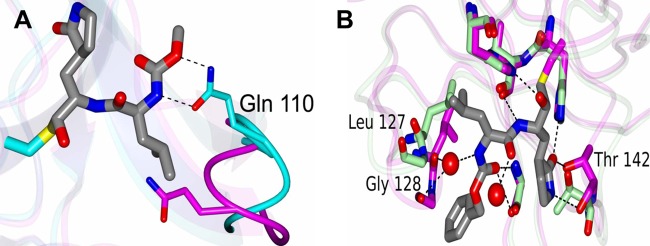
Conformational changes during binding of NV 3CLpro or PV 3Cpro and GC376. (A) Conformational changes in the loop containing Gln 110 in NV 3CLpro that occur upon inhibitor binding (see Movie S1 in the supplemental material for details). The apo and inhibitor-bound forms are colored magenta and cyan, respectively. The hydrogen bonds that form between Gln 110 and the inhibitor are indicated by dashed lines. (B) Conformational changes in the loops containing Leu 127, Gly 128, and Thr 142 of PV 3Cpro that occur upon ligand binding (see Movie S2 for details). The apo and ligand-bound forms are colored green and magenta, respectively. Leu 127 and Gly 128 undergo a conformational change to accommodate a water-mediated hydrogen bond. The red spheres are water molecules.
Crystal structures of PV 3Cpro and TGEV 3CLpro in complex with GC376, refined to 1.6-Å and 2.25-Å resolution, respectively.
The proteases of a picornavirus (PV 3Cpro) and a coronavirus (TGEV 3CLpro) were selected to study the interaction with GC376 in comparison to that with NV 3CLpro (Fig. 3D to I). Examination of the active site revealed a prominent difference in electron density (Fo − Fc) in each subunit (>3σ) that was consistent with GC376 (Fig. 3D and G). Like the case with NV 3CLpro, the glutamine surrogate ring and Leu of GC376 fit into the S1 and S2 sites, respectively (Fig. 3E and H), as expected. The GC376-bound structures of PV 3Cpro and TGEV 3CLpro are similar overall to the corresponding apo crystal forms (PV 3Cpro [PDB accession no. 1L1N] and TGEV 3CLpro [PDB accession no. 2AMP]). Hydrogen bonding interactions between PV 3Cpro or TGEV 3CLpro and GC376 are shown in Fig. 3F and I. For the PV 3Cpro-GC376 complex, the hydrogen bond interactions between amino acid residues His 40, Leu 127, Gly 128, Thr 142, His 161, Val 162, and Gly 164 in the protease and the compound are shown in Fig. 3F. Superposition of residues Ala 7 to Leu 174 of apo-PV 3Cpro with those of the PV 3Cpro-GC376 complex yielded an RMSD of 0.83 Å between the Cα atoms. The largest differences that occur upon ligand binding were observed in the loops containing Leu 127, Gly 128, and Thr 142 (Fig. 4B). Leu 127 and Gly 128 undergo a conformational change to accommodate a water-mediated hydrogen bond with the compound, and Thr 142 moves to form a hydrogen bond with the pyrrolidine ring of GC376 (Fig. 4B; see Movie S2 in the supplemental material). For the TGEV 3CLpro-GC376 complex, the hydrogen bond interactions between amino acid residues Thr 47, Phe 139, His 162, His 163, and Glu 165 in the protease and the compound are shown in Fig. 3I. The interactions between PV 3Cpro and GC376 also lead to conformational changes in loops containing Leu 127, Gly 128, and Gly 164 to accommodate hydrogen bonding. Like NV 3CLpro, the inhibitor was converted to the aldehyde form, which created a covalent bond with Cys 147 or Cys 144 in PV 3Cpro or TGEV 3CLpro, respectively. Water-mediated contacts between the protein and inhibitor were observed for PV 3Cpro and TGEV 3CLpro, as shown in Fig. 3. The B factors for these water molecules were 28.7 Å2 and 36.5 Å2 for PV 3Cpro, similar to the average B factor for all atoms in the model (22.7 Å2). For TGEV 3CLpro, a single water-mediated contact was observed in a similar position in 3 of the 4 subunits. The B factors for these water molecules were 34.8 Å2, 35.3 Å2, and 30.8 Å2 (subunits A, B, and C, respectively), comparable to the average B factors for all atoms (31.9 Å2). In addition, the water molecules fit well to the electron density maps, with no residual positive or negative density observed in the Fo − Fc map following refinement.
DISCUSSION
Viral proteases have been an important target for the development of antiviral drugs. Examples include several protease inhibitors currently on the market or under development for the treatment of viral infections, including infections with human immunodeficiency virus and hepatitis C virus (11, 27). The viruses in the picornavirus-like supercluster are characterized by the conserved viral genes, including those coding for 3Cpro or 3CLpro (see Fig. S2A and B in the supplemental material). The 3Cpro and 3CLpro possess common characteristics, such as conserved active sites with a Cys residue as a nucleophile, which may serve as useful targets for the design of antiviral drug development. Previously, we reported a series of dipeptidyl transition state inhibitors of NV 3CLpro (28, 40). Since NV 3CLpro shares sequence and structural similarities in the active site with those of picornaviruses and coronaviruses, we evaluated the dipeptidyl inhibitors for activity against various viruses in the picornavirus-like supercluster in a protease assay and/or cell-based system. In addition, we generated an inhibitor possessing a bisulfite adduct warhead (GC376) with the same backbone as that of previously generated dipeptidyl inhibitors and examined its broad-spectrum antiviral activity.
The viruses selected for evaluation in our study are important human and animal pathogens in the Caliciviridae, Coronaviridae, and Picornaviridae families. The Caliciviridae family includes 4 genera, namely, Vesivirus, Lagovirus, Norovirus, and Sapovirus. The NV and MD145 strains belong to norovirus genotypes I and II, respectively. Noroviruses of these genotypes are associated with the majority of norovirus outbreaks in humans. Viruses in the Coronaviridae family are divided into groups I, II, and III, and tentatively classified group IV (SARS-CoV), based upon antigenic properties and sequence homology. Coronaviruses in groups I and II infect mammals. The viruses or viral proteases tested in our study include those from groups I (229E, TGEV, and FIPV) and II (MHV and BCV) and SARS-CoV. The coronavirus 229E is a common cause of common cold in humans. SARS-CoV causes severe acute respiratory syndrome, with a high fatality rate, and was responsible for more than 800 fatalities in global outbreaks of SARS in 2003 (34), which accelerated studies on the development of effective antivirals against coronaviruses. TGEV, BCV, MHC, and FIPV are important animal pathogens with significant economic consequences or high fatality rates. Members of the Picornaviridae family are among the most diverse viruses and the most common causes of human viral infections. Picornaviruses have 12 distinct genera, including Enterovirus (PV and HRV), Cardiovirus, Aphthovirus (FMDV), Hepatovirus (HAV), and Teschovirus (PTV). These viruses exhibit a marked range of diversity in areas such as hosts, symptoms, and viral antigenicity. For example, HRV is a major cause of common cold in humans, with more than 100 distinct serotypes. PV is the causative agent of poliomyelitis, a devastating disease in humans. HAV is responsible for an acute hepatitis, affecting tens of millions of individuals each year in areas with poor hygiene standards (21). FMDV is highly contagious in domestic livestock, and FMDV outbreaks can result in devastating economic losses and international trade embargoes (17). Despite the diversity of these viruses in diseases and hosts, the advancement of refined structural elucidation of their 3C or 3CL proteases offers an opportunity for exploring inhibitors that are applicable to diverse viruses possessing these viral proteases.
The structures of the active sites and the substrate specificity of 3Cpro or 3CLpro are well elucidated. In most 3Cpro or 3CLpro, the P1 and P2 positions at the cleavage sites in the viral polyproteins are most commonly occupied by Gln (or Glu) and a hydrophobic residue, respectively (41). The inhibitors used in this study are dipeptidyl compounds with a Gln surrogate structure in a position that corresponds to the P1 position and a Leu in the P2 position (Fig. 1). Our dipeptidyl compounds of this particular structure exhibit broad antiviral activity against diverse viruses across multiple virus families possessing 3Cpro or 3CLpro with key conserved sites. In our enzyme- and/or cell-based studies, both GC376 and GC373 showed strong inhibition against viral replication or viral protease activity of most viruses tested that belong to the Caliciviridae, Coronaviridae, and Picornaviridae families, with IC50s in the low micromolar or high nanomolar range, except for HAV and FCV. The results indicate that dipeptidyl structures consisting of P1 and P2 may be sufficient for recognition and inhibition of the targeted proteases. It should be noted that GC373 and GC376 exhibited no or low potency at inhibiting the protease activity or replication of HAV or FCV. The available crystallographic structure studies on HAV 3Cpro showed that the side chains of amino acids that form the S2 pocket allow a P2 residue with relatively small side chains, such as Ser, Thr, or Ala residues, found in all confirmed P2 positions in the HAV cleavage sites (5, 45). Sequence analysis of the cleavage sites of FCV polyproteins seems to indicate that small residues such as Ser or Ala are preferred in the S2 position, rather than bulkier residues such as Leu or Ile. Thus, the space constraints in the S2 pocket in the protease of HAV or FCV may explain the lack of or minimal activity of the compounds against HAV or FCV. The compound GC375 was as potent as or more potent than GC373 and GC376 against the majority of coronaviruses and picornaviruses tested, including HAV. It is possible that the active sites of picornavirus 3Cpro and coronavirus 3CLpro have relatively broader shapes than that of calicivirus 3CLpro, which may allow easier accommodation of the bulkier ketoamide warhead. In contrast, GC375 was less effective than GC373 and GC376 against NV replication, and even less effective against the replication of FCV and MNV-1. It is possible that the ketoamide warhead is too bulky to fit well into the active sites of FCV and MNV-1 proteases, which may explain the lack of activity of the compound on the replication of those viruses.
Next, we established a binding model based on the X-ray crystal structures of NV 3CLpro-, PV 3Cpro-, and TGEV 3CLpro-GC376 complexes to examine the inhibition mechanism of GC376. The binding of GC376 to NV 3CLpro resembled that of acetyl-Glu-Phe-Gln-Leu-Gln-CHCHCOO− to Southampton norovirus 3CLpro, with irreversible covalent binding to Cys 139 (19). The cocrystal structure of the TGEV 3CLpro-GC376 complex also demonstrated similar binding interactions between the protease and the compound (Fig. 3G to I). These results confirm the conserved key sites of 3Cpro or 3CLpro as attractive targets for the design of broad-spectrum antivirals. The unexpected finding from our X-ray crystal structures was that the bisulfite group of GC376 was removed from the compound and GC376 was converted to the aldehyde form, which subsequently led to reversible covalent bond formation with Cys 139, Cys 147, or Cys 144 of NV 3CLpro, PV 3Cpro, or TGEV 3CLpro, respectively. This finding is consistent with our results from preliminary studies related to the facile transformation of aldehyde bisulfite (GC376) to aldehyde (GC373) when the drug was administered to animals (rats) via intravenous and oral routes. These results suggest that the aldehyde bisulfite adduct compound GC376 behaves as a prodrug of the aldehyde counterpart (Fig. 5). The bisulfite adduct warhead of GC376 was synthesized from the precursor aldehyde by reaction with sodium bisulfite and was intended to function as a transition state mimic (20, 32). The high aqueous solubility and pH-dependent equilibria between the precursor carbonyl compound and bisulfite adduct were also envisaged to have a significant effect on absorption, distribution, metabolism, excretion, and toxicity (ADMET) and on pharmacokinetics (PK). Solubility, as well as permeability and metabolic stability, affects oral bioavailability of a drug in vivo and also contributes to accurate and reliable in vitro assays. It is possible that a bisulfite adduct compound behaves as a prodrug of the precursor aldehyde, and this warrants further studies on its effects on pharmacokinetics and pharmacodynamics.
Fig 5.
Inhibition of viral 3Cpro and 3CLpro by GC376. (A) Design of transition state inhibitors for viral 3CLpro or 3Cpro. (B) Suggested mechanism of the aldehyde bisulfite adduct (GC376) as an inhibitor of viral 3CLpro or 3Cpro. It may act as a prodrug of the aldehyde inhibitor (GC373).
The range of antiviral activities of these compounds highlights the potential for the development of broad-spectrum antiviral compounds for multiple viruses. In recent years, compounds effective against multiple viruses have been explored based on advanced knowledge of the common features of viral proteins or host factors involved in the replication of viruses. Some enveloped viruses, including influenza virus, vaccinia virus, vesicular stomatitis virus, and cytomegalovirus, which belong to different virus families, utilize phosphatidylserine present in the host plasma membrane for survival in host cells (43). Targeting the phospholipid was proposed to be an effective strategy for the management of infections by these enveloped viruses and proved effective in an animal model of Lassa fever virus infection (43). As an irreversible peptidomimetic viral protease inhibitor, rupintrivir was originally developed for HRV, utilizing structure-based design of 3Cpro (13, 14). Rupintrivir and its derivatives were later found to have significant antiviral activity against other related picornaviruses and coronaviruses, as well as HRV, in cell culture (6, 12, 44). These examples suggest that the identification of common targets may lead to antiviral therapeutics effective against various important human and animal viruses. Since rupintrivir was shown to be effective against 3Cpro or 3CLpro, rupintrivir was used as a reference control in our studies (Tables 2 and 3). The compounds in this study were found to be generally as potent as rupintrivir against caliciviruses and picornaviruses and displayed greater potency against coronaviruses. Rupintrivir and the compounds in this study share a glutamine surrogate structure in a position corresponding to P1. However, the compounds in this study are dipeptidyl compounds with lower molecular masses, i.e., 403 (GC373), 489 (GC375), and 508 kDa (GC376), than the 599-kDa mass of rupintrivir, a tripeptidyl compound. Rupintrivir possesses an α,β-unsaturated ester as a warhead that interacts with the thiol group of the active site Cys in an irreversible way (33). In contrast, our preliminary study suggested that the compounds in this study may bind to the active site Cys by forming a reversible covalent adduct (our unpublished data). The low molecular masses and reversible binding mechanism of the compounds in this study may translate into better potency, efficacy, and pharmacological properties in vivo. Currently, we are examining their in vitro ADMET profiles as well as in vivo pharmacokinetics, including oral availability. In conclusion, this series of compounds target 3Cpro or 3CLpro and are effective against diverse viruses that belong to the picornavirus-like supercluster in cell culture and/or protease assays. The high efficacy and low toxicity of the compounds described herein suggest that they may be suitable candidates for further development as antivirals against one or more viruses or could serve as a starting point for the development of broad-spectrum antivirals.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH grant AI081891. Use of IMCA-CAT beamline 17-ID at the Advanced Photon Source was supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with Hauptman-Woodward Medical Research Institute. Use of the Advanced Photon Source was supported by the U.S. Department of Energy under contract DE-AC02-06CH11357. Use of the KU COBRE-PSF Protein Structure Laboratory was supported by NIH grant P20 RR-17708.
We thank David George for technical assistance and Joe Andersen at the Diagnostic Lab at KSU for providing virus samples.
Footnotes
Published ahead of print 22 August 2012
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1. Adams PD, et al. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66:213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Anand K, et al. 2002. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha-helical domain. EMBO J. 21:3213–3224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. 2003. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science 300:1763–1767 [DOI] [PubMed] [Google Scholar]
- 4. Anderson J, Schiffer C, Lee SK, Swanstrom R. 2009. Viral protease inhibitors. Handb. Exp. Pharmacol. 2009:85–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bergmann EM, Mosimann SC, Chernaia MM, Malcolm BA, James MN. 1997. The refined crystal structure of the 3C gene product from hepatitis A virus: specific proteinase activity and RNA recognition. J. Virol. 71:2436–2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Binford SL, et al. 2005. Conservation of amino acids in human rhinovirus 3C protease correlates with broad-spectrum antiviral activity of rupintrivir, a novel human rhinovirus 3C protease inhibitor. Antimicrob. Agents Chemother. 49:619–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Birtley JR, et al. 2005. Crystal structure of foot-and-mouth disease virus 3C protease. New insights into catalytic mechanism and cleavage specificity. J. Biol. Chem. 280:11520–11527 [DOI] [PubMed] [Google Scholar]
- 8. Chang KO, Sosnovtsev SV, Belliot G, King AD, Green KY. 2006. Stable expression of a Norwalk virus RNA replicon in a human hepatoma cell line. Virology 353:463–473 [DOI] [PubMed] [Google Scholar]
- 9. Chang KO, Takahashi D, Prakash O, Kim Y. 2012. Characterization of proteases from norovirus genogroup I and II with the fluorescence resonance energy transfer assay. Virology 423:125–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Collaborative Computational Project Number 4 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50:760–763 [DOI] [PubMed] [Google Scholar]
- 11. De Clercq E. 2010. Antiviral therapy: quo vadis? Future Med. Chem. 2:1049–1053 [DOI] [PubMed] [Google Scholar]
- 12. De Palma AM, et al. 2008. Potential use of antiviral agents in polio eradication. Emerg. Infect. Dis. 14:545–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dragovich PS, et al. 1999. Structure-based design, synthesis, and biological evaluation of irreversible human rhinovirus 3C protease inhibitors. 3. Structure-activity studies of ketomethylene-containing peptidomimetics. J. Med. Chem. 42:1203–1212 [DOI] [PubMed] [Google Scholar]
- 14. Dragovich PS, et al. 1999. Structure-based design, synthesis, and biological evaluation of irreversible human rhinovirus 3C protease inhibitors. 4. Incorporation of P1 lactam moieties as l-glutamine replacements. J. Med. Chem. 42:1213–1224 [DOI] [PubMed] [Google Scholar]
- 15. Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66:486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fauquet CM, et al. 2005. Virus taxonomy: classification and nomenclature of viruses. Eighth report of the International Committee on Taxonomy of Viruses Academic Press, San Diego, CA [Google Scholar]
- 17. Grubman MJ, Baxt B. 2004. Foot-and-mouth disease. Clin. Microbiol. Rev. 17:465–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hoffmann HH, Kunz A, Simon VA, Palese P, Shaw ML. 2011. Broad-spectrum antiviral that interferes with de novo pyrimidine biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 108:5777–5782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hussey RJ, et al. 2011. A structural study of norovirus 3C protease specificity: binding of a designed active site-directed peptide inhibitor. Biochemistry 50:240–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Iizuka K, et al. 1988. New human renin inhibitors containing an unnatural amino acid, norstatine. J. Med. Chem. 31:701–704 [DOI] [PubMed] [Google Scholar]
- 21. Jacobsen KH, Wiersma ST. 2010. Hepatitis A virus seroprevalence by age and world region, 1990 and 2005. Vaccine 28:6653–6657 [DOI] [PubMed] [Google Scholar]
- 22. Jaulent AM, et al. 2007. A continuous assay for foot-and-mouth disease virus 3C protease activity. Anal. Biochem. 368:130–137 [DOI] [PubMed] [Google Scholar]
- 23. Krissinel E, Henrick K. 2004. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60:2256–2268 [DOI] [PubMed] [Google Scholar]
- 24. Kuo CJ, Chi YH, Hsu JT, Liang PH. 2004. Characterization of SARS main protease and inhibitor assay using a fluorogenic substrate. Biochem. Biophys. Res. Commun. 318:862–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kuo CJ, et al. 2009. Individual and common inhibitors of coronavirus and picornavirus main proteases. FEBS Lett. 583:549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee CC, et al. 2009. Structural basis of inhibition specificities of 3C and 3C-like proteases by zinc-coordinating and peptidomimetic compounds. J. Biol. Chem. 284:7646–7655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liapakis A, Jacobson I. 2011. Telaprevir user's guide. Clin. Liver Dis. 15:555–571 [DOI] [PubMed] [Google Scholar]
- 28. Mandadapu SR, et al. Potent inhibition of Norwalk virus 3C protease by peptidyl α-ketoamides and α-ketoheterocycles. Bioorg. Med. Chem. 22:4820–4826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Matthews DA, et al. 1994. Structure of human rhinovirus 3C protease reveals a trypsin-like polypeptide fold, RNA-binding site, and means for cleaving precursor polyprotein. Cell 77:761–771 [DOI] [PubMed] [Google Scholar]
- 30. Nakamura K, et al. 2005. A norovirus protease structure provides insights into active and substrate binding site integrity. J. Virol. 79:13685–13693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nicklin MJ, Harris KS, Pallai PV, Wimmer E. 1988. Poliovirus proteinase 3C: large-scale expression, purification, and specific cleavage activity on natural and synthetic substrates in vitro. J. Virol. 62:4586–4593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Patel DV, et al. 1995. α-Hydroxy phosphinyl-based inhibitors of human renin. J. Med. Chem. 38:4557–4569 [DOI] [PubMed] [Google Scholar]
- 33. Patick AK, et al. 1999. In vitro antiviral activity of AG7088, a potent inhibitor of human rhinovirus 3C protease. Antimicrob. Agents Chemother. 43:2444–2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Perlman S, Netland J. 2009. Coronaviruses post-SARS: update on replication and pathogenesis. Nat. Rev. Microbiol. 7:439–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Reed LJ, Muench H. 1938. A simple method of estimating fifty percent endpoints. Am. J. Hyg. 27:493–497 [Google Scholar]
- 36. Schechter I, Berger A. 1967. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 27:157–162 [DOI] [PubMed] [Google Scholar]
- 37. Schultheiss T, Sommergruber W, Kusov Y, Gauss-Muller V. 1995. Cleavage specificity of purified recombinant hepatitis A virus 3C proteinase on natural substrates. J. Virol. 69:1727–1733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Soares MM, King SW, Thorpe PE. 2008. Targeting inside-out phosphatidylserine as a therapeutic strategy for viral diseases. Nat. Med. 14:1357–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Takahashi D, Kim Y, Chang KO, Anbanandam A, Prakash O. 2012. Backbone and side-chain (1)H, (15)N, and (13)C resonance assignments of Norwalk virus protease. Biomol. NMR Assign. 85:12570–12577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tiew KC, et al. 2011. Design, synthesis, and evaluation of inhibitors of Norwalk virus 3C protease. Bioorg. Med. Chem. Lett. 21:5315–5319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tong L. 2002. Viral proteases. Chem. Rev. 102:4609–4626 [DOI] [PubMed] [Google Scholar]
- 42. Vagin A, Teplyakov A. 2010. Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66:22–25 [DOI] [PubMed] [Google Scholar]
- 43. Wolf MC, et al. 2010. A broad-spectrum antiviral targeting entry of enveloped viruses. Proc. Natl. Acad. Sci. U. S. A. 107:3157–3162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yang H, et al. 2005. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 3:e324 doi:10.1371/journal.pbio.0030324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yin J, et al. 2006. An episulfide cation (thiiranium ring) trapped in the active site of HAV 3C proteinase inactivated by peptide-based ketone inhibitors. J. Mol. Biol. 361:673–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zeitler CE, Estes MK, Venkataram Prasad BV. 2006. X-ray crystallographic structure of the Norwalk virus protease at 1.5-A resolution. J. Virol. 80:5050–5058 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.