

Abstract
A major goal in rabies virus (RV) research is to develop a single-dose postexposure prophylaxis (PEP) that would simplify vaccination protocols, reduce costs associated with rabies prevention in humans, and save lives. Live replication-deficient RV-based vaccines are emerging as promising single-dose vaccines to replace currently licensed inactivated RV-based vaccines. Nonetheless, little is known about how effective B cells develop in response to live RV-based vaccination. Understanding this fundamental property of rabies immunology may help in developing a single-dose RV vaccine. Typically, vaccines induce B cells secreting high-affinity, class-switched antibodies during germinal center (GC) reactions; however, there is a lag time between vaccination and the generation of GC B cells. In this report, we show that RV-specific antibodies are detected in mice immunized with live but not inactivated RV-based vaccines before B cells displaying a GC B cell phenotype (B220+GL7hiCD95hi) are formed, indicating a potential role for T cell-independent and early extrafollicular T cell-dependent antibody responses in the protection against RV infection. Using two mouse models of CD4+ T cell deficiency, we show that B cells secreting virus-neutralizing antibodies (VNAs) are induced via T cell-independent mechanisms within 4 days postimmunization with a replication-deficient RV-based vaccine. Importantly, mice that are completely devoid of T cells (B6.129P2-Tcrβtm1Mom Tcrδtm1Mom/J) show protection against pathogenic challenge shortly after immunization with a live replication-deficient RV-based vaccine. We show that vaccines that can exploit early pathways of B cell activation and development may hold the key for the development of a single-dose RV vaccine wherein the rapid induction of VNA is critical.
INTRODUCTION
The prototypical vaccine used in postexposure settings was developed over a century ago for use following exposure to a potentially rabid animal (9, 14). Postexposure vaccination remains the worldwide standard for the prevention of rabies infections of humans. Despite the long history of rabies vaccine use as a postexposure treatment and the facts that over two-thirds of the world population live in regions where rabies is endemic and that over 55,000 people die every year due to RV infections, little information is available regarding the development of effective B cell responses early postvaccination, which may influence the outcome of postexposure vaccine efficacy. Current vaccines for human use are based on inactivated RV strains. Therefore, vaccine-induced protection against RV has long been described as being dependent solely on CD4+ T cell help for the induction of protective antibody responses (9). These observations are supported by experiments carried out in RV-vaccinated nude mice that did not develop anti-RV antibody responses and were not protected against pathogenic challenge in preexposure settings (29). Similarly, Mifune et al. (16) showed that vaccinated athymic nude mice did not produce anti-RV antibodies in postexposure experiments. Furthermore, neutralizing antibodies directed against RV antigens were shown to be produced only with the assistance of T-helper cells (1). Finally, T cell depletion studies further showed the importance of CD4+ T cells in generating neutralizing, protective antibodies in mice after RV infection (21). Together, these experiments showed that current inactivated RV-based vaccines rely on T cell help for the elicitation of effective antibody responses associated with protection against pathogenic challenge.
Vaccine-induced immunity is a complex process involving innate and adaptive immune responses. During the development of typical vaccine-induced immunity, antigen-primed T cells migrate to the T and B cell borders of secondary lymphoid organs, where they interact with their cognate antigen-specific B cells. After activation, B cells differentiate into early, short-lived extrafollicular plasma cells, germinal center (GC) B cells, or early unswitched memory B cells that can recirculate (7). Within GCs, B cells differentiate into memory B cells or long-lived plasma cells that secrete high-affinity, postswitched antibodies. In most cases, fully formed GC-derived memory B cells and plasma cells can take from days to weeks to develop. From a traditional vaccination standpoint, this lag time between vaccination and GC B cell development is required and usually acceptable, since GC B cells are critical for long-term B cell responses to protect against future exposure to the pathogen. However, vaccines are also used in postexposure settings after exposure to pathogens, such as for RV. Since the lag time between vaccination and the generation of effective GC B cell responses might be too long, administration of postexposure vaccines for the prevention of rabies infection may not be optimal. Understanding how B cells develop in response to experimental rabies vaccines may help to develop vaccines that reduce the time from vaccination to protective immunity, thereby increasing the utility of RV vaccines.
Previously, we described the safety and immunogenicity in mice and nonhuman primates of a replication-deficient RV-based vaccine vector that lacked the matrix (M) gene (rRV-ΔM) (2). In mice, VNA titers were observed as early as 5 days postimmunization with 105 focus forming units (FFU) of rRV-ΔM/mouse, which was the earliest time point tested. Furthermore, only 103 FFU/mouse was needed to induce VNAs capable of protecting 100% of mice against pathogenic challenge in preexposure settings. In nonhuman primates, rRV-ΔM induced more rapid and more potent type 1 T helper antibody responses than those induced in animals that received a commercially available human diploid cell vaccine (HDCV). Together, a kinetics analysis in two relevant animal models indicates that rRV-ΔM induces very rapid and potent anti-RV antibody responses, which makes rRV-ΔM a useful tool to characterize pathways in B cell development responsible for effective anti-RV antibody responses. In the study described here, we characterized B cell responses induced by live RV-based vaccines and demonstrated that VNAs produced by T cell-independent and -dependent mechanisms were associated with the control of rabies infections while GC B cells were still developing within the lymph nodes of immunized hosts. The speed at which B cells developed and secreted RV-specific VNAs and the efficacy of these B cell populations in protection against an RV challenge indicated that early events in B cell development can be exploited to increase the efficacy of vaccines used in postexposure settings such as rabies or other infectious diseases that require a rapid antibody response for protection.
MATERIALS AND METHODS
Immunization and early pathogenic challenge in wild-type mice.
Groups of 5 female C57BL/6 mice aged 6 to 8 weeks were immunized intramuscularly (i.m.) with a single dose of 106 FFU/mouse of rRV-ΔM, rRV, or rRV-UV (rRV-ΔM and rRV were constructed as described elsewhere and were previously named SPBN-ΔM and SPBN, respectively [2, 15, 24]). All vectors were derived from a molecular clone of the SAD-B19 vaccine strain of RV (2, 15, 24). Inactivation of rRV by UV light was completed as described previously, and inactivation was verified by inoculating BSR cells with an aliquot of UV-treated virus followed by analysis by immunostaining for the detection of RV nucleoprotein 48 h postinoculation (3). A separate group of 5 mice were immunized with phosphate-buffered saline (PBS) alone and used as controls. Immunized mice were challenged i.m. with 105 FFU/mouse of pathogenic challenge virus strain N2c (CVS-N2c), which is a mouse-adapted subclone of CVS-24 RV (2, 3, 19) at 1, 2, 3, 4, 5, or 6 days postimmunization. Mice were observed for 3 weeks for clinical signs of rabies and euthanized at the onset of neurological symptoms. Two independent experiments were completed (n = 10/group).
B cell development in immunized mice.
Groups of 5 female C57BL/6 mice aged 6 to 8 weeks were immunized i.m. with a single dose of 106 FFU/mouse of rRV-ΔM, rRV, rRV-UV, or PBS. On days 5, 7, 10, 14, and 21 postimmunization, draining lymph nodes and spleens were removed, single cell suspensions prepared, and erythrocytes present in splenocyte cultures were lysed with ACK lysing buffer. A total of 106 cells were washed in fluorescence-activated cell sorter (FACS) buffer (PBS supplemented with 2% fetal bovine serum) and then incubated with rat anti-mouse CD16/32 (1 μg/106 cells; Fc Block; Pharmingen) for 1 h on ice. Cells were washed twice in FACS buffer and incubated with 0.2 μg/106 cells of the following antibodies for 30 min at room temperature (RT) in the dark (all antibodies were purchased from BD Biosciences): APC-Cy7-(or PerCP)-B220/CD45R (RA3-6B2), FITC-GL-7 (GL7), and PE-Cy7-FAS/CD95 (Jo2). Cells were washed twice in FACS buffer, resuspended in 200 μl of 2% paraformaldehyde in PBS, and analyzed by FACScan (BD LSRII). FlowJo software was used to analyze the data. Three independent experiments were completed (n ≥ 10/group).
Evaluation of early antibody responses by ELISA and RFFIT.
Groups of 5 female C57BL/6 mice aged 6 to 8 weeks were immunized i.m. with a single dose of 106 FFU/mouse of rRV-ΔM, rRV, rRV-UV, or PBS. On days 3, 4, and 5 postimmunization, blood was collected and sera were isolated for analysis. RV glycoprotein (G)-specific IgG, IgG1, IgG2c, IgG3, and IgM antibodies were determined by enzyme-linked immunosorbent assay (ELISA) and reported at 1:50 dilution as described previously (2, 3), and virus-neutralizing antibody titers were determined using the rapid fluorescent focus inhibition test (RFFIT) as described previously (2, 3) (n ≥ 5).
Antibody responses in mice depleted of CD4+ T cells.
The B cell hybridoma expressing rat IgG2b specific for mouse CD4 (TIB-207; GK1.5; ATCC) was grown in Iscove's modified Dulbecco's medium (ATCC) containing 5% Absolute Low IgG fetal bovine serum (FBS) (Gemini Bio-Products). Supernatants were collected and antibodies precipitated with 50% (final) ammonium sulfate and then purified using a Melon Gel IgG purification kit as described by the manufacturer (Thermo Scientific, Waltham, MA). C57BL/6 mice were immunized on day 0 with rRV-ΔM. One group of mice were depleted of CD4+ T cells by injection of purified GK1.5 antibodies intraperitoneally (i.p.) on day −2 (200 μg), −1 (200 μg), 0 (200 μg), +2 (100 μg), +4 (100 μg), and + 6 (200 μg). On days 3, 5, and 7 postimmunization, blood and spleens were collected and sera analyzed by ELISA for IgG, IgG2c, IgG3, and IgM antibody titers and RFFIT to establish VNA titers as described above. Single cell suspensions of splenocytes were tested for CD4+ T cell depletion by flow cytometry and determined to be 95% to 99% depleted for the duration of the study timeline.
Immunogenicity and protection in TCRβδ−/− mice.
Groups of 5 female 6- to 8-week-old C57BL/6 or TCRβδ−/− mice were immunized i.m. with 106 FFU/mouse with rRV-ΔM. Groups of 3 female C57BL/6 or TCRβδ−/− mice immunized with PBS served as controls. Blood was collected on days 3, 5, 7, 10, 14, and 21, and serum was analyzed by RFFIT for VNA titers as described above. In two separate, independent experiments, female 6- to 8-week-old C57BL/6 (total of n = 15) or TCRβδ−/− (total of n = 15) mice were immunized with rRV-ΔΜ and then challenged 5 days later near the peak of VNA titers (detected above) with 105 FFU/mouse CVS-N2c. As controls, female 6- to 8-week-old C57BL/6 (total of n = 10) or TCRβδ−/− (total of n = 5) mice were mock immunized with PBS. Mice were observed for 4 weeks for clinical signs of rabies and euthanized at the onset of neurological symptoms.
Statistical analysis.
Kaplan-Meier survival curves were analyzed by the log rank test; P values of <0.001, 0.001 to 0.01, and 0.01 to 0.05 indicate levels of significance between two immunization groups (2, 3). To compare two groups of data for antibody responses, we used an unpaired, two-tailed t test, with P values of <0.05 indicating significance between two data points (2, 3, 5).
RESULTS
Live RV-based vaccines protect mice against RV challenge soon after immunization.
While it is known that antibodies directed against the RV glycoprotein (G) are critical for protection against RV infections and the WHO has defined VNA titers of ≥0.5 international units (IU)/ml to be indicative of a satisfactory immunization in humans and pets (14, 18, 22, 28, 30), little information exists regarding B cell responses early after immunization in the context of protection against RV. Therefore, we utilized a rabies infection mouse model to investigate the role of these early vaccine-induced antibody responses in protection against rabies. Challenge virus strain N2c is a highly neurotropic mouse-adapted RV strain that typically kills naïve mice within 8 days postinfection. The speed with which CVS-N2c kills mice makes it difficult to protect mice soon after vaccination, since protective antibody responses are slow to develop following immunization with conventional inactivated vaccines or with most experimental RV vaccines. However, the speed at which mice succumb to infection makes CVS-N2c an excellent challenge virus strain to evaluate the effects of vaccine-induced early antibodies on protection. To examine the role for antibodies generated early postvaccination, groups of mice were immunized with either rRV-ΔM, a replication-competent RV-based vaccine (rRV), a UV-inactivated RV-based vaccine to represent the rabies virus vaccine currently used in humans (rRV-UV), or PBS and then challenged 1, 2, 3, 4, 5, or 6 days postimmunization. As expected, most mice mock immunized with PBS died within 8 days postchallenge regardless of postimmunization challenge time (Fig. 1). Similarly, no significant protection was observed in rRV-UV-immunized mice compared to mice immunized with PBS alone at any infection time point. However, protection was observed early in mice immunized with rRV or rRV-ΔM. Mice immunized with rRV were significantly protected (90% protection) against challenge virus compared to mice immunized with rRV-UV or PBS alone 4 days postimmunization. Mice immunized with rRV-ΔM were significantly protected when immunized 1 day prior to challenge compared to mice immunized with PBS (P = 0.0161), rRV-UV (P = 0.0237), or rRV (P = 0.0269). Survival of rRV-ΔM-immunized mice challenged with CVS-N2c was statistically significant at all infection time points postimmunization tested compared to mice immunized with PBS or rRV-UV. To rule out the potential for inflammatory responses at the local site of inoculation for the clearance of CVS-N2c in rRV-ΔM-immunized mice, we immunized mice in the right hind leg and then challenged them in the left hind leg. Similar to the results presented in Fig. 1 (Day 1), 80% of mice survived challenge, indicating that vaccination with rRV-ΔM did not induce a local inflammatory response that was responsible for the protection observed when mice were immunized 1 day prior to challenge (data not shown). Together, rRV-ΔM induces an immune response that rapidly provides protection against pathogenic challenge. Furthermore, the rate at which protective immunity was elicited following rRV-ΔM vaccination makes this an excellent model for defining the nature of protective B cell responses associated with beneficial outcomes early following an RV infection.
Fig 1.

Protection against pathogenic RV challenge soon after immunization with RV-based vaccines. On day 0, mice were immunized with 106 FFU/mouse with rRV-ΔM, rRV, rRV-UV, or PBS and then challenged with 105 FFU/mouse challenge virus strain N2c (CVS-N2c) on day 1, 2, 3, 4, 5, or 6 postimmunization. Data shown are a combination of 2 independent experiments consisting of 5 mice per experiment (n = 10 per group). Kaplan-Meier survival curves were analyzed by the log rank test (*, P < 0.05). The data indicate that live RV-based vaccines elicited protective immune responses within days of immunization, while inactivated RV-based vaccines, which are the vaccines currently used to prevent rabies in humans, do not.
Kinetics analysis of the induction of GC B cells by live rRV and rRV-ΔM vaccines.
B cell responses are critical for protection against rabies (14, 22, 28, 30); however, the development of RV-specific vaccine-induced B cell responses remains largely unknown. The speed with which rRV-ΔM induced protective immunity early postchallenge (Fig. 1) is not consistent with B cell development kinetics within GCs. Indeed, despite an increase in total B220+ B cells in mice 5 days postimmunization with rRV-ΔM or rRV (data not shown), GC B cells, defined phenotypically as B220+GL7hiCD95hi (11, 12), were not detected until 7 days postimmunization with either vaccine (Fig. 2). An increase in GC B cells was also detected in mice immunized with rRV-ΔM or rRV 10, 14, and 21 days postimmunization compared to rRV-UV- or PBS-immunized mice. Throughout the time points tested, low levels of GC B cells were detected in the lymph nodes of mice immunized with rRV-UV; however, these levels were not significantly different from those of GC B cells observed in PBS-immunized mice. Consistent with the B220+ cell population, only transient and sporadic increases in GC B cells were detected in spleens from immunized mice, supporting the notion that B cell activation and development occur largely in the lymph nodes and not the spleens of RV-based vaccine-immunized mice. In addition, protection elicited soon after immunization with rRV-ΔM did not appear to be dependent on GC B cell formation since CVS-N2c-infected mice survived following challenge 1 day postimmunization, even though GC B cells did not develop until after 5 days postimmunization. This indicated that B cell subsets that are present before B cell development is complete in the GC play a role in conferring protection following rRV-ΔM vaccination. Together, these data indicated that B cell activation occurs in the draining lymph nodes and not the spleen following immunization with the respective live RV-based vaccines tested. Furthermore, the early expansion of B cells observed postimmunization with rRV-ΔM or rRV (but not in mice immunized with rRV-UV) might help explain the efficacy of the rRV-ΔM and rRV formulations in eliciting protective immunity against CVS-N2c challenge early postimmunization.
Fig 2.

Kinetics of GC B cell development in the lymph nodes and spleens of mice immunized with RV-based vaccines. Groups of 5 female C57BL/6 mice aged 6 to 8 weeks were immunized intramuscularly with a single dose of 106 FFU/mouse of rRV-ΔM, rRV, rRV-UV, or PBS. On the indicated days postimmunization, draining (inguinal) lymph nodes and spleens were removed, single cell suspensions prepared, and cells immunostained to determine the number of GC B cells. (A) Representative gating strategy of live cells gated for total B cells (B220+) from mice immunized with rRV (top) or PBS (bottom). (B) Representative gating strategy of total B cells (B220+) gated for GL7 and CD95 from mice immunized with rRV (top) or PBS (bottom). (C) Numbers of GC B cells in mice immunized with different RV-based vaccines per 100,000 total live cells in the lymph node. Significant numbers of GC B cells were not detected until 7 days postimmunization with rRV or rRV-ΔM vaccines. Although an increase in the number of GC B cells was detected in mice immunized with rRV-UV by day 7 postimmunization, these numbers were not significantly different from counts observed in mice immunized with PBS alone. (D) Number of GC B cells in mice immunized with the different RV-based vaccines per 100,000 live cells in the spleen. Consistent with the lack of B cell expansion in the spleens of immunized mice (not shown), the number of GC B cells forming in the spleen was minimal, supporting the notion that B cell activation occurs in the lymph node and not the spleen of mice immunized with RV-based vaccines. n ≥ 10/group; to compare two groups of data, we used an unpaired, two-tailed t test; *, P < 0.05.
Presence of broad-based antibody isotypes and IgG subclasses early after immunization with rRV-ΔM and rRV.
IgM-to-IgG class switching occurs in the perifollicular regions during the early stages of B cell activation and development (7), suggesting that rRV-ΔM-induced isotype switching most likely occurs before GC formation. To confirm this and to define the antibody isotype and IgG subclass profiles associated with the early postimmunization immune response, a kinetic analysis examining total IgG and IgM, in addition to the IgG subclasses (IgG1, IgG2c, and IgG3), was conducted using mice immunized with the different RV-based vaccines. As shown in Fig. 3A, significant levels of RV G-specific IgG were detected in rRV-ΔM- and rRV-immunized mice as early as 4 days postimmunization. Of note, these antibodies were detected prior to the detection of B cells displaying a GC phenotype (Fig. 2). Antibodies directed against RV G were not detected in rRV-UV-immunized mice within the first 5 days, consistent with the lack of protection observed when challenge occurred early postvaccination (Fig. 1). IgG3 (Fig. 3B), which is an antibody produced in a T cell-independent manner (6, 17, 26), and IgG2c (Fig. 3C), which can be produced in either a T cell-dependent or -independent manner, were detected in significant quantities as early as 4 days postimmunization with rRV-ΔM or rRV compared to levels observed in mice immunized with rRV-UV or PBS. Low levels of IgG2c antibodies were detected in mice 5 days following rRV-UV immunization; however, these levels were not significantly elevated above background levels observed in PBS-immunized mice. Low, but significant, levels of anti-RV G IgG1 antibodies were detected in mice immunized with the live RV-based (but not inactivated RV-based) vaccines within the first 5 days postimmunization (Fig. 3D). Compared to PBS-immunized mice, all RV-based-vaccine-immunized mice developed an IgM response as early as 3 days postimmunization (Fig. 3E).
Fig 3.

Early antibody titers induced by RV-based vaccines. Groups of 5 female C57BL/6 mice aged 6 to 8 weeks were immunized intramuscularly with a single dose of 106 FFU/mouse of rRV-ΔM, rRV, rRV-UV, or PBS. On the indicated times postimmunization, RV glycoprotein (G)-specific IgG (A), IgG3 (B), IgG2c (C), IgG1 (D), or IgM (E) antibodies were determined by ELISA. Natural (IgM) and class-switched antibodies were detected as early as 3 to 4 days postimmunization with live RV-based vaccines; however, only IgM was detected in significant amounts in mice immunized with rRV-UV compared to mice immunized with PBS alone at these early time points. n = 5/group; to compare two groups of data, we used an unpaired, two-tailed t test; *, P < 0.05. OD490, optical density at 490 nm.
The antibodies detected by ELISA showed neutralizing activity as early as 3 days postimmunization, and this activity increased significantly by day 5 postimmunization (Fig. 4). Of note, VNA titers detected 3 days postimmunization with rRV-ΔM or rRV were approximately 2-fold greater than the level indicative of a satisfactory immunization (i.e., 0.5 IU/ml) (14, 22, 28, 30), and by day 5 postimmunization, we observed 22- and 12-fold-higher VNA titers in mice immunized with rRV-ΔM and rRV, respectively, compared to the 0.5-IU/ml minimum titer. Only low VNA titers were detected in mice 5 days postimmunization with rRV-UV, and VNAs were not observed in PBS-immunized mice. The data collected do not allow us to discern the specific antibody subclass(es) or isotype(s) responsible for the neutralization measured. Nonetheless, these data indicated that antibody responses indicative of a satisfactory immunization could be induced in rRV-immunized or rRV-ΔM-immunized mice before GC B cells are formed, suggesting a role for protective, anti-RV T cell-independent or T cell-dependent extrafollicular antibody responses early postimmunization.
Fig 4.
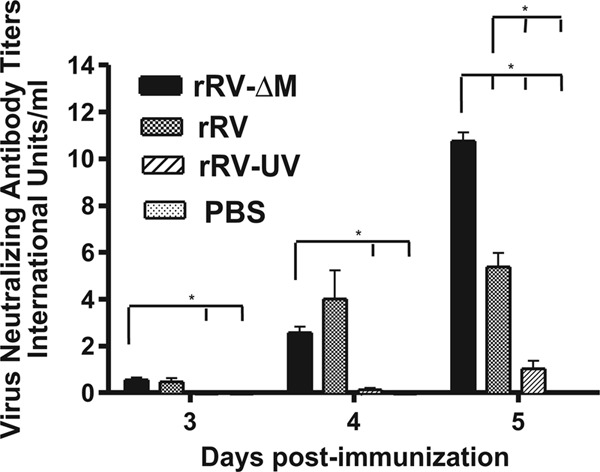
Early VNA responses in mice immunized with RV-based vaccines. The experiment illustrated in Fig. 3 was repeated, except VNA titers were determined by the rapid fluorescent focus inhibition test (RFFIT) from individual immunized mice. Neutralization titers, defined as the inverse of the highest serum dilution that neutralizes 50% of the challenge virus (challenge virus strain 11), were normalized to international units/ml (IU/ml) using the WHO anti-RV antibody reference standard. Virus-neutralizing antibodies were detected in mice immunized with live RV-based vaccines as early as 3 days postimmunization.
VNAs are detected in 2 different mouse models of CD4+ T cell deficiency.
Depletion of CD4+ T cells (Fig. 5A) resulted in a dramatic decrease in antibodies produced in immunized mice (Fig. 5B). However, significant levels of RV-specific total IgG (Fig. 5B) and IgM (Fig. 5C) antibodies directed against RV G could still be detected in CD4+ T cell-depleted mice compared to control mice. Of the IgG subclasses identified, IgG3 (Fig. 5D) and IgG2c (Fig. 5E) were detected in the absence of CD4+ T cells. Importantly, neutralizing antibodies were detected at titers above 0.5 IU/ml by day 5 postimmunization (Fig. 5F), suggesting the WHO minimum criterion for a satisfactory immunization was met in mice that lack CD4+ T cells. The possibility existed, however, that residual CD4+ T cells provided help to B cells for the generation of VNAs in the CD4+ T cell-depleted mice. Furthermore, while it has been reported that specific doses of antibodies deplete CD4+ T cells in the blood and secondary lymphoid organs (25), we tested for depletion in the spleen only and not in the lymph node. The possibility exists that residual CD4+ T cells remained in the draining lymph node, where we propose that anti-RV immunity develops. In addition, T-helper functions can be provided by CD4−CD8−TCRαβ+ T cells in the absence of CD4+ T cells. To confirm that antibodies induced early following rRV-ΔM immunization were generated in a CD4+ T cell-independent manner, we repeated the experiment using genetically modified mice lacking αβ and γδ T cells (Tcrβtm1MomTcrδtm1Mom/J, referred to here as TCRβδ−/−). Importantly, TCRβδ−/− mice lack all T cells, including CD8+ T cells, leaving B cells as the major adaptive immune cell population. As shown in Fig. 6A, VNA titers were detected in TCRβδ−/− mice as early as 3 days postimmunization with rRV-ΔM at levels above the WHO standard indicative of a satisfactory immunization. VNA titers increased through day 7, reaching titers over 4 times the base limit associated with a satisfactory immunization. VNA titers then decreased by day 10, consistent with short-lived B cells stimulated to produce antibodies in a T cell-independent manner (4, 20, 27). Upon reexposure to antigen in the form of a boost inoculation with rRV-ΔM, a recall response was not observed in TCRβδ−/− mice primed with rRV-ΔM (data not shown). These experiments confirmed that T cell-independent responses were short-lived and did not induce memory cells. Similar protection was observed in wild-type and TCRβδ−/− mice immunized with rRV-ΔM when challenged 5 days later with CVS-N2c (P = 0.079) (Fig. 6B). The protection observed in TCRβδ−/− mice was consistent with the levels of VNA detected in both models of CD4+ T cell deficiency.
Fig 5.

Antibody responses in rRV-ΔM-immunized mice depleted of CD4+ T cells. C57BL/6 mice were depleted of CD4+ T cells using anti-CD4 antibody (GK1.5) as described in Materials and Methods and then immunized intramuscularly with 106 FFU/mouse rRV-ΔM. Sera from immunized mice were collected on the indicated days. Spleens from CD4+ T cell-depleted and wild-type mice were collected throughout the sample period and tested for CD4+ T cell depletion. (A) Depletion of CD4+ T cell-depleted mice (right panel) at all time points, including day 7 postimmunization, was determined to be 95% to 99% compared to nondepleted mice (left panel) as shown by representative analysis of CD4 T cells. (B through E) On the indicated times postimmunization, RV glycoprotein (G)-specific IgG (B), IgM (C), IgG3 (D), and IgG2c (E) antibodies were determined by ELISA. OD490, optical density at 490 nm. (F) Virus-neutralizing antibodies were also determined as described in the legend to Fig. 4. While wild-type mice had higher VNA titers than CD4+ T cell-depleted mice, the latter showed antibody titers indicative of a satisfactory immunization (i.e., >0.5 IU/ml) as early as 4 days postimmunization in the absence of CD4+ T cell help. IgG (of all subclasses tested) increased over time in CD4+ T cell-depleted mice, and IgM antibodies were also detected. n = 5/group of CD4+ T cell-depleted mice; n = 4/group of wild-type mice; to compare two groups of data, we used an unpaired, two-tailed t test; *, P < 0.05.
Fig 6.
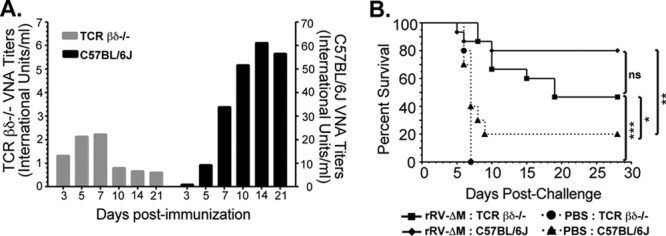
Anti-RV antibody responses and protection against pathogenic challenge in TCRβδ−/− mice. (A) Groups of 5 female 6- to 8-week-old C57BL/6 or TCRβδ−/− mice were immunized intramuscularly with 106 FFU/mouse with rRV-ΔM. Groups of 3 female C57BL/6 or TCRβδ−/− mice immunized with PBS served as controls. Blood was collected on the indicated days and sera analyzed by RFFIT for VNA titers as described for Fig. 4. (B) In two separate, independent experiments, female 6- to 8-week-old C57BL/6 (total of n = 15) or TCRβδ−/− (total of n = 15) mice were immunized with rRV-ΔΜ. As controls, female 6- to 8-week-old C57BL/6 (total of n = 10) or TCRβδ−/− (total of n = 5) mice were mock immunized with PBS. Mice were then challenged with 105 FFU/mouse CVS-N2c 5 days later near the peak of VNA titers (detected in TCRβδ−/− mice as described for panel A). Mice were observed for 4 weeks for clinical signs of rabies and euthanized at the onset of neurological symptoms. ***, P < 0.001; **, P = 0.001 to 0.01; *, P = 0.01 to 0.05.
Together, protection and/or immunogenicity data generated using two different CD4+ T cell-deficient mouse models confirmed that immunization with rRV-ΔM elicits the production of T cell-independent VNAs with the potential for influencing disease outcome following challenge early postvaccination, a property that is critical to the success of postexposure prophylaxis vaccines. Of note, a comparison between VNAs induced in wild-type mice and those induced in TCRβδ−/− mice (Fig. 6A) indicates that wild-type mice induce potent VNA titers in the presence of T cell help before GC B cells are formed. It appears that, in addition to T cell-independent antibody responses, T cell-dependent extrafollicular antibody responses are also induced by live rabies vaccination and may have contributed to the protection observed in the experiment illustrated in Fig. 1.
DISCUSSION
Previously, we described the safety and immunogenicity of rRV-ΔM in mice and nonhuman primates and showed that this replication-deficient RV-based vaccine vector induces rapid and potent antibody responses in two models of rabies immunogenicity. The purpose of this study was to characterize the early B cell responses detected in our previous studies. Our experiments showed that immunization with either rRV or rRV-ΔM (but not rRV-UV) elicited protection in mice challenged with a virulent RV strain within days of immunization. Consistent with the early protection observed following vaccination with either rRV or rRV-ΔM, the total B cell population was expanded in the lymph nodes 5 days postimmunization. This expansion in the total B cell population was not observed in mice immunized with rRV-UV until 14 days postimmunization (data not shown). Class-switched and virus-neutralizing antibodies were detected at levels indicative of a satisfactory immunization (i.e., >0.5 IU/ml) as early as 3 days postimmunization with rRV or rRV-ΔM. Importantly, the early B cell expansion and the detection of VNAs by day 3 postimmunization with rRV-ΔM or rRV did not coincide with an increase in B cells displaying a typical GC B cell phenotype (B220+GL7hiCD95hi). Based on these findings, we tested the ability of rRV-ΔM to induce protective antibodies in 2 models of CD4+ T cell deficiency, that is, mice with antibody-mediated CD4+ T cell depletion or TCRβδ−/− mice that lack all T cells. In both models, class-switched antibodies and VNA titers were detected above 0.5 IU/ml in rRV-ΔM-immunized mice as early as 3 to 4 days postimmunization. Consistent with antibody responses produced in T cell-independent mechanisms, the early T cell-independent response peaked around 5 to 7 days postimmunization and subsequently declined. In addition, immunized TCRβδ−/− mice were rested for 4 weeks and then immunized again with rRV-ΔM. A primary response, but not an anamnestic response, was detected, indicating that the T cell-independent B cell responses did not generate memory B cells (data not shown). In addition, TCRβδ−/− mice (completely devoid of T cells) were protected at levels similar to those of wild-type mice when challenged shortly after immunization. Together, these data indicated that antibodies generated in a T cell-independent manner provided protection against RV infection and influenced the outcome of vaccination early after immunization (i.e., before GC B cells are formed). To our knowledge, this is the first report of the induction of rabies-specific VNAs in a T cell-independent manner that influenced disease progression following challenge with pathogenic RVs.
Immunization of T cell-deficient mice with rRV-ΔM resulted in the production of VNA titers consisting of either IgG or IgM. However, previous work demonstrated that IgG, but not IgM, was associated with protection against RV infections (10, 28). In these earlier studies, even though IgM production was induced in mice immunized with inactivated RV-based vaccines, passive transfer of these antibodies was not protective in mouse models of RV pathogenicity (28). It should be noted that an IgM antibody with partial protective ability in vitro was recently described in humans (13), and therefore, the role of IgM following rRV-ΔM immunization in protection against RV challenge in our mouse models cannot be discounted. Nonetheless, we believe that IgM antibodies played little if any role in the protection observed in the vaccination experiments described here. The size of pentameric IgM hinders this isotype from traversing vascular endothelial layers into interstitial tissues, and therefore, IgM resides mostly in the blood (23). The challenge virus used (CVS-N2c) is highly neurotropic, and there is no current evidence suggesting that RV disseminates via the bloodstream (8); therefore, IgM in the blood likely did not play an important role in virus clearance/protection as described previously for rabies virus protection (28). IgG antibodies, either of the IgG3 or IgG2c subclass, appear to be responsible for the VNAs observed in vivo in the 2 models of CD4+ T cell deficiency. Nonetheless, additional work will be needed to identify the antibody isotypes and subclass(es) associated with protective T cell-independent responses.
Immunization with rRV-ΔM protected mice against pathogenic RV when challenged as early as 1 day postimmunization. In contrast, rRV-immunized mice were not significantly protected unless challenged at least 4 days postimmunization. However, the kinetics of B cell activation and antibody production were similar in mice immunized with rRV-ΔM and rRV. The role for T cell-independent help in the context of immunity elicited by rRV was not tested here since the focus for the current study was on rRV-ΔM due to the levels of protection observed when challenged 1 day postimmunization. However, based on the kinetics of B cell activation and antibody production, we speculate that rRV can also induce antibodies in a CD4+ T cell-independent manner; however, it is possible that immunization with rRV was unable to elicit VNA titers capable of inactivating both a replicating viral vaccine and a challenge virus. In addition, we cannot rule out potential differences between the innate immune responses following immunization with rRV-ΔM and rRV. The possibility exists that immunization with rRV-ΔM induced local danger signals at the injection site not associated with rRV immunization. However, this is an unlikely scenario given the requirement for B cells and antibodies in the clearance of RV, and mice were equally protected when immunized and then challenged in opposite legs (data not shown). Furthermore, TCRβδ−/− mice were protected when challenged 5 days postimmunization, a time when it would be expected that initial innate responses would have subsided. Nonetheless, the roles of soluble factors, including interferon (IFN), and cells of the innate immune system in protection remain to be determined in this model.
In addition to the contribution of T cell-independent B cell responses in protection against RV described here, early extrafollicular T cell-dependent B cell responses also appeared to be important for protection early postimmunization. Significantly higher RV G-specific antibodies (Fig. 5) and VNA titers (Fig. 6) were observed in T cell-competent mice than in T cell-deficient mice as early as 3 days postimmunization with rRV-ΔM. As described for the T cell-independent responses, these T cell-dependent responses developed before the expansion of B cells displaying a GC phenotype could be detected (Fig. 2), suggesting that early extrafollicular T cell-dependent antibody responses were also induced following immunization with live RV-based vaccines. Extrafollicular T cell-dependent B cell responses play an important role in the protection against microbial infections. In the context of postexposure vaccination, the combination of T cell-independent and -dependent B cell responses may hinder the spread of pathogenic RV after exposure while plasma cells secreting high-affinity antibodies are being formed in GC reactions.
Traditional rabies vaccines are developed with the intent of inducing potent and long-lived B cell responses following their development in GCs. However, protection against infection with many pathogens requires the development of immune responses at rates not associated with conventional vaccines. To that end, vaccines that can induce rapid and potent immune responses against certain pathogens may serve to reduce virus spread while B cells are being developed in GCs to secrete high-affinity antibodies.
ACKNOWLEDGMENTS
This work was supported by NIH/NIAID grant R01AI079211 to J.P.M.
We thank Matthew Farabaugh of the Kimmel Cancer Center Flow Cytometry Facility for assistance.
Footnotes
Published ahead of print 15 August 2012
REFERENCES
- 1. Bunschoten H, et al. 1990. Rabies virus cross-reactive murine T cell clones: analysis of helper and delayed-type hypersensitivity function. Viral Immunol. 3:41–53 [DOI] [PubMed] [Google Scholar]
- 2. Cenna J, et al. 2009. Replication-deficient rabies virus-based vaccines are safe and immunogenic in mice and nonhuman primates. J. Infect. Dis. 200:1251–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cenna J, et al. 2008. Immune modulating effect by a phosphoprotein-deleted rabies virus vaccine vector expressing two copies of the rabies virus glycoproteins. Vaccine 26:6405–6414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Erickson LD, et al. 2000. B cell immunopoiesis: visualizing the impact of CD40 engagement on the course of T cell-independent immune responses in an Ig transgenic system. Eur. J. Immunol. 30:3121–3131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Faul EJ, et al. 2010. Rabies virus infection induces type I interferon production in an IPS-1 dependent manner while dendritic cell activation relies on IFNAR signaling. PLoS Pathog. 6:e1001016 doi:10.1371/journal.ppat.1001016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gavin AL, Barnes N, Dijstelbloem HM, Hogarth PM. 1998. Identification of the mouse IgG3 receptor: implications for antibody effector function at the interface between innate and adaptive immunity. J. Immunol. 160:20–23 [PubMed] [Google Scholar]
- 7. Goodnow CC, Vinuesa CG, Randall KL, Mackay F, Brink R. 2010. Control systems and decision making for antibody production. Nat. Immunol. 11:681–688 [DOI] [PubMed] [Google Scholar]
- 8. Jackson AC. 2011. Update on rabies. Res. Rep. Trop. Med. 2011(2):31–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jackson AC, Wunner W. (ed). 2007. Rabies, 2nd ed Elsevier Academic Press, Amsterdam, Netherlands [Google Scholar]
- 10. Johnson N, Cunningham AF, Fooks AR. 2010. The immune response to rabies virus infection and vaccination. Vaccine 28:3896–3901 [DOI] [PubMed] [Google Scholar]
- 11. Linterman MA, et al. 2010. IL-21 acts directly on B cells to regulate Bcl-6 expression and germinal center responses. J. Exp. Med. 207:353–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Linterman MA, et al. 2009. Follicular helper T cells are required for systemic autoimmunity. J. Exp. Med. 206:561–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Matsumoto T, et al. 2010. Isolation and characterization of novel human monoclonal antibodies possessing neutralizing ability against rabies virus. Microbiol. Immunol. 54:673–683 [DOI] [PubMed] [Google Scholar]
- 14. McGettigan JP. 2011. Experimental rabies vaccines for humans. Exp. Rev. Vaccines 9:1177–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McGettigan JP, et al. 2003. Functional human immunodeficiency virus type 1 (HIV-1) Gag-Pol or HIV-1 Gag-Pol and env expressed from a single rhabdovirus-based vaccine vector genome. J. Virol. 77:10889–10899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mifune K, Takeuchi E, Napiorkowski PA, Yamada A, Sakamoto K. 1981. Essential role of T cells in the postexposure prophylaxis of rabies in mice. Microbiol. Immunol. 25:895–904 [DOI] [PubMed] [Google Scholar]
- 17. Minami M, Usui M, Kanno T, Tamura N, Matuhasi T. 1978. Demonstration of two types of helper T cells for different IgG subclass responses to dinitrophenylated flagellin polymer. J. Immunol. 120:1195–1200 [PubMed] [Google Scholar]
- 18. Moore SM, Hanlon CA. 2010. Rabies-specific antibodies: measuring surrogates of protection against a fatal disease. PLoS Negl. Trop. Dis. 4:e595 doi:10.1371/journal.pntd.0000595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Morimoto K, et al. 1998. Rabies virus quasispecies: implications for pathogenesis. Proc. Natl. Acad. Sci. U. S. A. 95:3152–3156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. O'Connor BP, Gleeson MW, Noelle RJ, Erickson LD. 2003. The rise and fall of long-lived humoral immunity: terminal differentiation of plasma cells in health and disease. Immunol. Rev. 194:61–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Perry LL, Lodmell DL. 1991. Role of CD4+ and CD8+ T cells in murine resistance to street rabies virus. J. Virol. 65:3429–3434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Plotkin SA. 2008. Vaccines: correlates of vaccine-induced immunity. Clin. Infect. Dis. 47:401–409 [DOI] [PubMed] [Google Scholar]
- 23. Pone EJ, et al. 2010. Toll-like receptors and B-cell receptors synergize to induce immunoglobulin class-switch DNA recombination: relevance to microbial antibody responses. Crit. Rev. Immunol. 30:1–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schnell MJ, et al. 2000. Recombinant rabies virus as potential live-viral vaccines for HIV-1. Proc. Natl. Acad. Sci. U. S. A. 97:3544–3549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shellito J, et al. 1990. A new model of Pneumocystis carinii infection in mice selectively depleted of helper T lymphocytes. J. Clin. Invest. 85:1686–1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Slack J, Der-Balian GP, Nahm M, Davie JM. 1980. Subclass restriction of murine antibodies. II. The IgG plaque-forming cell response to thymus-independent type 1 and type 2 antigens in normal mice and mice expressing an X-linked immunodeficiency. J. Exp. Med. 151:853–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sze DM, Toellner KM, Garcia de Vinuesa C, Taylor DR, MacLennan IC. 2000. Intrinsic constraint on plasmablast growth and extrinsic limits of plasma cell survival. J. Exp. Med. 192:813–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Turner GS. 1978. Immunoglobulin (IgG) and (IgM) antibody responses to rabies vaccine. J. Gen. Virol. 40:595–604 [DOI] [PubMed] [Google Scholar]
- 29. Turner GS. 1976. Thymus dependence of rabies vaccine. J. Gen. Virol. 33:535–538 [DOI] [PubMed] [Google Scholar]
- 30. WHO 1997. Recommendations on rabies post-exposure treatment and the correct technique of intradermal immunization against rabies. World Health Organization, Geneva, Switzerland [Google Scholar]