

Abstract
Human cytomegalovirus (HCMV) encodes four putative G protein-coupled receptors, including pUL78, whose rodent orthologues are known to be important for replication and spread in their hosts. To investigate the mechanism by which pUL78 contributes to viral replication and pathogenesis, we generated a derivative of the TB40/E clinical isolate of HCMV that is unable to express the receptor. Consistent with previous findings using laboratory strains of the virus, the mutant replicated normally in fibroblasts. Although laboratory strains are restricted to growth in fibroblasts, clinical isolates grow in many cell types, including epithelial and endothelial cells, in which the pUL78-deficient TB40/E derivative exhibited a growth defect. Infection with the mutant virus resulted in a significant decrease in viral RNA and protein expression. Although there was no difference in binding of the virus to the cell, we detected a delay in the entry and subsequent delivery of virion DNA and protein to the nuclei of epithelial cells following infection with the UL78 mutant virus. Taken together, our results demonstrate that pUL78 supports infection at a point after binding but before entry in epithelial cells, a cell type important for in vivo viral replication and spread.
INTRODUCTION
Human cytomegalovirus (HCMV) is a betaherpesvirus that establishes a life-long latent infection in its host. Although infection is usually asymptomatic in healthy children and adults, it can cause serious disease in immunologically immature or compromised hosts (6). HCMV carries approximately 200 proteins, including 4 proteins with homology to G protein-coupled receptors (GPCRs): pUL33, pUL78, pUS27, and pUS28 (1, 8). GPCRs are a family of seven-transmembrane-domain proteins which are typically found on the cell surface and possess extracellular domains that interact with specific ligands. This interaction causes the receptor to recruit and activate a heterotrimeric G protein, and the combination of GPCR and G protein leads to the production of specific second messengers, e.g., cyclic AMP (cAMP), calcium, and phosphoinositides, that induce downstream signaling cascades (12). GPCR signaling modulates numerous cellular functions, including adhesion, migration, proliferation, differentiation, apoptosis, cell survival, chemotaxis, and cytoskeletal rearrangement.
GPCRs are defined by distinct structural and sequence motifs (22). HCMV pUL33, pUS27, and pUS28 possess these motifs. In contrast, although HCMV pUL78 satisfies the minimal requirements for inclusion in this family, it shows the lowest similarity to other known GPCRs. Thus, HCMV-encoded pUL78 and its primate and rodent CMV orthologues comprise a family of orphan GPCRs (3, 4). To date, no signaling properties have been attributed to HCMV pUL78 or its relatives encoded by nonhuman viruses.
The rodent orthologues of HCMV pUL78 have been studied in cultured cells and within their respective hosts (3, 18). Rat CMV (RCMV)-encoded R78 is needed for efficient viral growth in smooth muscle cells and fibroblasts but is dispensable for growth in rat monocytes, macrophages, and endothelial cells. Infection of animals with an R78-deficient virus results in reduced pathogenesis and increased survival compared to those of animals infected with wild-type rat CMV (3). A mutant derivative of murine CMV (MCMV) unable to express M78 exhibits a modest (2- to 3-fold) growth defect in cultured murine embryonic fibroblasts and a somewhat greater defect in murine macrophages (about 10-fold), and similar to the case with RCMV, mice infected with the M78-deficient mutant virus display increased survival compared to mice infected with the parental virus (18). Taken together, these data argue that pUL78 homologues play a significant role during infection, and their requirement for successful replication is greater in some cell types than in others.
Mutational analysis has shown that HCMV pUL78 is not required for normal viral growth in fibroblasts or in a renal artery tissue culture model (15). To investigate the possibility that the viral GPCR is needed for replication in other cell types, we generated a pUL78-deficient derivative of a clinical strain of HCMV with broad host cell tropism. We report that HCMV pUL78 is needed for efficient viral growth in both endothelial and epithelial cells. Although not required for binding, pUL78 is critical for timely entry into epithelial cells and for delivery of virion constituents to the nuclei of infected epithelial cells.
MATERIALS AND METHODS
Cells and viruses.
Primary human foreskin fibroblasts (HFFs; passages 9 to 13) were maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 0.1 mM nonessential amino acids, 10 mM HEPES, 1 mM sodium pyruvate, 2 mM l-glutamine, and 100 U/ml each of penicillin and streptomycin. Human umbilical vein endothelial cells (HUVECs; passages 3 to 5) (26) were cultured in Primaria tissue culture plates (BD Falcon) and maintained in EBM-2 medium containing 2% FBS with EGM-2 supplements (Lonza). Primary retinal pigment epithelial cells (ARPE19; passages 28 to 35) (ATCC) were maintained in 1:1 DMEM–Ham's F-12 medium containing 10% FBS, 2.5 mM l-glutamine, 0.5 mM sodium pyruvate, 15 mM HEPES, 1.2 g/liter NaHCO3, and 100 U/ml each of penicillin and streptomycin.
A clinical, endothelial cell-tropic, bacterial artificial chromosome (BAC)-derived HCMV isolate, TB40/E-BAC (clone 4), was utilized in this study (21). The construct was engineered to express mCherry to facilitate the monitoring of viral infection (17). This derivative, TB40/Ewt-mCherry, was used to generate a recombinant virus expressing a pUL78 C-terminal FLAG tag (TB40/E-mCherry-inUL78-3xF). The method to construct the epitope-tagged pUL78 protein has been described elsewhere (17). In brief, the epitope and Kan-frt cassette were amplified by PCR from pGTE-3xFLAG-Kan-frt (17) by use of the following primers, where the underlined sequences are complementary to the pGTE-3xFLAG-Kan-frt template: forward, 5′-TGCATCGACGGCGAAAACACCGTCGCGTCCGACGCGACGGTGACGGCATTATTAGATTATAAAGATGATGATGATAAA-3′; and reverse, 5′-TAACGTGATTTATCTGCCACTTTTCTCCCCGCTGCCGTACAGCGCCGCCGCGGCCGCGGGAATTCGAAGTT-3′. This product was subsequently used to generate TB40/E-mCherry-inUL78-3xF by using linear recombination to substitute the tag for the Kan-frt cassette (13, 17).
TB40/E-mCherry-inUL78-3xF was then used to generate two independent UL78 deletion mutants by galK recombineering techniques as previously described (16, 17, 29). In brief, the galK gene was amplified by PCR using the primers 5′-GTCCCCGGAGAGGGTATATTCGTTCGGCGAGAGCGGGCGGCGGTGGTGGGTCCTGTTGACAATTAATCATCGGCA-3′ (forward) and 5′-TAACGTGATTTATCTGCCACTTTTCTCCCCGCTGCCGTACAGCGCCGCCGCTCAGCACTGTCCTGCTCCTT-3′ (reverse) (the underlined sequences are complementary to the galK template). Recombination-competent Escherichia coli SW105 containing TB40/E-mCherry-inUL78-3xF was then transformed with the resulting PCR product, and clones positive for galK were selected and electroporated with an annealed complementary oligonucleotide (5′-GGGTATATTCGTTCGGCGAGAGCGGGCGGCGGTGGTGGGTGCGGCGGCGCTGTACGGCAGCGGGGAGAAAAGTGGCAGAT-3′). Mutants were then counterselected against galK. Two mutants, TB40/E-mCherry-dlUL78-1 and TB40/E-mCherry-dlUL78-2, were validated by sequencing.
Additionally, these methods and primers were used to generate similar recombinant viruses from a second clinical, BAC-derived strain, BFXwtGFP (16). As described above, BFX-GFP-inUL78-3xF was produced first and was then used to delete the entire UL78 open reading frame (ORF). We generated two independent deletion mutants, BFX-GFP-dlUL78-1 and BFX-GFP-dlUL78-2, and validated their mutations by sequence analysis.
Two independently derived deletion mutants were produced for each of the two clinical isolates to ensure that the manipulations did not introduce off-site mutations. Each mutant contains a deletion of the entire open reading frame, from the start to the stop codon. No difference in phenotype was evident for the two deletion mutants in either background, so we present data for TB40/E-mCherry-dlUL78-1 and BFX-GFP-dlUL78-2 throughout this article.
Viruses were propagated by transfecting primary fibroblasts with viral BAC DNA. The virus in medium and the cells were harvested when complete cytopathic effect was observed. Cells were disrupted by three freeze-thaw cycles, and then the virus was partially purified by centrifugation through a 20% sorbitol cushion. Virus stocks were stored in DMEM containing 5% FBS and 1.5% bovine serum albumin (BSA) at −80°C. Virus stocks were titrated on primary fibroblasts by 50% tissue culture infective dose (TCID50) assay.
Multistep growth assays were performed on fibroblasts and HUVECs by infecting cells at a multiplicity of infection of 0.01 TCID50/cell. Multistep and single-step growth properties in ARPE19 epithelial cells were analyzed by infecting cells at multiplicities of infection of 0.1 and 1.0 TCID50/cell, respectively. Cells and medium were collected at various times after infection, after which cells were disrupted by three freeze-thaw cycles and cell debris was pelleted by centrifugation. Infectious virus was titrated on primary fibroblasts by monitoring the production of IE1-positive cells at 24 h postinfection (hpi), using an IE1-specific monoclonal antibody (clone 1B12) (30) and a fluorescently labeled secondary antibody (Molecular Probes). Three random fields were quantified for positive cells, as previously described (17, 23).
Viral RNA and protein assays.
To assess viral RNA expression, ARPE19 cells were infected at a multiplicity of infection of 0.5 TCID50/cell. At various times after infection, cells were washed and harvested and RNA was extracted with TriReagent (Sigma). cDNA was generated from 1 μg of RNA by use of TaqMan reverse transcription (RT) reagents (Roche). Equal volumes of cDNA were then used for quantitative PCR (qPCR), using primers specific for UL123 (forward, 5′-GCCTTCCCTAAGACCACCAAT-3′; and reverse, 5′-ATTTTCTGGGCATAAGCCATAATC-3′), UL44 (forward, 5′-TACAACAGCGTGTCGTGCTCCG-3′; and reverse, 5′-GGCGTGAAAAACATGCGTATCAAC-3′), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (forward, 5′-ACCCACTCCTCCACCTTTGAC-3′; and reverse, 5′-CTGTTGCTGTAGCCAAATTCGT-3′). Samples were analyzed in triplicate and normalized to GAPDH levels.
To monitor the expression of viral proteins by Western blot assay, fibroblasts, ARPE19 cells, or HUVECs were infected with either wild-type or mutant virus at a multiplicity of infection of 0.5 TCID50/cell. Where indicated, cells were maintained in medium containing 100 μg/ml phosphonoacetic acid (PAA) from the end of the adsorption period until lysates were prepared. At the indicated times after infection, cells were harvested and lysed in RIPA buffer, and equal amounts of protein were analyzed by Western blotting. The primary antibodies used included anti-IE1 (clone 1B12) (30), anti-pUL99 (clone 10B4-29) (20), and anti-pp65 (clone 8A8) (2), all diluted 1:100; anti-UL44 (Virusys), diluted 1:2,500; anti-tubulin (Sigma), diluted 1:5,000; and anti-FLAG M2 (Sigma), diluted 1:7,500. A horseradish peroxidase (HRP)-conjugated goat anti-mouse secondary antibody was used in all immunoblots, diluted 1:10,000 (Jackson Laboratory).
Protein localization was analyzed by immunofluorescence assay of fibroblasts or epithelial cells grown on coverslips and infected at a multiplicity of infection of 0.5 TCID50/cell or 10 TCID50/cell, as indicated (17). At various times postinfection, cells were washed with phosphate-buffered saline (PBS), fixed with 2% paraformaldehyde, and permeabilized with 0.1% Triton X-100. Slides were blocked at 4°C in 2% BSA in PBS plus 0.2% Tween until samples from all time points were collected, and then the slides were probed with anti-FLAG M2 (Sigma) diluted 1:500 in blocking buffer. Samples were next treated with Alexa 488-conjugated anti-mouse secondary antibody (Molecular Probes) diluted 1:1,000 and with Hoechst dye (Sigma) diluted 1:10,000. Slides were mounted with SlowFade reagent (Molecular Probes), and images were collected using a Zeiss LSM 510 confocal microscope.
Assay for virion binding and entry and nuclear localization of input virion components.
To determine relative efficiencies with which wild-type and mutant viruses bind cells, ARPE19 cells were preincubated in medium comprised of equal portions of DMEM and Ham's F-12 medium without serum for 1 h at 4°C. Virus (0.1 TCID50/cell) was added to cells and adsorbed for 1 h at 4°C. Cultures were then washed twice with PBS at 4°C, harvested by scraping, pelleted by centrifugation, and stored at −80°C until assayed. As a control, separate infected cultures were washed twice with PBS at 4°C and immediately incubated with trypsin (100 μg/ml) for 15 min at 37°C (27). Soybean trypsin inhibitor (100 μg/ml; Sigma) was then added, and the cells were harvested, pelleted, washed, and stored at −80°C.
To assess entry, cells were infected as described above at 4°C, washed twice with PBS at 4°C, fed with fresh medium, and incubated at 37°C. After 15 min, 45 min, or 120 min, cultures were washed twice with PBS, followed by trypsin treatment as described above, and cell pellets were collected and subsequently stored at −80°C. As a control, separate infected cultures were treated with trypsin immediately after adsorption at 4°C, washed twice with PBS at room temperature, replated, and then, 5 h later, washed twice with PBS, harvested, pelleted, and stored at −80°C.
Binding and entry were also monitored by thin-section electron microscopy (TEM). ARPE19 cells were infected (50 TCID50/cell) at 4°C for 1 h, washed twice with PBS and once with serum-free medium at 4°C, and then incubated in serum-free medium at 37°C for 15 min to allow entry (27). Cells were then washed once in PBS at 37°C and fixed for 5 min at 37°C, followed by fixation overnight at 4°C in 4% paraformaldehyde–2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer. Samples were then washed with sodium cacodylate buffer (0.2 M, pH 7.3) three times, after which they were incubated for 1 h at 4°C in 1% osmium tetroxide. Samples were washed as described above, rinsed with maleate buffer (pH 5.1), and then stained with 1% uranyl acetate in maleate buffer for 1 h. The samples were next dehydrated in ethanol, washed three times with propylene oxide, and infiltrated in 1:1 propylene oxide and Eponate-12 epoxy resin (Ted Pella, Inc.) at room temperature overnight, after which the medium was exchanged for Eponate-12 epoxy resin (Ted Pella, Inc.) for an additional 5 h at room temperature. The samples were then embedded and polymerized overnight. Ultrathin sections of 85 nm were cut with a diamond knife and stained with uranyl acetate and lead citrate. Images were collected on a Philips CS12/STEM electron microscope (FEI Company) operated at 60 kV and fitted with an SC100W fiber-optically charge-coupled device (CCD) camera capable of 11 megapixels (Gatan, Inc.).
To assess the association and entry of the viral genome in cells, DNA was extracted using a blood and cell culture DNA miniprep kit (Qiagen), and viral DNA was quantified by qPCRs using primers for UL123 (see above) and the cellular MDM2 gene (forward, 5′-CCCCTTCCATCACATTGCA-3′; and reverse, 5′-AGTTTGGCTTTCTCAGAGATTTCC-3′). All samples were analyzed in triplicate, and the number of viral genomes was normalized to the MDM2 level.
Delivery of the virion tegument protein pUL83 (pp65) to the nucleus was assayed by infecting ARPE19 cells or fibroblasts at a multiplicity of infection of 0.1 TCID50/cell. Virus was adsorbed for 1 h at 37°C, after which cultures were washed twice with PBS and fed with fresh medium. The number of pUL83-positive nuclei was assayed by immunofluorescence assay as previously described (2). Cells were fixed in ice-cold methanol and stained using an anti-pUL83 antibody (clone 8A8) (2) diluted 1:10 followed by a DyLight-488-conjugated goat anti-mouse secondary antibody (Pierce) diluted 1:1,000 and Hoechst dye (Sigma) diluted 1:10,000. Five random fields were visualized to quantify the number of pUL83-positive nuclei.
In parallel, ARPE19 cells were infected with either wild-type or mutant virus at a multiplicity of infection of 0.5 TCID50/cell and harvested at 10 hpi, and nuclei were isolated. Cell pellets were resuspended in buffer A (10 mM HEPES, pH 7.5; 5 mM MgCl2; 15 mM KCl; 1 mM phenylmethylsulfonyl fluoride [PMSF]) and lysed three times by freeze-thawing. The suspension was passed through a 27-gauge needle, and a 0.5× volume of 50% sucrose was added to the homogenate. The suspension was centrifuged at 16,000 × g, the nuclei were then resuspended in buffer A and sonicated on ice, and the debris was pelleted at 16,000 × g. Viral genomes were then quantified by qPCRs using primers for UL123 and the cellular MDM2 gene. Each sample was analyzed in triplicate, and viral genome copies were normalized to the MDM2 level.
RESULTS
pUL78 localizes to distinct structures late after infection and copurifies with virions.
To characterize the expression and localization of pUL78, we generated a derivative of TB40/Ewt-mCherry (17) expressing a pUL78 fusion protein. The variant, termed TB40/E-mCherry-inUL78-3xF, expresses pUL78 with three tandem FLAG epitopes fused to its C terminus (pUL78-3xF) (see Fig. 2A). We infected fibroblasts at a multiplicity of infection of 0.5 TCID50/cell, harvested cells after various time intervals, and analyzed the expression of the viral GPCR by immunoblot assay for the FLAG epitope. pUL78-3xF expression was maximal at late times after infection, and the majority of the protein migrated as an approximately 45-kDa band, with minor amounts of higher-molecular-mass species that likely represent differential glycosylation (Fig. 1A). Additionally, at later times, a smaller band of approximately 28 kDa was evident (Fig. 1A and 2C). Our lab previously reported a similar banding pattern for murine cytomegalovirus pM78, the mouse orthologue of HCMV pUL78 (18). We also observed the more rapidly migrating band for the HCMV FIX clinical isolate (Fig. 1B), although the pUL78 proteins migrated differently for TB40/E compared to FIX (pUL78 differs at 3 amino acids between the two strains). This smaller species is pUL78 specific, as it is absent from viruses lacking the UL78 ORF (Fig. 2C). It might be an inactive C-terminal breakdown product, or it could perform an as yet unknown function. Accumulation of pUL78-3xF and the smaller, 28-kDa species was sensitive to PAA (Fig. 1A), which inhibits viral DNA replication. Earlier work had judged UL78 RNA to accumulate with early kinetics (7). Subsequent transcriptional mapping showed that the pUL78 coding region is contained within 1.8- and 5.5-kb RNAs (28). The smaller RNA is present beginning early after infection and is resistant to PAA, whereas the longer species is found during the late phase and is sensitive to PAA (15, 28). Our data demonstrate that pUL78 accumulation is sensitive to PAA, and therefore the protein accumulates with late kinetics. In light of the earlier data, we anticipate that pUL78 is made exclusively from the larger RNA species spanning the UL78 open reading frame.
Fig 2.
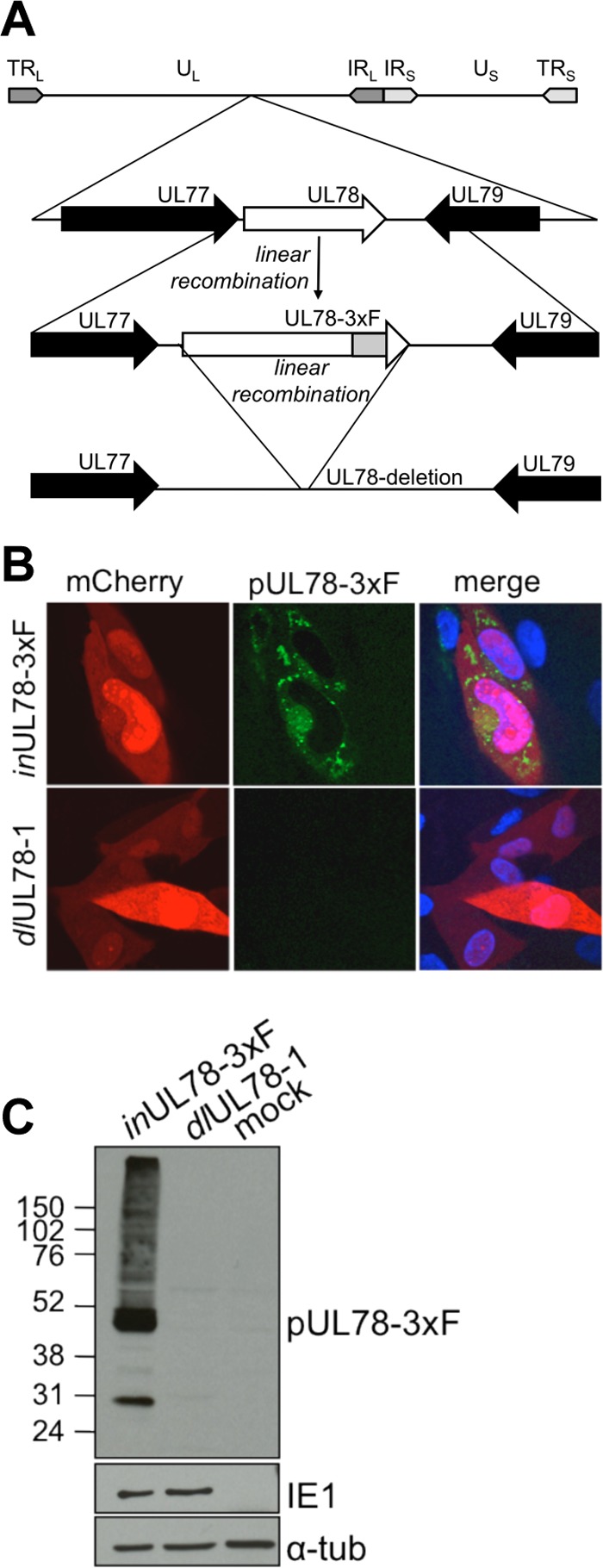
pUL78 expression is ablated in TB40/E-mCherry-dlUL78. (A) Diagram showing the wild-type UL78 locus of TB40/Ewt-mCherry (UL78), the locus in TB40/E-mCherry-inUL78-3xF expressing FLAG-tagged pUL78 (inUL78-3xF), and the deleted region in TB40/E-mCherry-dlUL78-1 (dlUL78). TR, terminal repeat; IR, internal repeat; U, unique; L and S, long and short, respectively. (B and C) Loss of pUL78-3xF expression in TB40/E-mCherry-dlUL78-1 as assayed by immunofluorescence assay (B) and Western blotting (C). Fibroblasts were infected (0.5 TCID50/cell) with either inUL78-3xF or dlUL78-1 and analyzed at 96 hpi. In the immunofluorescence images, pUL78-3xF (green) is identified via its FLAG epitope, mCherry (red) is shown as a marker for infection, and the nuclei (blue) are stained with Hoechst dye. For the immunoblot, lysates were additionally assessed for expression of a viral protein (IE1) and α-tubulin as loading controls.
Fig 1.

pUL78 is a late protein that is partially localized to juxtanuclear structures in fibroblasts and copurifies with virions. (A) FLAG-tagged pUL78 accumulates during the late phase of TB40/E-mCherry-inUL78-3xF infection, and its accumulation is dependent on viral DNA replication. Fibroblasts were infected (0.5 TCID50/cell), and whole-cell lysates were immunoblotted for the FLAG epitope tag to detect pUL78-3xF. A separate culture was treated with PAA after adsorption and collected at 72 hpi. To ensure equal loading, lysates were assayed for α-tubulin (α-tub). (B) Expression of FLAG-tagged pUL78 after infection with a different HCMV clinical isolate. Fibroblasts were infected (0.5 TCID50/cell) with either TB40/E-mCherry-inUL78-3xF (T) or BFX-GFP-inUL78-3xF (F). Lysates were collected at 96 hpi and immunoblotted for the FLAG epitope. α-Tubulin is shown as a loading control. M, mock. (C) Immunofluorescence detection of pUL78-3xF following TB40/E-mCherry-inUL78-3xF infection. Fibroblasts were infected (0.5 TCID50/cell) with TB40/E-mCherry-inUL78-3xF and then fixed and permeabilized after various time intervals. mCherry (red) is shown as a marker of infection. pUL78-3xF expression was assayed with an antibody directed to the FLAG epitope (green), and nuclei were stained with Hoechst dye (blue). (D) pUL78 copurifies with virions. TB40/Ewt-mCherry (TB40/Ewt) or TB40/E-mCherry-inUL78-3xF (inUL78-3xF) virions were partially purified by centrifugation through a 20% sorbitol cushion. The presence of pUL78-3xF was assessed by immunoblot assay with an antibody to the FLAG epitope. The tegument protein pUL83 is shown in the lower panels as a loading control.
We next examined the localization of the protein by immunofluorescence assay after infection of fibroblasts with TB40/E-mCherry-inUL78-3xF. Early after infection, pUL78-3xF displayed a diffuse staining pattern throughout the cell (Fig. 1C). However, by 96 hpi, a substantial portion of pUL78 localized to distinct cellular structures that appeared to partially coincide with the viral assembly zone (20). We did not observe staining for pUL78 on the cell surface of the infected fibroblasts. This finding is consistent with previous data from our laboratory detailing the localization of another HCMV GPCR, pUS27 (17), as well as with reports from other investigators who have described the rapid internalization of the viral GPCRs (11, 25), including pUL78 (24). Importantly, we do not believe that the addition of the epitope tag altered pUL78 function, as the growth of TB40/E-mCherry-inUL78-3xF was similar to that of TB40/Ewt-mCherry in a variety of cell types (Fig. 3A, lower right panel).
Fig 3.

Growth of pUL78-deficient virus is attenuated in HUVECs and ARPE19 cells but not in fibroblasts. (A) The growth of TB40 viruses was assayed. HFFs (upper left) and HUVECs (upper right) were infected (0.01 TCID50/cell) with TB40/Ewt-mCherry (TB40/Ewt) or TB40/E-mCherry-dlUL78-1 (dlUL78-1). Supernatants were collected at the indicated times to assess viral titers. ARPE19 cells were infected (0.1 TCID50/cell in lower left panel and 1.0 TCID50/cell in lower right panel) with the indicated viruses. Infected cells and supernatants were collected to assess viral titers at the indicated times. For all assays, titers were determined in triplicate by infecting fibroblasts and quantifying IE1-positive cells at 24 hpi. IU, infectious units. (B) The growth of FIX viruses was assayed. HFFs (left) or ARPE19 cells (right) were infected (0.01 TCID50/cell or 0.1 TCID50/cell, respectively) with either BFXwtGFP, BFXinUL78-3xF, or BFXdlUL78-2, and at the indicated times, infected cells and supernatant were collected. Viral titers were assessed by TCID50 assay.
The finding that pUL78 partially localizes to a juxtanuclear structure late during infection is consistent with its presence in the assembly compartment and predicts that the GPCR could be incorporated into the mature virion. Similar to what was observed with the MCMV M78 orthologue (18), HCMV pUL78 was associated with partially purified preparations of virus (Fig. 1D). The major isoform of pUL78 that copurified with virions was the 45-kDa species; only a small amount of the 28-kDa moiety was present.
pUL78-deficient virus displays growth defects in epithelial and endothelial cells.
To assess the requirement for pUL78 during infection, we generated two independent mutants in which we deleted the entire UL78 ORF, termed TB40/E-mCherry-dlUL78-1 and TB40/E-mCherry-dlUL78-2 (Fig. 2A). The production and analysis of two independently derived mutants reduced the likelihood of off-target mutations contributing to observed phenotypes. The two mutants produced identical results in all of our tests, so we present data for just one of them, TB40/E-mCherry-dlUL78-1. The mutants were generated in the TB40/E-mCherry-inUL78-3xF background, allowing us to readily confirm the loss of pUL78 accumulation by using a FLAG tag-specific antibody to monitor expression of the protein by immunofluorescence and immunoblot assays (Fig. 2B and C). To evaluate the growth of the mutant in fibroblasts, cells were infected (0.01 TCID50/cell) with mutant and wild-type viruses, and the production of infectious progeny was monitored. The pUL78-deficient mutant grew to titers similar to those of its wild-type counterpart (Fig. 3A, upper left panel), demonstrating, in agreement with an earlier report (15), that the expression of pUL78 is not required for normal replication in fibroblasts.
To determine if pUL78 was required for efficient virus replication in other cell types, endothelial (HUVECs) and epithelial (ARPE19) cells were infected (0.01 TCID50/cell and 0.1 TCID50/cell, respectively). The loss of pUL78 reduced the production of virus in both cell types. We observed a 5-fold defect for the mutant in endothelial cells (Fig. 3A, upper right panel), with a greater defect, approaching 100-fold, in epithelial cells (Fig. 3A, lower left panel). To test the effect of input multiplicity on the growth defect, we infected ARPE19 cells at 1.0 TCID50/cell. Under these conditions, the mutant virus displayed a 10-fold reduction in yield (Fig. 3A, lower right panel), indicating that its growth defect is multiplicity dependent in ARPE19 cells. Importantly, this growth phenotype is not strain dependent. We generated BFX-GFP-inUL78-3xF and BFX-GFP-dlUL78 recombinant viruses from BFXwtGFP. We then infected HFFs and ARPE19 cells with these constructs, at a multiplicity of infection of 0.01 TCID50/cell or 0.1 TCID50/cell, and confirmed that the pUL78 deletion virus grows to wild-type titers in fibroblasts (Fig. 3B, left panel) but displays a defect in viral production in epithelial cells (Fig. 3B, right panel). These results confirm a pUL78 requirement for efficient replication within cells known to be important for HCMV replication and spread in vivo.
pUL78 is required for efficient accumulation of viral proteins and RNA in epithelial and endothelial cells.
We next asked if pUL78 is required for the accumulation of other viral proteins. To this end, we infected fibroblasts, HUVECs, or ARPE19 epithelial cells (0.5 TCID50/cell) and assayed viral proteins by Western blotting after various time intervals. Consistent with the growth analysis, the viral proteins tested accumulated normally in fibroblasts infected with the pUL78-deficient virus (Fig. 4A). However, we observed a substantial decrease in both IE1 protein (immediate-early protein) and pUL44 (early protein) for TB40/E-mCherry-dlUL78-1 compared to its wild-type parent in HUVECs (Fig. 4B) and ARPE19 cells (Fig. 4C).
Fig 4.

pUL78 is needed for normal viral protein accumulation in HUVECs and ARPE19 cells but not in fibroblasts. Fibroblasts (A), HUVECs (B), and ARPE19 cells (C) were infected (0.5 TCID50/cell) with TB40/Ewt-mCherry or TB40/E-mCherry-dlUL78-1 (dlUL78-1). Whole-cell lysates were prepared at the indicated times after infection, and representative viral proteins (IE1 and pUL44) were assessed by immunoblot analyses. α-Tubulin (α-tub) was assayed as a loading control.
Next, ARPE19 cells were infected with mutant and wild-type viruses (0.5 TCID50/cell), RNAs were isolated at various times after infection, and the accumulation of UL123 (IE1; immediate early) and UL44 (early) RNAs was assayed by qRT-PCR. The pUL78-deficient virus yielded reduced UL123 and UL44 RNA levels compared to those of the wild-type virus at all times tested (Fig. 5A and B), arguing that pUL78 functions during infection, prior to the accumulation of immediate-early viral RNA.
Fig 5.

pUL78 is needed for optimal viral mRNA accumulation in epithelial cells. ARPE19 cells were infected (0.5 TCID50/cell) with TB40/Ewt-mCherry (TB40/Ewt) or TB40/E-mCherry-dlUL78-1 (dlUL78-1), and at the indicated times, total RNA was prepared and analyzed by qRT-PCR for expression of UL123 RNA (A) and UL44 RNA (B). All samples were analyzed in triplicate and normalized to the amount of cellular GAPDH transcript. AU, arbitrary units.
pUL78 functions after binding and before entry into epithelial cells.
Since pUL78 is a membrane protein and copurifies with virions (Fig. 1D), we hypothesized that it might be involved in particle binding and/or entry into ARPE19 cells. To test for binding, we adsorbed virus to ARPE19 cells (0.1 TCID50/cell) at 4°C for 1 h, washed the cells twice with PBS at 4°C, isolated DNA, and assayed for viral DNA by qPCR. There was no difference in the amounts of mutant and wild-type virion DNAs that became cell bound (Fig. 6A). As a control, a duplicate culture infected at 4°C for 1 h with wild-type virus was treated with trypsin prior to isolation of DNA. This treatment reduced the amount of viral DNA by a factor of about 8, confirming that the preponderance of viral DNA was present in virions that had not yet entered the cell and supporting the conclusion that pUL78 is not required for efficient adsorption of HCMV to epithelial cells.
Fig 6.

pUL78 is required for viral entry into ARPE19 cells. ARPE19 cells were infected (0.1 TCID50/cell) at 4°C with TB40/Ewt-mCherry (TB40/Ewt) or TB40/E-mCherry-dlUL78-1 (dlUL78-1) for 1 h and then washed twice with PBS at 4°C. (A) Cell-associated viral DNA was assessed by qPCR as a measure of the relative number of particles bound to the cells. As a control, TB40/Ewt-infected cells were treated with trypsin to remove surface-exposed virus. (B) To monitor viral entry, infected cultures were shifted to 37°C for various time intervals following the 4°C washes. Virions that did not enter the cell were removed by trypsin treatment. As a control, cells were treated immediately following the 4°C incubation and returned to 37°C for 5 h to ensure that the trypsin treatment efficiently removed bound virus. Samples were assayed by qPCR in triplicate and normalized to the level of the cellular MDM2 gene.
Next, the 4°C binding protocol was repeated and followed by a 15-, 45-, or 120-min incubation at 37°C to allow entry of virus bound to the cell surface. Cells were then treated with trypsin to remove virions remaining exposed on the cell surface, and DNA was isolated and assayed for internalized viral DNA by qPCR. At 15 and 45 min, about 50% less viral DNA was cell associated after infection with the mutant than after infection with wild-type virus, and at 2 h the amounts of cell-associated viral DNA were similar for the two viruses (Fig. 6B). Importantly, when cells were treated with trypsin immediately after the 4°C adsorption period and then warmed to 37°C for 5 h before isolation of DNA, about 80-fold less viral DNA remained cell associated when either the mutant or wild-type virus was assayed (Fig. 6B, control bars). This control confirmed the ability of the trypsin treatment to remove virions that were bound to the cell surface but not internalized. We conclude that the association of the virion with the cell is altered between 45 min, when a portion of virus particles can still be removed by trypsin, and 120 min, when virions are no longer removed by the protease. Presumably, virions have entered the cell by 120 min, but it is also possible that the association of the virion with the cell surface changes with time so that virions on the cell surface cannot be removed.
We employed electron microscopy as a second approach to monitor the fate of mutant virus particles at the start of infection (Fig. 7). ARPE19 cells were infected at 4°C for 1 h (50 TCID50/cell) and then washed, fed with medium at 37°C, and held for an additional 15 min. Although numerous wild-type virions were evident within intracellular vesicles, mutant particles remained outside the cell, in the vicinity of the plasma membrane, supporting the conclusion that internalization of virus particles is delayed when pUL78 is not present in the virion envelope.
Fig 7.

Intracellular virions are evident in ARPE19 cells shortly after infection with wild-type virus but not with dlUL78-1. Virus was bound to cells at 4°C for 1 h, and then cultures were shifted to 37°C for 15 min to allow virus entry before processing for microscopy. Two representative images are shown for each virus. Chevrons, intracellular wild-type particles; arrows, extracellular mutant particles; C, cytoplasm; N, nucleus. Bars, 1 μm.
If entry of mutant particles is delayed, then the nuclear accumulation of virion tegument proteins and DNA should also be delayed. To test this prediction, ARPE19 cells were infected (0.1 TCID50/cell) with wild-type or pUL78-deficient virus and fixed after various time intervals, and the localization of pUL83 was assayed by immunofluorescence assay. This abundant constituent of virions rapidly moves to the nucleus after infection (2). Although there was no difference in the delivery of the protein to the nuclei of fibroblasts by mutant or wild-type particles (Fig. 8A), we found a significant delay in ARPE19 cells (Fig. 8B). To confirm that we were assaying the localization of input pUL83 (a late protein) before newly synthesized pUL83 accumulated within infected epithelial cells, we infected ARPE19 cells with wild-type or mutant virus and assessed total viral DNA in the presence and absence of the HCMV DNA replication inhibitor PAA. The total amounts of DNA were similar in the presence and absence of the drug (Fig. 8C), and the drug was active, as it inhibited expression of a late protein (Fig. 1A). This experiment demonstrates that neither viral DNA nor de novo pUL83 accumulation had begun. Therefore, the lower levels of pUL83 nuclear accumulation in ARPE19 cells can be attributed to a delay in accumulation of input levels of this tegument protein (Fig. 8B).
Fig 8.

pUL78 is required for efficient delivery of virion DNA and protein to the nuclei of infected ARPE19 cells but not fibroblasts. Cells were infected (0.1 TCID50/cell) with TB40/Ewt-mCherry (TB40/Ewt) or TB40/E-mCherry-dlUL78-1 (dlUL78-1). HFFs (A) and ARPE19 cells (B) were fixed at the indicated times and stained for pUL83, and the number of pUL83-positive nuclei as well as the total cell number in each of five fields was counted. Whole-cell (C) or nuclear (D) DNA was prepared from ARPE19 cells at the indicated times after infection, DNA was extracted, and the relative number of viral genomes was quantified by qPCR using primers specific for UL123. Each sample was analyzed in triplicate and normalized to the MDM2 level. pUL78-3xF displayed perinuclear staining at 2 (E) and 72 (F) hpi in fibroblasts and epithelial cells. HFFs and ARPE19 epithelial cells were infected (10 TCID50/cell for 2-hpi samples and 0.5 TCID50/cell for 72-hpi samples) with TB40/E-mCherry-inUL78-3xF (inUL78-3xF). pUL78-3xF expression was assessed by an immunofluorescence assay using an antibody for the FLAG epitope. mCherry could not be detected at 2 hpi. Green, FLAG; blue, Hoechst dye.
We also assayed the efficiency with which virion DNA reached the nucleus in the absence of pUL78. ARPE19 cells were infected (0.1 TCID50/cell) with TB40/E-mCherry-dlUL78-1 or its wild-type parent, and nuclei were prepared 10 h later. Nuclei prepared from cells infected with the mutant contained less than half the amount of viral DNA seen in nuclei from wild-type virus-infected cells (Fig. 8D), although total cell-associated viral DNA remained at wild-type levels (Fig. 8C).
Finally, to ensure that this phenotype was not due to localization differences of pUL78 in epithelial cells, we compared the cellular distribution of inUL78-3xF in fibroblasts and epithelial cells at early and late times postinfection and confirmed that pUL78 displayed similar cellular localizations in the two cell types at 2 (Fig. 8E) and 72 (Fig. 8F) hpi.
Taken together, our results argue for a role of pUL78 in the entry of virion constituents into epithelial cells.
DISCUSSION
The establishment of an efficient and productive HCMV infection requires a coordinated series of events, from binding and entry at the host cell surface to initiation of the cascade of viral gene transcription, subsequent translation of viral proteins, and assembly of infectious particles. The primary finding in this study is a requirement for pUL78 for timely entry of virion constituents into epithelial cells and subsequent delivery of a virion protein and viral DNA to the nuclei of epithelial cells.
There are multiple instances where an HCMV mutant lacks an observable phenotype in one cell type but displays a significant growth defect in cells of a different tissue origin (10, 17, 26). Consistent with this precedent, we have identified a cell type-specific role for pUL78. A pUL78-deficient derivative of the TB40/E clinical isolate generated a 5-fold-reduced yield in endothelial cells and 100-fold less virus in epithelial cells than those seen for wild-type virus (Fig. 3). The protein acts early during infection, as it is required for efficient accumulation of immediate-early (IE1) RNA (Fig. 5) and protein (Fig. 4). Although pUL78 copurifies with viral particles (Fig. 1D), it is not required for binding to epithelial cells (Fig. 6A). However, pUL78 is needed for the efficient and timely entry of the virion into epithelial cells (Fig. 6B and 7), as well as for the timely delivery of at least one virion protein, pUL83, and the viral genome to the nuclei of epithelial cells (Fig. 8).
Another HCMV-encoded GPCR, pUS27, also exhibits cell type-dependent growth properties (17). However, in contrast to pUL78, pUS27 functions late during infection to support virus spread by the extracellular route. It is important for growth in fibroblasts and endothelial cells, where the virus spreads mainly through the extracellular route, but dispensable for growth in epithelial cells, where the main route of transmission is cell-to-cell spread.
How does pUL78 facilitate entry into epithelial cells, and why is it not needed in fibroblasts? HCMV enters fibroblasts and epithelial cells by different mechanisms. The enveloped virus enters fibroblasts by fusing with the plasma membrane (9), but it enters epithelial cells by endocytosis (5, 19, 27). Within epithelial cells, the virion escapes from the endosome into the cytoplasm via a poorly understood pH-dependent membrane fusion event (19, 27). Since pUL78 is not required in fibroblasts but is needed in epithelial cells, it might function after binding at the cell surface to stimulate endocytosis, a complex process involving multiple pathways that is known to be modulated by numerous enveloped and nonenveloped viruses (14). Indeed, pUL78 (24) and the other HCMV GPCRs (11) are rapidly endocytosed from the cell surface. This interpretation is consistent with the failure of cell surface-associated mutant virions to enter epithelial cells as rapidly as wild-type particles (Fig. 6B and 7).
It is noteworthy that after 2 h at 37°C, pUL78-deficient virions became as resistant as wild-type virus to removal from cells by trypsin treatment (Fig. 6B), while the mutant continued to exhibit a delay in nuclear accumulation of virion DNA and protein at 10 hpi (Fig. 8B and D). If the virion has become internalized by 2 hpi, why are delays in nuclear accumulation of virion constituents evident at much later times? It is possible that resistance to trypsin treatment does not necessarily reflect internalization of virus particles, but rather results from a change in accessibility to trypsin of particles that remain outside the cell. If this is the case, then the entry of mutant particles could be delayed for a longer period than that suggested by the trypsin treatment, explaining the longer delay observed for nuclear accumulation of virion constituents. Alternatively, pUL78-deficient particles might enter cells aberrantly and, as a result, suffer delays in additional very early events. Finally, pUL78 might function at multiple stages of the entry process, perhaps initially stimulating endocytosis and subsequently facilitating movement of the tegumented capsid from the endosome to the cytoplasm. pUL78 could function directly, likely in concert with other viral glycoproteins, to execute the fusion event. Alternatively, the protein might function by signaling, even though no signaling capability has yet been attributed to the GPCR homologue. It could be deposited on the cell surface through endosomal recycling after the protein has been deposited into the endosomal membrane by the fusion event. pUL78 signaling could then facilitate movement of viral proteins and DNA to the nucleus.
In conclusion, we have demonstrated a cell type-dependent role for pUL78 at the start of infection, and our results provide a framework for further dissection of the very earliest stages of the infectious process.
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health (CA82396 and AI63335). C.M.O. was supported by fellowships from the New Jersey Commission on Cancer Research (09-1960-CCR-EO) and the American Cancer Society (PF-10-164-01-MPC).
We thank C. Sinzger (Eberhard Karls Universität, Tübingen, Germany) for the generous gift of TB40/E and Mei Yin and Judith A. Drazba of the Cleveland Clinic Foundation's Imaging Core for assistance with electron microscopy.
Footnotes
Published ahead of print 22 August 2012
REFERENCES
- 1. Attwood TK, Findlay JB. 1994. Fingerprinting G-protein-coupled receptors. Protein Eng. 7:195–203 [DOI] [PubMed] [Google Scholar]
- 2. Bechtel JT, Shenk T. 2002. Human cytomegalovirus UL47 tegument protein functions after entry and before immediate-early gene expression. J. Virol. 76:1043–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beisser PS, Grauls G, Bruggeman CA, Vink C. 1999. Deletion of the R78 G protein-coupled receptor gene from rat cytomegalovirus results in an attenuated, syncytium-inducing mutant strain. J. Virol. 73:7218–7230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beisser PS, Lavreysen H, Bruggeman CA, Vink C. 2008. Chemokines and chemokine receptors encoded by cytomegaloviruses. Curr. Top. Microbiol. Immunol. 325:221–242 [DOI] [PubMed] [Google Scholar]
- 5. Bodaghi B, et al. 1999. Role of IFN-gamma-induced indoleamine 2,3 dioxygenase and inducible nitric oxide synthase in the replication of human cytomegalovirus in retinal pigment epithelial cells. J. Immunol. 162:957–964 [PubMed] [Google Scholar]
- 6. Britt W. 2008. Manifestations of human cytomegalovirus infection: proposed mechanisms of acute and chronic disease. Curr. Top. Microbiol. Immunol. 325:417–470 [DOI] [PubMed] [Google Scholar]
- 7. Chambers J, et al. 1999. DNA microarrays of the complex human cytomegalovirus genome: profiling kinetic class with drug sensitivity of viral gene expression. J. Virol. 73:5757–5766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chee MS, et al. 1990. Analysis of the protein-coding content of the sequence of human cytomegalovirus strain AD169. Curr. Top. Microbiol. Immunol. 154:125–169 [DOI] [PubMed] [Google Scholar]
- 9. Compton T, Nepomuceno RR, Nowlin DM. 1992. Human cytomegalovirus penetrates host cells by pH-independent fusion at the cell surface. Virology 191:387–395 [DOI] [PubMed] [Google Scholar]
- 10. Dunn W, et al. 2003. Functional profiling of a human cytomegalovirus genome. Proc. Natl. Acad. Sci. U. S. A. 100:14223–14228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fraile-Ramos A, et al. 2002. Localization of HCMV UL33 and US27 in endocytic compartments and viral membranes. Traffic 3:218–232 [DOI] [PubMed] [Google Scholar]
- 12. Heitzler D, et al. 2009. Towards a systems biology approach of G protein-coupled receptor signalling: challenges and expectations. C. R. Biol. 332:947–957 [DOI] [PubMed] [Google Scholar]
- 13. Lee EC, et al. 2001. A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics 73:56–65 [DOI] [PubMed] [Google Scholar]
- 14. Marsh M, Helenius A. 2006. Virus entry: open sesame. Cell 124:729–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Michel D, et al. 2005. The human cytomegalovirus UL78 gene is highly conserved among clinical isolates, but is dispensable for replication in fibroblasts and a renal artery organ-culture system. J. Gen. Virol. 86:297–306 [DOI] [PubMed] [Google Scholar]
- 16. Murphy E, Vanicek J, Robins H, Shenk T, Levine AJ. 2008. Suppression of immediate-early viral gene expression by herpesvirus-coded microRNAs: implications for latency. Proc. Natl. Acad. Sci. U. S. A. 105:5453–5458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. O'Connor CM, Shenk T. 2011. Human cytomegalovirus pUS27 G protein-coupled receptor homologue is required for efficient spread by the extracellular route but not for direct cell-to-cell spread. J. Virol. 85:3700–3707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Oliveira SA, Shenk TE. 2001. Murine cytomegalovirus M78 protein, a G protein-coupled receptor homologue, is a constituent of the virion and facilitates accumulation of immediate-early viral mRNA. Proc. Natl. Acad. Sci. U. S. A. 98:3237–3242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ryckman BJ, Jarvis MA, Drummond DD, Nelson JA, Johnson DC. 2006. Human cytomegalovirus entry into epithelial and endothelial cells depends on genes UL128 to UL150 and occurs by endocytosis and low-pH fusion. J. Virol. 80:710–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Silva MC, Yu QC, Enquist L, Shenk T. 2003. Human cytomegalovirus UL99-encoded pp28 is required for the cytoplasmic envelopment of tegument-associated capsids. J. Virol. 77:10594–10605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sinzger C, et al. 2008. Cloning and sequencing of a highly productive, endotheliotropic virus strain derived from human cytomegalovirus TB40/E. J. Gen. Virol. 89:359–368 [DOI] [PubMed] [Google Scholar]
- 22. Sodhi A, Montaner S, Gutkind JS. 2004. Viral hijacking of G-protein-coupled-receptor signalling networks. Nat. Rev. Mol. Cell Biol. 5:998–1012 [DOI] [PubMed] [Google Scholar]
- 23. Terhune S, et al. 2007. Human cytomegalovirus UL38 protein blocks apoptosis. J. Virol. 81:3109–3123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wagner S, et al. 2012. The 7-transmembrane protein homologue UL78 of the human cytomegalovirus forms oligomers and traffics between the plasma membrane and different intracellular compartments. Arch. Virol. 157:935–949 [DOI] [PubMed] [Google Scholar]
- 25. Waldhoer M, Kledal TN, Farrell H, Schwartz TW. 2002. Murine cytomegalovirus (CMV) M33 and human CMV US28 receptors exhibit similar constitutive signaling activities. J. Virol. 76:8161–8168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang D, Shenk T. 2005. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc. Natl. Acad. Sci. U. S. A. 102:18153–18158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang D, Yu QC, Schroer J, Murphy E, Shenk T. 2007. Human cytomegalovirus uses two distinct pathways to enter retinal pigmented epithelial cells. Proc. Natl. Acad. Sci. U. S. A. 104:20037–20042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang SK, Duh CY, Wu CW. 2004. Human cytomegalovirus UL76 encodes a novel virion-associated protein that is able to inhibit viral replication. J. Virol. 78:9750–9762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. 2005. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 33:e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhu H, Shen Y, Shenk T. 1995. Human cytomegalovirus IE1 and IE2 proteins block apoptosis. J. Virol. 69:7960–7970 [DOI] [PMC free article] [PubMed] [Google Scholar]