

Abstract
Programmed −1 ribosomal frameshifting is widely used in the expression of RNA virus replicases and represents a potential target for antiviral intervention. There is interest in determining the extent to which frameshifting efficiency can be modulated before virus replication is compromised, and we have addressed this question using the alpharetrovirus Rous sarcoma virus (RSV) as a model system. In RSV, frameshifting is essential in the production of the Gag-Pol polyprotein from the overlapping gag and pol coding sequences. The frameshift signal is composed of two elements, a heptanucleotide slippery sequence and, just downstream, a stimulatory RNA structure that has been proposed to be an RNA pseudoknot. Point mutations were introduced into the frameshift signal of an infectious RSV clone, and virus replication was monitored following transfection and subsequent infection of susceptible cells. The introduced mutations were designed to generate a range of frameshifting efficiencies, yet with minimal impact on encoded amino acids. Our results reveal that point mutations leading to a 3-fold decrease in frameshifting efficiency noticeably reduce virus replication and that further reduction is severely inhibitory. In contrast, a 3-fold stimulation of frameshifting is well tolerated. These observations suggest that small-molecule inhibitors of frameshifting are likely to have potential as agents for antiviral intervention. During the course of this work, we were able to confirm, for the first time in vivo, that the RSV stimulatory RNA is indeed an RNA pseudoknot but that the pseudoknot per se is not absolutely required for virus viability.
INTRODUCTION
Akey event in the replication cycle of retroviruses is expression of the pol gene, encoding reverse transcriptase (RT), integrase, and RNase H. In all retroviruses except spumaviruses (15), Pol is expressed initially as a Gag-Pol fusion from which the enzymes are subsequently cleaved by the virus-encoded protease (PR). The synthesis of the Gag-Pol polyprotein is achieved through one of two translational mechanisms. In most retroviruses, the gag and pol coding sequences overlap and translation of Gag-Pol requires programmed −1 ribosomal frameshifting within the overlap region (7, 25). Translation of the full-length retroviral mRNA generates predominantly the Gag polyprotein, but about 5% of ribosomes frameshift prior to encountering the gag stop codon and continue translation of the −1 reading frame (encoding pol), producing the Gag-Pol polyprotein and generating an intracellular Gag/Gag-Pol ratio of approximately 20:1. In the gammaretroviruses, typified by Moloney murine leukemia virus, gag and pol are in the same reading frame, separated by a UAG stop codon. In this virus group, Gag-Pol expression requires termination codon readthrough (51) rather than frameshifting, but the efficiency of the process is similar and generates a comparable Gag/Gag-Pol ratio in infected cells (16). The expression of Pol as part of a Gag-Pol fusion is known to be crucial in targeting the replicative enzymes to virions. In HIV-1, for example, expression of Gag alone is sufficient for assembly and release of virus-like particles but yields noninfectious virions lacking indispensable viral enzymes (19). Expression of Gag-Pol alone is also detrimental, in the case of HIV-1 due to intracellular protease activation and inhibition of assembly and budding (9, 28, 39). It is becoming clear that maintenance of a defined Gag/Gag-Pol ratio is critical to retrovirus replication. Indeed, subtle modulation of this ratio is known to have profound effects on HIV-1 infectivity, based on studies of cells transfected with plasmids expressing Gag and Gag-Pol from separate vectors (44) and of viruses with mutations in and around the frameshift region (5, 13, 21, 46). Viruses with a reduction in frameshifting of about 65% have decreased infectivity compared to the wild type (46), and the overexpression of Gag-Pol to generate a Gag/Gag-Pol ratio of approximately 1:1 (equivalent to an ∼10-fold stimulation of frameshifting efficiency) also reduced HIV-1 infectivity some 250- to 1,000-fold (44). The effect on virus replication of modulating frameshifting efficiency has also been studied in the double-stranded RNA virus L-A of Saccharomyces cerevisiae (10) and in the coronavirus associated with severe acute respiratory syndrome (37, 41). As in retroviruses, it has been shown that virus replication can be severely affected by mutations that distort the natural ratio of nonframeshifted and frameshifted species.
Much of our understanding of the relationship between frameshifting and virus replication has been derived from studies of HIV-1, but in this virus, the gag/pol overlap is a relatively long dual-coding region, and it has proven difficult in some cases to be certain that the phenotypes of the frameshift site mutations are not in fact a consequence of changes to the amino acid sequences of proteins encoded by the overlap region, particularly in gag (p1) or gag/pol (trans-frame protein, p6*) (discussed in reference 31). Nevertheless, the experimental evidence to date indicates that maintaining the precise ratio of frameshifting is essential for virus replication, although some modulation in frameshift efficiency may be tolerated (46), at least as regards a reduction in frameshifting. What is unclear is exactly how far the frameshift efficiency can deviate from the wild-type level before infectivity is compromised. If frameshifting is to be a valid antiviral target (see Discussion), this question is of considerable importance. In order to further investigate the upper and lower limits of frameshifting for retroviral infectivity, we have focused on creating a range of mutations that stimulate or reduce frameshift efficiency in the alpharetrovirus Rous sarcoma virus (RSV) and testing their effect on virus replication. Our choice of RSV as a model system was based primarily on the fact that the overlap between gag and pol is very short, comprising just 7 nucleotides, which makes the introduction of synonymous mutations more straightforward than in a virus with a longer overlap such as HIV-1. RSV also differs from HIV-1 (and most other retroviruses) in that PR is at the C terminus of Gag (as opposed to the N terminus of Pol) and is thus expressed stoichiometrically (with respect to Gag) rather than catalytically. It was therefore of interest to ascertain whether this would affect the sensitivity of virus replication to modulation of frameshifting efficiency. Like most −1 frameshifting signals, the RSV signal has two components, a heptanucleotide slippery sequence, AAAUUUA, where the ribosome changes reading frame, and a stimulatory RNA structure just downstream (Fig. 1). Here, we manipulated these elements to generate a series of RSV variants in which frameshifting was stimulated or reduced by various amounts. In carefully controlled infectivity assays, it was found that reducing the RSV frameshifting efficiency 3-fold results in a modest or severe replication defect and that an ∼8-fold reduction essentially abolishes replication. However, up to a 3-fold stimulation of frameshifting is well tolerated by the virus. During the course of the work, we were also able to confirm for the first time that the RSV pseudoknot forms in virus-infected cells and is essential for frameshifting.
Fig 1.
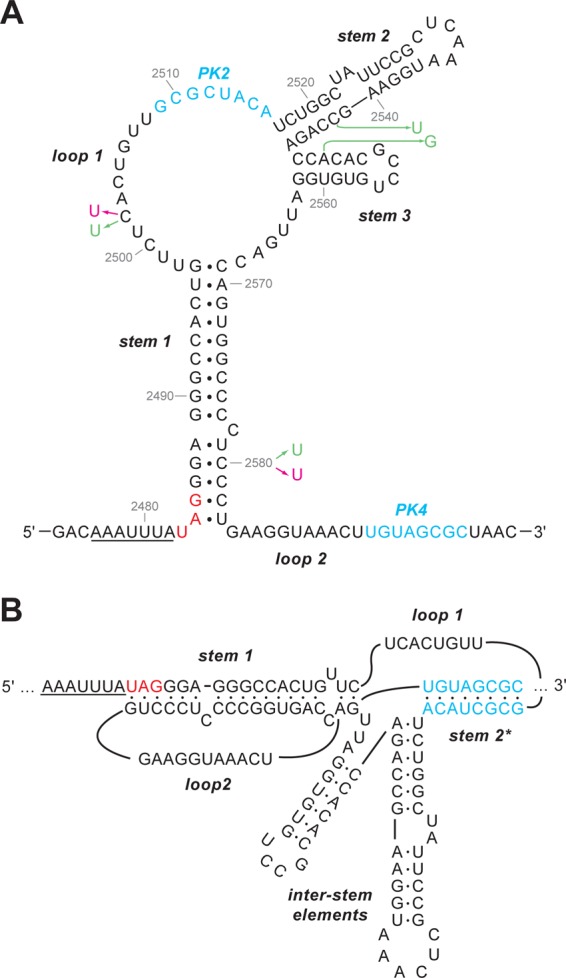
Secondary structure of the Rous sarcoma virus frameshift site (33). (A) The slippery sequence is underlined, and the gag stop codon is shown in red. The complementary PK2 and PK4 regions involved in forming the pseudoknot are in blue. The sequence shown is that of the Prague C strain (PrC) (43). Nucleotides that differ in ALV are in pink, and those that differ in the pRCAS proviral clone, which is based on the Schmidt-Ruppin strain (subgroup A), are in green. (B) An alternative view shows the pseudoknot formation more clearly, with stems 2 and 3 as interstem elements. In this representation, base pairing of PK2 and PK4 leads to the formation of pseudoknot stem 2*, which is marked with an asterisk to distinguish it from the unrelated stem 2 in the representation shown in panel A.
MATERIALS AND METHODS
RSV proviral clones.
Four plasmids harboring infectious proviral clones were utilized in this work: pLADI (4), containing a highly infectious avian leukosis virus (ALV) subgroup A strain (kind gift of J.-L. Darlix, Ecole normale supérieure de Lyon); pATV8 (29), containing the RSV Prague C (PrC) strain (from ATCC); pRCAS (22), containing the RSV Schmidt-Ruppin subgroup proviral genome lacking v-src, with the pol gene replaced by that of the RSV Bryan high-titer strain; and RCAS-AP, a variant of pRCAS with a heat-stable human placental alkaline phosphatase (AP) gene cloned into a unique ClaI site and positionally replacing v-src (both plasmids the kind gift of V. M. Vogt, Cornell University).
Plasmids.
The assessment of in vitro frameshift efficiencies and site-directed mutagenesis of the RSV frameshift signal was carried out using plasmids derived from pKT1+ (47). This vector contains the influenza A/PR8/34 PB1 gene under the control of bacteriophage T7 and SP6 promoters. Unique SacII and HpaI sites were introduced at nucleotide position 493 within the PB1 gene (generating plasmid pMV1) to facilitate insertion of the RSV frameshift region as a 924-bp SacII/HpaI fragment derived from infectious clone plasmid pLADI, pATV8, or pRCAS, generating plasmids pMV-ALV, pMV-RSV, and pMV-RCAS, respectively. In these plasmids, the frameshift region is cloned such that gag is in frame with the upstream portion of the PB1 gene and pol with the downstream portion, in the −1 frame with respect to gag. Following mutagenesis of the frameshift region within pMV plasmids, modified 924-bp SacII/HpaI fragments were cloned back into the pRCAS infectious clone to generate the pRCAS series of mutants. Some plasmids of the pRCAS series were subsequently digested with ClaI and the human placental alkaline phosphatase gene (derived from ClaI digestion of pRCAS-AP) inserted to generate the pRCAS-AP series (it was not convenient to insert the SacII/HpaI fragments directly into pRCAS-AP because the AP gene has a SacII site). For assessment of frameshifting efficiencies in transfected tissue culture cells, we employed the dual-luciferase frameshift reporter vector of Grentzmann and colleagues (20). DNA fragments of 171 bp spanning the RSV frameshift region were derived by PCR amplification from pRCAS or mutant derivatives using primers tailed with SalI and BamHI restriction enzyme sites and ligated into appropriately cleaved p2luc. All plasmid sequences were confirmed by dideoxy sequencing.
In vitro transcription and translation.
Frameshift reporter plasmids were linearized with AflII, and capped runoff transcripts were generated using SP6 RNA polymerase as described previously (8). mRNAs were recovered by a single extraction with phenol-chloroform (1:1, vol/vol) followed by ethanol precipitation. Remaining unincorporated nucleotides were removed by gel filtration through a NucAway spin column (Ambion). The eluate was concentrated by ethanol precipitation, and the mRNA was resuspended in water, checked for integrity by agarose gel electrophoresis, and quantified by spectrophotometry.
mRNAs were translated in nuclease-treated rabbit reticulocyte lysate (RRL) (Promega) programmed with ∼50 μg/ml template mRNA. A typical reaction mixture had a volume of 10 μl and was composed of 90% (vol/vol) RRL, 20 μM amino acids (lacking methionine), and 0.2 MBq [35S]methionine. Reaction mixtures were incubated for 1 h at 30°C and stopped by the addition of an equal volume of 10 mM EDTA–100 μg/ml RNase A followed by incubation at room temperature for 20 min. Samples were prepared for SDS-PAGE by adding 10 volumes of 2× Laemmli's sample buffer (30) and boiling for 3 min, and proteins were resolved on 12% SDS-PAGE gels. Dried gels were exposed to a Cyclone Plus Storage Phosphor Screen (PerkinElmer), the screen was scanned using a Typhoon TRIO Variable Mode Imager (GE Healthcare) in storage phosphor autoradiography mode, and bands were quantified using ImageQuantTL software (GE Healthcare). The calculations of frameshifting efficiency take into account the methionine content of the various products.
Frameshifting assays in tissue culture.
DF1 cells were maintained in Dulbecco's modification of Eagle's medium supplemented with 10% (vol/vol) fetal calf serum (DMEM-FCS). Plasmids were transfected using a commercial liposome method (FuGENE6; Roche). Cells were seeded in dishes of a 24-well plate and grown for 18 to 24 h until 80% confluence was reached. Transfection mixtures (containing plasmid DNA, serum-free medium [Opti-MEM; Gibco-BRL] and FuGENE) were set up as recommended by the manufacturer and added directly (dropwise) to the tissue culture cell growth medium. The cells were harvested 24 h posttransfection, and reporter gene expression was determined using a dual-luciferase assay system kit (Promega). Each data point represents the mean value (± standard deviation) from six separate transfections.
Virus assays.
RSV replication was assessed by transfection of proviral clone plasmids and subsequent infection of DF1 cells. Monolayers at 40% confluence were prepared in 6-cm dishes and transfected in duplicate with pRCAS, pRCAS-AP, or mutant derivative, using FuGENE6. To assess transfection efficiency, one set of dishes were harvested at 12 h posttransfection and the cell lysates subjected to SDS-PAGE and Western blotting for viral proteins and a cellular protein (GAPDH) used as a loading control. Two days posttransfection, the medium from the remaining set of dishes was removed and replaced with 4 ml fresh medium, which was harvested 24 h later and filtered through a 0.45-μm filter ready for infection of a fresh monolayer. The indicated amount (see Results) of filtered medium from transfected cells was added directly to the new monolayers, and the volume was adjusted to 4 ml with fresh medium. Cells were incubated at 39°C and 10% CO2 for a total of 3 days, with the culture medium replaced 2 days postinfection. Cells were harvested in phosphate-buffered saline (PBS), pelleted, lysed in cell lysis buffer (1% [vol/vol] Triton X-100, 10 mM β-glycerophosphate [pH 7.4], 50 mM Tris [pH 7.5], 1 mM EDTA [pH 8], 1 mM EGTA, 0.1% [vol/vol] β-mercaptoethanol, and protease inhibitors [EDTA-free; Roche; 1 tablet per 10 ml]), and proteins were analyzed by Western blotting. Culture medium was sterile filtered and used for further rounds of infection, Western blotting, or reverse transcriptase (RT) assays. In RT assays, virus was pelleted from 1 ml medium at 154,000 × g for 1 h at 4°C, and the pellet was gently resuspended in 5 μl of 10 mM Tris (pH 7.5) and incubated for 2 h at 37°C with 20 μl Vogt RT assay buffer, consisting of 50 mM Tris (pH 8), 60 mM NaCl, 6 mM MgCl2, 20 mM dithiothreitol (DTT), 0.05% (vol/vol) NP-40 substitute, 10 μg/ml poly(A), 5 μg/ml oligo(dT), and 10 μM dTTP, and with 8 μCi [α-32P]dTTP. Reaction aliquots (5 μl) were pipetted onto DE81 paper in grid formation and allowed to dry. The DE81 paper was washed in 0.3 M NaCl–30 mM trisodium citrate (4 × 5 min) and in 100% ethanol (2 × 1 min) and allowed to dry completely before exposure to X-ray film. Incorporation of [α-32P]dTTP was monitored by scintillation counting of individual grid squares. Virus replication was also assessed by 50% tissue culture infective dose (TCID50) assay. One day prior to infection, DF1 cells were seeded in 96-well plates at 4 × 103 cells/well in a final volume of 100 μl/well. Sterile-filtered culture medium was harvested 3 days posttransfection from a 6-cm dish transfected with a pRCAS derivative plasmid, and then the medium was serially diluted from 10−1 to 10−10. An aliquot of each diluted sample (100 μl/well) was added to each of 5 wells of a 96-well plate containing DF1 cells seeded as above. Two days postinfection, the culture medium was replaced by100 μl Opti-MEM, and incubation continued for a further 24 h. The cells were washed with PBS and fixed in 4% paraformaldehyde (50 μl/well) for 20 min at room temperature. After two further washes with PBS, the cells were permeabilized by incubation in a solution of 0.2% Triton X-100 in PBS (50 μl/well) for precisely 5 min at room temperature. After two more PBS washes, nonspecific antibody binding sites were blocked by incubation in 100 μl IF blocking buffer (1× PBS containing 1% [vol/vol] newborn calf serum) for 1 h at room temperature, in a humid environment to prevent evaporation. The cells were incubated for 1 h at room temperature on a rocking platform with 50 μl/well of a 1:1,000 dilution of primary antibody (rabbit anti-RSV p27 hyperimmune serum, α-p27; Life Sciences Inc.). After washing in IF blocking buffer (2 × 1 min, followed by 5 min on a rocking platform), cells were incubated for 1 h at room temperature in 50 μl/well of a 1:10,000 dilution of secondary antibody (Alexa Fluor goat α-rabbit IgG; Invitrogen). Cells were washed in PBS and visualized under a magnification of ×10, using an Olympus IX70 fluorescence microscope. Wells containing any brightly stained red cells were scored as positive. For each virus, the 50% endpoint titer was calculated according to the method of Reed and Muench (42).
Western blotting.
Proteins were separated by SDS-PAGE and transferred to nitrocellulose, and the membrane was blocked for 1 h at room temperature with gentle agitation, using a solution of 5% powdered milk (Marvel) in 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.5 mM KH2PO4 (pH 6.7), and 0.1% Tween 20 (PBST). The filter was incubated with α-p27 diluted 1:1,000 in Marvel-PBST for 1 h at room temperature, and the antibody was removed by washing with PBST (3 × 15 min). Incubation with a secondary antibody in PBST then proceeded for 1 h at room temperature, followed by 5 × 5-min washes in PBST. For IRDye-conjugated secondary antibodies, the secondary antibody incubation and subsequent washing steps were performed in the dark and were followed by washing for 5 min in 1× PBS. Blots were scanned and bands quantified using an Odyssey Infrared Imaging System (Licor). For horseradish peroxidase (HRP)-conjugated secondary antibodies, bands were detected by chemiluminescence using ECL Plus Western blotting detection reagents (Amersham, United Kingdom) as per the manufacturer's instructions, followed by exposure of the membrane to X-ray film for various lengths of time.
Alkaline phosphatase staining.
Cell monolayers were washed twice in 1× PBS and fixed with 0.5% glutaraldehyde in 1× PBS for 15 min at room temperature. The fix was removed, and the cells were overlaid with 1 ml of 1× PBS and placed on a plastic tray at 65°C for 2 h to heat inactivate endogenous alkaline phosphatase. The PBS was removed, and the cells were overlaid with 5 ml (per 6-cm dish) of AP staining solution (0.4 mM nitrotetrazolium blue chloride [NBT], 0.5 mM 5-bromo-4-chloro-3-indoxyl phosphate [X-phos] in 100 mM Tris [pH 9.5], 100 mM NaCl, 0.5 mM MgCl2). After 2 h, this solution was removed and the cells were rinsed twice in 1× PBS and photographed.
RESULTS
Generation of RSV frameshift signal mutants.
Four plasmids harboring infectious proviral clones were utilized in this work: pLADI (4), containing a highly infectious avian leukosis virus (ALV) subgroup A strain; pATV8 (29), containing the RSV Prague C (PrC) strain; pRCAS (22), containing the RSV Schmidt-Ruppin subgroup proviral genome lacking v-src, with the pol gene replaced by that of the RSV Bryan high-titer strain; and RCAS-AP, a variant of pRCAS with a heat-stable human placental alkaline phosphatase (AP) gene cloned into a unique ClaI site and positionally replacing v-src. The four proviral clones have close sequence similarity in the frameshift region (Fig. 1 and 2). To investigate the role of frameshifting in RSV replication, we mutated the signal of the RCAS strain to reduce or stimulate frameshifting (Fig. 2A), confirmed the effects in in vitro translation reactions, and reintroduced the mutations into the pRCAS infectious clone for replication assays. A primary consideration was that the mutations be silent with respect to amino acid sequence or, at the very least, that any amino acid changes be shown to be tolerated by the virus. Synonymous mutations that reduce frameshifting (Fig. 2A) were prepared by destabilizing the pseudoknot stems through point mutations in PK1, PK2, and PK4 (RCAS-9, -11, and -12, respectively). A “Gag-only” construct was also made (RCAS-20) by inserting a U residue toward the bottom of the main stem of the pseudoknot that introduced a premature stop codon at the start of pol. The generation of frameshift-stimulatory changes was more challenging. In our earlier studies of RSV Prague C (33), we observed that a mutation that destabilized stem 2 and another in which both stems 2 and 3 were deleted led to a stimulation of frameshifting of about 1.5-fold in vitro, suggesting that these structures were somewhat inhibitory to frameshifting, perhaps by compromising pseudoknot formation through the PK2-PK4 interaction. For this reason, we introduced mutations that were predicted to destabilize stem 2 (RCAS-S2) and stem 3 (RCAS-S3) independently. As an alternative approach, a series of point mutations were introduced in the loop regions at the top of stem 1 such that the length of stem 1 would be extended by three (RCAS-22) or four (RCAS-25) base pairs (Fig. 2B). We postulated that the extended stem would prove to be a more substantial barrier to elongating ribosomes and would lead to increased frameshifting efficiencies. A limitation of this approach was that we were unable to achieve this with synonymous mutations; thus, in both RCAS-22 and RCAS-25, an amino acid change (valine to glycine) is present in the pol frame. This change falls within a seven-amino-acid spacer region between the protease and reverse transcriptase that has no assigned function, except to allow unhindered cleavage by the viral protease at the PR-RT cleavage site (C2500UU↓ACU [Fig. 2C]). Amino acid changes and insertions at this site have been shown to have no effect on Gag-Pol processing, although extreme changes can lead to an alternative site at the N-terminal end of the seven-amino-acid spacer being used instead (45). It was thus postulated that a change in the amino acid adjacent to the P1 cleavage position would not adversely affect processing and, since the spacer has no other known function, the substitution was not expected to hinder virus replication. However, as a control, RCAS-23, in which the amino acid change was present but only a very modest extension to stem 1 was predicted (one base pair), was prepared in the hope that a modest stabilization would not influence frameshifting significantly. As a template for mutagenesis, a 924-bp fragment encompassing the RCAS frameshift region was subcloned into the in vitro frameshift assay plasmid pMV1 (see Materials and Methods) to generate pMV-RCAS. In this plasmid, the RCAS gag/pol overlap is cloned within the influenza virus PB1 gene such that the 5′ section of the PB1 gene is in frame with gag and the 3′ section is in frame with pol. In vitro transcripts generated using SP6 RNA polymerase, when translated in rabbit reticulocyte lysates, were predicted to produce a 40-kDa nonframeshifted product and a 54-kDa −1-frameshifted product. For the wild-type RCAS (RCAS-WT) signal (4.0% frameshifting), both products were seen (Fig. 2D), although they migrated somewhat more slowly than predicted because of the highly basic nature of the PB1 protein. The frameshifting efficiencies for the various mutants are shown in Fig. 2D and in Table 1. As expected, destabilization of the pseudoknot stems reduced frameshifting, dramatically for stem 1 (pMV-RCAS-9; 0.2%), less so for the PK2 and PK4 (stem 2*) mutants (pMV-RCAS-11 and -12; 1.6%). Increasing the length of stem 1 indeed led to a stimulation of frameshifting; as stem 1 was increased by 1 bp (pMV-RCAS-23), 3 bp (pMV-RCAS-22), or 4 bp (pMV-RCAS-25), frameshifting increased to 6.0%, 7.6%, and 9.4%, respectively. Contrary to what we had observed with RSV Prague C (33), destabilization of RCAS stem 2 and stem 3 did not lead to a stimulation of frameshifting; in fact, no effect was seen with pMV-RCAS-S2, and frameshifting was actually reduced with pMV-RCAS-S3 (1.1%) (Table 1). This was unexpected but may reflect a difference in folding of the major loop region in Prague C and RCAS (see Discussion). In addition to the mutants above, we also cloned into pMV1 the frameshifting signals of the PrC and ALV strains (to generate pMV-ALV and pMV-PrC) and a version of ALV in which U2502 (conserved in RCAS) was changed to C (to mimic the situation in PrC [Fig. 1]). The frameshift efficiencies measured in RRL were similar for pMV-RCAS-WT, pMV-ALV-C, and pMV-RCAS-WT, but pMV-ALV-WT was approximately twice as active (7.8%), perhaps reflecting an influence of U2502 on the conformation of the major loop. In summary, we were able to identify a series of mutations, either synonymous or causing an amino acid substitution likely to be tolerated, that generate in vitro a range of frameshifting efficiencies from 0.2% to 9.4% with a wild-type value of 4%.
Fig 2.

Mutational analysis of the RCAS frameshifting signal. (A) Mutations were introduced into pMV-RCAS by site-directed mutagenesis and are color coded to indicate the predicted effect on pseudoknot stability in orange (stabilization), blue (destabilization), or green (little effect/uncertain effect). Genomic sequence differences between RCAS and ALV/PrC are shown in gray. The valine codon (GUU) mutated in RCAS-22, -23, and -25 is boxed. The wild-type slippery sequence is underlined, and a variant with slippery sequence UUUAAAC is shown (IBV). The PK2 and PK4 regions, which form stem 2* of the pseudoknot, are in bold. The stop codon of the gag ORF is in red. (B) Predicted effect of the RCAS-23 and -25 mutations on the length of stem 1. The wild-type stem 1 is outlined in gray, and the mutated nucleotides are shown in the same colors as in panel A. Potential Watson-Crick base pairs are indicated by a dashed gray line. The valine codon, which in RCAS-22, -23, and -25 is mutated to a glycine codon, is boxed. (C) The protein sequences of Gag (green) and Pol (blue) in the overlap region are shown. The cleavage site at the C-terminal end of the seven-amino-acid spacer region is indicated. The two U residues mutated in RCAS-23 and RCAS-25 (see panel A), which change valine to glycine, are shown in red. Hydrophobic residues at position a (circled) and d (boxed) of a putative leucine zipper motif (45) (see the text) are indicated. (D) Ribosomal frameshift assays of pMV-RCAS variants. mRNAs derived from AflII-cut pMV-RCAS and derivatives were translated in RRL (at ∼50 μg/ml) for 1 hour, and the products resolved by 10% SDS-PAGE and visualized by autoradiography. Molecular size markers (in kDa) were also run on the gel (M). The 40-kDa nonframeshift (stop) and 54-kDa −1 frameshift products (FS) are indicated. The frameshifting efficiency measured for each signal is indicated below the relevant lanes and takes into account the number of methionines present in each product (stop, 11; FS, 11).
Table 1.
Frameshift efficiencies measured in RRL for pMV-RCAS and mutant derivatives at 30°C or 39°Ca
| Construct | Assay details | Frameshift efficiency (%) |
|---|---|---|
| RCAS (WT) | Wild-type RCAS frameshift signal present | 4.0 |
| 9 | Destabilized PK through modification of PK1 | 0.2 |
| 11 | Destabilized PK through modification of PK2 | 1.6 |
| 12 | Destabilized PK through modification of PK4 | 1.6 |
| 20 | Inserted stop codon in pol frame (Gag only) | 0.0 |
| 22 | Modulation of stem 1 (3-bp extension) | 7.6 |
| 23 | Modulation of stem 1 (1-bp extension) | 6.0 |
| 25 | Modulation of stem 1 (4-bp extension) | 9.4 |
| ALV | Wild-type ALV frameshift signal present | 7.8 |
| ALV-C | ALV signal with U2502 changed to C2502 | 3.5 |
| RSV | Wild-type RSV frameshift signal present | 3.4 |
| S2 | Destabilized stem 2 (part of ISE) | 3.6 |
| S3 | Destabilized stem 3 (part of ISE) | 1.1 |
| 12-IBV | SS changed to UUUAAAC in context of RCAS-12 | 4.2 |
| 25-IBV | SS changed to UUUAAAC in context of RCAS-25 | 15.0 |
| WT | Wild-type RCAS frameshift signal present | 4.4 (4.6) |
| 12 | Destabilized PK through modification of PK4 | 1.9 (1.4) |
| 25 | Modulation of stem 1 (4-bp extension) | 12.8 (7.8) |
| SRV | SS changed to AAAUUUU | 8.3 (7.5) |
| 12-SRV | SS changed to AAAUUUU in context of RCAS-12 | 4.3 (3.0) |
| 25-SRV | SS changed to AAAUUUU in context of RCAS-25 | 17.4 (13.5) |
| IBV2 | AAC inserted immediately downstream of natural SS to generate IBV SS | 6.5 (6.1) |
| 12-IBV2 | AAC inserted immediately downstream of natural SS to generate IBV SS in context of RCAS-12 | 4.4 (2.0) |
| 25-IBV2 | AAC inserted immediately downstream of natural SS to generate IBV SS in context of RCAS-25 | 18.2 (16.8) |
Values in parentheses are for translations that were carried out at 39°C. SS, slippery sequence.
Effect of modulating frameshifting efficiency on RSV replication.
The mutated frameshift signals were introduced into the pRCAS-AP vector, and replication was assessed in an infectivity assay. This vector was chosen initially because it contains a heat-stable alkaline phosphatase (AP) gene in place of the v-src oncogene, allowing direct visualization of virus spread by staining for the expressed enzyme (see Materials and Methods). Monolayers of DF1 cells were transfected in triplicate and even transfection efficiency was confirmed by harvesting one set of dishes after 12 h (when only input virus gene products are being expressed) and subjecting cell lysates to SDS-PAGE and blotting for Pr76Gag (Gag) using a polyclonal anti-p27 (CA) antibody (which also detects Pr180Gag-Pol [Gag-Pol]) (data not shown). Two days posttransfection, growth medium from the other dishes was removed and replaced with 4 ml fresh medium, which was harvested 24 h later, filtered, and used to inoculate fresh subconfluent (30%) DF1 monolayers. Cells were incubated at 39°C for a total of 3 days, with the culture medium replaced 2 days postinfection. One set of monolayers were stained for alkaline phosphatase expression and the other analyzed by Western blotting. Culture medium was also harvested and sterile filtered, and aliquots were analyzed by Western blotting. As shown in Fig. 3A, the “down” mutants (pRCAS-AP-9, -11, -12, and -20) were found to be severely attenuated. In transfected cells (3 days posttransfection), some Gag expression was evident, but little or no virus was released into the medium. In infected cells, there was no evidence for replication of these mutants; thus, it seems that RSV replication is intolerant to a reduction in frameshift efficiency of ≥60%, based on the in vitro frameshift assays. However, analysis of the “up” mutants (pRCAS-AP-22, -23, -25, and -ALV) failed to identify an upper boundary to frameshifting efficiency, as all of the clones appeared to be viable, judged by the presence of Gag and Gag-Pol (and some processed forms) in infected cells and p27 in the medium (released virus). This conclusion is supported by unambiguous AP staining of cells infected with the “up” mutants (Fig. 3B). Even in transfected cells, there was evident replication of these viruses. (Fig. 3A). It was also clear that replacing the RCAS frameshift signal with that of other “wild-type” RSV variants (pRCAS, pRCAS-PrC, pRCAS-ALV, and pRCAS-ALV-C) generated viruses that replicated in a fashion similar to that of pRCAS-AP in this assay. We also tested the stem destabilization mutants (Fig. 3C). pRCAS-AP-S2 replicated like the WT strain, but pRCAS-AP-S3 was clearly attenuated, consistent with the measured frameshifting efficiencies (S2, 3.6%; S3, 1.1%).
Fig 3.

Infectivity of pRCAS-AP and derivatives. (A) DF1 cells were transfected with pRCAS-WT or mutant derivatives (Transfection), and the released virus was used subsequently to infect new cells (Infection) as detailed in Materials and Methods. Cell lysates (upper panels) and supernatant virus (lower panels) were analyzed by SDS-PAGE (8% and 15% gels, respectively) and Western blotting using a polyclonal anti-CA (p27) serum. (B) Duplicate dishes from the transfection illustrated in panel A were stained for alkaline phosphate activity. (C) Analysis of the replication of pseudoknot stem 2 (S2) and stem 3 (S3) destabilization mutants. Cells and virus were processed and analyzed as described for panel A.
Extending the frameshift gradient.
As an alternative strategy to enhance frameshifting levels, we replaced the natural slippery sequence of RSV (AAAUUUA) with that of the coronavirus infectious bronchitis virus (IBV) (UUUAAAC), since previous work had revealed that it was a more effective stimulator of frameshifting than the native RSV sequence (33). Two new viruses were prepared, pRCAS-AP-12-IBV and -25-IBV, containing the UUUAAAC slippery sequence in the context of the pRCAS-AP-12 (destabilized pseudoknot) and pRCAS-AP-25 (increased stem 1 length) mutations. We postulated that pRCAS-AP-25-IBV would be a very efficient frameshift signal and hoped that pRCAS-AP-12-IBV would act as a control for the amino acid changes introduced by the slippery sequence mutation, with the increased activity in frameshifting caused by this mutation expected to compensate for the deficit in pseudoknot formation. Indeed, in in vitro translations, these expectations were realized, with pMV-RCAS-25-IBV engendering some 15% frameshifting (a 4-fold stimulation over pRCAS-WT) and pMV-RCAS-12-IBV effecting 4.2% frameshifting, very similar to that measured with the wild-type signal (Table 1). However, as can be seen in Fig. 4, these viruses showed little or no replication in comparison to pRCAS-AP-WT or pRCAS-AP-25, and material pelleted from tissue culture medium had very little RT activity (Fig. 4C). While the failure of pRCAS-AP-25-IBV to replicate could be ascribed to the heightened frameshifting efficiency, in this particular case, this seems unlikely. In pRCAS-AP-12-IBV-transfected cell culture medium, there was evidence for secreted, unprocessed Gag but little free p27, indicating that introduction of the IBV slippery sequence had impacted negatively on PR activity. The RSV protease is encoded at the 3′ end of the gag open reading frame (ORF), and the slippery sequence contains the codons for the two C-terminal residues of the cleaved enzyme (Asn-Leu) (Fig. 5B). From the structure of the RSV protease (27, 35), it is evident that the penultimate amino acid (Asn123) is critical for protease function (50), and its replacement by leucine in RCAS derivatives with the IBV slippery sequence likely disrupts dimerization and thus proteolytic catalysis (2).
Fig 4.
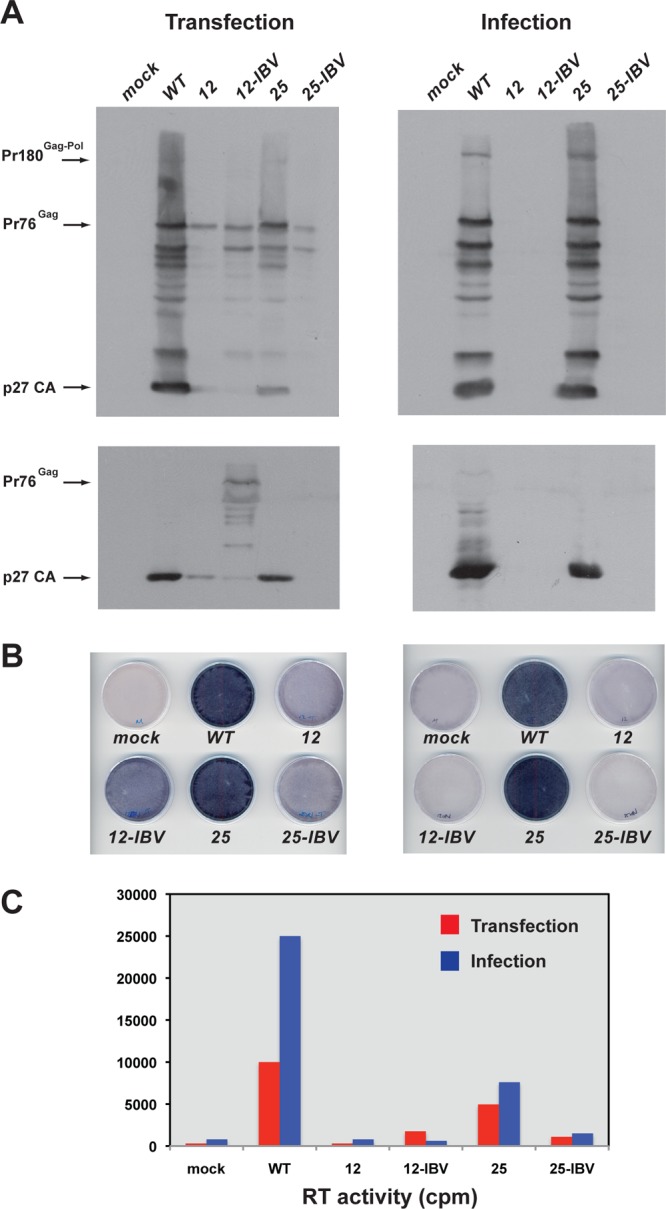
Infectivity of pRCAS-AP slippery sequence variants. (A) DF1 cells were transfected with pRCAS-WT or mutant derivatives (Transfection), and released virus was used subsequently to infect new cells (Infection) as detailed in Materials and Methods. Cell lysates (upper panels) and supernatant virus (lower panels) were analyzed by SDS-PAGE (8% and 15% gels, respectively) and Western blotting using a polyclonal anti-CA (p27) serum. (B) Duplicate dishes from the transfection illustrated in panel A were stained for alkaline phosphate activity. (C) Virus particles were harvested from 1 ml culture medium by ultracentrifugation, and reverse transcriptase activities were assayed. Incorporation of [α-32P]dTTP was visualized by scintillation counting.
Fig 5.

Enhancement of RSV frameshifting by combinatorial changes. (A) Mutations in pMV-RCAS are shown, with pseudoknot mutations color coded as described for Fig. 2A. The wild-type slippery sequence is underlined, and variants with slippery sequences UUUAAAC (IBV), AAAUUUU (SRV), and AAAUUUAAAC (IBV2) are indicated. (B) Amino acid changes in Gag and Pol generated by slippery sequence mutations. The critical asparagine involved in protease dimerization is outlined in red. The amino acid sequence given for the IBV2 mutant is based on the assumption that the frameshift event occurs at the IBV slippery sequence (UUUAAAC). Frameshifting may also occur at the original RSV slippery sequence (AAAUUUA), which is maintained, and this would give an additional leucine at the N terminus of Pol. The amino acid changes in Pol lie in a seven-amino-acid spacer region thought to tolerate point mutations (45) (Fig. 2C). (C) Ribosomal frameshift assays of pMV-RCAS variants. mRNAs were prepared, translated, and analyzed as described for Fig. 2. (D) Comparison of ribosomal frameshifting efficiencies measured for pMV-RCAS-derived mRNAs translated at 30°C or 39°C.
On the basis of these observations, an alternative strategy of slippery sequence modulation was implemented, with the aim of stimulating frameshifting while retaining PR function (Fig. 5A and B). In the first virus, the RSV slippery sequence was modified by an A-to-U point mutation such that it resembled that of the simian retrovirus 1 pro/pol slippery sequence (AAAUUUU; pRCAS-SRV). Substitution of this sequence at the RSV slip site has been shown to generate a 2-fold increase in frameshifting, probably because all three anticodon positions are paired in the −1 reading frame (26). This mutation changes only the terminal leucine to phenylalanine, and as this maintains the hydrophobic nature of the C-terminal residue, which is not actively involved in dimerization, we expected no impairment to protease function. In a second virus, an additional AAC codon was inserted after the RSV slippery sequence to append the IBV slippery sequence (UUUAAAC; pRCAS-IBV2) one codon after that of RSV. This mutant would retain the residues involved in dimerization but would have an additional asparagine at the C terminus. Given that the side chain of the terminal leucine in the native protein is displaced from the plane of the C-terminal β-sheet and found on the surface of the protein (50), it was reasonable to assume that an additional amino acid at this point would not affect dimerization. These mutations were introduced into pMV-RCAS and also in the background of pMV-RCAS-12 and pMV-RCAS-25 (for the reasons discussed above) and assayed for frameshifting in RRL (Fig. 5C and D). In these experiments, the translations were carried out both at the standard temperature (30°C) and at 39°C, the temperature used in the infectivity assays for DF1 cell culture. At 30°C, we found that the SRV and IBV2 mutations had a stimulatory effect on frameshifting in all cases, either alone or in combination with the RCAS-12 and RCAS-25 mutations, peaking at 18.2% (25-IBV2) and 17.4% (25-SRV). As before, the combination of destabilized pseudoknot mutation (RCAS-12) and more active slippery sequence engendered in vitro frameshift efficiencies close to the wild-type value. The pattern of frameshifting at 39°C was similar, although frameshifting efficiency was somewhat reduced for some mRNAs. Nevertheless, the RCAS-25-SRV1 and RCAS-25-IBV2 mutations gave approximately a 3- and a 4-fold stimulation, respectively, of frameshift efficiency at both 30°C and 39°C.
To assess the effects of these mutations on virus viability, we introduced them into the pRCAS infectious clone and carried out infectivity assays. We chose pRCAS rather than pRCAS-AP in this instance, since in an effort to discriminate between viruses of potentially similar replication capacity, we wished to conduct multiple rounds of infectivity and inserted genes (such as the AP gene) may be lost on serial passage (23; L. M. King and I. Brierley, unpublished observation). At the end of the infectivity assays, RT-PCR and DNA sequencing were carried out to confirm that the original mutation(s) was retained and that no other mutations had arisen within the frameshift region. Following transfection of DF1 cells as before, the infectious potential of released particles was tested by infecting new monolayers with a set volume (500 μl  1:10 dilution, or 50 μl 1:100 dilution) of sterile-filtered, p27-containing culture medium from transfected cells (infection round 1). After 3 days, supernatant virus was again used (at the same two dilutions) to infect cells, and this was repeated once more, so that in total three rounds of infection were completed. Analysis of cell-associated and supernatant virus from all three rounds of infection was carried out by Western blotting and RT assays, and a selection of the data from the third round is shown in Fig. 6. The two Western blots in Fig. 6 are from passages of virus at the highest (Fig. 6A) and lowest (Fig. 6B) concentrations; we therefore expected that any effect of the various mutations on viability would be most evident in Fig. 6B. As can be seen in Fig. 6B, only two viruses, pRCAS-SRV1 and pRCAS-IBV2, replicated as well as the wild-type virus, and these viruses retained wild-type levels of RT activity (Fig. 6C). Throughout the infectivity assay, viruses containing a combination of slippery sequence and pseudoknot changes (pRCAS-25-SRV, -12-IBV2, and -25-IBV2) were able to replicate to some extent depending on the passage dilution, except pRCAS-12-SRV, which did not replicate at all. As observed before, individual mutations destabilizing the pseudoknot (pRCAS-9 and -12) also prevented replication. To obtain a more quantitative assessment of replication capacity, we carried out TCID50 assays (Table 2). From this analysis, it was possible to group the viruses into three categories: nonreplicating (pRCAS-9 and -12), with a TCID50 some 4 to 5 log lower than the wild-type virus; moderate replication (pRCAS-12-IBV2, -25, -25-IBV2, and -25-SRV) with TCID50 reduced by 1 to 2 log; and fully replicating (pRCAS, -IBV2, and -SRV). The pRCAS-12-SRV clone was nonreplicating in the infectivity assay and had a TCID50 titer some 2 to 3 log lower than the wild-type virus, indicating that this is the lower limit of sensitivity of the infectivity assay. That pRCAS-IBV2 and -SRV were not attenuated in cell culture indicates that a 1.5- to 2-fold stimulation in frameshifting efficiency is not detrimental to RSV replication. It also demonstrates that the amino acid changes at the C terminus of the viral protease are tolerated, as we had predicted (Fig. 5). In light of the latter observation, the reduced replication seen with pRCAS-25-IBV2 and -25-SRV could therefore be ascribed either to the change in frameshift efficiency or to the amino acid change associated with the RCAS-25 mutation. To test this, we compared the replication of pRCAS-25 with that of pRCAS-23, which retains the amino acid change but was less stimulatory to frameshifting in RRL (Fig. 2D). In three rounds of infection, we found little difference between the two viruses (data not shown). Both replicated less well than pRCAS-WT, with reduced supernatant p27 and RT activity, and this was reflected in the TCID50 (Table 2). These results reveal that the valine-to-glycine mutation in the spacer region between PR and RT results in a 10-fold reduction in virus replication and that the reduced fitness of pRCAS-25 and -25-SRV is almost certainly due to this change rather than an effect on frameshifting. Based on the in vitro frameshift assays, it appears, therefore, that RSV replication can tolerate up to a 3- to 4-fold stimulation of frameshifting without a detrimental effect on replication.
1:10 dilution, or 50 μl 1:100 dilution) of sterile-filtered, p27-containing culture medium from transfected cells (infection round 1). After 3 days, supernatant virus was again used (at the same two dilutions) to infect cells, and this was repeated once more, so that in total three rounds of infection were completed. Analysis of cell-associated and supernatant virus from all three rounds of infection was carried out by Western blotting and RT assays, and a selection of the data from the third round is shown in Fig. 6. The two Western blots in Fig. 6 are from passages of virus at the highest (Fig. 6A) and lowest (Fig. 6B) concentrations; we therefore expected that any effect of the various mutations on viability would be most evident in Fig. 6B. As can be seen in Fig. 6B, only two viruses, pRCAS-SRV1 and pRCAS-IBV2, replicated as well as the wild-type virus, and these viruses retained wild-type levels of RT activity (Fig. 6C). Throughout the infectivity assay, viruses containing a combination of slippery sequence and pseudoknot changes (pRCAS-25-SRV, -12-IBV2, and -25-IBV2) were able to replicate to some extent depending on the passage dilution, except pRCAS-12-SRV, which did not replicate at all. As observed before, individual mutations destabilizing the pseudoknot (pRCAS-9 and -12) also prevented replication. To obtain a more quantitative assessment of replication capacity, we carried out TCID50 assays (Table 2). From this analysis, it was possible to group the viruses into three categories: nonreplicating (pRCAS-9 and -12), with a TCID50 some 4 to 5 log lower than the wild-type virus; moderate replication (pRCAS-12-IBV2, -25, -25-IBV2, and -25-SRV) with TCID50 reduced by 1 to 2 log; and fully replicating (pRCAS, -IBV2, and -SRV). The pRCAS-12-SRV clone was nonreplicating in the infectivity assay and had a TCID50 titer some 2 to 3 log lower than the wild-type virus, indicating that this is the lower limit of sensitivity of the infectivity assay. That pRCAS-IBV2 and -SRV were not attenuated in cell culture indicates that a 1.5- to 2-fold stimulation in frameshifting efficiency is not detrimental to RSV replication. It also demonstrates that the amino acid changes at the C terminus of the viral protease are tolerated, as we had predicted (Fig. 5). In light of the latter observation, the reduced replication seen with pRCAS-25-IBV2 and -25-SRV could therefore be ascribed either to the change in frameshift efficiency or to the amino acid change associated with the RCAS-25 mutation. To test this, we compared the replication of pRCAS-25 with that of pRCAS-23, which retains the amino acid change but was less stimulatory to frameshifting in RRL (Fig. 2D). In three rounds of infection, we found little difference between the two viruses (data not shown). Both replicated less well than pRCAS-WT, with reduced supernatant p27 and RT activity, and this was reflected in the TCID50 (Table 2). These results reveal that the valine-to-glycine mutation in the spacer region between PR and RT results in a 10-fold reduction in virus replication and that the reduced fitness of pRCAS-25 and -25-SRV is almost certainly due to this change rather than an effect on frameshifting. Based on the in vitro frameshift assays, it appears, therefore, that RSV replication can tolerate up to a 3- to 4-fold stimulation of frameshifting without a detrimental effect on replication.
Fig 6.
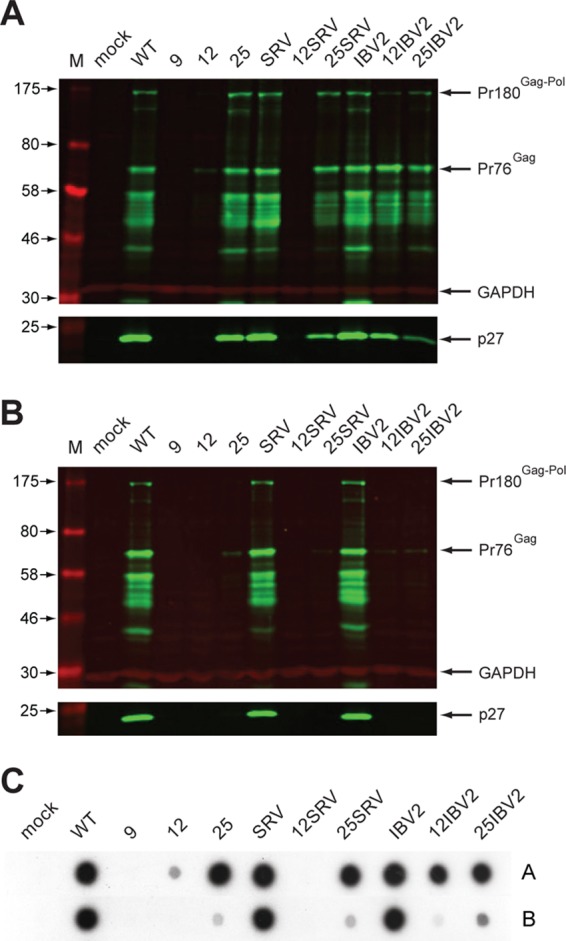
Multipassage infectivity assay. (A, B) DF1 cells were transfected with pRCAS-WT or mutant derivatives, and released virus was used subsequently to infect new cells as detailed in Materials and Methods. Three sequential rounds of infection were carried out. In round 1, two different volumes (500 μl or 50 μl, corresponding to a 1:10 or 1:100 dilution, respectively) of medium from transfected cells were used to infect two new dishes of cells. Three days later, the procedure was repeated, generating four dishes in infection round 2, and the process was repeated again, generating eight infected-cell dishes in round 3 (dishes 3.1 to 3.8). Cell lysates (upper panel) and supernatant virus (lower panel) from the eight dishes were analyzed by SDS-PAGE (8% and 15% gels, respectively) and Western blotting using a polyclonal anti-CA (p27) serum. Molecular sizes (in kDa) are indicated on the left. Shown in panel A is the Western blot from dish 3.1, infected with medium that had received the larger volume of medium at each round of the infectivity assay (500 μl for each of the three rounds), and in panel B, that from dish 3.8, which received the lowest (50 μl for each of the three rounds). GAPDH was used as a loading control. All viral proteins were detected with a green fluorescent secondary antibody, and GAPDH with a red fluorescent secondary antibody. (C) Reverse transcriptase activities of supernatant virus harvested from dishes 3.1 (row A) and 3.8 (row B). Virus particles were harvested from 1 ml culture medium by ultracentrifugation, and reverse transcriptase activity was assayed. Incorporation of [α-32P]dTTP was visualized by autoradiography.
Table 2.
Infectivity assay results for pRCAS and mutant derivativesa
| Construct | Log TCID50/ml |
Behavior in infectivity assayb | Relative −1 FS efficiency in DF1 cellsc | |
|---|---|---|---|---|
| Expt 1 | Expt 2 | |||
| pRCAS | −7.50 | −7.83 | ++++ | 100 |
| pRCAS-9 | −2.50 | −2.50 | − | 13 |
| pRCAS-12 | −3.50 | −4.17 | − | 13 |
| pRCAS-23 | −6.83 | −7.17 | ++ | 138 |
| pRCAS-25 | −6.38 | −6.63 | ++ | 146 |
| pRCAS-SRV | −7.83 | −7.63 | ++++ | 151 |
| pRCAS-12-SRV | −5.16 | −5.20 | − | 19 |
| pRCAS-25-SRV | −6.63 | −6.56 | + | 229 |
| pRCAS-IBV2 | −7.25 | −7.56 | ++++ | 225 |
| pRCAS-12-IBV2 | −6.50 | −6.56 | + | 35 |
| pRCAS-25-IBV2 | −6.50 | −6.63 | + | 306 |
| pRCAS-11 | −3.38 | −3.17 | − | ND |
| pRCAS-13 | −3.17 | −3.17 | − | ND |
| pRCAS-14 | −6.50 | −6.63 | ++++ | ND |
| pRCAS-15 | −7.17 | −7.50 | ++++ | ND |
| pRCAS-16 | −7.50 | −6.83 | ++++ | ND |
| pRCAS-17 | −6.83 | −7.25 | ++++ | ND |
Values are averages from the two data sets illustrated in Fig. 7. FS, frameshifting; ND, not determined.
Qualitative estimate of the behavior of the viruses in multiple-passage infectivity assays.
Shown for comparison, with the wild-type value set to 100.
Ribosomal frameshifting efficiencies in vivo.
The comparisons of replication capacity and frameshift efficiency above are based on frameshift values measured in vitro. However, from these data, we could not account for the reduced replication of pRCAS-12-SRV, since this mutant showed activity in RRL (at 39°C) close to that of the wild-type signal. To permit a more appropriate comparison, we determined the frameshifting efficiency of the mutations in transfected DF1 cells, following subcloning of the frameshift region between the Renilla and firefly luciferase genes of the dual-luciferase frameshift reporter plasmid p2luc (20). As can be seen in Fig. 7, the pattern of frameshift efficiencies measured in vivo was the same as that seen in RRL, but there were some quantitative differences. Most noticeably, those signals containing the pseudoknot destabilization mutation (RCAS-12) had lower efficiencies, particular pRCAS-12-SRV, which retained only about one-quarter of the wild-type frameshift efficiency in DF1 cells. The reduced frameshift activity in vivo almost certainly accounts for the reduced fitness of this virus. With the stimulatory mutations, the maximum values observed were a little lower than those seen in RRL at 39°C but were still around two to three times that of the wild-type signal. From this, we can conclude that RSV can tolerate at least a 2-fold stimulation in frameshifting (e.g., pRCAS-IBV2 [Table 2]). For pRCAS-25-IBV2, while the aforementioned amino acid change associated with the “25” mutation is a major factor in the reduced infectivity of this virus, we cannot rule out that the 3-fold increase in frameshifting seen with this virus may also contribute to it, as in infectivity assays, pRCAS-25-IBV2 generally replicated less well than pRCAS-25, pRCAS-IBV2, and pRCAS-SRV. It should be noted that in DF1 cells, the frameshift efficiencies of RCAS-23 and RCAS-25 (Fig. 7; Table 2) were very similar, in contrast to what was found in RRL (Fig. 2D; Table 1). Thus, the stimulatory effect of the predicted extension to pseudoknot stem 1 in RCAS-25 appears to be less marked in vivo (see Discussion).
Fig 7.
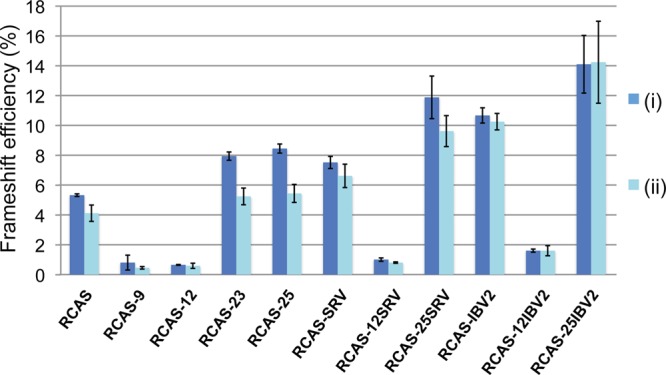
Ribosomal frameshifting efficiencies in DF-1 cells. DF1 cells were transfected with p2lucRCAS or a mutant derivative, and 24 h later, lysates were prepared and assayed for Renilla and firefly luciferase. Frameshift efficiencies were determined by comparing luciferase activities to an in-frame control construct. Each data point represents the mean value (± standard deviation) from six separate transfections. Two separate experiments are shown (i and ii).
We also addressed the question of whether an enhanced frameshifting efficiency led to an increased proportion of Gag-Pol in virions. In this experiment, we focused on RCAS-IBV2 and RCAS-25-IBV2, which showed a stimulation of frameshifting in DF1 cells of 2- and 3-fold, respectively. The RT/p27 ratios of released virions were calculated using data from quantitation of Western blots to calculate p27 levels and RT assays to estimate the level of released RT. To minimize any potential artifacts arising from signal nonlinearity (Western blots) or dose-response behavior (RT assays), serial 2-fold dilutions were tested for each sample and the experiment was performed in triplicate. As can be seen in Fig. 8, when the RT/p27 ratio data were normalized to the wild-type virus, virions from RCAS-IBV2 and RCAS-25-IBV2-infected cells contained, respectively, some 1.37- and 3.66-fold more RT. These data suggest that the assembly process in RSV does not preclude incorporation of excess Gag-Pol.
Fig 8.

Estimation of RT content of virions. (A) DF1 cells were infected with pRCASwt, pRCASIBV2, or pRCAS25IBV2, supernatant virus was harvested by centrifugation, and serial dilutions were subjected to Western blotting with the anti-p27 antibody (left) and to RT activity assays (right). The graphs show quantification of p27 and RT levels for neat virus and three dilutions. (B) The RT/p27 ratio at each dilution was calculated for each virus (left) and is shown normalized to the ratio for pRCASwt (right).
Role of the pseudoknot in replication.
To confirm a requirement for the RSV pseudoknot in virus replication, complementary point mutations were introduced into PK2 or PK4 to destabilize stem 2* (RCAS-11 and RCAS-13), or both mutations were made, which should be compensatory and reform stem 2 (RCAS-14 [Fig. 9]). These changes were made in the context of pMV-RCAS for in vitro translation analysis and in the infectious RCAS clone. In the case of RCAS-11, the introduced mutations were silent with respect to the encoded amino acids, but with RCAS-13, two amino acid substitutions were present (V to E and A to G). As can be seen in Fig. 9, the individual mutations reduced frameshifting some 2- to 3-fold in RRL and blocked virus replication, yet in the double mutant constructs, frameshifting was at a level close to that of the wild type, and virus replication was restored. Thus, the pseudoknot forms in virus-infected cells and is required for replication. Additionally, the amino acid substitutions present in RCAS-13 and RCAS-14 are clearly tolerated by the virus. We also investigated whether the lower region of stem 1 of the pseudoknot was required for frameshifting and replication in vivo. In RSV, the bases immediately 3′ of the slippery sequence are thought to be base paired, but given that this region in most frameshift sites normally acts as a spacer region separating the slippery sequence and stimulatory RNA, it is questionable whether such base pairing has functional relevance. To investigate this, we introduced complementary and compensatory changes into the GU pair within this region (Fig. 9A), chosen to retain the same amino acid sequence, and tested frameshifting and replication as above. We found that the mutations had no effect on frameshifting or virus replication, indicating that base pairing in this region is not required for function.
Fig 9.

Mutational analysis of the RCAS frameshifting signal. (A) Mutations in pMV-RCAS are shown, with pseudoknot mutations color coded as described for Fig. 2A. The PK2 and PK4 regions, which form stem 2* of the pseudoknot, are in bold. The stop codon of the gag ORF is in red. (B) Ribosomal frameshift assays of pMV-RCAS variants were carried out in RRL as described in the legend to Fig. 2. Molecular sizes (in kDa) are indicated on the left. (C) DF1 cells were transfected with pRCAS-WT or mutant derivatives, and released virus was used subsequently to infect new cells. Cell lysates (upper panel) and supernatant virus (lower panel) were analyzed by SDS-PAGE (8% and 15% gels, respectively) and Western blotting using a polyclonal anti-CA (p27) serum. (D) Reverse transcriptase activities were assayed as described in Materials and Methods and quantified by scintillation counting.
DISCUSSION
This study investigated the effect on RSV replication of modulating ribosomal frameshifting frequency. Based on infectivity assays and frameshift activity measurements in DF1 cells, it was found that an ∼3-fold reduction in frameshifting generated a severe replication defect and an ∼8-fold reduction essentially abolished replication. That a decrease in frameshifting is detrimental to replication is unsurprising. Although reducing Gag-Pol levels would not affect particle assembly and release (49), it would decrease Gag-Pol incorporation into virions with consequent effects on subsequent rounds of infection. In RSV, the 3-fold decrease in frameshift efficiency led to a marked (10-fold) reduction in infectivity, and this has also been observed with HIV-1 (5, 46). Regarding stimulation of frameshifting, we found that RSV replication was largely unaffected by a 3-fold increase in Gag-Pol levels. In contrast, in the original study of the role of frameshifting in virus replication, Dinman and Wickner (10) observed that a 2-fold stimulation in frameshifting in the yeast double-stranded RNA totivirus L-A abolished replication. However, given the differences in the mechanisms of RNA packaging and capsid formation between the totivirus and retrovirus families (17), this is perhaps unsurprising. Similar to what is observed in RSV, an ∼3.5-fold increase in the ratio of HIV-1 Gag-Pol to Gag (generated by cotransfection of plasmids expressing Gag and Gag-Pol separately) had little effect on virus replication, although higher levels of Gag-Pol were detrimental (44). At the level of 3.5-fold stimulation of HIV-1 Gag-Pol levels, no obvious change was seen in the proportion of Gag and Gag-Pol in virions, with the phenotypic effect of increased Gag-Pol/Gag ratios being attributed to reduced formation of stable virion RNA dimers (44). In contrast, we found that RSV virions derived from up mutants RCAS-25 and RCAS-25-IBV2 had an increased Gag-Pol (RT) content, arguing against a specific mechanism that selects a precise Gag/Gag-Pol ratio at the assembly step. However, further work will be required to substantiate this observation, including the development of strategies to further increase the frameshifting efficiency and a more detailed characterization of virion content and morphology. A factor that restricted our ability to define an upper limit to frameshift efficiency in RSV was that the frameshift-stimulatory effect of the RCAS-25 mutation was not as evident at higher temperatures (in RRL) or in transfected cells, whether this mutation was introduced alone or in the background of the frameshift-stimulatory slippery sequence changes. Different thermodynamic stability or refolding kinetics of engineered mutants at the temperatures or ionic conditions found in cultured cells could account for this (see below).
An interesting observation was the mildly detrimental effect on virus replication of the amino acid substitution present in RCAS-23 and RCAS-25 (and derivatives), a valine-to-glycine substitution within the seven-amino-acid spacer between protease and reverse transcriptase (Fig. 2C). Stewart and Vogt (45) have proposed that the C-terminal region of Gag containing this seven-amino-acid spacer contains a leucine zipper-like motif of hydrophobic repeats favorable for formation of a coiled coil—but such a coiled coil was not found in active protease dimers. Stewart and Vogt hypothesized that before virion maturation, Gag-Pol polyproteins in contact with one another or with Gag could be held in an extended conformation through coiled-coil interactions, preventing folding of the beta sheet required for protease dimerization and activation (27, 50). This might be a mechanism used by RSV to prevent intracellular activation of the high levels of viral protease. In RCAS-23 and RCAS-25, the valine adjacent to one of the leucine residues crucial in the formation of the putative coiled coil is mutated to glycine, and the loss of hydrophobicity or a conformational difference caused by the lack of a bulky side chain could potentially disrupt coiled-coil formation or dimerization to some extent, leading to premature activation of the protease. While this could account for the replication deficit of pRCAS-23 and pRCAS-25, the patterns of intracellular processing observed in viruses containing this mutation did not differ noticeably from that of the wild-type virus; thus, there may be an alternative explanation for the phenotypic effect of this mutation.
While RSV has the distinction of being the first frameshifting signal of this class to be described (25), the stimulatory RNA is one of the more complex. Initial studies characterized it as a stem-loop with substructures within the loop region, although 3′ sequences also appeared to contribute to frameshifting (26). It was later proposed that a downstream region (PK4) could form a pseudoknot via interaction with the sequence in loop 1 designated PK2 (Fig. 1) (6), and this was subsequently confirmed in in vitro translation reactions and by RNA structure mapping (33). In the present work, we show for the first time that the pseudoknot forms in virus-infected cells and is essential for frameshifting. We also found, unexpectedly, that the formation of stem 3 (but not stem 2) within the main loop is important for efficient frameshifting in vitro and virus viability in infected cells. Previous studies of the Prague C strain of RSV had indicated that neither stem 2 nor stem 3 was important for frameshifting and that their destabilization (stem 2) or deletion (both stem 2 and stem 3) actually leads to a 50% stimulation of −1 frameshifting in vitro (33). In the Schmidt-Ruppin strain studied here (RCAS) and also in ALV (data not shown), the destabilization of stem 2 leads, perversely, to a reduction in frameshifting. This contrasting effect was unexpected, as the frameshift regions of RCAS, Prague C, and ALV differ by only four nucleotides, three of which appear to participate in phylogenetically conserved base pairing within the stems. The fourth variant base, located at position 2502 in the genome within loop 1, is a U in RCAS and ALV, yet a C in Prague C. It is possible, therefore, that the identity of this base could account for the differential effects of stem 2 destabilization in the various strains, perhaps through interactions across the main loop. If the RSV pseudoknot folds in a manner similar to that shown in Fig. 1B, the residue at position 2502 and stems 2 and 3 could be in close proximity at the junction of the main pseudoknot stems. It may be significant that mutation of the adjacent base C2500 to G stimulates frameshifting some 1.5-fold in vitro in the Prague C background (33) but the same change has no effect in ALV (data not shown). Similarly, changing U2502 to C2502 in the ALV background reduces frameshifting about 2-fold (Table 1). Together, these experiments hint at the potential for complex and contributory interactions between major loop elements of the RSV pseudoknot. Further support comes from the studies of Stewart and Vogt (45), who observed that short insertions within the major loop (loop 1 in Fig. 1) were detrimental to frameshifting, presumably by distorting the global architecture. Despite this complexity, it is clear that the formation of the pseudoknot is not absolutely required for virus viability if frameshifting is enhanced by an alternative route. In RCAS-12-IBV2, containing the IBV slippery sequence in combination with a pseudoknot destabilization mutation, the reduction of infectivity was only 1 log (Table 2), indicating that the more active slippery sequence can compensate for the reduced activity of the stimulatory RNA in this case.
It has long been proposed that frameshifting could be exploited as a target for antiviral intervention, and various small-molecule modulators have been described (1, 3, 11, 12, 14, 18, 24, 32, 34, 36, 38, 40, 48, 52). At present, the point at which increasing frameshifting compromises replication is not known, but at least a 3-fold stimulation (in RSV and HIV-1) would seem to be required, and no current small-molecule stimulator is sufficiently active to stimulate frameshifting to these levels (11, 24, 36). Taking into account the present data and earlier studies, it is clear that small-molecule inhibitors of frameshifting would seem to be the favored class, since even a 2-fold effect would be likely to severely compromise replication.
ACKNOWLEDGMENTS
This work was supported by grants from the Medical Research Council UK, the Biotechnology and Biological Sciences Research Council UK (to I.B.), and the Wellcome Trust (to N.I.).
We thank Volker Vogt for providing the RCAS/RCAS-AP vectors, anti-RSV sera, and advice, Jean-Luc Darlix for the pLADI vector, and Myriam Scherer for help in pilot experiments.
Footnotes
Published ahead of print 15 August 2012
REFERENCES
- 1. Aupeix-Scheidler K, et al. 2000. Inhibition of in vitro and ex vivo translation by a transplatin-modified oligo (2′-O-methylribonucleotide) directed against the HIV-1 gag-pol frameshift signal. Nucleic Acids Res. 28:438–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Babé LM, Pichuantes S, Craik CS. 1991. Inhibition of HIV protease activity by heterodimer formation. Biochemistry 30:106–111 [DOI] [PubMed] [Google Scholar]
- 3. Bidou L, Rousset JP, Namy O. 2010. Translational errors: from yeast to new therapeutic targets. FEMS Yeast Res. 10:1070–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bieth E, Darlix JL. 1992. Complete nucleotide sequence of a highly infectious avian leukosis virus. Nucleic Acids Res. 20:367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Biswas P, Jiang X, Pacchia AL, Dougherty JP, Peltz SW. 2004. The human immunodeficiency virus type 1 ribosomal frameshifting site is an invariant sequence determinant and an important target for antiviral therapy. J. Virol. 78:2082–2087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brierley I, Digard P, Inglis SC. 1989. Characterization of an efficient coronavirus ribosomal frameshifting signal: requirement for an RNA pseudoknot. Cell 57:537–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brierley I, Gilbert RJC, Pennell S. 2010. Pseudoknot-dependent programmed -1 ribosomal frameshifting: structures, mechanisms and models, p 149–174 In Atkins JF, Gesteland RF. (ed), Recoding: expansion of decoding rules enriches gene expression. Springer, New York, NY [Google Scholar]
- 8. Brierley I, Jenner AJ, Inglis SC. 1992. Mutational analysis of the “slippery-sequence” component of a coronavirus ribosomal frameshifting signal. J. Mol. Biol. 227:463–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cherry E, et al. 1998. Characterization of human immunodeficiency virus type-1 (HIV-1) particles that express protease-reverse transcriptase fusion proteins. J. Mol. Biol. 284:43–56 [DOI] [PubMed] [Google Scholar]
- 10. Dinman JD, Wickner RB. 1992. Ribosomal frameshifting efficiency and Gag/Gag-Pol ratio are critical for yeast M1 double-stranded RNA virus propagation. J. Virol. 66:3669–3676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dinman JD, Kinzy TG. 1997. Translational misreading: mutations in translation elongation factor 1alpha differentially affect programmed ribosomal frameshifting and drug sensitivity. RNA 3:870–881 [PMC free article] [PubMed] [Google Scholar]
- 12. Dinman JD, Ruiz-Echevarria MJ, Peltz SW. 1998. Translating old drugs into new treatments: ribosomal frameshifting as a target for antiviral agents. Trends Biotechnol. 16:190–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dulude D, Berchiche YA, Gendron K, Brakier-Gingras L, Heveker N. 2006. Decreasing the frameshift efficiency translates into an equivalent reduction of the replication of the human immunodeficiency virus type 1. Virology 345:127–136 [DOI] [PubMed] [Google Scholar]
- 14. Dulude D, Théberge-Julien G, Brakier-Gingras L, Heveker N. 2008. Selection of peptides interfering with a ribosomal frameshift in the human immunodeficiency virus type 1. RNA 14:981–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Enssle J, Jordan I, Mauer B, Rethwilm A. 1996. Foamy virus reverse transcriptase is expressed independently from the Gag protein. Proc. Natl. Acad. Sci. U. S. A. 93:4137–4141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Felsenstein KM, Goff SP. 1988. Expression of the Gag-Pol fusion protein of Moloney murine leukemia virus without Gag protein does not induce virion formation or proteolytic processing. J. Virol. 62:2179–2182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fujimura T, Esteban R, Esteban LM, Wickner RB. 1990. Portable encapsidation signal of the L-A double-stranded RNA virus of S. cerevisiae. Cell 62:819–828 [DOI] [PubMed] [Google Scholar]
- 18. Gareiss PC, Miller BL. 2009. Ribosomal frameshifting: an emerging drug target for HIV. Curr. Opin. Investig. Drugs 10:121–128 [PubMed] [Google Scholar]
- 19. Gheysen D, et al. 1989. Assembly and release of HIV-1 precursor Pr55gag virus-like particles from recombinant baculovirus-infected insect cells. Cell 59:103–112 [DOI] [PubMed] [Google Scholar]
- 20. Grentzmann G, Ingram JA, Kelly PJ, Gesteland RF, Atkins JF. 1998. A dual-luciferase reporter system for studying recoding signals. RNA 4:479–486 [PMC free article] [PubMed] [Google Scholar]
- 21. Hill MK, Shehu-Xhilaga M, Crowe SM, Mak J. 2002. Proline residues within spacer peptide p1 are important for human immunodeficiency virus type 1 infectivity, protein processing, and genomic RNA dimer stability. J. Virol. 76:11245–11253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hughes SH, Greenhouse JJ, Petropoulos CJ, Sutrave P. 1987. Adaptor plasmids simplify the insertion of foreign DNA into helper-independent retroviral vectors. J. Virol. 61:3004–3012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hughes SH, et al. 2008. The RCAS system. http://home.ncifcrf.gov/hivdrp/RCAS/index.html
- 24. Hung M, Patel P, Davis S, Green SR. 1998. Importance of ribosomal frameshifting for human immunodeficiency virus type 1 particle assembly and replication. J. Virol. 72:4819–4824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jacks T, Varmus HE. 1985. Expression of the Rous sarcoma virus pol gene by ribosomal frameshifting. Science 230:1237–1242 [DOI] [PubMed] [Google Scholar]
- 26. Jacks T, Madhani HD, Masiarz FR, Varmus HE. 1988. Signals for ribosomal frameshifting in the Rous sarcoma virus gag-pol region. Cell 55:447–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jaskólski M, Miller M, Rao JK, Leis J, Wlodawer A. 1990. Structure of the aspartic protease from Rous sarcoma retrovirus refined at 2Å resolution. Biochemistry 29:5889–5898 [DOI] [PubMed] [Google Scholar]
- 28. Karacostas V, Wolffe EJ, Nagashima K, Gonda MA, Moss B. 1993. Overexpression of the HIV-1 Gag-Pol polyprotein results in intracellular activation of HIV-1 protease and inhibition of assembly and budding of virus-like particles. Virology 193:661–671 [DOI] [PubMed] [Google Scholar]
- 29. Katz RA, et al. 1982. Restriction endonuclease and nucleotide sequence analyses of molecularly cloned unintegrated avian tumor virus DNA: structure of large terminal repeats in circle junctions. J. Virol. 42:346–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- 31. Leiherer A, Ludwig C, Wagner R. 2009. Uncoupling human immunodeficiency virus type 1 Gag and Pol reading frames: role of the transframe protein p6* in viral replication. J. Virol. 283:7210–7220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Marcheschi RJ, Mouzakis KD, Butcher SE. 2009. Selection and characterization of small molecules that bind the HIV-1 frameshift site RNA. ACS Chem. Biol. 4:844–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Marczinke B, Fisher R, Vidaković M, Bloys AJ, Brierley I. 1998. Secondary structure and mutational analysis of the ribosomal frameshift signal of Rous sarcoma virus. J. Mol. Biol. 284:205–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McNaughton BR, Gareiss PC, Miller BL. 2007. Identification of a selective small-molecule ligand for HIV-1 frameshift-inducing stem-loop RNA from an 11,325 member resin bound dynamic combinatorial library. J. Am. Chem. Soc. 129:11306–11307 [DOI] [PubMed] [Google Scholar]
- 35. Miller M, Jaskólski M, Rao JK, Leis J, Wlodawer A. 1989. Crystal structure of a retroviral protease proves relationship to aspartic protease family. Nature 337:576–579 [DOI] [PubMed] [Google Scholar]
- 36. Miyauchi K, et al. 2006. Rapid propagation of low-fitness drug-resistant mutants of human immunodeficiency virus type 1 by a streptococcal metabolite sparsomycin. Antivir. Chem. Chemother. 17:167–174 [DOI] [PubMed] [Google Scholar]
- 37. Neuman BW, et al. 2005. Inhibition, escape, and attenuated growth of severe acute respiratory syndrome coronavirus treated with antisense morpholino oligomers. J. Virol. 79:9665–9676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Palde PB, Ofori LO, Gareiss PC, Lerea J, Miller BL. 2010. Strategies for recognition of stem-loop RNA structures by synthetic ligands: application to the HIV-1 frameshift stimulatory sequence. J. Med. Chem. 53:6018–6027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Park J, Morrow CD. 1991. Overexpression of the Gag-Pol precursor from human immunodeficiency virus type 1 proviral genomes results in efficient proteolytic processing in the absence of virion production. J. Virol. 65:5111–5117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Park SJ, Kim YG, Park HJ. 2011. Identification of RNA pseudoknot-binding ligand that inhibits the -1 ribosomal frameshifting of SARS-coronavirus by structure-based virtual screening. J. Am. Chem. Soc. 133:10094–10100 [DOI] [PubMed] [Google Scholar]
- 41. Plant EP, Rakauskaitė R, Taylor DR, Dinman JD. 2010. Achieving a golden mean: mechanisms by which coronaviruses ensure synthesis of the correct stoichiometric ratios of viral proteins. J. Virol. 84:4330–4340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Reed LJ, Muench H. 1938. A simple method of estimating fifty percent endpoints. Am. J. Epidemiol. 27:493–497 [Google Scholar]
- 43. Schwartz DE, Tizard R, Gilbert W. 1983. Nucleotide sequence of Rous sarcoma virus. Cell 32:853–869 [DOI] [PubMed] [Google Scholar]
- 44. Shehu-Xhilaga M, Crowe SM, Mak J. 2001. Maintenance of the Gag/Gag-Pol ratio is important for human immunodeficiency virus type 1 RNA dimerization and viral infectivity. J. Virol. 75:1834–1841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Stewart L, Vogt VM. 1994. Proteolytic cleavage at the Gag-Pol junction in avian leukosis virus: differences in vitro and in vivo. Virology 204:45–59 [DOI] [PubMed] [Google Scholar]
- 46. Telenti A, et al. 2002. Analysis of natural variants of the human immunodeficiency virus type 1 gag-pol frameshift stem-loop structure. J. Virol. 76:7868–7873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tibbles KW, Brierley I, Cavanagh D, Brown TD. 1995. A region of the coronavirus infectious bronchitis virus 1a polyprotein encoding the 3C-like protease domain is subject to rapid turnover when expressed in rabbit reticulocyte lysate. J. Gen. Virol. 76:3059–3070 [DOI] [PubMed] [Google Scholar]
- 48. Vickers TA, Ecker DJ. 1992. Enhancement of ribosomal frameshifting by oligonucleotides targeted to the HIV gag-pol region. Nucleic Acids Res. 20:3945–3953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Voynow SL, Coffin JM. 1985. Truncated Gag-related proteins are produced by large deletion mutants of Rous sarcoma virus and form virus particles. J. Virol. 55:79–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Weber IT. 1990. Comparison of the crystal structures and intersubunit interactions of human immunodeficiency and Rous sarcoma virus proteases. J. Biol. Chem. 265:10492–10496 [PubMed] [Google Scholar]
- 51. Yoshinaka Y, Katoh I, Copeland TD, Oroszlan S. 1985. Murine leukemia virus protease is encoded by the gag-pol gene and is synthesized through suppression of an amber termination codon. Proc. Natl. Acad. Sci. U. S. A. 82:1618–1622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yu CH, Noteborn MH, Olsthoorn RC. 2010. Stimulation of ribosomal frameshifting by antisense LNA. Nucleic Acids Res. 38:8277–8283 [DOI] [PMC free article] [PubMed] [Google Scholar]