

Abstract
Mammalian prions are thought to consist of misfolded aggregates (protease-resistant isoform of the prion protein [PrPres]) of the cellular prion protein (PrPC). Transmissible spongiform encephalopathy (TSE) can be induced in animals inoculated with recombinant PrP (rPrP) amyloid fibrils lacking mammalian posttranslational modifications, but this induction is inefficient in hamsters or transgenic mice overexpressing glycosylphosphatidylinositol (GPI)-anchored PrPC. Here we show that TSE can be initiated by inoculation of misfolded rPrP into mice that express wild-type (wt) levels of PrPC and that synthetic prion strain propagation and selection can be affected by GPI anchoring of the host's PrPC. To create prions de novo, we fibrillized mouse rPrP in the absence of molecular cofactors, generating fibrils with a PrPres-like protease-resistant banding profile. These fibrils induced the formation of PrPres deposits in transgenic mice coexpressing wt and GPI-anchorless PrPC (wt/GPI−) at a combined level comparable to that of PrPC expression in wt mice. Secondary passage into mice expressing wt, GPI−, or wt plus GPI− PrPC induced TSE disease with novel clinical, histopathological, and biochemical phenotypes. Contrary to laboratory-adapted mouse scrapie strains, the synthetic prion agents exhibited a preference for conversion of GPI− PrPC and, in one case, caused disease only in GPI− mice. Our data show that novel TSE agents can be generated de novo solely from purified mouse rPrP after amplification in mice coexpressing normal levels of wt and anchorless PrPC. These observations provide insight into the minimal elements required to create prions in vitro and suggest that the PrPC GPI anchor can modulate the propagation of synthetic TSE strains.
INTRODUCTION
Transmissible spongiform encephalopathies (TSEs), otherwise known as prion diseases, are fatal neurodegenerative diseases of mammals that include Creutzfeldt-Jakob disease (CJD) and scrapie. The nature of the infectious agent of TSEs, called the prion, has long been a subject of debate. In 1967, Griffith first proposed the concept that TSEs may be caused by a self-propagating form of a host protein (26). Years later, Prusiner later refined this concept and coined the term “prion” for the infectious agent, which had been enriched from scrapie-infected hamster brain (45). Subsequent studies showed the scrapie agent preparations primarily consisted of a detergent-insoluble, protease-resistant isoform of the prion protein (termed PrP27-30, PrPSc, or PrPres), suggesting that this novel infectious agent was a corrupted version of a normal host protein referred to as cellular prion protein (PrPC) (6, 14, 41, 46). Tremendous advances have been made since then to improve our understanding of TSE agents, including new transgenic (Tg) mouse models that spontaneously give rise to transmissible agents (28, 52). However, the most convincing evidence arguing for a protein-based agent is based on the development of improved systems for cell-free generation of TSE infectivity.
The most commonly used system at present is protein misfolding cyclic amplification (PMCA) (11, 47, 48). PMCA relies on propagation of PrPres through intermittent cycles of sonication and rest. Sonication is proposed to fragment large PrPres particles into smaller particles, each of which can direct the conversion of more PrPC molecules, thereby amplifying the number of seeding units in the reaction (3, 34, 35, 55). The energy introduced by sonication also likely induces partial unfolding of the PrPC substrate, which could assist the conversion process in seeded reactions initiated by PrPres from TSE-infected material and is a driving force in unseeded reactions involving de novo conversion of PrPC without the aid of preformed PrPres as a template. Polyanion cofactors also play a role in refolding of PrPC to assist conversion (20, 24). The exceptional efficiency of PMCA applied in a serial reaction format was instrumental to the first reports of seeded cell-free amplification of hamster TSE infectivity (11, 47). Formation of infectivity in PMCA reactions has since been reproduced using other TSE strains (10, 25, 27, 38, 40, 50). Prolonged rounds of serial PMCA can lead to de novo formation of TSE infectivity in unseeded reactions (5, 16, 20), but such experiments require extreme care to exclude cross-contamination (19). A recent study showed evidence of high infectivity titers in PMCA reactions with the hyper strain of transmissible mink encephalopathy, although incubation times per infectious unit were longer than those observed for brain-derived infectivity (51). A related report of a study performing careful quantitative comparisons of the PMCA-generated infectivity titer versus the amount of PrPres has drawn attention to the fact that a significant fraction of PrPres generated in PMCA reactions may not be infectious (32). Consistent with this observation, albeit in the context of mutant PrPC molecules, PMCA seeding activity can be present in samples with limited infectivity (39). This indicates that more work is needed to better characterize the products of PMCA reactions.
A limiting factor with classic PMCA reactions is the use of brain homogenate as a source of PrPC substrate, which has a complex molecular composition and thus complicates efforts to identify the essential components required to create infectivity. Deleault and coworkers broke this barrier with the use of partially purified brain-derived PrPC as a substrate and polyanions (plus lipids copurifying with the PrPC), reporting that these constituted the minimal components necessary for formation of infectivity (20). More recent studies have shown that the preferred cofactors differ between species (21) and that phosphatidylethanolamine is a promiscuous solitary cofactor active across diverse species (22). On the other hand, seeded PMCA reactions using highly purified recombinant PrP (rPrP) from bacteria have shown that infectivity can be propagated in the absence of any cofactors (31). When conducted in the presence of lipids and polyanions with rPrPC substrate, serial PMCA was reported to create high levels of PrPres and the reaction products induced TSE disease in wild-type (wt) mice with a high attack rate and short incubation period (60). Thus, there are conflicting reports on the importance of cofactors for generating TSE infectivity in vitro. The most likely interpretation may be that cofactors are not essential but can greatly improve the formation of infectivity. This highlights the fact that questions remain about the nature and molecular composition of TSE agents.
An alternative approach to cell-free creation of infectivity has involved the incubation of purified rPrP in the presence of denaturants that promote partial unfolding and spontaneous conversion to β-sheet-rich amyloid fibrils. This is intended to mimic events presumed to occur in sporadic CJD, where spontaneous misfolding of PrPC generates a very rare minimal seed of PrPres that is able to propagate itself before degradation by the cellular protein quality control machinery. Legname et al. (2004) were the first to report success with this experimental paradigm (33). Inoculation of rPrP fibril preparations induced TSE disease after a prolonged incubation period in transgenic mice vastly overexpressing (16×) N-terminally truncated PrPC, but many questions remained, in addition to concerns about the transgenic mouse model used (12, 54). Subsequent studies have shown that a myriad of synthetic prion strains can be generated with properties modulated by the reaction conditions, albeit still via primary passage in transgenic mice overexpressing mouse PrPC (17, 18). Potential issues associated with PrPC overexpression have been addressed in a hamster system, where inoculation of hamster rPrP fibrils subjected to a separate “annealing” procedure with heat, detergent, and normal brain homogenate allowed detection of infectivity after two serial passages in hamsters (35). Later, these authors reported that fibrils “annealed” in the presence of bovine serum albumin (BSA) instead of brain homogenate can also induce TSE disease but only after three serial passages, which required an evolution of the agent in hamsters in a process termed “deformed templating” (36). It is unclear how this high-temperature “annealing” would be reproduced by physiological processes in cells. In a very recent paper, TSE disease also arose after serial passage of one of three fibril preparations generated without annealing (37).
The present study was initiated shortly after the publication of the work of Legname et al. (33). To investigate methods of creating prions de novo solely from rPrP, we implemented three novel modifications. First, we developed new conditions for amyloid fibril formation using full-length mouse rPrP (rMoPrP) that produced fibrils with a PrPres-like protease-resistant profile. Second, since rPrP fibrils tend to be less protease resistant than brain-derived PrPres and thus might undergo more rapid clearance after inoculation, we tested the effect of conjugation of the fibrils to magnetic beads prior to inoculation in an attempt to enhance the persistence of the fibrils in vivo. Finally, for the initial passage we used transgenic mice coexpressing wt and glycosylphosphatidylinositol (GPI)-anchorless (GPI−) PrPC (wt/GPI− mice) but, importantly, at a combined level comparable to the level of PrPC expression in wt mice. This was based on the rationale that the rPrP fibrils might more efficiently convert GPI-anchorless PrPC since anchorless PrPC is molecularly similar as it is also largely unglycosylated. In addition, anchorless PrPC preferentially forms amyloid fibrils during TSE infection (15). Coexpression of wt PrPC allowed the option for conversion of wt PrPC in parallel. These experiments led to the generation of novel TSE strains with an increased preference for conversion of anchorless PrPC compared with that for known lab-adapted strains, suggesting that GPI anchoring of PrPC can influence the selection of synthetic TSE strains.
MATERIALS AND METHODS
Cloning of rMoPrP from residues 23 to 230.
DNA primers (forward primer AAGGAGATATACATATGAAAAAGCGGCCAAAGCCTG, reverse primer AATACGAATTCAAGCTTCTAGGATCTTCTCCCGTCGTAA) were designed according to the mouse Prnp sequence obtained from GenBank (accession number BC006703.1) and used to amplify a fragment encoding residues 23 to 230 from a cloned template of wt mouse Prnp. The amplicant was ligated into a pET41 (Novagen) vector as an NdeI/HindIII insert such that PrP residues 1 to 22 were replaced by methionine and a stop codon was inserted after serine 230. The insert was verified by sequencing.
Purification of rMoPrP (residues 23 to 230).
Expression and purification of rMoPrP was performed as described elsewhere, with the exception that the inclusion bodies were washed with 5 mM Tris, pH 7.5, 5 mM EDTA, 0.5% Triton X-100, followed by a solution consisting of BugBuster diluted 10-fold with water (1). Chromatography was performed using an Äkta Explorer (GE Healthcare) instrument and column located in a laboratory that has never been exposed to prions. The purified protein was stored at 4°C. The purity of the preparation was verified by SDS-PAGE and mass spectrometry (see Fig. S1 in the supplemental material). The infrared spectrum of the purified protein was consistent with an α-helix-rich structure, as expected for natively folded rPrP (data not shown).
Fibrillization reactions.
Fibrillization reaction setup and manipulation of reaction products to prepare rMoPrP inocula were performed in a newly renovated laboratory not previously used for prion research using new pipettors and centrifuge rotors. Aerosol-barrier pipette tips were used for all procedures. Purified rMoPrP was concentrated to ∼0.9 mg/ml using a Millipore Ultrafree 15 centrifugal filter device immediately before use. The reaction mixtures for H fibrils (fibrils derived from reaction conditions containing higher concentrations of denaturants) consisted of 300 μl of concentrated rMoPrP and 300 μl of 2 M guanidine hydrochloride (GdnHCl)–4.8 M urea in 50 mM phosphate buffer (pH 6.5). The reaction mixtures for L fibrils (fibrils derived from reaction conditions containing lower concentrations of denaturants) consisted of 300 μl of concentrated rMoPrP, 150 μl of 2 M GdnHCl–4.8 M urea in 50 mM phosphate buffer (pH 7.3), and 150 μl of 50 mM phosphate buffer (pH 6.5). H- and L-fibril reactions were then shaken continuously at 1,400 rpm and 37°C in an Eppendorf thermal mixer for 16 to 24 h. For L-fibril reaction mixtures only, the tubes were incubated for an additional 24 h at 37°C without shaking. To generate sufficient material for infection studies, 5 to 6 replicate reactions were conducted in parallel and the reaction mixtures were pooled.
TEM.
Samples were analyzed by transmission electron microscopy (TEM) at 80 kV essentially as described elsewhere (4), with the exception that 300-mesh copper grids coated with 3% Parlodion (Ted Pella) were also used. Samples were spotted directly onto freshly glow-discharged grids.
Conjugation of rMoPrP to magnetic beads.
Pooled fibrillization reactions were diluted 5-fold with phosphate-buffered saline (PBS; pH 7.4) and centrifuged in a Beckman TL100.3 rotor at 100,000 rpm for 30 min to pellet rMoPrP fibrils. The pellets were thoroughly resuspended in PBS and centrifuged again as described above. The final pellet combined from four reaction mixtures was resuspended in 500 μl of PBS with brief cup horn sonication. Tosyl-activated Dynal M-280 magnetic beads (0.5 ml) were washed with PBS, resuspended in 400 μl of PBS, and then mixed with 100 μl of washed fibrils (estimated to contain 100 μg rMoPrP) and incubated on a rotisserie at 37°C for 48 h. A Monomer control sample contained magnetic beads incubated with ∼100 μg of purified rMoPrP under the same conditions. Beads were collected with a magnet, washed 3 times with PBS, resuspended in 0.5 ml of PBS, and stored at 4°C. An additional control sample to test the effect of binding of PrP fibrils to the beads consisted of magnetic beads (0.5 ml, PBS washed) incubated as described above in 400 μl of 20 mM Tris, pH 8.0, 150 mM NaCl to preblock the primary amine-reactive tosyl functional groups and prevent covalent binding to PrP. These preblocked beads were then mixed with 100 μl of washed L fibrils immediately prior to inoculation to minimize the opportunity for the fibrils to associate with the beads by any mechanism. This control was designated “L fibrils (free).”
Animal inoculations.
Brain homogenates (10% in PBS) were diluted to the desired final concentration in phosphate-buffered balanced saline supplemented with 2% fetal bovine serum. Four- to 6-week-old mice were inoculated intracerebrally (i.c.) with 50 μl of diluted brain homogenate or 50 μl of freshly vortexed magnetic bead preparation. All magnetic bead inoculations were performed in a single session beginning with Monomer-infected control animals. Mouse strains used included Tga20 (which overexpress wt PrPC 8-fold) (23), C57BL/10, tg44+/+ (homozygous for GPI-anchorless PrPC on a C57BL/10 wt PrPC-knockout background) (13), and wt/GPI− (F1 of C57BL/10 crossed with tg44+/+). Animals were observed daily for signs of neurological disease and euthanized on confirmation of progressive neurological disease or at designated time points.
Protease digestions and immunoblotting.
To assay for PrPres in fibril reactions, 100 μl of the fibrillization reaction was first diluted into 400 μl of 50 mM Tris, pH 8.0, 150 mM NaCl (TN buffer) and centrifuged in a TLA120.1 rotor at 100,000 rpm for 30 min at 15°C. The pellets were resuspended with brief sonication in 273 μl of TN buffer. Proteinase K (PK; Calbiochem) digests were performed with 20-μl reaction mixtures using 19 μl of washed fibrils per digest and adjusted to 1% Sarkosyl plus various concentrations of PK, as indicated. Monomer control reactions contained a similar quantity of purified rMoPrP. After incubation at 37°C for 60 min, digests were terminated by addition of 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride (Pefabloc; final concentration, 5 mM). The samples were then mixed with 1 volume of 2× sample buffer and boiled for 10 min.
To assay for PrPres in brain, 10% homogenates in PBS were first prepared by either Dounce homogenization (with new Dounce devices dedicated to each sample) or an Omni-mixer with disposable generators (a new generator was used for every sample). To allow normalization between samples, the protein concentration in each homogenate was determined by bicinchoninic acid assay in the presence of 1% SDS. Samples were adjusted to 4 mg/ml protein (with PBS), 1% Sarkosyl, 50 U/ml Benzonase nuclease (from 500 U/ml stock in 10 mM Tris, 20 mM MgCl2, 20 mM NaCl, pH 8.0) and incubated at 37°C for 30 min. The samples were then PK digested (12 μg/ml PK) at 37°C for 60 min. PK digestion was terminated by addition of Pefabloc (4 mM) and thyroglobulin (20 μg). PrPres was enriched by phosphotungstic acid (PTA) precipitation as described elsewhere (49, 59) with two modifications. First, after incubation with PTA and prior to centrifugation, EDTA was added to a final concentration of 1 mM and the mixture was incubated at room temperature for 5 min. Second, the PTA-precipitated pellets were washed once with PTA wash buffer (2% Sarkosyl, 2 mM EDTA in PBS) to remove salt. Final pellets were resuspended in 1× sample buffer with cup horn sonication.
All samples described above were analyzed by SDS-PAGE and immunoblotting as described elsewhere (4, 53). D13 anti-PrP antibody (62) was obtained from InPro Biotechnology (South San Francisco, CA). 31C6 anti-PrP antibody was a generous gift from Motohiro Horiuchi (30). Treatment of polyvinylidene difluoride membranes with 0.2 M NaOH posttransfer was found to improve detection of rMoPrP, anchorless PrP, and low-molecular-weight (low-MW) PrP fragments.
Glycosidase and phospholipase digestions.
Brain homogenates were first PK digested and PTA precipitated as described above. Pellets were resuspended in peptide-N-glycosidase F (PNGase F) denaturing buffer (NEB) and denatured by boiling for 10 min. After adjustment to 1% NP-40 to neutralize the SDS, the samples were treated with PNGase F and/or phosphatidylinositol-specific phospholipase C (PIPLC; MP Biomedicals), where indicated, for 7 h at 37°C. Finally, the samples were methanol precipitated in the presence of 20 μg of bovine thyroglobulin as a carrier protein, resuspended in 1× sample buffer, boiled, and analyzed by SDS-PAGE and immunoblotting (4, 53).
Pathology and immunohistochemistry.
Mice were euthanized, brains were removed, and the sagittal half contralateral to the site of inoculation (except for primary passage of rMoPrP fibrils) was placed in 50 ml of 3.7% phosphate-buffered formalin for 3 to 5 days before dehydration and embedding in paraffin. New dissection tools were used for each inoculum to prevent laboratory scrapie contamination or cross-contamination between inoculum groups. Serial 4-μm sections were cut using a standard Leica microtome, placed on positively charged glass slides, and dried overnight at 56°C.
Detection of PrPres was done as follows. Antigen retrieval was done in a Ventana automated Discovery XT stainer by incubation for 60 min at 100°C in CC1 buffer (Ventana) containing Tris-borate-EDTA, pH 8.0, followed by staining of PrPres with monoclonal human anti-mouse PrP antibody D13 at a 1:500 dilution at 37°C for 2 h. Biotinylated anti-human IgG (1:250 dilution; Jackson Immuno Research, West Grove, PA) was used as the secondary antibody, followed by either streptavidin-alkaline phosphatase with Fast Red chromogen (initial passage mice) or avidin-horseradish peroxidase with diaminobenzidine as the chromogen (subsequent passage mice). Amyloid staining was done with a 1% (wt/vol) solution of thioflavin S (MP Biomedicals) in 50% ethanol (EtOH). Slides were rinsed in 50% EtOH twice, followed by two water washes prior to coverslipping. Astrocyte detection used a mild antigen retrieval step (22 min at 100°C) and staining with polyclonal rabbit anti-glial fibrillary acidic protein (anti-GFAP; Dako) at a 1:3,500 dilution for 16 min and secondary antibody goat biotinylated anti-rabbit IgG for 16 min, followed by streptavidin-alkaline phosphatase using Fast Red as the chromogen. Slides were stained with a standard protocol of hematoxylin-eosin (H&E) for observation of overall pathology.
All histopathology slides were grouped by mouse strain and analyzed blinded using an Olympus BX51 microscope or as high-resolution scanned slide images visualized on computer monitors (Scanscope XT; Aperio). Images for figures were obtained using Microsuite FIVE software on the microscope or Imagescope software on the computer.
RESULTS
Formation and biochemical characterization of rMoPrP amyloid fibrils.
At the time that these studies were initiated, there were no reports demonstrating spontaneous conversion of full-length recombinant PrP into amyloid fibrils in vitro. A survey led to the identification of buffer and incubation conditions that promoted the spontaneous formation of full-length recombinant hamster or mouse PrP amyloid fibrils, as determined by thioflavin T (ThT) fluorescence, infrared (IR) spectroscopy, and electron microscopy (EM) (R. Kodali, unpublished data). Ultimately, we focused on reactions using rMoPrP with two reaction conditions that primarily differed in the concentration of denaturants (see Materials and Methods). Fibrils generated from these reactions are designated H or L to denote whether they were derived from reaction conditions containing higher or lower concentrations of denaturants, respectively.
H- and L-fibril reaction mixtures showed a strong increase in ThT fluorescence after 16 to 21 h incubation (L fibrils, from 23 ± 6 to 481 ± 77 fluorescence units [mean ± standard deviation {SD}, n = 4]; H fibrils, from 22 ± 4 to 323 ± 61 fluorescence units [mean ± SD, n = 5]). Immunoblot analysis showed that both H and L fibrils exhibited partial resistance to PK digestion (Fig. 1A). The pattern of PK-resistant bands was dependent upon the anti-PrP antibody used. A major PK-resistant band of ∼17 kDa was detected by the D13 antibody (epitope residues 96 to 104) (Fig. 1A, lanes 6 to 8 and 9 to 11), reminiscent of the PK truncation of PrPres from TSE-infected animals and similar to that reported previously for rPrP fibrils made under different conditions (8). This band was more pronounced in L fibrils and persisted under digestion conditions with a high level of PK (20 μg/ml) (Fig. 1A, lanes 8 and 9). Additional lower-molecular-mass bands of ∼10 to 13 kDa were detected in immunoblots using an antibody directed against the C terminus (R20; residues 219 to 232) (Fig. 1A, lanes 18 to 23, bracket). L fibrils exhibited two lower-molecular-mass bands, while H fibrils had three (Fig. 1A, compare lanes 18 and 23, bracket), providing further evidence of conformational differences between some of the aggregates present in the L and H fibril preparations. The ultrastructure of H and L fibrils consisted of large, densely matted accumulations of fibrils, possibly a result of the aggressive shaking conditions used for the reactions (Fig. 1B). No fibrils were present in rMoPrP monomer and buffer control samples (Fig. 1B). Together, the above observations establish that H and L fibril reactions generated PK-resistant amyloid fibrils with evidence of conformational differences between them.
Fig 1.

Characterization of rMoPrP fibrils. (A) Immunoblot analysis of rMoPrP fibrils or monomer control treated with the indicated concentrations of PK. PrP was detected using either D13 (epitope hamster residues 96 to 104; left) or R20 (epitope hamster residues 219 to 232; right) antibody. Arrowhead, full-length rMoPrP; arrow, ∼17-kDa, PK-truncated fragment; bracket, low-MW PK-resistant bands detected with R20 antibody. Numbers on the left indicate apparent molecular masses (in kDa). (B) Ultrastructure of rMoPrP fibrils. Aliquots of H or L fibril reaction mixtures were directly spotted onto EM grids, washed, and stained with ammonium molybdate. Control grids were prepared using purified rMoPrP monomer (rMoPrP control) or reaction buffer used to form H fibrils (buffer control). Bars, 100 nm (lower panels of H and L fibrils) and 2 μm (all other panels). (C) Conjugation of rMoPrP preparations to magnetic beads. Aliquots (1/100 equivalents) from the unbound fractions (lanes 3, 7, and 11), first bead wash fractions (lanes 2, 6 and 10), and second bead wash fractions (lanes 1, 5, and 9) were analyzed. Bead-associated rMoPrP was eluted by boiling an aliquot of beads (1/100 equivalents) in sample buffer (lanes 4, 8, and 12). (D) Immunoblot quantitation of bead-associated rMoPrP. Bead-bound rMoPrP was eluted by boiling in sample buffer (lanes 6 to 8). Various amounts of purified rMoPrP (lanes 1 to 5) were loaded as standards. The vertical white line indicates the removal of irrelevant lanes. For panels C and D, rMoPrP was detected by immunoblotting with D13 anti-PrP antibody. Arrows, full-length rMoPrP monomer.
rMoPrP fibrils induce PrPres formation in mice coexpressing wild-type and GPI-anchorless PrP.
To determine if rMoPrP fibrils could induce TSE disease in animals, we performed the primary passage in mice coexpressing wt and GPI-anchorless PrP. The mice were created by crossing C57BL/10 mice with tg44+/+ mice, which are homozygous for anchorless Prnp and lack wt PrPC (13). This resulted in mice hemizygous for wt and anchorless Prnp genes (herein termed wt/GPI− mice) that express, at most, wt levels of total PrPC (13, 15; D. K. Offerdahl, G. S. Baron, and B. Race, unpublished observations). We attempted to enhance the persistence of rMoPrP fibrils after inoculation by conjugation to magnetic beads. Native, monomeric rMoPrP was also conjugated to magnetic beads as a control. Equal amounts of H fibrils, L fibrils, or normally folded monomeric rMoPrP were associated with the magnetic beads, as determined by immunoblot analysis of rMoPrP eluted from the beads by boiling in SDS-PAGE sample buffer (Fig. 1C and D). Two additional control samples were tested. One included mice inoculated with a 1/10 dilution of bead-conjugated L fibrils (L fibrils [0.1×]) to provide some indication of the titer of infectivity present. To evaluate the effect of bead conjugation, the second group (L fibrils [free]) consisted of L fibrils mixed with Tris buffer-blocked magnetic beads immediately prior to inoculation (i.e., within 2 to 4 min), conditions which should limit the opportunity for fibrils to bind to the beads. For all five groups, equal amounts of total rMoPrP (∼4 μg) were inoculated by the i.c. route. We euthanized all animals by 693 days postinfection (dpi), since no evidence of clinical disease was observed in any inoculated animals.
To test for evidence of subclinical infection, brains from 693 dpi animals were divided in half, with one hemisphere subjected to biochemical analysis and the other subjected to histopathological analysis. Surprisingly, the brains of all fibril-inoculated animals tested showed evidence of protease-resistant PrP by immunoblotting (Fig. 2A, top, lanes 4 to 13, bracket). The PK-resistant PrP was not due to persistence of the inocula, as we did not detect any PrPres in the brains of animals tested at 615 dpi (Fig. 2B, lanes 3 to 10). In most cases, the apparent molecular mass of PrPres and its banding pattern was similar to that of PrPres derived from scrapie infection of transgenic mice expressing only GPI-anchorless PrPC (13, 15), suggesting there may have been preferential conversion of anchorless PrPC in the fibril-inoculated mice. The one exception was H fibril-inoculated mouse C413, which showed three PrPres bands with a higher apparent molecular mass and an increased proportion of middle and upper bands versus the lowest band (bracket in Fig. 2A, top, lane 13; B, lane 14; and C, lane 5). This prompted consideration that conversion of wt PrPC may have occurred in mouse C413 since anchorless PrPres is largely unglycosylated (15). In addition to the PrPres bands described above, additional low-molecular-mass PK-resistant bands of ∼12 kDa (Fig. 2A, arrowhead) and ∼8 kDa (Fig. 2A, top, arrow, lane 4) were detected. The 12-kDa band was partially obscured by a comigrating autofluorescent species and was more clearly visualized on immunoblots with a reduced load of total protein for those samples in which this band was most abundant (Fig. 2A, bottom). The 8-kDa band was similar to a band associated with amyloid-associated prion diseases such as Gerstmann-Sträussler-Scheinker syndrome (GSS) (42, 43, 58) and variably protease-sensitive prionopathy (VPSPr) (63). The overall banding patterns (PrPres and low molecular mass) differed from those detected in wt/GPI− mice with scrapie (15), providing evidence that the fibril-induced PrPres was not attributed to contamination of the inocula with laboratory mouse scrapie. Many other lines of evidence presented below, in addition to precautions taken during sample preparation (see Materials and Methods), argue that there is no evidence for contamination with laboratory scrapie in these studies.
Fig 2.

rMoPrP fibrils induce PrPres formation in mice coexpressing wt and GPI− PrPC. (A) Immunoblot assay for PrPres in rMoPrP fibril-inoculated mice at 693 dpi. Lanes 2 to 13, brain homogenates from wt/GPI− mice inoculated with the indicated rMoPrP preparations. L fibrils (0.1×), animals received 1/10 dose of bead-conjugated L fibrils; L fibrils (free), L fibrils mixed with preblocked beads immediately prior to inoculation. Brain homogenates were PK digested and precipitated with PTA (top) or methanol (bottom). Each lane contains 600 μg (top) or 38 μg (bottom) total protein equivalents. The 90 to 231 fragment of purified recombinant hamster PrP (0.2 ng) was loaded on the upper gel for comparison (lane 1). Closed arrowhead and arrow, low-molecular-mass PK-resistant bands in fibril-inoculated animals; open arrowhead, detection of methanol-precipitated PK due to antibody cross-reactivity. PrPres IHC, detection of PrPres by immunohistochemistry (see Fig. S2 in the supplemental material; data not shown). (B) Immunoblot assay for PrPres in rMoPrP fibril-inoculated mice at 615 dpi. Brain homogenates from wt/GPI− mice inoculated with the indicated rMoPrP preparations and euthanized at 615 dpi (lanes 1 to 10) or 693 dpi (lanes 11 to 14). Each lane contains 600 μg total protein equivalents. Bands in lane 10 are likely due to residual mouse IgG in the brain homogenate. (C) rMoPrP fibril-induced PrPres lacks a GPI anchor. After PK digestion and PTA precipitation, brain homogenate samples (lanes 2 to 10) were denatured and treated with PNGase F (PNG-F) and PIPLC, as indicated. 22L-infected Tga20 mouse brain was included as a wt PrPres control (lanes 8 to 10). Lanes 2 to 7, 600 μg total protein equivalents per lane; lanes 8 to 10, 15 μg total protein equivalents per lane. The 90 to 231 fragment of purified recombinant hamster PrP (1 ng) was loaded for comparison (lanes 1 and 11). Asterisks, bands shifted by PIPLC treatment (lane 9 versus lane 10). For panels A to C (except where indicated), all samples were PK treated, PTA precipitated, and probed with anti-PrP mouse monoclonal antibody 6D11.
Consistent with clinical observations, there was no evidence of significant pathology on the basis of histological analysis of the brains of 693 dpi animals (see Fig. S2 in the supplemental material). However, occasional plaque-like deposits of PrPres (see Fig. S2 in the supplemental material; data not shown) were detected in many but not all fibril-inoculated animals, as summarized in Fig. 2A (PrPres immunohistochemistry [IHC]). Discordance between PrPres detection by immunoblotting versus immunohistochemistry (e.g., L fibrils [0.1×]) may be attributed to insufficient tissue sampling and/or differences between the brain hemispheres. The PrPres deposits (see Fig. S2, arrows, in the supplemental material) were most abundant near the corpus callosum and were in the vicinity of, but not overlapping with, magnetic beads (see Fig. S2, arrowheads, in the supplemental material). Rare deposits were also present in the hippocampus or near the anterior commissure. No PrPres deposits were detected in any control animal inoculated with bead-bound normal rMoPrPC. Together with the immunoblotting data presented above, these observations demonstrate that inoculation of rMoPrP fibrils induced a low, subclinical level of new PrPres formation.
rMoPrP fibril-induced PrPres lacks a GPI anchor.
Since the host mice express both wt and anchorless PrPC, it was possible that the PrPres detected resulted from conversion of either type of PrPC. To investigate this question, samples were denatured after PK digestion and treated with phosphatidylinositol-specific phospholipase C (PIPLC) to cleave GPI anchors, if present. Samples were also deglycosylated to simplify the banding profile and improve the resolution of migration differences. Even after denaturation it was difficult to achieve complete deglycosylation of PrPres. Nevertheless, in wt PrPres control samples from Tga20 mice infected with 22L mouse scrapie, there was a clear PIPLC-dependent increase in apparent molecular mass (Fig. 2C, lane 10, asterisks) of PrPres bands indicative of GPI anchor removal (56). In contrast, there was no evidence of PIPLC-dependent differences in apparent molecular mass for PrPres associated with mouse C413 or D061, indicating that the majority of the PrPres in both samples lacked a GPI anchor (Fig. 2C, lanes 2 to 7). These data show that both L and H fibrils preferentially induced conversion of anchorless PrPC.
Induction of TSE disease on serial passage of rMoPrP fibril-induced PrPres.
To determine if the rMoPrP fibril-induced PrPres on primary passage in wt/GPI− mice was associated with the generation of a TSE agent, we conducted serial passage studies. Brain homogenates from two animals in each of the five groups (Monomer, animals D092 and D114; L fibrils, animals D013 and D061; L fibrils [0.1×], animals D090 and D098; L fibrils [free], animals D018 and D063; H fibrils, animals C411 and C413) were inoculated i.c. into four different strains of mice expressing wt PrPC, GPI− PrPC, or both (wt/GPI−) to assess the effect of host PrPC type and expression level on agent propagation and pathogenesis (Fig. 3). Four different inocula, all derived from initial inoculation of bead-conjugated L (D013, D061) or H (C411, C413) fibrils, induced a TSE disease upon serial passage (Table 1), based on clinical signs, histopathological analysis, and detection of PrPres, as described below.
Fig 3.

Overview of serial passage studies of rMoPrP fibrils in various mouse strains. After primary passage of rMoPrP fibrils in mice coexpressing wt and wt/GPI−, subsequent passages were conducted in wt/GPI−, Tga20 (which overexpresses wt MoPrPC 8-fold) (23), C57BL/10, and tg44+/+ (homozygous for GPI− Prnp transgene on C57BL/10 [wt] PrP-knockout background) mice. Incubation time and attack rate data corresponding to secondary and tertiary passage studies are shown in Table 1.
Table 1.
Incubation periods and TSE disease attack rates in each mouse strain on serial passage of brain homogenates from fibril-treated wt/GPI− micea

BH, brain homogenate; ND, not done; dpi, days postinoculation; NA, not applicable; neg, negative for neurological signs of disease and for detection of PrPres by immunohistochemistry (IHC) and/or immunoblotting; p1, p2, and p3, passages 1, 2, and 3, respectively.
bThe TSE attack rate is given as the number of clinical animals/total number of animals in the group. Groups of 6 to 8 animals were used for passages of all inocula tested. Smaller group sizes for some inocula were due to intercurrent deaths and/or premature euthanasia to provide age-matched control samples for immunoblot or histopathology analyses (individuals marked by an asterisk). Data for animals lost to intercurrent deaths were not included in these data. Intercurrent deaths were defined as death without neurological signs prior to the date that the first animal from any inoculum into a given mouse strain was determined to be TSE positive by either neurological signs or brain PrPres (as determined by IHC and/or immunoblotting). For groups with clinically ill animals, numbers in parentheses indicate the mean dpi ± SD. Also shown are the mean dpi to euthanasia ± SD for animals that were negative in these groups. For groups without clinically positive animals (e.g., Monomer controls), numbers in parentheses indicate incubation times for individual animals (or multiple animals, as indicated in brackets) prior to euthanasia. Where possible for the D061 and C413 inocula, 1% brain homogenates from two animals per group in passage 1 (Tga20, C57BL/10) or passage 2 (wt/GPI−) were passaged again in the respective mouse strains. Bold italicized values for a given combination of inoculum and mouse strain are dilutions of the same brain homogenate from an individual animal.
cAny surviving mice were euthanized at the following dpi for each of the mouse strains used: Tga20, 455 (passage 1) and 300 (passage 2); C57BL/10, 701 (passage 1) and 469 (passage 2); wt/GPI−, 726 (passage 2) and 467 (passage 3); and tg44+/+ , 717 (passage 1) and 500 (passage 2).
dMice euthanized prematurely due to ulcerative dermatitis.
eIncubation periods for individual D013-inoculated tg44+/+ mice, 387, 513, 559, and 594 (3) dpi. Brain homogenate from the 387 dpi mouse was used for passage 2 in tg44+/+ mice.
fAnimals from passage of wt/GPI− brain homogenates from mice originally inoculated with beads conjugated to monomeric mouse rPrP as a negative control.
gControl samples analyzed for Fig. 7 and Fig. S3 in the supplemental material and used for passage 2 (Tga20, C57BL/10) or passage 3 (wt/GPI−) experiments were from these designated mice.
Of the four TSE-inducing inocula, the inoculum from H fibril-infected mouse C413 was most efficient, as it induced TSE disease with 100% attack rates and the most rapid incubation periods across all mouse strains except tg44+/+, suggesting that a higher titer or a more virulent agent was present compared to the other inocula (Table 1). The D061 inoculum also induced disease in all mouse strains, but with incomplete attack rates in animals expressing only wt PrPC (Tga20, C57BL/10) and 100% attack rates in animals expressing anchorless PrPC (wt/GPI−, tg44+/+). Also, since the mean incubation period of the D061 agent in tg44+/+ mice was significantly shorter (469 ± 38 dpi) than that observed for the C413 agent (589 ± 10 dpi), anchorless PrPC may promote the propagation of the D061 agent. An extreme example of this effect was the D013 inoculum (derived from L fibrils as for D061), which caused disease only in tg44+/+ mice with a 100% attack rate, suggesting a strong preference for host expression of anchorless PrPC. Inocula derived from D090 and D098, animals that were initially treated with a 10-fold lower dose of bead-conjugated L fibrils than animals D013 or D061, did not induce TSE disease on secondary passage in any mouse strain. This showed that the titer of agent present in the bead-conjugated L fibril preparation was very low. Although they did not induce clinical disease, the L-fibril (0.1×) (D090, D098) and L fibril (free) (D018, D063) inocula all induced low levels of PrPres deposition in wt/GPI− and tg44+/+, mice as discussed below.
Tertiary-passage studies were conducted on representative samples from each 2nd-passage TSE-positive inoculum-mouse strain combination, as depicted in Fig. 3. Brain homogenates from age-matched (or older) 2nd-passage Monomer-inoculated control animals were inoculated in parallel as negative controls. In all cases, 3rd-passage incubation periods for animals injected with 1% brain homogenates of TSE-positive 2nd-passage inocula were dramatically reduced (Table 1). With the exception of tg44+/+ mice, incubation periods converged on values longer than those associated with typical mouse scrapie in the respective mouse strains (13, 15, 29). Remarkably, the average incubation period for the 2nd passage of the D013 agent in tg44+/+ mice (234 ± 19 dpi) was approximately 100 days faster than that for primary passage of 22L or Chandler in the same mouse strain (13). All animals inoculated with 0.1% brain homogenates maintained a 100% attack rate, indicating that the titer of infectivity present was significantly above the endpoint.
The clinical signs of neurological disease observed were indistinguishable for all inocula and independent of the mouse strain inoculated. Early signs of disease were termination of nesting behavior, malaise, and hyperresponsiveness to physical or auditory stimulation. Over time, these signs were accompanied by reduced grooming behavior and development of progressive neurological disease that occurred in the following sequence: (i) a prolonged period of frequent jerking and intermittent tremors, (ii) kyphosis, and (iii) a short period of difficulty eating and drinking, at which point the animals were euthanized. Most of the animals also exhibited an unusual bladder dysfunction, indicated by the occurrence of full urinary bladders. This phenotype seemed to be neurological in nature, since bladder evacuation occurred immediately upon euthanization and there was no histopathological evidence of abnormalities in the urinary tract (data not shown). The length of the clinical phase was very prolonged (2 to 3 weeks in Tga20 mice, 4 to 6 weeks in all other mouse strains). Aside from reduced incubation periods, all clinical features were otherwise unchanged on tertiary passage of the rMoPrP fibril-induced agents (Table 1). In contrast, no evidence of disease was ever observed in any mouse strain inoculated with bead-conjugated normal rMoPrP control samples, even after three serial passages and observation for extended incubation periods covering the natural life span of the mice (Table 1, Monomer controls). The overall clinical spectrum induced by the rMoPrP fibril agents was readily distinguished from that induced by known scrapie strains in our laboratory, indicating that the rMoPrP fibril preparations had given rise to at least two novel TSE agents.
Unique neuropathology induced by rMoPrP fibril-induced TSE agents.
We characterized the neuropathology associated with the fibril-induced TSE agents using standard histological stains. The neuropathological features observed varied between agents and mouse strains, but all diseased mice infected with rMoPrP fibril agents could be distinguished from positive-control animals infected with 22L or Chandler mouse scrapie. Astrogliosis (GFAP staining), a common feature of TSE pathology, was present in every mouse strain, with the distribution usually mirroring PrPres deposition patterns (Fig. 4 and data not shown). rMoPrP TSE agents induced intense, highly punctate PrPres deposition in the thalamus of mice expressing only wt PrPC (C57BL/10, Tga20) (Fig. 4 and data not shown). In contrast, PrPres deposits in 22L-infected C57BL/10 mice were much more abundant and widespread throughout the brain (Fig. 4). The PrPres distribution in Chandler-infected C57BL/10 mice was similar to that in rMoPrP agent-infected animals but could be differentiated on the basis of PrPres in the preoptic area and a lack of cerebellar involvement (data not shown). All samples were negative for amyloid staining with thioflavin S (data not shown). Spongiosis was most prominent in the thalamus of both C57BL/10 and Tga20 mice infected with fibrils (Fig. 4 and data not shown). In summary, the pathological features associated with infection of mice expressing wt PrPC with the rMoPrP fibril agents were similar among the three samples tested (from animals D061, C411, and C413) but differed from those induced by infection with typical laboratory mouse scrapie strains.
Fig 4.

Histopathology of C57BL/10 mice infected with rMoPrP fibril agents from wt/GPI− PrP mice. Brain homogenates (1%) from 693 dpi wt/GPI− mice treated with rMoPrP fibril preparations or monomer control were passaged into C57BL/10 mice. Brain sagittal sections were stained for PrPres with D13 antibody (PrPres), GFAP, or H&E, as indicated. Small panels correspond to the thalamus. All fibril agent-treated animals that developed TSE-like neurological disease showed similar PrPres deposition, astrocytosis, and spongiform change (arrows). A C413 agent-treated animal is shown as a representative example. Note the differences in distribution (large panels) and nature (punctate for fibril versus diffuse for 22L; small panels) of PrPres deposits induced by the fibril agent compared with that induced by the 22L scrapie strain control. A Monomer control-inoculated animal (D114, 441 dpi) is shown for comparison. Bars, 2 mm (large panels) and 50 μm (small panels).
Neuropathological differences between rMoPrP TSE agents and typical scrapie strains were more pronounced in infections of wt/GPI− and tg44+/+ mice. Infection of wt/GPI− mice with 22L (Fig. 5) or Chandler (data not shown) led to both diffuse nonamyloid PrPres and thioflavin S-positive amyloid plaque PrPres, which corresponds to conversion of both wt and anchorless PrPC to PrPres (15). Only the D061 and C413 agents induced clinical disease in wt/GPI− mice, which was readily distinguished from that induced by 22L and Chandler on the basis of the collective observations in D061/C413 agent-treated mice that included an abundance of widely distributed uniform plaques in both white and gray matter and consistent involvement of the cerebellum, a lack of diffuse PrPres, and white matter vacuolation (Fig. 5 and data not shown).
Fig 5.

Histopathology of rMoPrP fibril agent passaged in wt/GPI− and tg44+/+ mice. Brain homogenates (1%) from 693 dpi wt/GPI− mice treated with rMoPrP fibril preparations or Monomer control were passaged into wt/GPI− or tg44+/+ mice. Brain sagittal sections were stained for PrPres with D13 antibody (PrPres), thioflavin S, or H&E, as indicated. Images for wt/GPI− mice correspond to the thalamus. In wt/GPI− mice, note the diffuse PrPres deposits (arrow) in the 22L-infected control mouse that are absent from the C413-infected mouse. Thioflavin S-positive amyloid plaques and spongiform pathology (arrows) are present in C413- and 22L-infected control mice. tg44+/+ images correspond to cerebral cortex (columns 1 to 3) or corpus callosum (column 4). Note differences in the size, morphology (the D061 agent is multifocal), and distribution of plaques in D061 agent- and D013 agent-infected tg44+/+ mice versus Chandler-infected control mice. Although aged tg44+/+ mice (D114, 574 dpi and uninfected controls [data not shown]) show some white matter spongiosis, vacuoles (arrows) were more abundant in D061 agent-, D013 agent-, and Chandler-infected mice, especially around plaques (arrowheads). Bars, 2 mm (column 1) and 100 μm (columns 2 to 4).
In tg44+/+ mice, striking differences were observed in the distribution, abundance, and morphology of PrPres deposits compared to those in mice with typical mouse scrapie. Plaques in D013 agent-infected tg44+/+ mice were widespread in both white and gray matter (Fig. 5). The PrPres distribution in D061 agent-infected (not shown) and C413 agent-infected mice was similar to that in D013 agent-infected mice but also included the thalamus and midbrain. In addition, D061/C413 agent-induced plaques had a unique morphology with multiple small, dense foci which we have termed “multifocal” (Fig. 5). In contrast, plaques in Chandler-infected tg44+/+ mice were extremely large, not multifocal, and exclusively perivascular, and although they were widespread, they were rarely present in the cerebellum (Fig. 5). Even greater differences were observed compared to 22L-infected mice (data not shown), with PrPres deposits primarily found on the meningeal and ventricular surfaces and the cerebellum. Thus, the rMoPrP fibril-induced agents segregated into two groups (the D013 and D061/C413 agents), each of which caused pathology that was distinct from the pathology associated with Chandler or 22L infection in control animals. This supported the proposition that at least two novel strains of TSE agent were created in this study.
Subclinical propagation of rMoPrP fibril agents in wt/GPI− and tg44+/+ mice.
Given the preferential induction of disease in tg44+/+ mice by the D013 agent, we examined the brains of D013 agent-inoculated wt/GPI− mice to determine if the D013 agent propagation efficiency correlated with the GPI− Prnp gene dosage. Despite the lack of clinical disease during the life span of D013 agent-inoculated wt/GPI− mice (714 dpi), all five mice tested displayed PrPres deposits (Fig. 6). As in D061 agent- and C413 agent-inoculated wt/GPI− mice, PrPres deposits in D013 agent-inoculated mice were strictly in the form of plaques, although the plaques were much less abundant in D013 agent-inoculated animals (Fig. 5 and 6). The plaque load was also much lower than that observed in D013 agent-inoculated tg44+/+ mice, providing strong evidence of the GPI− PrPC tropism of the D013 agent and consistent with the lack of clinical disease in wt/GPI− mice (Fig. 5 and 6). Reinforcing this point, after careful examination of D013 agent-infected Tga20 (3/3 mice aged 401 to 455 dpi) and C57BL/10 mice (2/2 mice, aged 701 dpi), we found only one small cluster of dense PrPres-positive plaques in a single location near the thalamus in one C57BL/10 mouse (data not shown).
Fig 6.
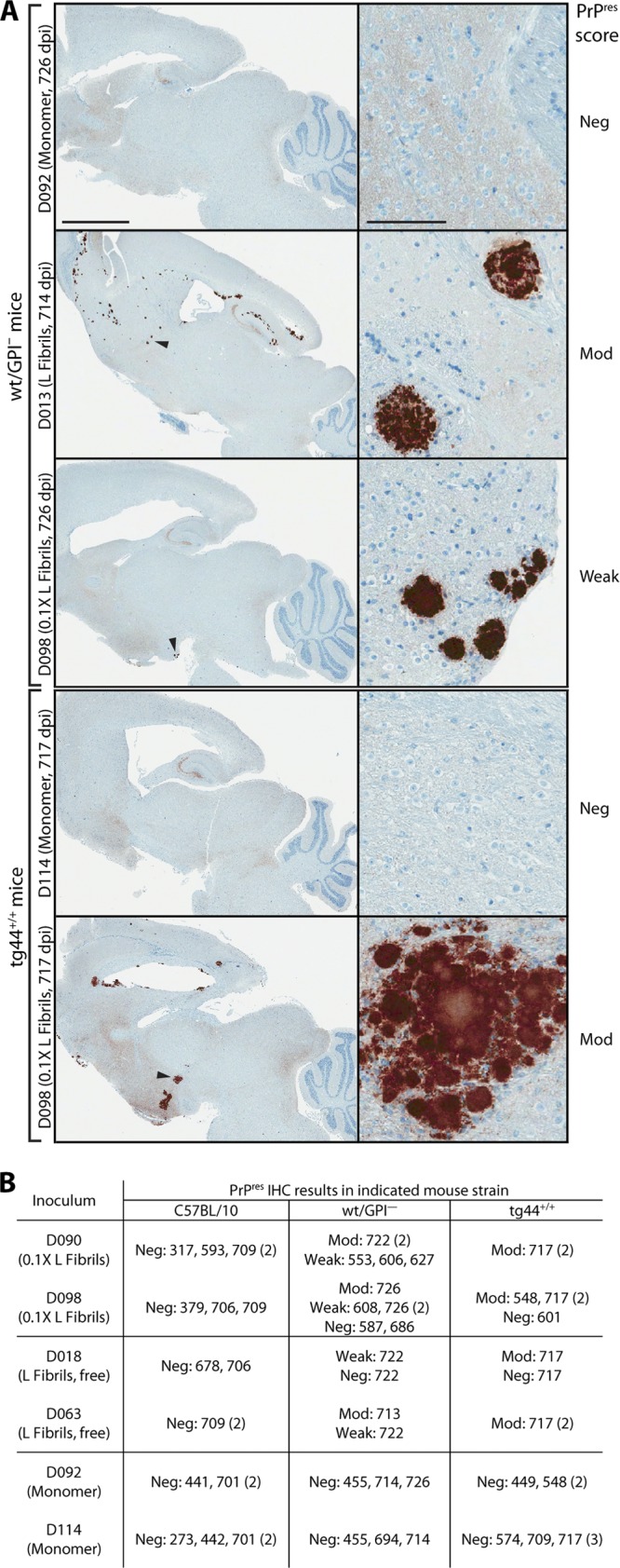
Histopathology of subclinical wt/GPI− mice infected with secondary-passage rMoPrP fibril agents. Brain homogenates (1%) from 693 dpi wt/GPI− mice treated with rMoPrP fibril preparations or Monomer control underwent secondary passage in wt/GPI− or tg44+/+ mice. (A) Brain sagittal sections were stained for PrPres with D13 antibody. Representative examples are shown. Arrowheads, PrPres plaques shown at a higher magnification on the right. PrPres plaque score: Neg, negative, Weak, weak positive (≤8 plaques); Mod, moderate positive (>8 plaques). Bars, 2 mm (column 1) and 100 μm (column 2). (B) Summary of PrPres immunohistochemistry. Numbers indicate the dpi on which individual animals were euthanized. Values in parentheses indicate the number of animals with the same dpi of euthanization.
The observations presented above prompted further investigation of the secondary-passage animals that received rMoPrP fibril-derived inocula from animals initially given a 0.1× dose of bead-conjugated L fibrils (animals D090 and D098) or L fibrils without bead conjugation (animals D018 and D063). None of these animals showed any clinical signs and were euthanized near the end of their natural life span. Surprisingly, PrPres plaques were present but only in wt/GPI− and tg44+/+ mice (Fig. 6). There was a trend toward larger plaques with a morphology resembling a multifocal morphology and a higher plaque load score among positive animals for tg44+/+ versus wt/GPI− mice (Fig. 6). Altogether, this suggested that as with the D013 and D061 L fibril agents, GPI− PrPC may facilitate the propagation of these subclinical agents. No plaques were observed in any strain of age-matched mice treated with the corresponding serially passaged inocula of bead-bound rPrP monomer controls (from animals D092 and D114), proving that the plaques were not age related. These data demonstrate that all rMoPrP fibril inoculations in this study induced self-propagating PrPres formation in vivo but did so at various efficiencies that correlated with the ability to induce clinical disease. Moreover, these observations provide further evidence that anchorless PrPC can promote the propagation of rMoPrP fibril-induced agents.
Novel low-MW PK-resistant PrP banding profiles associated with rMoPrP fibril agents.
To further compare the rMoPrP fibril agents with known scrapie strains, we analyzed the PrPres banding profiles by immunoblotting. Consistent with the observations presented above, the fibril-induced agents had profiles that differed from those of the Chandler and 22L controls in all mouse strains examined. The clearest differences occurred in the pattern of low-MW, PK-resistant bands and were present across all mouse strains. In all mouse strains infected with the rMoPrP agents, there were higher levels of an ∼8-kDa band versus 22L-, Chandler-, and ME7-infected control animals (Fig. 7; see Fig. S3, arrow, in the supplemental material). In Tga20 mice, those inoculated with rMoPrP agents showed lower levels of an ∼12-kDa band than Chandler- and 22L-inoculated controls (Fig. 7A, lanes 11 to 18 versus lanes 19 to 20, arrowhead). Among rMoPrP agents in tg44+/+ mice, mice inoculated with the D013 agent had higher levels of the 8-kDa band (i.e., compare the ratio of the 8-kDa band [arrow] to the PrPres bands [bracket] in Fig. 7C [lanes 3 to 6] and Fig. S3B in the supplemental material [lanes 3 to 8 and 15 to 20]) and lower overall levels of PrPres (Fig. 7C; see Fig. S3B in the supplemental material; compare the D013, D061, and C413 PrPres band intensities [brackets] with those for the ME7, Chandler, and 22L controls on the respective gels). When probed with 6D11 antibody (anti-residues 93 to 109), the intensity of the 12-kDa band varied between individual tg44+/+ mice inoculated with the D013 or C413 agents (see Fig. S3B, lanes 3 to 8 and 15 to 17, arrowhead, in the supplemental material). At least for D013 agent-treated mice, this seemed to be attributable to ragged N-terminal cleavage by PK, as when the samples were probed with an antibody directed against a more internal epitope (31C6, anti-residues 143 to 149), we saw comparable reactivity across the three samples tested (see Fig. S3C, lanes 11 to 13, arrowhead, in the supplemental material). The 31C6 blot also showed limited reactivity with the 8-kDa band (see Fig. S3C, lanes 11 to 13, arrow, in the supplemental material). This suggests that the 8-kDa band roughly maps to at least residues ∼93 to 149 and raises the possibility that it may be similar to the fragment from approximately residues 89 to 145 described in certain forms of amyloid-associated CJD (42, 43, 58, 63).
Fig 7.

Transmission of PrPres propagation on secondary passage. PrPres profiles in brain homogenates from inoculation of Tga20 (A), wt/GPI− (B), or tg44+/+ (C) mice with the indicated inocula from primary passage in wt/GPI− mice in Fig. 2A. Monomer control samples were derived from age-matched (usually older) mice (Table 1). Where available, samples from Tga20, wt/GPI−, and tg44+/+ mice infected with established mouse-adapted scrapie strains (22L, Chandler, ME7) were included for comparison, as indicated. Brackets indicate bands corresponding to PrPres monomers. SDS-resistant oligomers are also visible. Darker exposure panels are a darker exposure of the corresponding panel above. Arrowhead and arrow, ∼12-kDa and ∼8-kDa PK-resistant bands, respectively; vertical white lines, removal of irrelevant lanes. All samples were PK treated, PTA precipitated, and probed with 6D11 antibody. The total protein equivalents loaded per lane were as follows: 30 μg in lanes 1 to 9 and 300 μg in lanes 10 to 22 in panel A; 150 μg in lanes 1 to 7 and 15 μg in lanes 8 to 14 in panel B; 200 μg in panel C.
DISCUSSION
In the current study, we have shown that inoculation of rMoPrP amyloid fibrils into mice expressing wt levels of PrPC gives rise to novel TSE agents. Using transgenic mice coexpressing wt and GPI-anchorless PrPC for initial passage of rMoPrP fibrils, we detected low levels of newly induced PrPres after a long incubation period but without clinical signs (Fig. 2A; see Fig. S2 in the supplemental material). The low levels of PrPres and limited brain pathology were consistent with the absence of a clinical phenotype. However, TSE agents were present in at least four animals, as shown by induction of TSE disease on secondary passage into various mouse strains expressing wt PrPC, anchorless PrPC, or both (Table 1). In contrast, control animals inoculated with an equal amount of normal α-helix-rich rMoPrP failed to exhibit PrPres, central nervous system pathology, or evidence of clinical signs at any time during their life span even after three serial passages of brain homogenates from aged mice. Serial passage of tissue from such control animals allowed the opportunity for amplification of any infectious agent that might initially have been present at very low levels. Our application of serial passage to this important control was unprecedented among papers reporting the generation of infectivity from rMoPrP and provides strong evidence that the infectivity observed was attributed to the inoculation of rMoPrP fibrils as opposed to contamination with laboratory scrapie or induction of infectivity in transgenic animals by inoculation of any form of rMoPrP. Other lines of evidence include the generation of novel TSE strains with clinical features, PK-resistant PrP immunoblot banding profiles, and brain PrPres deposition patterns that are distinct from those for known laboratory scrapie strains inoculated into the same mouse strains. As described in Materials and Methods, extensive precautions were taken in our experiments to prevent the possibility of contamination of the samples with laboratory scrapie. Collectively, our data indicate that the TSE infectivity reported here was induced by our rMoPrP fibril preparations.
One group has reported the spontaneous generation of prions in transgenic mice expressing anchorless PrPC (57). This phenomenon was specific to animals expressing myc-tagged anchorless hamster PrPC at levels much higher than those present in our transgenic mice. Transgenic lines expressing low levels of anchorless hamster PrPC with or without coexpression of wt PrPC, which are more comparable to those used in this study, did not spontaneously develop prions or brain pathology (57). Thus, it is unlikely that the spontaneous anchorless prions described by Stöhr and coworkers (57) can account for our observations.
One notable feature common to all our synthetic TSE strains was a prolonged clinical phase. The phenotype of a prolonged clinical phase remained constant even between serial passages, with substantial differences in overall incubation period (e.g., Table 1, strain D013 passaged in tg44+/+ mice), indicating that a prolonged clinical phase is an inherent property of our rMoPrP TSE strains. Among synthetic TSE strains described to date, the prolonged clinical phase is reminiscent of but clearly distinct from that for rPrP-induced TSE in hamsters reported elsewhere, as the hamsters exhibited an extremely long clinical phase (on the order of several months) but showed clinical signs not observed in our experiments, such as obesity and hair loss (35, 36). This might be attributed to many factors, such as species differences, effects of GPI-anchorless PrPC, and the use of very different reaction conditions to create rPrP fibrils, as discussed below. Regardless of the explanation, our system facilitated the formation of a novel class of TSE agents.
It is difficult to provide a mechanism to account for the prolonged clinical phase associated with the rMoPrP TSE agents. Immunoblot assays showed that PrPres levels in the brains of animals infected with our synthetic TSE agents were as high as or greater than those present in the brains of control animals inoculated with known mouse scrapie strains (Fig. 7; see Fig. S3A and B in the supplemental material). However, the kinetics of accumulation of pathological PrP isoforms in clinical target areas may differ between wt and synthetic TSE strains, which could contribute to the differences in clinical periods, as has been proposed for other scrapie strains (2). Alternatively, there were striking differences in the nature and distribution of PrPres. For example, in Tga20 and C57BL/10 mice, the rMoPrP TSE strains primarily targeted the thalamus in the form of punctate PrPres deposits, while 22L and Chandler control strains affected other important regions and formed more diffuse PrPres deposits (Fig. 4). In wt/GPI− mice, the synthetic TSE agents (D061 and C413) exclusively induced amyloid plaque deposits (Fig. 5) that had a PrPres banding pattern consistent with conversion of anchorless PrPC (Fig. 7B). In contrast, the 22L control strain induced a combination of diffuse and plaque PrPres (Fig. 5), which has been shown to correspond to conversion of both wt and GPI− PrPC (15). Hence, it is also possible that the abnormal PrP induced by infection with the rMoPrP strains is less toxic than that induced by the control scrapie strains. Further studies are needed to determine the underlying basis for the strain-dependent differences in the clinical phase.
To develop improved reaction conditions for de novo formation of infectivity from rMoPrP, we focused on optimizing the production of a PK-resistant species exhibiting a PrPres-like 6- to 7-kDa shift in size after PK digestion, since this is one of the best, albeit imperfect, biochemical markers for infectivity. Both the L and H fibril reaction conditions allowed the detection of a PrPres-like ∼17-kDa PK-resistant fragment when probed with an antibody (D13) directed against an epitope near the N terminus of the PK-resistant core of TSE-associated PrPres. The 17-kDa band was more pronounced in L fibrils than H fibrils and was detected even under stringent PK digestion conditions (20 μg/ml) (Fig. 1A). The presence of a D13-reactive, PK-resistant band was of great interest as this region contains dramatic structural differences between poorly infectious amyloid rPrP fibril preparations and authentic brain-derived PrPres (53). Although we were able to induce TSE disease in mice after inoculation of L and H fibrils, the requirement for two serial passages to induce disease suggests that the infectivity titer was low. As suggested by results from other systems, it is possible that an additional cofactor(s) is required to generate higher titers of infectivity from rPrP (20, 22, 60). We cannot exclude the possibility that a cofactor is recruited to H and L fibrils after inoculation. If it occurs, this cofactor recruitment postinoculation may be either less efficient or less likely to associate with rPrP fibrils in a manner that induces an infectious conformation than when the cofactor is present during the rPrP fibrillization reaction. Nevertheless, our data still show that rMoPrP fibrils formed without seeding are sufficient to induce the generation of TSE infectivity in a host animal and thus provide evidence in support of the protein-only prion hypothesis.
One comparable reaction condition (i.e., one in which the reaction contained only purified α-rPrP plus salts) generated a similarly robust PK-resistant D13-reactive ∼16- to 17-kDa band (9). This required a separate “annealing” step by heating preformed mouse or hamster rPrP fibrils to 80°C in the presence of Triton X-100, a procedure that resulted in β-sheet-rich fibrils with a PK-resistant core extended to residue Gln-97 and an altered β-sheet structure compared to untreated fibrils (9). In addition to the annealing treatment, the initial fibrillization conditions employed were dramatically different from ours with respect to denaturant concentration, incubation time, ionic strength, pH, and shaking speed (9). When performed in the presence of brain homogenate plus Triton X-100, annealed hamster rPrP fibrils induced TSE disease after two serial passages in hamsters (35). Since the authors have not reported infection studies with fibrils annealed with Triton X-100 alone, the contribution of brain homogenate components to the infectivity they observed remains unclear. In annealing reactions performed with BSA in place of brain homogenate, TSE disease was observed only after three serial passages through a process termed “deformed templating” (36). Deformed templating was also reported to account for TSE infectivity that arose after serial passage of fibrils created without annealing in 0.5 M GdnHCl (37). Two additional fibril preparations made in 2 M GdnHCl with either strong or modest agitation did not induce TSE disease or PrPC misfolding in hamsters, at least after one passage (37). We saw no evidence to suggest that deformed templating occurred in our studies, based in part on immunoblotting with R20 antibody (anti-residues 219 to 232), which would be expected to detect the atypical PrPres associated with deformed templating (data not shown). Clearly, the annealing-based system is much more complex than that described in the present study, highlighting an advantage of our simple reaction conditions. Taken together with our work, it would appear that there is a correlation between the presence of an ∼17-kDa PK-resistant band in rPrP preparations and more efficient induction of TSE infectivity in vivo, although the infectivity titer is obviously lower than that in PMCA systems that rely on cofactors (60).
In contrast to rMoPrP preparations, the relationship between infectivity and protease-resistant PrP was more complicated with respect to analysis of brain homogenates after primary passage of rMoPrP fibrils in wt/GPI− mice. Although levels were very low in some cases and variable between individuals, PK-resistant PrP was detected in the brains of all rMoPrP fibril-inoculated animals tested, regardless of the reaction conditions (L versus H fibrils), dose, and conjugation to magnetic beads (Fig. 2A). Of the eight brain homogenates from fibril-treated animals that were subjected to secondary passage studies (two representative animals per group), only the four homogenates from animals that received a 1× dose of bead-conjugated L (animals D013 and D061) or H (animals C411 and C413) fibrils induced TSE disease in at least one mouse strain (Table 1). Some homogenates that had at least as much protease-resistant PrP as those described above (Fig. 2A, lanes 7 to 10 versus lanes 5, 12, and 13) did not induce TSE disease on secondary passage, indicating a discordance between PrPres levels and infectivity. This raises the possibility that conjugating rMoPrP to magnetic beads assisted the in vivo propagation of rMoPrP TSE infectivity. Such a result would be consistent with experiments reporting an enhancement of PMCA-derived infectivity per amount of PrPres by preadsorbing PMCA reaction products to nitrocellulose beads, a procedure that delayed the in vivo clearance of radiolabeled cell lysate proteins after intracerebral inoculation (61). In addition to stabilizing rMoPrP fibrils, we cannot exclude the possibility that bead association induced conformational changes in rMoPrP fibrils that facilitated the formation of an infectious conformation or that the infectious species are a minor component of the samples.
Another interpretation is that the PrPres detected after primary passage in wt/GPI− mice actually represented at least two categories of agent. The category 1 agent (e.g., the D013, D090, or D063 agents) is characterized by higher levels of PK-resistant PrP on primary passage, a strong preference for anchorless PrPC, high amyloidogenicity, and a lower specific infectivity (infectivity per amount of PrPres), perhaps due to a tendency to seed formation of PrPres conformers with greater stability. The category 2 agent (e.g., the C413 agent) is characterized by low to intermediate levels of PK-resistant PrP on initial passage, higher specific infectivity, lower amyloidogenicity, and increased compatibility with wt PrPC, thereby allowing efficient propagation in mice expressing either anchorless or wt PrPC. The second category also templates PrPres conformers with lower stability that may correspond to agents with increased fitness (7, 17, 34). The relationship between fitness and stability is under debate for hamster scrapie strains (3). Nevertheless, these concepts are consistent with those mentioned in a recent paper describing a dichotomy in the seeding behavior of poorly infectious rPrP fibrils compared to PMCA-induced infectious recombinant PrPSc (rPrPSc) preparations (44). The authors found that rPrP amyloid fibrils showed poor seeding activity in PMCA reactions using wt PrPC but efficiently seeded amyloid formation in fibrillization reactions with rPrP, a behavior consistent with the hypothetical category 1 agent described above. Conversely, rPrPSc efficiently seeded PMCA reactions but inhibited rPrP fibrillization, akin to the category 2 agent. The present data do not allow us to distinguish between these different possibilities but do suggest that anchorless PrPC could greatly facilitate the detection of category 1 agents.
Indeed, the data point to an important contribution from anchorless PrPC in agent propagation, especially in the case of L fibrils. On primary passage in wt/GPI− mice, rMoPrP L and H fibrils preferentially induced conversion of anchorless PrPC to PrPres (Fig. 2C). This preference for anchorless PrPC was apparently maintained for L and H fibril agents on secondary passage in wt/GPI− mice (Fig. 5 and 7B), and in the case of the D013 agent, it was so strong as to induce disease only in tg44+/+ mice. As shown in Fig. 6, four brain homogenates from L-fibril-treated animals (D090, D098, D018, D063) induced subclinical levels of PrPres formation on secondary passage but only in mice expressing anchorless PrPC (wt/GPI− or tg44+/+). Whether the D013 preference for GPI− PrPC is absolute will require additional passage studies. Since GPI− PrPC is largely unglycosylated, it may be that the D013 agent is a novel TSE strain with a strong preference for unglycosylated PrPC as opposed to the fully glycosylated PrPC that predominates in wt animals. Alternatively, D013 may be a category 1 agent with a limited ability to induce conversion of membrane-bound PrPC, perhaps due to a tendency to seed the formation of amyloid PrP, as occurs with anchorless PrPC, rather than the nonamyloid type of PrPres often formed by membrane-bound PrPC. We also noted that in tg44+/+ mice D013 agent plaques were much more likely to be perivascular than D061 or C413 agent plaques. Hence, another possibility is that D013 PrPres propagation is assisted by cofactors (e.g., glycosaminoglycans) that are enriched on perivascular basement membranes, as proposed for 22L and Chandler (13), but that the D061 and C413 agents are less dependent on these cofactors. Additional studies are required to determine the basis for these phenomena.
Our data indicate that at least two different TSE agents were generated in the present study. It is remarkable that two very different TSE agents (D013 and D061) emerged from the same rMoPrP fibril preparation. This suggests that a single rPrP fibrillization reaction condition may encipher multiple PrPres conformations. This is consistent with the results of a previous study characterizing synthetic prions (34). This may not be surprising, given the nature of our rMoPrP fibrillization reactions, which rely on spontaneous formation of rMoPrP fibrils. Hence, it is conceivable that multiple seeds of different conformation could spontaneously arise and propagate themselves to ultimately create a mixture of fibrils that segregate into a limited number of conformations. Unfortunately, the extremely low levels of PrPres and insufficient amounts of brain tissue precluded conformational studies after initial primary passage of rMoPrP fibrils in wt/GPI− mice. We are currently in the process of accumulating sufficient tissue to allow comprehensive conformational comparisons of PrPres associated with the different agents. On the basis of the PrPres immunoblot banding profiles and histopathology, the D061 and C413 agents were indistinguishable, suggesting that they may be similar agents. However, there were differences with respect to attack rates in mice expressing only wt PrPC (that for C413 was 100% with tightly grouped incubation periods between individuals, and that for D061 was incomplete with longer incubation periods) and with respect to incubation period in tg44+/+ mice that express only GPI− PrPC (that for D061 was >100 days shorter than that for C413) (Table 1). The observations from wt PrPC-expressing mice would suggest that the titer of the C413 agent was greater than that of the D061 agent, but this interpretation is inconsistent with the tg44+/+ infection data. Although inconclusive, these observations suggest that D061 may in fact be a different strain of agent than C413 or that D061 may be a mixture of strains with D013-like and C413-like properties. Thus, it is possible that three different TSE agents have been created. In summary, our data clearly show that TSE infectivity can be generated solely from rMoPrP fibril preparations and that the lack of a GPI anchor on the host PrPC can assist the discovery of novel TSE agents induced by fibril preparations.
Supplementary Material
ACKNOWLEDGMENTS
Animal experiments were performed in accordance with animal welfare guidelines under an animal study protocol approved by the Animal Care and Use Committee of the Rocky Mountain Laboratories, National Institute of Allergy and Infectious Diseases, National Institutes of Health.
This research was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
We thank Motohiro Horiuchi (Hokkaido University, Japan) for the kind gift of 31C6 antibody. We thank Byron Caughey for helpful discussions and critical reading of the manuscript. We are grateful for excellent animal care by Jeff Severson and graphics assistance from Heather Murphy and Anita Mora. We thank Lori Lubke and Nancy Kurtz for tissue preparation and histochemical staining.
Footnotes
Published ahead of print 22 August 2012
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1. Atarashi R, et al. 2007. Ultrasensitive detection of scrapie prion protein using seeded conversion of recombinant prion protein. Nat. Methods 4:645–650 [DOI] [PubMed] [Google Scholar]
- 2. Ayers JI, Kincaid AE, Bartz JC. 2009. Prion strain targeting independent of strain-specific neuronal tropism. J. Virol. 83:81–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ayers JI, et al. 2011. The strain-encoded relationship between PrP replication, stability and processing in neurons is predictive of the incubation period of disease. PLoS Pathog. 7:e1001317 doi:10.1371/journal.ppat.1001317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Baron GS, et al. 2011. Effect of glycans and the glycophosphatidylinositol anchor on strain dependent conformations of scrapie prion protein: improved purifications and infrared spectra. Biochemistry 50:4479–4490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Barria MA, Mukherjee A, Gonzalez-Romero D, Morales R, Soto C. 2009. De novo generation of infectious prions in vitro produces a new disease phenotype. PLoS Pathog. 5:e1000421 doi:10.1371/journal.ppat.1000421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Basler K, et al. 1986. Scrapie and cellular PrP isoforms are encoded by the same chromosomal gene. Cell 46:417–428 [DOI] [PubMed] [Google Scholar]
- 7. Bett C, et al. 2012. Biochemical properties of highly neuroinvasive prion strains. PLoS Pathog. 8:e1002522 doi:10.1371/journal.ppat.1002522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bocharova OV, Breydo L, Parfenov AS, Salnikov VV, Baskakov IV. 2005. In vitro conversion of full-length mammalian prion protein produces amyloid form with physical properties of PrP(Sc). J. Mol. Biol. 346:645–659 [DOI] [PubMed] [Google Scholar]
- 9. Bocharova OV, et al. 2006. Annealing prion protein amyloid fibrils at high temperature results in extension of a proteinase K-resistant core. J. Biol. Chem. 281:2373–2379 [DOI] [PubMed] [Google Scholar]
- 10. Castilla J, et al. 2008. Crossing the species barrier by PrP(Sc) replication in vitro generates unique infectious prions. Cell 134:757–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Castilla J, Saa P, Hetz C, Soto C. 2005. In vitro generation of infectious scrapie prions. Cell 121:195–206 [DOI] [PubMed] [Google Scholar]
- 12. Caughey B, Baron GS, Chesebro B, Jeffrey M. 2009. Getting a grip on prions: oligomers, amyloids, anchors and pathological membrane interactions. Annu. Rev. Biochem. 78:177–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chesebro B, et al. 2010. Fatal transmissible amyloid encephalopathy: a new type of prion disease associated with lack of prion protein membrane anchoring. PLoS Pathog. 6:e1000800 doi:10.1371/journal.ppat.1000800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chesebro B, et al. 1985. Identification of scrapie prion protein-specific mRNA in scrapie-infected and uninfected brain. Nature 315:331–333 [DOI] [PubMed] [Google Scholar]
- 15. Chesebro B, et al. 2005. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 308:1435–1439 [DOI] [PubMed] [Google Scholar]
- 16. Chianini F, et al. 2012. Rabbits are not resistant to prion infection. Proc. Natl. Acad. Sci. U. S. A. 109:5080–5085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Colby DW, et al. 2009. Design and construction of diverse mammalian prion strains. Proc. Natl. Acad. Sci. U. S. A. 106:20417–20422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Colby DW, et al. 2010. Protease-sensitive synthetic prions. PLoS Pathog. 6:e1000736 doi:10.1371/journal.ppat.1000736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cosseddu GM, et al. 2011. Ultra-efficient PrP(Sc) amplification highlights potentialities and pitfalls of PMCA technology. PLoS Pathog. 7:e1002370 doi:10.1371/journal.ppat.1002370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Deleault NR, Harris BT, Rees JR, Supattapone S. 2007. Formation of native prions from minimal components in vitro. Proc. Natl. Acad. Sci. U. S. A. 104:9741–9746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Deleault NR, Kascsak R, Geoghegan JC, Supattapone S. 2010. Species-dependent differences in cofactor utilization for formation of the protease-resistant prion protein in vitro. Biochemistry 49:3928–3934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Deleault NR, et al. 2012. Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc. Natl. Acad. Sci. U. S. A. 109:E1938–E1946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fischer M, et al. 1996. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J. 15:1255–1264 [PMC free article] [PubMed] [Google Scholar]
- 24. Geoghegan JC, et al. 2007. Selective incorporation of polyanionic molecules into hamster prions. J. Biol. Chem. 282:36341–36353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Green KM, et al. 2008. Accelerated high fidelity prion amplification within and across prion species barriers. PLoS Pathog. 4:e1000139 doi:10.1371/journal.ppat.1000139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Griffith JS. 1967. Self-replication and scrapie. Nature 215:1043–1044 [DOI] [PubMed] [Google Scholar]
- 27. Imamura M, et al. 2011. Glycosylphosphatidylinositol anchor-dependent stimulation pathway required for generation of baculovirus-derived recombinant scrapie prion protein. J. Virol. 85:2582–2588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jackson WS, et al. 2009. Spontaneous generation of prion infectivity in fatal familial insomnia knockin mice. Neuron 63:438–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Karapetyan YE, et al. 2009. Prion strain discrimination based on rapid in vivo amplification and analysis by the cell panel assay. PLoS One 4:e5730 doi:10.1371/journal.pone.0005730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim CL, et al. 2004. Cell-surface retention of PrPC by anti-PrP antibody prevents protease-resistant PrP formation. J. Gen. Virol. 85:3473–3482 [DOI] [PubMed] [Google Scholar]
- 31. Kim JI, et al. 2010. Mammalian prions generated from bacterially expressed prion protein in the absence of any mammalian cofactors. J. Biol. Chem. 285:14083–14087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Klingeborn M, Race B, Meade-White KD, Chesebro B. 2011. Lower specific infectivity of protease-resistant prion protein generated in cell-free reactions. Proc. Natl. Acad. Sci. U. S. A. 108:E1244–E1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Legname G, et al. 2004. Synthetic mammalian prions. Science 305:673–676 [DOI] [PubMed] [Google Scholar]
- 34. Legname G, et al. 2006. Continuum of prion protein structures enciphers a multitude of prion isolate-specified phenotypes. Proc. Natl. Acad. Sci. U. S. A. 103:19105–19110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Makarava N, et al. 2010. Recombinant prion protein induces a new transmissible prion disease in wild-type animals. Acta Neuropathol. 119:177–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Makarava N, et al. 2011. Genesis of mammalian prions: from non-infectious amyloid fibrils to a transmissible prion disease. PLoS Pathog. 7:e1002419 doi:10.1371/journal.ppat.1002419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Makarava N, et al. 2012. A new mechanism for transmissible prion diseases. J. Neurosci. 32:7345–7355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Meyerett C, et al. 2008. In vitro strain adaptation of CWD prions by serial protein misfolding cyclic amplification. Virology 382:267–276 [DOI] [PubMed] [Google Scholar]
- 39. Miller MB, Geoghegan JC, Supattapone S. 2011. Dissociation of infectivity from seeding ability in prions with alternate docking mechanism. PLoS Pathog. 7:e1002128 doi:10.1371/journal.ppat.1002128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Murayama Y, et al. 2010. Sulfated dextrans enhance in vitro amplification of bovine spongiform encephalopathy PrP(Sc) and enable ultrasensitive detection of bovine PrP(Sc). PLoS One 5:e13152 doi:10.1371/journal.pone.0013152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Oesch B, et al. 1985. A cellular gene encodes scrapie PrP 27-30 protein. Cell 40:735–746 [DOI] [PubMed] [Google Scholar]
- 42. Parchi P, et al. 1998. Different patterns of truncated prion protein fragments correlate with distinct phenotypes in P102L Gerstmann-Straussler-Scheinker disease. Proc. Natl. Acad. Sci. U. S. A. 95:8322–8327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Piccardo P, et al. 2001. Prion proteins with different conformations accumulate in Gerstmann-Straussler-Scheinker disease caused by A117V and F198S mutations. Am. J. Pathol. 158:2201–2207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Piro JR, et al. 2011. Seeding specificity and ultrastructural characteristics of infectious recombinant prions. Biochemistry 50:7111–7116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Prusiner SB. 1982. Novel proteinaceous infectious particles cause scrapie. Science 216:136–144 [DOI] [PubMed] [Google Scholar]
- 46. Prusiner SB, Groth DF, Bolton DC, Kent SB, Hood LE. 1984. Purification and structural studies of a major scrapie prion protein. Cell 38:127–134 [DOI] [PubMed] [Google Scholar]
- 47. Saa P, Castilla J, Soto C. 2006. Ultra-efficient replication of infectious prions by automated protein misfolding cyclic amplification. J. Biol. Chem. 281:35245–35252 [DOI] [PubMed] [Google Scholar]
- 48. Saborio GP, Permanne B, Soto C. 2001. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 411:810–813 [DOI] [PubMed] [Google Scholar]
- 49. Safar J, et al. 1998. Eight prion strains have PrP(Sc) molecules with different conformations. Nat. Med. 4:1157–1165 [DOI] [PubMed] [Google Scholar]
- 50. Shikiya RA, Ayers JI, Schutt CR, Kincaid AE, Bartz JC. 2010. Coinfecting prion strains compete for a limiting cellular resource. J. Virol. 84:5706–5714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shikiya RA, Bartz JC. 2011. In vitro generation of high-titer prions. J. Virol. 85:13439–13442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sigurdson CJ, et al. 2009. De novo generation of a transmissible spongiform encephalopathy by mouse transgenesis. Proc. Natl. Acad. Sci. U. S. A. 106:304–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Smirnovas V, et al. 2011. Structural organization of brain-derived mammalian prions examined by hydrogen-deuterium exchange. Nat. Struct. Mol. Biol. 18:504–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Soto C. 2011. Prion hypothesis: the end of the controversy? Trends Biochem. Sci. 36:151–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Soto C, Saborio GP, Anderes L. 2002. Cyclic amplification of protein misfolding: application to prion-related disorders and beyond. Trends Neurosci. 25:390–394 [DOI] [PubMed] [Google Scholar]
- 56. Stahl N, Borchelt DR, Hsiao K, Prusiner SB. 1987. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 51:229–240 [DOI] [PubMed] [Google Scholar]
- 57. Stöhr J, et al. 2011. Spontaneous generation of anchorless prions in transgenic mice. Proc. Natl. Acad. Sci. U. S. A. 108:21223–21228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tagliavini F, et al. 1994. Amyloid fibrils in Gerstmann-Straussler-Scheinker disease (Indiana and Swedish kindreds) express only PrP peptides encoded by the mutant allele. Cell 79:695–703 [DOI] [PubMed] [Google Scholar]
- 59. Wadsworth JD, et al. 2001. Tissue distribution of protease resistant prion protein in variant Creutzfeldt-Jakob disease using a highly sensitive immunoblotting assay. Lancet 358:171–180 [DOI] [PubMed] [Google Scholar]
- 60. Wang F, Wang X, Yuan CG, Ma J. 2010. Generating a prion with bacterially expressed recombinant prion protein. Science 327:1132–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Weber P, et al. 2007. Generation of genuine prion infectivity by serial PMCA. Vet. Microbiol. 123:346–357 [DOI] [PubMed] [Google Scholar]
- 62. Williamson RA, et al. 1998. Mapping the prion protein using recombinant antibodies. J. Virol. 72:9413–9418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zou WQ, et al. 2010. Variably protease-sensitive prionopathy: a new sporadic disease of the prion protein. Ann. Neurol. 68:162–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.