

Abstract
The HNH domain is found in many different proteins in all phylogenetic kingdoms and in many cases confers nuclease activity. We have found that the Bacillus subtilis hlpB (yisB) gene encodes a stand-alone HNH domain, homologs of which are present in several bacterial genomes. We show that the protein we term HlpB is essential for viability. The depletion of HlpB leads to growth arrest and to the generation of cells containing a single, decondensed nucleoid. This apparent condensation-segregation defect was cured by additional hlpB copies in trans. Purified HlpB showed cooperative binding to a variety of double-stranded and single-stranded DNA sequences, depending on the presence of zinc, nickel, or cobalt ions. Binding of HlpB was also influenced by pH and different metals, reminiscent of HNH domains. Lethality of the hlpB deletion was relieved in the absence of addA and of addAB, two genes encoding proteins forming a RecBCD-like end resection complex, but not of recJ, which is responsible for a second end-resectioning avenue. Like AddA-green fluorescent protein (AddA-GFP), functional HlpB-YFP or HlpB-FlAsH fusions were present throughout the cytosol in growing B. subtilis cells. Upon induction of DNA damage, HlpB-FlAsH formed a single focus on the nucleoid in a subset of cells, many of which colocalized with the replication machinery. Our data suggest that HlpB plays a role in DNA repair by rescuing AddAB-mediated recombination intermediates in B. subtilis and possibly also in many other bacteria.
INTRODUCTION
HNH domains exist in a broad spectrum of different organisms such as bacteria, phages, viruses, archaea, and eukaryotes (7). To date, more than 2,000 proteins containing an HNH motif have been identified. HNH stands for the three most conserved histidine and asparagine residues located within a nucleic acid-binding and cleavage site, which is composed of 30 to 40 amino acid (aa) residues (15). The HNH motif is found in a variety of different enzymes, including homing endonucleases, which initiate the insertion of mobile genetic elements into specific sites (such as self-splicing introns and inteins, e.g., yeast intron 1 protein), MutS homologs involved in DNA mismatch repair, bacterial toxins such as colicins and pyocins that degrade chromosomal DNA or rRNA (7, 23, 26), restriction endonucleases, e.g., methyl cytosine-specific restriction endonuclease MrcA of Escherichia coli (2), transposases, and DNA packaging factors (6). Immunity proteins are coexpressed with and tightly bound to colicins to avoid degradation of host DNA (18). In addition, HNH domains are found in proteins such as reverse transcriptases (34) and DNases. All these proteins play important roles in many different cellular processes, e.g., DNA repair, replication, and recombination and processes related to RNA (8).
HNH domains are best characterized in colicins, where they clearly provide nuclease activity. The first crystal structures of HNH domains were resolved in the nuclease domains of E. coli colicins E7 and E9 (4, 18, 19). The structures revealed that the E7 and E9 domains are metalloproteins 30 to 40 aa residues in length, containing a single tetrahedrally coordinated divalent transition metal, Zn2+ or Ni2+, respectively. The active site of HNH domains exhibits a highly conserved structure and is reminiscent of the Zn finger motif. The core consists of two antiparallel β-strands (β1 and β2) that are linked by a loop of varying lengths. A C-terminal α-helix flanks the β-strands in a way that allows binding of the metal ion in the center between these two structural elements. The metal ion is coordinated by three histidines and an additional soluble ligand (a phosphate ion in the crystals, which itself is usually coordinated by a fourth histidine) and links the β1-strand to the α-helix. The first H of the HNH motif stands for a strictly conserved histidine residue that is positioned at the end of the first β-strand and functions as a base in the DNA cleavage reaction. The N of HNH represents an asparagine residue that is responsible for the correct positioning of the two β-strands relative to one another. It is located on the long loop and is thus quite distant from the active site. But even though it has no catalytic function, it is the major force keeping the loop in the metal finger structure and the invariant histidine as well as the scissile phosphate in the correct orientation for the catalytic reaction. The last conserved residue of the HNH motif is a metal-binding histidine that is sometimes replaced by an additional asparagine (HNN) (10, 15).
Homing endonucleases can cleave one or both strands of the DNA double helix, while colicins generally cleave both strands (12). The nuclease domain of colicin E7 has been shown to bind to DNA as a dimer (4), which might explain the double-strand hydrolysis induced by this enzyme. Thus, HNH domains are very heterogeneous and apparently very flexible and adaptable to performing different tasks within their surrounding context.
Interestingly, although HNH domains show high similarities, the enzymes are not activated by the same metal ions (8, 9, 14, 22, 32, 33). Several metal ions have been reported to stabilize the protein or are necessary for either DNA binding or cleavage. To date, the discussions about which metal is the physiological cofactor for each enzyme are still ongoing.
Bacillus subtilis yisB (renamed hlpB) encodes a stand-alone HNH domain at the 3′ end of the add operon, located at 98° on the chromosome. This operon contains the addA and addB genes (analogs of E. coli recBCD genes) and sbcC and sbcD genes (homologs of eukaryotic genes encoding Mre11 and Rad50 proteins), all of which are implicated in DNA recombinational repair of double-strand breaks (DSB) (3, 5, 27). The gene is highly conserved in members of the Firmicutes and also present in many other bacterial phyla. In this report, we show that the hlpB product, HlpB, performs a vital function during cell growth, providing the first example of an HNH nuclease protein that confers an essential function to the cell. HlpB lethality was suppressed by addA and addB deletions, suggesting a function in DNA repair.
MATERIALS AND METHODS
Growth conditions.
Cloning was done in E. coli strains DH5α, XL1-Blue (Stratagene), and GM 48 (Stratagene). E. coli cells were grown in LB medium supplemented with 100 μg/ml ampicillin. Incubation occurred at 37°C and under conditions of constant shaking at 200 rpm. For protein overexpression, the cells were induced with 0.1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at an optical density at 600 nm (OD600) of 0.6 to 0.8. B. subtilis cells were cultivated in LB medium at 30°C with constant shaking at 200 rpm. Selection pressure was maintained by addition of antibiotics at needed concentrations (5 μg/ml chloramphenicol, 10 μg/ml kanamycin, 100 μg/ml spectinomycin), and promoters were induced with 0.5% xylose. For fluorescence microscopy studies, B. subtilis cells were grown in S750 medium (16). For induction of DNA double-strand breaks, cells were treated with 100 ng/ml mitomycin C (MMC) for 30 to 45 min. B. subtilis PY79 was used as the wild-type strain.
Construction of strains and plasmids.
To generate a version of the hlpB gene that was under the control of the inducible xylose promoter, the 3′ part of the upstream yirY gene was amplified by PCR using primers 5′-ATTCAAGCTTGCAAACTTGAAAACGAG-3′ and 5′-TCGGAATTCACCACCGCCCATCAACTCAAGTGATACCCG-3′ and cloned into pSG1164 (24) using HindIII and EcoRI sites. The resulting plasmid was integrated into the B. subtilis chromosome by single-crossover selection for chloramphenicol resistance, resulting in strain JN1 (pxyl-hlpB). To generate a copy of hlpB in trans, the hlpB gene was amplified with primers 5′-ATCAAGCTTGAGTTGATGTAAGGGAG-3′ and 5′GCTTGCATGCGACAAATTATATAGACCCC-3′ and was cloned into pDG148 autonomously replicating plasmid (Bacillus Genetic Stock Center [BGSC]) using HindIII and SphI sites. B. subtilis wild-type cells were transformed with the resulting pDGhlpB plasmid, selecting for kanamycin resistance, generating strain JM40 (pDGhlpB). The plasmid carries a spac promoter that can be induced with IPTG. Strain JM40 was transformed with chromosomal DNA from JN1, generating VK1 (pxyl-hlpB, pDGhlpB). For expression of a C-terminal yellow fluorescent protein (YFP) fusion of HlpB from the original gene locus, the hlpB gene was amplified with primers 5′-CCCCTCGAGTTAAGTTTTGATCAGCTTCTCAGG-3′ and 5′-TTGAGATCTAGTTTTGATCAGCTTCTCAGG-3′ and was cloned into pSG1187 (11) using EcoRI and ApaI sites. The resulting pVK4 plasmid was integrated into the B. subtilis chromosome via single-crossover selection for chloramphenicol [VK4 (hlpB-yfp)], such that the hlpB-yfp fusion was under the control of original promoter and HlpB-YFP was the only version of HlpB synthesized in the cells. For generation of a His6 tag fusion of HlpB protein, hlpB gene was amplified using the primers 5′-GTACCATGGCAAAGCAGATTGCC-3′ and 5′-TTGAGATCTTTTTTTCTTTTTTCGTTCATTTG-3′. The insertion was cloned into the NcoI and BglII sites of plasmid pQE60 (Qiagen), resulting in plasmid pVK5. E. coli M42 cells (Feingold 1970) were transformed with pVK5, generating strain VK05. To create a C-terminal FlAsH tag fusion of HlpB for single-crossover integration into the B. subtilis chromosome, the 3′ region of the gene was amplified using the primers 5′-TTAGAATTCAGCATCATCTTACACCGAAAG-3′ and 5′-CTTACTAGTTTACGGCTCCATGCAGCAGCCCGGGCAG-3′. The insertion was cloned into the EcoRI and SpeI sites of plasmid pSG1164, resulting in plasmid pMH04 (gfp gene replaced by hlpB FlAsH). PY79 was transformed with pMH04, resulting in strain MH23. For colocalization experiments with HlpB-FlAsH and DnaX-mCherry, competent cells of strain CDS4 were transformed with plasmid pMH04, generating strain MH37.
For the hlpB deletion, long flanking homology PCR (LFH-PCR) was used. Thereby, the gene was replaced by a tetracycline cassette as described by Wach (37). The tetracycline resistance cassette was obtained from plasmid pDG1515 (13).
Purification of HlpB.
Deep-frozen E. coli cells (strain VK05; see Table 1) were thawed on ice and resuspended in 15 ml Tris buffer. Phenylmethylsulfonyl fluoride (PMSF) (100 μM), 1 U of DNase, and 1 mg/ml (final concentration) lysozyme were added. The cells were disrupted using a French pressure cell (110 MPa, 3 repeats), and cell debris was sedimented by centrifugation (14,000 rpm, 30 min, 4°C). The supernatant was filtered using a sterile 0.45-μm-pore-size filter, and the cleared lysate was applied to a 1 ml HisTrap HP column (GE Healthcare) equilibrated with immobilized-metal affinity chromatography (IMAC) binding buffer (50 mM HEPES, 150 mM NaCl, 20 mM imidazole, pH 7.5). Affinity chromatography was performed at 4°C using an ÄKTAprime plus system (Amersham Biosciences). The loaded column was washed with binding buffer, and the protein was eluted using a 30-ml gradient ranging from 20 mM to 500 mM imidazole. The absorption maximum was analyzed via SDS-PAGE (12% SDS gels), and fractions showing sufficient purity were combined. The protein solution was concentrated via ultrafiltration (Vivaspin 20; Sartorius Stedim Biotech) (5 molecular weight cutoff [MWCO]) and afterward applied to a gel filtration column (Superdex 200; Amersham Biosciences) equilibrated in Tris buffer (pH 7.5). Gel filtration chromatography was performed at room temperature (RT) using an ÄKTA Fast Protein Liquid Chromatography (FPLC) system (Amersham Biosciences). Peak fractions were analyzed via SDS-PAGE, combined, and again concentrated via ultrafiltration. The protein concentration was determined photometrically (Implen NanoPhotometer).
Table 1.
List of bacterial strains
| Strain | Relevant genotypesa | Reference or source |
|---|---|---|
| PY79 | Prototrophic, Bacillus subtilis, wild type | 40 |
| CDS4 | dnaX mCherry Spcr | 35 |
| JN01 | pxyl-hlpB Cmr | This work |
| VK01 | pxyl-hlpB pDGhlpB Cmr Kanr | This work |
| VK04 | hlpB-yfp Cmr | This work |
| VK05 | hlpB His6 Ampr | This work |
| MH23 | hlpB FlAsH Cmr | This work |
| MH37 | hlpB FlAsH dnaX mCherry Cmr Spcr | This work |
| MH44 | hlpB::tet pDGhlpB | This work |
Amp, ampicillin; Cm, chloramphenicol; Kan, kanamycin; Spc, spectinomycin.
DNA binding assays.
Different DNA molecules (plasmid DNA [100 ng], 500-bp PCR fragment [0.5 μg], chromosomal DNA [0.5 μg], single-stranded DNA [ssDNA] oligonucleotide [0.5 μg]) were incubated in the presence of different constant concentrations of protein for 30 min at RT. The reaction was carried out in Tris buffer in a reaction volume of 15 μl. The samples were stained with DNA loading dye (containing Gel Red [Biotium]) and applied to a 0.8% agarose (in Tris buffer). Electrophoresis was performed at 40 mA, and the DNA and DNA-protein complexes were finally visualized using an UV transilluminator.
Gel shift assays using native polyacrylamide gels (electrophoretic mobility shift assay [EMSA]) were performed as follows. Increasing amounts of protein (0 μg, 1.5 μg, 2.5 μg, 3 μg, 3.5 μg, 4 μg, 4.5 μg) were incubated in the presence of 3 μg of a 70-bp DNA fragment in 1× binding buffer containing 0.5 mM Zn2+ for 30 min at RT. The samples were stained with DNA loading dye (containing Gel Red [Biotium]) and applied to a native polyacrylamide gradient gel (gradient ranging from 5% to 15% acrylamide-bisacrylamide). Electrophoresis was performed as described above. The DNA and DNA-protein complexes were finally visualized using a UV transilluminator. Annealing of the DNA oligonucleotides (3′-TTCAAAGCTTAGAAAGGAGATTCTTACCATGGGCGGGCCCGTCGACGCTAGCGGTACCGCATGCAAT-5′, 5′-ATTGCATGCGGTACCGCTAGCGTCGACGGGCCCGCCCATGGTAAGAATCTCCTTTCTAAGCTTTGAA-3′) for obtaining the 70-bp DNA fragment was achieved by mixing the two oligonucleotides in a molar ratio of 1:1 (final concentration, 5 μg/μl). This solution was further diluted in annealing buffer (10 mM Tris, 1 mM EDTA, 50 mM NaCl, pH 7.5) at a ratio of 1:5 (final concentration, 1 μg/μl). Subsequently, the mixture was heated to 95°C for 5 min and slowly cooled to RT for efficient annealing.
Image acquisition.
Fluorescence microscopy was performed on an Olympus AX70 microscope with a CoolSNAP ES2 camera (Photometrics) and a mercury-xenon light source (MT10; Olympus). Cells (3 to 4 μl) were mounted on thin agarose pads (1%, prepared in S750 minimal medium) on an object slide. Images were processed with Metamorph 6.3 or ImageJ 1.43 software. Microscopes were equipped with oil objectives with ×100 magnification and a numeric aperture of 1.45. DNA was stained with 4′,6-diamidino-2-phenylindole (DAPI; final concentration, 0.2 ng/ml), and membranes were stained with FM4-64 (final concentration, 1 nM). For FlAsH tag experiments, TC FlAsH II in-cell tetracysteine tag detection kits (Invitrogen) were used according to the instructions of the manufacturer.
RESULTS
The yisB-hlpB product, HlpB, performs an essential function in Bacillus subtilis cells.
yisB-hlpB is the last gene of the putative add operon in B. subtilis, located at the 98° location on the chromosome, comprising addA, addB, sbcD, and sbcC genes, all of which confer functions in DNA repair. yisB contains a typical rho-independent terminator structure downstream of its stop codon, and the genes downstream of yisB are transcribed in the opposite orientation, suggesting that it is the last gene of the operon. The gene encodes a 11.3-kDa polypeptide of 100 residues with a pI of 9.9 that shows significant homology to the HNH nuclease domain (see below). Because of its similarity to the HNH domain, we renamed the gene product HlpB (HNH-like protein of Bacillus) and the gene hlpB, which are the designations used here. During our work on this operon, we have found that it is not possible to insert a promoterless plasmid between the hlpB gene and its upstream gene, sbcC (yirY). It was possible to obtain an sbcC-gfp fusion construct only by using a plasmid that contains an inducible xylose promoter, and only in the presence of xylose (data not shown). Consistent with this, the removal of xylose from the medium of a culture of strain JN1 (pxyl-hlpB) led to an arrest in growth (Fig. 1A). The growth arrest took place slowly, only when cells grown in the absence of inducer were resuspended into fresh medium; the growth of the culture was arrested relative to that of cells grown in the presence of inducer (Fig. 1B). To verify that this effect was due to the absence of the hlpB transcript, we cloned the hlpB gene into the pDG148 autonomous replicating plasmid that contains an IPTG-inducible spac promoter. Strain VK1 (pxyl-hlpB, pDGhlpB), in contrast to strain JN1, was able to grow in the absence of xylose (Fig. 1A). To verify that hlpB is essential, we generated a long flanking sequence construct that replaced the hlpB gene with a tetracycline resistance cassette. This construct was able to generate an hlpB deletion in a strain carrying pDGhlpB but not in wild-type cells (Fig. 1C), showing that hlpB performs an essential function in Bacillus. The essential nature of hlpB was not seen in a systematic genome-wide deletion analysis, most likely because the spac promoter, which in our hands could not be repressed as strongly as the xylose promoter used in our study, was used for depletion studies.
Fig 1.

(A) Growth of Bacillus subtilis strains in the presence or absence of inducer (xylose) driving synthesis of HlpB. pxyl-hlpB, transcription of hlpB (encoding HlpB) depends on presence of xylose; pDGhlpB, plasmid expressing hlpB; wild type, JM11 (scpA-yfp) strain expressing a functional YFP fusion unrelated to HlpB that grows on plates containing chloramphenicol. (B) Growth curves of B. subtilis strain VK01 (pxyl-hlpB) in minimal medium (plus Casamino Acids) in the presence (black line) or absence (gray line) of xylose. Dashed gray line, cells grown in the absence of xylose resuspended into fresh medium lacking xylose. (C) Competent cells transformed with 0.01 μg of chromosomal DNA from MH44 (hlpB::tet, pDGhlpB), selecting for tet resistance. Note that all strains could be transformed by an unrelated marker (comEA::cat).
When cells depleted for HlpB were streaked on plates containing rich medium and xylose as the inducer, only 2% to 5% of the cells grew as colonies (as expected from their final optical density), indicating that depletion of HlpB is toxic for cells and leads to cell death.
A role of HlpB in chromosome replication-segregation or in DNA repair.
To investigate the role of hlpB, we resuspended exponentially growing JN1 (pxyl-hlpB) cells in medium containing or lacking xylose, the inducer for hlpB transcription. After several generations, growth was arrested in cells in which HlpB was depleted, and the culture mostly comprised single cells (Fig. 2B) or short chains (Fig. 2C), whereas cells growing in the presence of inducer were still present in longer chains characteristic of a growing B. subtilis PY79 culture (Fig. 2A). Staining of cells with DAPI DNA stain showed that HlpB-depleted cells contained decondensed nucleoids (Fig. 2B and C), in contrast to the more compact nucleoids in wild-type cells (Fig. 2A). In addition, in contrast to wild-type cells that contained one (smaller cells) or two (larger cells) nucleoids (Fig. 2A), HlpB-depleted cells almost exclusively contained single nucleoids (Fig. 2B and C). These data rule out a role in cell division, because a defect in this process results in filamentous cells having normally segregated chromosomes (1). A classical block in replication generates elongated cells with a single central nucleoid (36). However, the results with respect to the depletion of HlpB are compatible with a function in replication (or DNA repair, which can affect the restoration of collapsed or stalled replication forks) or in chromosome segregation.
Fig 2.

Fluorescence microscopy of B. subtilis strain VK1 (pxyl-hlpB). (A) Cells growing exponentially in the presence of xylose. (B and C) Single cells (B) and short chains (C) showing growth representing about 12 doubling times after removal of xylose (images show the state of the cells when growth has ceased). Note that growing B. subtilis cells grow in chains rather than as single cells. Membranes were stained with FM4-64, DNA with DAPI. White scale bars, 2 μm; all images are equal in scale.
HlpB binds nonspecifically and cooperatively to DNA in the presence of zinc, cobalt, or nickel.
To obtain information on the biochemical properties of HlpB, we purified the protein to apparent homogeneity by two-step nickel nitrilotriacetic acid (NTA) and gel filtration chromatography (Fig. 3A). HlpB eluted as a monomer (at a size of 14 kDa) from gel filtration analysis (Fig. 3B; note that the second peak represents the presence of imidazole but the absence of HlpB), in contrast to colicin E7, which has been shown to form a dimer in solution (4).
Fig 3.

Purification of HlpB after heterologous expression in E. coli. (A) Coomassie-stained SDS-PAGE of purification steps. Lane 1, marker; lane 2, cell extract from noninduced E. coli strain; lane 3, cell extract after induction of HlpB synthesis; lane 4, elution fraction after immobilized metal ion affinity chromatography (IMAC) on Ni2+-Sepharose; lane 5, elution fraction after gel filtration. (B) Elution profile of gel filtration chromatography on superose. Absorption at 280 nm is shown in black. Fractions containing HlpB are designated by the horizontal line; values representing elution of marker proteins are given above the graph.
HNH nuclease domains have been reported to require divalent ions, mostly magnesium, zinc, or nickel, for DNA binding and/or nuclease activity. We therefore performed agarose gel shift assays in the presence of different metals to test if HlpB has DNA binding activity. In the presence of zinc and at physiological pH (7.5), purified HlpB showed binding activity with respect to a circular plasmid (Fig. 4A and B). Induction of binding was very pronounced even with a relatively small change in ion concentration, while at 2 mM Zn2+, 100 nM HlpB was unable to bind to DNA; a 2-fold increase in the Zn2+ concentration led to complete binding of the DNA (Fig. 4C). Similar concentrations of divalent ions have been reported for DNA binding or DNase activity of HNH domains (9, 32). Therefore, HlpB is a metal-dependent DNA binding protein.
Fig 4.

Gel retardation experiments (0.8% agarose, except for panel E, for which a 5% to 10% native polyacrylamide gradient gel was employed) using purified HlpB. (A and B) Plasmid DNA (0.5 μg/6 nM) was incubated with different amounts of HlpB as indicated above the lanes in the presence of 5 mM ZnCl2. (C) Plasmid VK1 (0.5 μg/6 nM) was incubated with 100 nM HlpB and different concentrations of Zn2+ as indicated. (D) Different DNA molecules were incubated with 5 mM ZnCl2 and without (lanes 2 to 5) or with (lanes 6 to 9) 200 nM HlpB. Lane 1, marker DNA; lanes 2 and 6, pVK1 (0.5 μg); lanes 3 and 7, pQE60 (0.5 μg); lanes 4 and 8, B. subtilis chromosomal DNA (0.5 μg); lanes 5 and 9, 500-bp PCR fragment (0.5 μg). Note that the shifted band is not in but is underneath the wells of the gel. (E) A 70-bp dsDNA oligonucleotide (3.2 μg) was incubated with increasing amounts of HlpB. Lane 1, no HlpB; lane 2, 1.5 μg HlpB; lane 3, 2.5 μg HlpB; lane 4, 3 μg HlpB; lane 5, 3.5 μg HlpB; lane 6, 4 μg HlpB; lane 7, 4.5 μg HlpB. (F) Different DNA molecules were incubated without (lanes 1 to 4) or with (lanes 5 to 8) 200 nM HlpB and 0.5 mM Zn2+. Lane 9, PstI-digested DNA; lanes 4 and 8, plasmid DNA (100 ng); lanes 3 and 7, 500-bp PCR fragment (0.5 μg); lanes 2 and 6, chromosomal Bacillus subtilis DNA (0.5 μg); lanes 1 and 5, ssDNA oligonucleotide (0.5 μg). (G) Plasmid DNA was incubated with 2 mM ZnCl2, without or with 100 nM HlpB, and without or with 5 mM MgCl2 as indicated. (H) Plasmid DNA was incubated with or without 100 nM HlpB and with different metal ions (5 mM) as indicated above the lanes. (I) Plasmid DNA was incubated with or without 100 nM H22A mutant HlpB and with different metal ions (5 mM) as indicated above the lanes.
To test if HlpB binds to various forms of DNA, we used different DNA substrates and a fixed concentration of HlpB that leads to complete binding of plasmid DNA. HlpB showed stable binding to different circular plasmids (Fig. 4D, lanes 2 and 3 and lanes 6 and 7), as well as chromosomal DNA (lanes 4 and 8) and a linear double-stranded 500-bp DNA fragment (lanes 5 and 9). In all cases, HlpB formed very-high-molecular-weight complexes, irrespective of the nature of the DNA. Note that the shifted DNA band was migrating within the gel, just underneath the wells, so HlpB truly binds to DNA and does not simply lead to precipitation of DNA in the gel pockets. To support this notion, we tested binding of HlpB in a native PAGE. HlpB bound to the 30-bp double-stranded DNA (dsDNA) in the presence of Zn2+ and led to a shift within the gel, retarding all DNA molecules, showing that HlpB truly binds to DNA (Fig. 4E). Interestingly, suboptimal amounts of Zn2+ supported binding to plasmid and shorter linear DNA but no longer supported binding to chromosomal DNA (Fig. 4F, lanes 2 and 6), revealing unusual binding properties (i.e., a need for sufficient concentrations of Zn for binding to chromosomal DNA) that, however, do not depend on DNA supercoiling, as both PCR DNA and chromosomal DNA, in contrast to plasmid DNA, are not supercoiled.
At physiological pH, HlpB bound to DNA with highest affinity in the presence of Zn2+ (Fig. 4A) but also of Ni2+ or Co2+ (Fig. 4H). Efficient DNA binding was observed at 4.5 mM Ni2+ but not at lower concentrations (data not shown). Addition of 5 mM Mn2+ or of Mg2+ allowed only partial binding of DNA (Fig. 4H). Interestingly, at higher pH (pH 8.0 or 8.5), lesser amounts of Ni2+and Co2+ were required for optimal DNA binding of HlpB (data not shown). Note that Ni2+ and Co2+ did not result in a shift in DNA by themselves, in contrast to Cu2+ (data not shown). Zn2+, Co2+, and Mn2+ have been shown to bind to HNH domains (14), so HlpB has genuine HNH domain properties.
Binding of HlpB to dsDNA was highly cooperative. Although 10 nM protein did not result in any DNA binding in the presence of Zn2+ at an optimal concentration of 4 mM, plasmid DNA was both completely shifted at 30 nM HlpB and shifted about 90% at 20 nM (Fig. 4A). More amazingly, the DNA was not efficiently bound at 12 nM HlpB, but considerable binding occurred at 14 nM protein (Fig. 4B). Although some smearing of the DNA was apparent at concentrations of HlpB between 14 and 20 nM (Fig. 4B), a single discrete shifted band appeared at 30 mM HlpB, which can be most easily explained by highly cooperative binding of HlpB to DNA. It is interesting that HlpB bound to supercoiled (lower band of control DNA) and nicked (upper band) circular DNAs with similar levels of affinity. To test if Zn2+ itself has an effect in the shift assay, we incubated DNA with 5 mM Zn2+ in the presence or absence of HlpB. At pH 7.5, Zn2+ did not have an effect on the DNA, and a shift in DNA was accomplished only by addition of 100 nM HlpB (data not shown); thus, DNA binding as monitored in the assay is a property of HlpB. Interestingly, binding of HlpB to DNA was also influenced by Mg2+. In the presence of a suboptimal concentration of Zn2+ (2 mM), 100 nM HlpB completely shifted the DNA in the presence, but not in the absence, of 5 mM MgCl2 (Fig. 4G), showing that an excess of a nonpreferred metal can compensate for suboptimal concentrations of a preferred ion. We have not observed clear nuclease activity in HlpB in a variety of assays (data not shown), indicating that this putative activity may depend on additional factors.
An H22A variant loses high-affinity DNA binding.
HlpB contains all four of the characteristic histidine residues of HNH domains, two of which are highly conserved and two moderately conserved. We investigated if the mutation of one of these residues, H22, which is one of the three histidines coordinating the bound divalent ion (moderately conserved), leads to a change in DNA binding. In contrast to wild-type protein, the H22A HlpB mutant no longer bound to DNA with high efficiency in the presence of Co2+ or of Ni2+ (Fig. 4I). HlpB H22A was able to bind to DNA with low affinity, in the presence of Ni2+, Co2+, Mg2+, or Mn2+, and to a much lesser extent than was seen even with wild-type HlpB in the presence of Mn2+ or Mg2+ (Fig. 4H). Therefore, the loss of a critical ion ligand results in an almost complete loss of DNA binding of HlpB, further validating the hypothesis that it is a true HNH-like protein.
HlpB was dispersed throughout the cells and assembled at different sites, including replication forks, on the nucleoid after induction of DNA damage.
To obtain further information on the function of HlpB, we constructed different fusions (N and C terminal) of hlpB to the gene encoding YFP at the original gene locus on the chromosome. We succeeded in obtaining a C-terminal YFP fusion at the original hlpB locus, which is fully functional, because cells exclusively expressing hlpB-yfp grow indistinguishably from wild-type cells. Figure 5A shows that the HlpB-YFP fusion localized throughout the cell (showing rather faint fluorescence), with no apparent preferential staining of the nucleoids. These data indicate that HlpB is not a general DNA binding protein in vivo. We also generated an HlpB-FlAsH fusion, which can be specifically coupled to a green chromophore. Again, fluorescence was homogeneous throughout the cytosol in exponentially growing cells (Fig. 5B), while there was no fluorescence above normal background in PY79 cells. Interestingly, when mitomycin C (MMC) (which generates DNA adducts, cross-links, and, consequently, DNA double-strand breaks) was added, some cells showed clear foci on the nucleoids (Fig. 5C). Of 700 cells analyzed, 13% contained a single HlpB-FlAsH focus or, rarely, two foci, indicating that few molecules of HlpB assemble at a specific site on the nucleoids upon induction of DNA damage in a subset of the cells. Note that addition of the FlAsH substrate to MMC-treated cells lacking the HlpB-FlAsH fusion did not induce expression of any foci (Fig. 5G). HlpB-FlAsH foci were visible in only 0.2% of all cells during exponential growth (Fig. 5B), a result which may have corresponded to naturally occurring DNA damage. We did not observe any foci of HlpB-YFP after addition of MMC. Possibly, small numbers of assembled YFP-fused proteins would not have been visible against the dispersed HlpB proteins, in contrast to the brighter FlAsH tag.
Fig 5.

Fluorescence microscopy of Bacillus subtilis cells. (A) HlpB-YFP in exponentially growing cells. (B and C) Localization of HlpB-FlAsH in exponentially growing cells before (B) and 45 min after (C) induction with 100 ng/ml MMC. (D to F) Colocalization experiments with HlpB-FlAsH (green in the overlay) and DnaX-mCherry (red in the overlay) during exponential growth (D) and 45 min after induction with 100 ng/ml MMC (E and F). (G) PY79 cells (lacking HlpB-FlAsH) treated with FlAsH reagent 45 min after addition of MMC. White arrowheads indicate the localization of the fusion proteins. Red arrowheads indicate the localization of DnaX, green the localization of HlpB, and yellow the colocalization of both proteins. White bars, 2 μm.
Two patterns of protein assembly in response to DNA damage have been described so far. SbcC assembles specifically at replication forks after the addition of MMC (27), while RecN assembles at sites on the nucleoids away from replication forks (17) and recruits several proteins, including RecA, to these so-called repair centers. A mixed pattern was observed for SbcE protein, which assembles both at the replisome and at RCs after induction of DNA damage (21). Unfortunately, we were not able to colocalize RecN and HlpB, because the red chromophore ReAsH did not appear to enter Bacillus cells but rather generated nonspecific staining of the cell wall, and a RecN-mCherry fusion did not reveal clear foci. Therefore, we are not yet able to state if HlpB is recruited to RCs. However, we were able to visualize both HlpB-FlAsH and DnaX-mCherry, which represents the localization of the replisome. During exponential growth, one or, less frequently, two DnaX-mCherry foci were visible in each cell and completely dispersed HlpB-FlAsH fluorescence (Fig. 5D). Interestingly, after induction of DNA damage through the addition of MMC, 30% of the HlpB-FlAsH foci (of 70 cells that contained foci) colocalized with DnaX-mCherry, while 40% of the foci did not (650 cells analyzed) (Fig. 5E and F). In 30% of the cells having HlpB-FlAsH foci, no DnaX-mCherry foci were visible. Thus, HlpB is recruited to stalled replication forks in at least a subset of the cells having DNA damage.
The lethality of an hlpB deletion is suppressed in addA and addAB mutant cells.
It is possible that HlpB is important to remove toxic intermediates of certain DNA pathways. We therefore generated several strains with mutations in genes involved in homologous recombination (HR) that carry the plasmid expressing HlpB. Mutant cells with and without this plasmid were grown to competence and transformed with 0.01 to 0.1 ng of chromosomal DNA from the hlpB deletion strain. sbcC, recN, or recJ mutant cells carrying the plasmid, like wild-type cells, could be transformed with hlpB mutant DNA, but not plasmid-free cells (Fig. 1C). Conversely, addA or addAB mutant cells free of plasmid could be transformed to also contain the hlpB deletion (Fig. 1C). All strains could be transformed with chromosomal DNA from a comEA deletion strain, with only recN mutant cells showing transformation activity that was reduced 2- to 3-fold (data not shown). These experiments show that the genes encoding the AddAB complex involved in end resection suppress the lethality of the hlpB deletion. We tested wild-type cells with and without pDGhlpB and addA and addAB hlpB double-mutant cells (with and without the plasmid) for their sensitivity to MMC. All strains were roughly equally sensitive to 50 ng/ml or 75 ng/ml of MMC; addAB mutant cells were marginally more sensitive than wild-type cells, while recJ and, to an even greater degree, recN mutant cells showed considerable MMC sensitivity (Fig. 6). These experiments support the idea that HlpB removes intermediates of AddAB activity or is even essential for the proper activity of the complex whereas it has no effect on the RecJQS end resection pathway or on RecN or SbcC activity.
Fig 6.
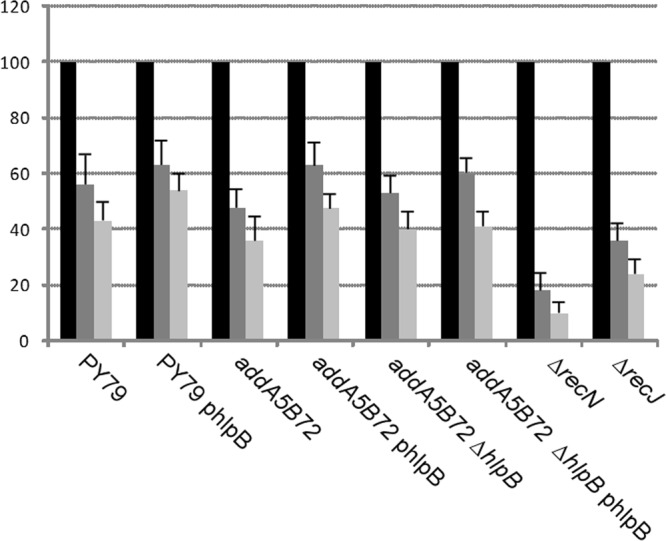
Survival assays. Strains were grown to mid-exponential phase (OD = 0.5) and were split into three cultures, one of which was treated with 50 ng/ml (dark gray bars), one of which was treated with 70 ng/ml (light gray bars), and one of which was left untreated (black bars). Survival is indicated in percentages of cells relative to the nontreated culture. Data are means of the results of three independent experiments. Note that addAB recJ double-mutant cells are much more sensitive to MMC treatment than recJ or addAB single-mutant cells, showing that AddAB proteins play an important role in DNA repair.
HNH domains contain a highly conserved HNH motif in which the first histidine is invariant, while the asparagines can be a histidine and the second histidine an asparagine residue (7). The first H is most frequently followed by an L, as is the N, and between these residues are an invariant P and two glycines (7). All these residues are conserved in HlpB (underlined in Fig. 7, with the HNH shaded in gray), except for the N (indicated in parentheses). The first H (corresponding to H22 of HlpB) is situated on the first β-strand of the ββα fold of HNH domains, while N (M38 in HlpB) is located on the loop between β1 and β2. The second H (H44) is situated on the α-helix. The Zn2+ or Ni2+ ion in the crystal structures obtained from colicin E7 and E9 is coordinated by three histidines (H4, H19, H20) corresponding to H21, H22, and H48 in HlpB and by a phosphate group that is in turn bonded to a histidine (H44). These residues are highly conserved in HlpB (Fig. 7). The N of the HNH motif is most likely important for folding and can be replaced by another stabilizing residue (10), and a mutation resulted in reduced but still substantial activity (38). It may therefore not be conserved in all HNH domains such as it is in HlpB. We performed PSI BLAST searches to find homologs of hlpB and found over 300 orthologs, in bacteria and also phages, which are between 100 and 125 amino acids long. HlpB is highly conserved in members of the Bacilliales (>60% sequence identity) and Firmicutes (>40%). HlpB from cyanobacteria (Synechococcus sp. PCC 7335) can have 43% sequence identity and more than 40% identity in members of the Deinococcales. The gene encoding HlpB is also found in Bacteriodes and in Proteobacteria (e.g., Magnetospirillum magnetotacticum MS-1 or Rhizobiales, with 39% sequence identity); it thus represents a member of a highly conserved bacterium-phage gene family. All proteins contain the HNH nuclease signature, although the N residue is changed to another residue, and also an unusual, highly positively charged C terminus. HlpB contains 8 positive side chains and 1 negative side chain within the last 14 residues and 12 positive and 2 negative residues within the last 27 amino acids (Fig. 7). Because a mutation in the ion binding site almost completely abolishes DNA binding (Fig. 4I), it is not clear to what degree the C terminus can be involved in DNA binding.
Fig 7.

HlpB sequence and consensus sequence of HlpB-like proteins from bacteria and phages. Invariant residues are shaded in black, conserved residues are shaded in gray. Plus signs corresponding to the consensus sequences indicate conserved types of amino acids. Residues usually conserved in the HNH domain are underlined in the consensus sequences, with the HNH motifs highlighted in gray. The conserved arginine is not found in HlpB-like proteins and is indicated in parentheses.
DISCUSSION
In this work, we describe a novel protein family of stand-alone HNH domain proteins, HLPs, whose member in B. subtilis confers a function that is essential for viability. HLPs have all residues characteristic of the HNH domain, which often confers nuclease activity in proteins, but also contain a highly positively charged C terminus. These proteins can be found in many bacterial genomes, as well as in phages. The genome-wide B. subtilis deletion screen obviously failed to identify HlpB as an essential protein. As for its detailed function, we have found several clues. First, HlpB is a DNA-binding protein that shows strong cooperativity in its binding and is also strongly influenced by the presence of distinct metal ions. Nickel, zinc, and cobalt ions confer high efficiency for DNA binding of HlpB to a variety of DNA molecules, including plasmid and linear DNA. It is possible that the function of HlpB within the cell is regulated by metal ion availability or by a cofactor that regulates metal ion binding. Second, however, HlpB does not assemble on the chromosome in live B. subtilis cells; rather, it is dispersed throughout the cell. Thus, it does not confer general DNA compaction activity like HBsu or SMC, which localize throughout the nucleoid or assemble at two distinct sites on the chromosome, respectively (20, 25, 28), but must play a more specific role. Third, interestingly, the depletion of HlpB gave rise to cells containing single decondensed nucleoids, suggesting that DNA replication and/or chromosome segregation, but not cell division, is compromised in the absence of HlpB. Most importantly, we found that HlpB lethality is suppressed by the deletion of either of the two genes encoding subunits of the AddAB complex, which is the analog of E. coli RecBCD. AddAB degrades one strand of the DNA duplex in a processive manner starting at the site of the DNA break (end resection) and thus generates the substrate for RecA. The other end resection pathway that also has both helicase and nuclease activity is composed of RecJ and RecQ or RecS. Lethality of HlpB was not suppressed by deletions in recJQS, or in sbcC, which is also within the add operon and forms a nuclease complex with SbcD. Viability was also not restored by the deletion of other genes whose products are involved in homologous recombination, e.g., by recN, so the suppression by addAB appears to be rather specific instead of a general effect of deletions of genes involved in HR. A possible function of HlpB may thus be the removal of intermediates generated by AddAB, which otherwise would be toxic to the cells. Such effects for enzymes involved in HR have been seen, for example, for the PcrA helicase, an essential protein involved in fork reestablishment (30). Even in normally growing cells, stalling of replication forks frequently arises during the replication cycle (39). One mechanism to repair stalled forks is to reverse the fork, generate a cut, and recombine the broken strand with the replicated sister strand, which after resolution of the recombination intermediate results in a new replication fork that can be reinitiated. Interestingly, a pcrA deletion can be suppressed by mutations in RecF and RecO that are involved in DNA recombination and DNA DSB repair (31). This finding suggests that the PcrA helicase might be required to remove or remodel recombination events after fork reversal and that, in the absence of PcrA, fork repair through RecFO-mediated recombination creates toxic intermediates. Likewise, DNA-binding RdgC protein counteracts toxic intermediates occurring during replication restart in priA mutant cells (29), highlighting the importance of proper fork rescue in normally growing cells. In analogy, AddAB may provide a function during fork reversal and HlpB may rescue abortive processes or remove toxic intermediates. Interestingly, an HlpB-FlAsH fusion assembled at distinct sites on the nucleoids upon induction of DNA damage, at least in a subset of the cells. To a considerable degree, HlpB colocalized with replication forks, supporting a function at this position. In a majority of the cells, HlpB was dispersed throughout the cells similarly to AddAB (27). It is important to elucidate the exact function of HlpB, because several other bacteria contain HlpB homologs (HLPs) that may also be essential proteins. In addition to the putative HNH motif, all HLP proteins contain a highly positively charged C terminus, whose function remains to be investigated.
In toto, HlpB is an interesting protein in several aspects. It is similar to HNH domain protein and widely conserved in bacteria and some phage genomes. It has metal-dependent DNA binding but no detectable endonuclease activity (but may have such activity in the presence of additional factors, such as AddAD), and it confers a vital function in the cell, apparently related to DNA repair, via the AddAB end resection pathway. Future work should shed more light on the mode of action of HLP proteins.
ACKNOWLEDGMENTS
We thank Vanessa Kaiser, Judita Mascarenhas, Nicole Scherr, and Clarisse Defeu Soufo for technical assistance.
This work was supported by the Deutsche Forschungsgemeinschaft and the Fonds der Chemischen Industrie.
Footnotes
Published ahead of print 14 September 2012
REFERENCES
- 1. Beall B, Lutkenhaus J. 1991. FtsZ in Bacillus subtilis is required for vegetative septation and for asymmetric septation during sporulation. Genes Dev. 5:447–455 [DOI] [PubMed] [Google Scholar]
- 2. Bujnicki JM, Radlinska M, Rychlewski L. 2000. Atomic model of the 5-methylcytosine-specific restriction enzyme McrA reveals an atypical zinc finger and structural similarity to betabetaalphaMe endonucleases. Mol. Microbiol. 37:1280–1281 [DOI] [PubMed] [Google Scholar]
- 3. Chédin F, Handa N, Dillingham MS, Kowalczykowski SC. 2006. The AddAB helicase/nuclease forms a stable complex with its cognate chi sequence during translocation. J. Biol. Chem. 281:18610–18617 [DOI] [PubMed] [Google Scholar]
- 4. Cheng YS, Hsia KC, Doudeva LG, Chak KF, Yuan HS. 2002. The crystal structure of the nuclease domain of colicin E7 suggests a mechanism for binding to double-stranded DNA by the H-N-H endonucleases. J. Mol. Biol. 324:227–236 [DOI] [PubMed] [Google Scholar]
- 5. Connelly JC, Kirkham LA, Leach DR. 1998. The SbcCD nuclease of Escherichia coli is a structural maintenance of chromosomes (SMC) family protein that cleaves hairpin DNA. Proc. Natl. Acad. Sci. U. S. A. 95:7969–7974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cymerman IA, Obarska A, Skowronek KJ, Lubys A, Bujnicki JM. 2006. Identification of a new subfamily of HNH nucleases and experimental characterization of a representative member, HphI restriction endonuclease. Proteins 65:867–876 [DOI] [PubMed] [Google Scholar]
- 7. Dalgaard JZ, et al. 1997. Statistical modeling and analysis of the LAGLIDADG family of site-specific endonucleases and identification of an intein that encodes a site-specific endonuclease of the HNH family. Nucleic Acids Res. 25:4626–4638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Doudeva LG, et al. 2006. Crystal structural analysis and metal-dependent stability and activity studies of the ColE7 endonuclease domain in complex with DNA/Zn2+ or inhibitor/Ni2+. Protein Sci. 15:269–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Drouin M, Lucas P, Otis C, Lemieux C, Turmel M. 2000. Biochemical characterization of I-CmoeI reveals that this H-N-H homing endonuclease shares functional similarities with H-N-H colicins. Nucleic Acids Res. 28:4566–4572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Eastberg JH, Eklund J, Monnat R, Jr, Stoddard BL. 2007. Mutability of an HNH nuclease imidazole general base and exchange of a deprotonation mechanism. Biochemistry 46:7215–7225 [DOI] [PubMed] [Google Scholar]
- 11. Feucht A, Lewis PJ. 2001. Improved plasmid vectors for the production of multiple fluorescent protein fusions in Bacillus subtilis. Gene 264:289–297 [DOI] [PubMed] [Google Scholar]
- 12. Galburt EA, Stoddard BL. 2002. Catalytic mechanisms of restriction and homing endonucleases. Biochemistry 41:13851–13860 [DOI] [PubMed] [Google Scholar]
- 13. Guérout-Fleury AM, Shazand K, Frandsen N, Stragier P. 1995. Antibiotic-resistance cassettes for Bacillus subtilis. Gene 167:335–336 [DOI] [PubMed] [Google Scholar]
- 14. Hannan JP, et al. 2000. NMR studies of metal ion binding to the Zn-finger-like HNH motif of colicin E9. J. Inorg. Biochem. 79:365–370 [DOI] [PubMed] [Google Scholar]
- 15. Huang H, Yuan HS. 2007. The conserved asparagine in the HNH motif serves an important structural role in metal finger endonucleases. J. Mol. Biol. 368:812–821 [DOI] [PubMed] [Google Scholar]
- 16. Jaacks KJ, Healy J, Losick R, Grossman AD. 1989. Identification and characterization of genes controlled by the sporulation regulatory gene spo0H in Bacillus subtilis. J. Bacteriol. 171:4121–4129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kidane D, Graumann PL. 2005. Dynamic formation of RecA filaments at DNA double strand break repair centers in live cells. J. Cell Biol. 170:357–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kleanthous C, et al. 1999. Structural and mechanistic basis of immunity toward endonuclease colicins. Nat. Struct. Biol. 6:243–252 [DOI] [PubMed] [Google Scholar]
- 19. Ko TP, Liao CC, Ku WY, Chak KF, Yuan HS. 1999. The crystal structure of the DNase domain of colicin E7 in complex with its inhibitor Im7 protein. Structure 7:91–102 [DOI] [PubMed] [Google Scholar]
- 20. Köhler P, Marahiel MA. 1997. Association of the histone-like protein HBsu with the nucleoid of Bacillus subtilis. J. Bacteriol. 179:2060–2064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Krishnamurthy M, Tadesse S, Rothmaier K, Graumann PL. 2010. A novel SMC-like protein, SbcE (YhaN), is involved in DNA double strand break repair and competence in Bacillus subtilis. Nucleic Acids Res. 38:455–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ku WY, et al. 2002. The zinc ion in the HNH motif of the endonuclease domain of colicin E7 is not required for DNA binding but is essential for DNA hydrolysis. Nucleic Acids Res. 30:1670–1678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Landthaler M, Begley U, Lau NC, Shub DA. 2002. Two self-splicing group I introns in the ribonucleotide reductase large subunit gene of Staphylococcus aureus phage Twort. Nucleic Acids Res. 30:1935–1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lewis PJ, Marston AL. 1999. GFP vectors for controlled expression and dual labelling of protein fusions in Bacillus subtilis. Gene 227:101–110 [DOI] [PubMed] [Google Scholar]
- 25. Lindow JC, Kuwano M, Moriya S, Grossman AD. 2002. Subcellular localization of the Bacillus subtilis structural maintenance of chromosomes (SMC) protein. Mol. Microbiol. 46:997–1009 [DOI] [PubMed] [Google Scholar]
- 26. Malik HS, Henikoff S. 2000. Dual recognition-incision enzymes might be involved in mismatch repair and meiosis. Trends Biochem. Sci. 25:414–418 [DOI] [PubMed] [Google Scholar]
- 27. Mascarenhas J, et al. 2006. Bacillus subtilis SbcC protein plays an important role in DNA inter-strand cross-link repair. BMC Mol. Biol. 7:20 doi:10.1186/1471-2199-7-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mascarenhas J, Soppa J, Strunnikov AV, Graumann PL. 2002. Cell cycle-dependent localization of two novel prokaryotic chromosome segregation and condensation proteins in Bacillus subtilis that interact with SMC protein. EMBO J. 21:3108–3118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Moore T, McGlynn P, Ngo HP, Sharples GJ, Lloyd RG. 2003. The RdgC protein of Escherichia coli binds DNA and counters a toxic effect of RecFOR in strains lacking the replication restart protein PriA. EMBO J. 22:735–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Petit MA, et al. 1998. PcrA is an essential DNA helicase of Bacillus subtilis fulfilling functions both in repair and rolling-circle replication. Mol. Microbiol. 29:261–273 [DOI] [PubMed] [Google Scholar]
- 31. Petit MA, Ehrlich D. 2002. Essential bacterial helicases that counteract the toxicity of recombination proteins. EMBO J. 21:3137–3147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pommer AJ, et al. 2001. Mechanism and cleavage specificity of the H-N-H endonuclease colicin E9. J. Mol. Biol. 314:735–749 [DOI] [PubMed] [Google Scholar]
- 33. Pommer AJ, et al. 1999. Homing in on the role of transition metals in the HNH motif of colicin endonucleases. J. Biol. Chem. 274:27153–27160 [DOI] [PubMed] [Google Scholar]
- 34. San Filippo J, Lambowitz AM. 2002. Characterization of the C-terminal DNA-binding/DNA endonuclease region of a group II intron-encoded protein. J. Mol. Biol. 324:933–951 [DOI] [PubMed] [Google Scholar]
- 35. Soufo CD, et al. 2008. Cell-cycle-dependent spatial sequestration of the DnaA replication initiator protein in Bacillus subtilis. Dev. Cell 15:935–941 [DOI] [PubMed] [Google Scholar]
- 36. Sun Q, Margolin W. 2001. Influence of the nucleoid on placement of FtsZ and MinE rings in Escherichia coli. J. Bacteriol. 183:1413–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wach A. 1996. PCR-synthesis of marker cassettes with long flanking homology regions for gene disruptions in S. cerevisiae. Yeast 12:259–265 [DOI] [PubMed] [Google Scholar]
- 38. Walker DC, et al. 2002. Mutagenic scan of the H-N-H motif of colicin E9: implications for the mechanistic enzymology of colicins, homing enzymes and apoptotic endonucleases. Nucleic Acids Res. 30:3225–3234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xu L, Marians KJ. 2003. PriA mediates DNA replication pathway choice at recombination intermediates. Mol. Cell 11:817–826 [DOI] [PubMed] [Google Scholar]
- 40. Youngman PJ, Perkins JB, Losick R. 1983. Genetic transposition and insertional mutagenesis in Bacillus subtilis with Streptococcus faecalis transposon Tn917. Proc. Natl. Acad. Sci. U. S. A. 80:2305–2309 [DOI] [PMC free article] [PubMed] [Google Scholar]