

Abstract
To evaluate vaccine efficacy in protecting against coxsackievirus A16 (CA16), which causes human hand, foot, and mouth disease (HFMD), we established the first neonatal mouse model. In this article, we report data concerning CA16-induced pathological changes, and we demonstrate that anti-CA16 antibody can protect mice against lethal challenge and that the neonatal mouse model could be used to evaluate vaccine efficacy. To establish a mouse model, a BJCA08/CA16 strain (at 260 50% lethal doses [LD50]) was isolated from a patient and used to intracerebrally (i.c.) inoculate neonatal mice. The infection resulted in wasting, hind-limb paralysis, and even death. Pathological examination and immunohistochemistry (IHC) staining indicated that BJCA08 had a strong tropism to muscle and caused severe necrosis in skeletal and cardiac muscles. We then found that BJCA08 pretreated with goat anti-G10/CA16 serum could significantly lose its lethal effect in neonatal mice. When the anti-G10 serum was intraperitoneally (i.p.) injected into the neonatal mice and, within 1 h, the same mice were intracerebrally inoculated with BJCA08, there was significant passive immunization protection. In a separate experiment, female mice were immunized with formaldehyde-inactivated G10/CA16 and BJCA08/CA16 and then allowed to mate 1 h after the first immunization. We found that there was significant protection against BJCA08 for neonatal mice born to the immunized dams. These data demonstrated that anti-CA16 antibody may block virus invasion and protect mice against lethal challenge, and that the neonatal mouse model was a viable tool for evaluating vaccine efficacy.
INTRODUCTION
Coxsackievirus A16 (CA16) belongs to the Enterovirus genus of the Picornaviridae family and is one of the major pathogens associated with human hand, foot, and mouth disease (HFMD) (4, 16, 19). CA16 was first isolated in 1951 (43). It is a single positive-stranded RNA virus and has an icosahedral symmetry structure. Its genome has approximately 7,410 nucleotides with one predominant serotype. Based on the VP4 nucleotide sequences, CA16 is classified into three distinct genetic lineages: A, B, and C. Before the 1990s, lineages A and B were the major epidemic strains in Asia (predominantly the B strain). After that, the CA16 gene gradually mutated to form lineage C, which replaced the B strain as the predominant epidemic strain (22).
Epidemics of HFMD have been reported in England, Australia, Japan, Germany, Malaysia, Singapore, mainland China, and Taiwan (2, 4, 11, 19, 23, 27, 32, 38, 41, 48). Recently, HFMD was highly epidemical in the west Pacific region, resulting in severe illness and fatalities (15, 23). The HFMD epidemics were mainly caused by CA16 and human enterovirus 71 (EV71), which circulated alternatively or together in the epidemic area (19, 22, 23, 25, 37). Because the most severe or fatal cases were caused by EV71, studies have mainly focused on EV71 but not CA16. However, in England, the largest HFMD outbreak (in 1994) was caused by CA16 (2). Similarly, in Taiwan the leading cause of HFMD from 1999 to 2006 was also CA16 (2,579 cases), followed by EV71 (1,760 cases) (http://www.cdc.gov.tw). From 2001 to 2007, surveillance data in Singapore showed that the predominantly circulating virus was CA16 for three epidemic years (2002, 2005, and 2007) and was EV71 for only 1 year (2006) (23). Recently, in mainland China the predominant circulating virus strain was also CA16 (28, 42).
While most CA16-associated HFMD infections present only mild symptoms, many recent reports show that CA16 infections might lead to severe health issues, such as aseptic meningitis, rhombencephalitis, cardiac and pericardial disease, pulmonary complications, spontaneous abortion, and even lethal myocarditis and pneumonia (14, 21, 23, 40, 44–46, 50). The coinfection of EV71 and CA16 makes it more complex and difficult to control epidemic HFMD (54). Also, the coinfections of EV71 and CA16 can easily cause serious complications in the central nervous system (CNS) with worse conditions, longer duration, and even higher critical illness transfer rates (54). Coinfection increases the chance of gene recombination of the RNA viruses. The rates of coinfection in different areas of mainland China vary from 0.62% (during 2008 to 2010 in Hu Nan) (17) to 14.3% (2009 in Hang Zhou) (52), 7.4% (2010 in Beijing) (20), and 9.3% (2010 in Fo Shan) (55). Therefore, epidemic HFMD could not be controlled by solely relying on an EV71-specific vaccine. Because the clinical symptoms of EV71 and CA16 infections are difficult to differentiate, there are further restrictions on the application of the EV71 vaccine. For these reasons, it is urgent to develop CA16-specific or combined CA16/EV71 vaccines.
Because most severe cases of HFMD were caused by EV71, studies on EV71 vaccines have progressed rapidly in recent years. Among them, the EV71 inactivated vaccines developed in mainland China and Taiwan have entered phase III and phase I trials, respectively (clinicaltrials.gov numbers NCT01508247, NCT01507857, NCT01569581, and NCT01268787). However, research work on CA16 has been neglected in the past. Recently, diseases induced by CA16 and the development of CA16 vaccine have received increasing attention, and several companies in mainland China have begun to launch CA16 vaccine development projects. The successful development of an EV71 vaccine benefits not only from other well-developed inactivated enterovirus vaccines but also from the establishment and application of a neonatal mouse model and a macaque model for evaluating the clinical or preclinical protection effects (1, 10, 33, 35). For CA16, a study using a CA16 animal experiment was first reported in 1951 (43). It indicated that CA16 causes paralysis and even death in newborn mice. However, until now no CA16 animal model has been established to further evaluate the efficacy of a potential vaccine. In the current study, we established a neonatal mouse model by applying a particular clinically isolated CA16 strain, BJCA08, and evaluated the efficacy of an inactivated CA16 vaccine to provide a useful tool for the research and development of CA16 vaccines.
MATERIALS AND METHODS
Cells and virus.
Vero cells (American Type Culture Collection, Manassas, VA) were maintained in Eagle's minimum essential medium (MEM) containing 2 or 10% fetal bovine serum (FBS) plus 2 mM l-glutamine, 100 IU of penicillin, and 100 μg of streptomycin per ml. A local isolate, BJCA08/CA16 (GenBank accession no. JX481738), was taken from a throat swab sample of a 3-year-old boy with hand, foot, and mouth disease (HFMD) in Beijing in 2008. Viral RNA was extracted from the BJCA08/CA16 isolate, and reverse transcription-PCR (RT-PCR) was used to obtain the sequence (51). Phylogenetic trees established from the alignment of the VP1 and VP4 regions indicated that BJCA08 belonged to a C1 subtype. The titer of BJCA08 (9.6 × 105 PFU/ml) was determined using a plaque assay on Vero cells. The Institute of Medical Biology, Chinese Academy of Medical Science, kindly provided G10/CA16, the first prototype of a CA16 strain (South Africa/51; GenBank accession no. U05876). G10 belongs to the A subtype of CA16 and was grown in Vero cells to a titer of 3.0 × 106 PFU/ml. All stock viruses were grown in Vero cells, subjected to three freeze-thaw cycles, clarified by centrifugation at 3,900 × g for 10 min at 4°C, filtered through a syringe filter (0.2 μm; Pall Corporation, Germany), and stored at −80°C.
Mouse infectious experiments.
Outbred, specific-pathogen-free ICR mice (Vital River Lab Animal Technology Co., Ltd., Beijing, China) were used to develop an animal model. All institutional (National Institute for Food and Drug Control) guidelines for animal care and use were strictly followed. The mice in an age-dependent experiment (at 1, 3, 5, 7, and 14 days of age) were selected (n = 8 to ∼10 per age group) and intracerebrally (i.c.) inoculated with BJCA08 (1.9 × 104 PFU/mouse). For the dose-dependent experiment and the 50% lethal dose (LD50) study, 1-day-old ICR mice (n = 8 to 10 per group) were intracerebrally inoculated with 8-fold serial dilutions of BJCA08 (0.3, 2.4, 1.9 × 10, 3.0 × 102, and 1.9 × 104 PFU/mouse). The control mice were given an uninfected culture medium and kept in a separate cage from the infected mice. Mice were observed daily for clinical illness and death until 21 days postinoculation (dpi). The grade of clinical disease was scored as follows: 0, healthy; 1, lethargy and inactivity; 2, wasting; 3, limb-shake weakness; 4, hind-limb paralysis; and 5, morbidity and death (Table 1) (26). The control mice were healthy throughout the experiments. The LD50 was calculated as described by Reed and Muench (39).
Table 1.
Grading score for clinical symptoms of BJCA08/CA16-infected mice
| Grade | Clinical sign(s) |
|---|---|
| 0 | Healthy |
| 1 | Lethargy and inactivity |
| 2 | Wasting |
| 3 | Limb-shake weakness |
| 4 | Hind limb paralysis |
| 5 | Moribund and death |
Histopathologic and immunohistochemical staining.
Five days after intracerebral inoculation with BJCA08 (300 PFU/mouse) or uninfected culture medium, 12 experimental animals (grades 3 to 5) and 6 control animals (grade 0) were subjected to histopathologic and immunohistochemical examination. After anesthetization, samples were taken of blood, brain, spine, heart, lung, thymus, liver, spleen, kidney, intestines, and hind-limb and spinal skeletal muscles. The mouse tissues were fixed by immersion in 10% neutral buffered formalin for at least 2 days. Tissues then were bisected and embedded in paraffin. For histopathologic examination, tissue sections were stained with hematoxylin and eosin. For immunohistochemical testing, tissue sections were dewaxed, dehydrated, and microwaved for 14 min at 92 to ∼99°C in a citrate buffer. Monoclonal mouse anti-CA16 VP1 antibody (MAb) T26 (1:32,000 dilution; a gift from Beijing Wantai Biological Pharmacy Enterprise Co., Ltd., Beijing, China) was applied for 1 h at 37°C. A peroxidase-conjugated secondary Ab (1:2 dilution; Fuzhou Maixin Biotechnology Development Co., Ltd., Fuzhou, China) was added for 30 min at room temperature, followed by 3,3′-diaminobenzidine tetrahydrochloride (Fuzhou Maixin Biotechnology Development Co., Ltd., Fuzhou, China) chromogen. Tissues were counterstained with hematoxylin. Control sections were incubated with normal goat serum instead of anti-CA16 MAb.
Virus loads in infant mouse tissues postchallenge.
After intracerebral inoculation with BJCA08 (300 PFU/mouse) or uninfected culture medium, 24 experimental mice and 3 control mice were subjected to virus load assays.
After the heart was punctured, blood samples were collected and stored at −80°C. Tissue samples (from the brain, heart, lung, liver, spleen, kidney, intestines, and hind-limb skeletal muscles) were aseptically removed, weighed, and stored at −80°C. Tissues and blood samples from experimental mice (n = 3 per time point) were collected at 1, 6, 12, 24, 48, 72, 96, and 120 h postinfection. Samples from control mice (n = 3) were collected at 0 h postinoculation. The tissue samples were homogenized in sterile phosphate-buffered saline (10%, wt/vol), disrupted by three freeze-thaw cycles, and centrifuged. Virus loads in clarified supernatants and blood were determined by real-time quantitative reverse transcriptase PCR (qRT-PCR) and expressed as log10 copies/mg of tissue or log10 copies/ml of blood.
qRT-PCR.
At selected intervals postinfection, the mice in the experimental and negative-control groups were sacrificed. Blood and homogenized tissues were harvested for RNA by a Mag Max viral isolation kit (Ambion Inc., Austin, TX) by following the manufacturer's recommended protocols. According to the instructions of the Ag Path-ID One-Step RT-PCR kit (Ambion Inc., Austin, TX), cDNA was synthesized from RNA by reverse transcription for 30 min at 42°C and subsequently amplified for 40 cycles at 95°C for 5 s, 55°C for 35 s, and 72°C for 2 min. RNA from the negative-control group was run simultaneously in each qRT-PCR.
Protective efficacy of neutralization antibody.
To confirm the protective role of the humoral immune response, three experiments were carried out. First, by using goat anti-G10/CA16 serum (neutralizing antibody [NTAb] titer, 1,280; a gift from the Institute of Medical Biology, Chinese Academy of Medical Science), a passive immunization to protect pups against a CA16 challenge was studied in vivo. One-day-old ICR mice (n = 8 to 10 per group) were intraperitoneally (i.p.) inoculated with 50 μl of 10-fold serially diluted goat anti-G10 serum (10- to 10,000-fold dilutions) and medium. Within 1 h after inoculation, each mouse was intracerebrally challenged with 300 PFU of BJCA08. The mortality and clinical symptoms were then monitored and recorded daily after infection until 21 days after inoculation. The 50% protective dose obtained by a passive immunization protection test (ED50I) was calculated as described by Reed and Muench (39).
In the second experiment, BJCA08 was first neutralized by serially diluted anti-G10 serum in vitro and then intracerebrally inoculated into the pups to test the neutralizing effect of antiserum. Serially diluted anti-G10/CA16 serum (10- to 10,000-fold dilutions) and medium were incubated with 300 PFU of BJCA08 at 37°C for an hour. One-day-old ICR mice (n = 8 to 10 per group) were intracerebrally inoculated with the mixture described above. All mice were monitored daily for clinical symptoms and death until 21 days after inoculation, and the 50% protective dose obtained by the in vivo neutralizing experiment (ED50II) was calculated as described by Reed and Muench (39).
Lastly, maternal antibody protection was studied. BJCA08 (9.6 × 105 PFU/ml) and G10 (3.0 × 106 PFU/ml) were inactivated by adding 37% formaldehyde (Sinopharm Group, Beijing, China) to the suspensions of virus to a final formaldehyde concentration of 1/4,000. The viral suspension was then incubated at 37°C for 3 days (31). The inactivated suspensions were filtered as described above and stored at −80°C. No live viruses were detected after repeated development of Vero cell cultures for a period of up to 3 weeks. Eight-week-old female ICR mice (n = 2 per group) were intraperitoneally injected twice at 2-week intervals with 0.5 ml formaldehyde-inactivated G10, formaldehyde-inactivated BJCA08, or medium. The mice were allowed to mate 1 h after the first injection. After delivery (about 5 to 10 days after the boost), pups were intracerebrally challenged with BJCA08 (300 PFU/mouse) on postnatal day 1. Body weight, clinical symptoms, and death of the challenged suckling mice were monitored as described above. Dams were euthanized and sera were collected and stored at −80°C until use.
Statistical analysis.
The clinical scores and virus load values were analyzed by the nonparametric one-way analysis of variance (ANOVA) and unpaired t test. Survival rates were evaluated by the Mantel-Cox log-rank test. The body weight was compared using Dunn's multiple-comparison test. The results were expressed as means and standard deviations (SD) from the means. LD50, ED50I, and ED50II were calculated as described by Reed and Muench (39). A P value of <0.05 was considered statistically significant.
Nucleotide sequence accession number.
The sequence of local isolate, BJCA08/CA16, has been deposited in GenBank under accession no. JX481738.
RESULTS
Intracerebral inoculation of BJCA08/CA16 results in age-related disease and mortality.
To examine the sensitivities to BJCA08/CA16 infection of neonatal mice of different ages (in days), neonatal mice at 1 to 14 days old were intracerebrally inoculated with BJCA08 (1.9 × 104 PFU/mouse). The results showed that various grades of clinical diseases, such as wasting, hind-limb paralysis, and even death occurred in the neonatal mice (inoculated at 1, 3, 5, and 7 days), but all mice inoculated at 14 days survived (Fig. 1). The severity of clinical symptoms, from mild to severe, was scored as five grades (Table 1). Figure 1 shows that clinical diseases of 1-day-old mice first occurred at 4 days postinfection (dpi) with a mean clinical score of grade 3, and all were dead at 6 dpi. Both 3-day-old mice and 5-day-old mice began to get sick at 5 dpi at grade 3, and these mice were dead at the 13th and 11th day postinfection, respectively. Seven-day-old mice started to exhibit clinical symptoms at 5 dpi at grade 2, and five of them gradually recovered starting at 10 dpi, with a reduced mortality rate of 55.6%. No clinical incidents or mortality was observed among 14-day-old mice. These data indicated that 1-day-old neonatal mice are the most sensitive to BJCA08 infection. The symptoms and mortality rates steadily decreased as age increased. The Mantel-Cox log-rank test found that there was a statistically significant difference in survival rate between 1-day-old mice and other age groups (P < 0.0001). Therefore, neonatal 1-day-old mice were selected as the target animal in the current study.
Fig 1.
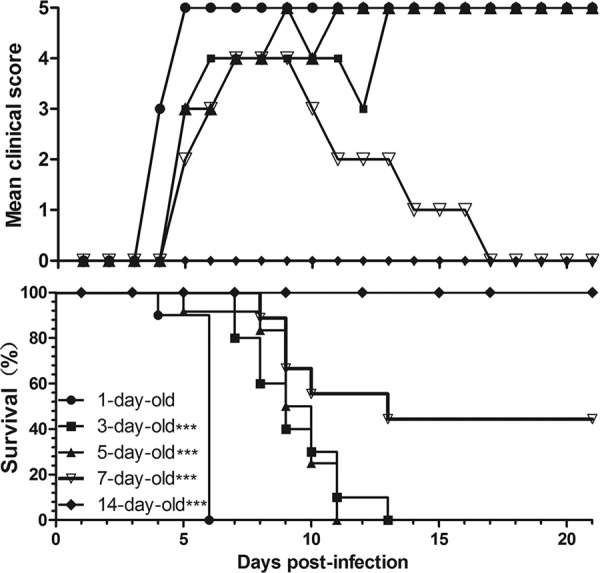
Intracerebral inoculation of BJCA08/CA16 results in age-related disease and mortality. ICR mice (n = 8 to 10 per age) were intracerebrally inoculated with BJCA08 (1.9 × 104 PFU/mouse) at 1, 3, 5, 7, or 14 days of age. BJCA08-induced mean score of clinical disease and BJCA08-induced mortality were monitored and recorded daily after infection. Representative results from two similar experiments are shown. The Mantel-Cox log-rank test was used to compare the survival rates of pups between each age group and the 1-day-old group at 21 days postinfection. ***, P < 0.0001.
Intracerebral inoculation of BJCA08/CA16 results in dose-related disease and mortality.
To determine the dose-response effects and LD50, 1-day-old mice were inoculated intracerebrally with serially diluted BJCA08. Figure 2 shows that the mice that were i.c. inoculated with doses of 1.9 × 104 and 3.0 × 102 PFU/mouse became sick at 4 dpi at grade 3, and mice infected with a dose of 1.9 × 10 PFU/mouse became sick at 7 dpi at grade 4. The three infective doses mentioned above (1.9 × 104, 3.0 × 102, and 1.9 × 10 PFU/mouse) caused all mice to die at different days postinfection (6, 9, and 12 dpi, respectively). With infection doses of 2.4 and 0.3 PFU/mouse, the mortality rates were 62.5 and 25%, respectively, with some mice progressively recovering after infection. These results demonstrated significant dose-response effects between BJCA08 infective dosages and the mortality rates of neonatal mice (the calculated LD50 was 1.2 PFU/mouse). To guarantee the reproducibility of the animal model, a dose of 3.0 × 102 PFU/mouse (i.e., 260 LD50) was given as a lethal challenge. Repetitive experiments showed that under this dosage, all 1-day-old mice became sick within 4 to 5 dpi and all were dead between 6 and 10 dpi. The morbidity time and mortality rates were stable and had good reproducibility (see Table S1 in the supplemental material).
Fig 2.
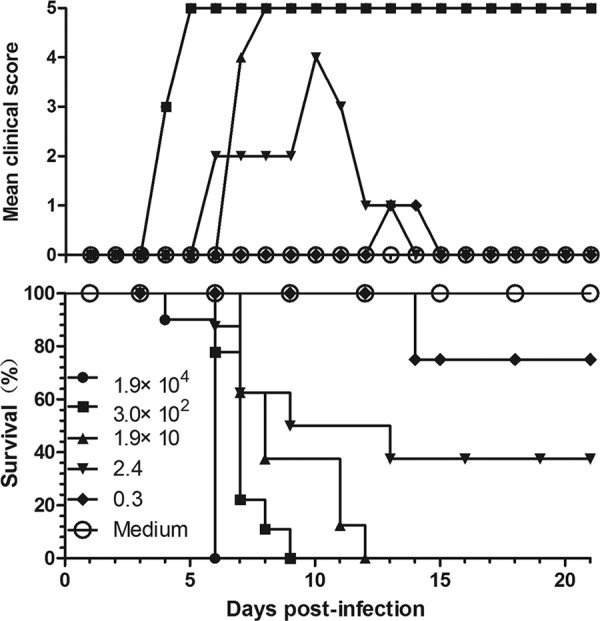
Intracerebral inoculation of BJCA08/CA16 results in dose-related disease and mortality. One-day-old ICR mice (n = 8 to 10 per group) were intracerebrally inoculated with increasing doses of BJCA08 (0.3, 2.4, 1.9 × 10, 3.0 × 102, and 1.9 × 104 PFU/mouse). Control animals were given medium instead of a virus. Mortality and clinical disease were monitored and recorded daily after infection. One representative from two independent experiments is shown. The LD50 was calculated as 1.2 PFU/mouse, and 3.0 × 102 PFU/mouse (260 LD50) was chosen as the challenge dose.
Pathological changes in infected mice after intracerebral challenge with a lethal dose of BJCA08/CA16.
Although mice with different challenge ages have different disease onset times (Fig. 1), the typical clinical signs were the same, i.e., they all exhibited wasting, hind-limb paralysis, and so on. Therefore, in this study we selected the representative endpoints, including wasting and hind-limb paralysis, from 1-day-old mice infected by BJCA08 (260 LD50) at 8 dpi (Fig. 3a and b). To understand the distribution of virus and pathological changes, 1-day-old mice infected by BJCA08 at grade levels 3 to 5 were sacrificed for pathological and immunohistochemistry (IHC) examinations. The results showed that the hind-limb skeletal muscle fibers exhibited severe necrosis (Fig. 3c); IHC staining indicated that widespread CA16 antigen was observed in the corresponding areas of lesions (Fig. 4a). Moreover, severe necrosis and CA16 antigen were also observed in the juxtaspinal skeletal muscle fibers (Fig. 3d and 4c), while on the same IHC section, intramedullary nerve cells surrounded by necrotic skeletal muscles were normal with no viral antigen (Fig. 4d). This indicated that BJCA08 had a strong tropism to muscle rather than to nervous tissues and also indicated how it differed from EV71. Within the encephalocoele (the place of inoculation), limited CA16 antigen was observed only in the ependymal epithelium and the choroid plexus epithelium of the lateral ventricle (Fig. 4g), but no obviously pathological change was found (Fig. 3g). Also, on the same IHC section, no obviously positive reaction to viral antigen was found in the neurocytes of the brain (Fig. 4h). In addition, for moribund neonatal mice (grade 5), necrosis in muscle fiber was observed in focal cardiac tissues (Fig. 3e), and the relevant IHC staining showed viral antigen (Fig. 4e). No viral antigen or pathological change was detected in lymphoid organs, including the spleen and thymus of the BJCA08-infected neonatal mice. In the control group treated with the culture medium, all detected tissues had no observable pathological change, and the IHC staining also showed negative reactions (Fig. 3f and h and 4b and f).
Fig 3.
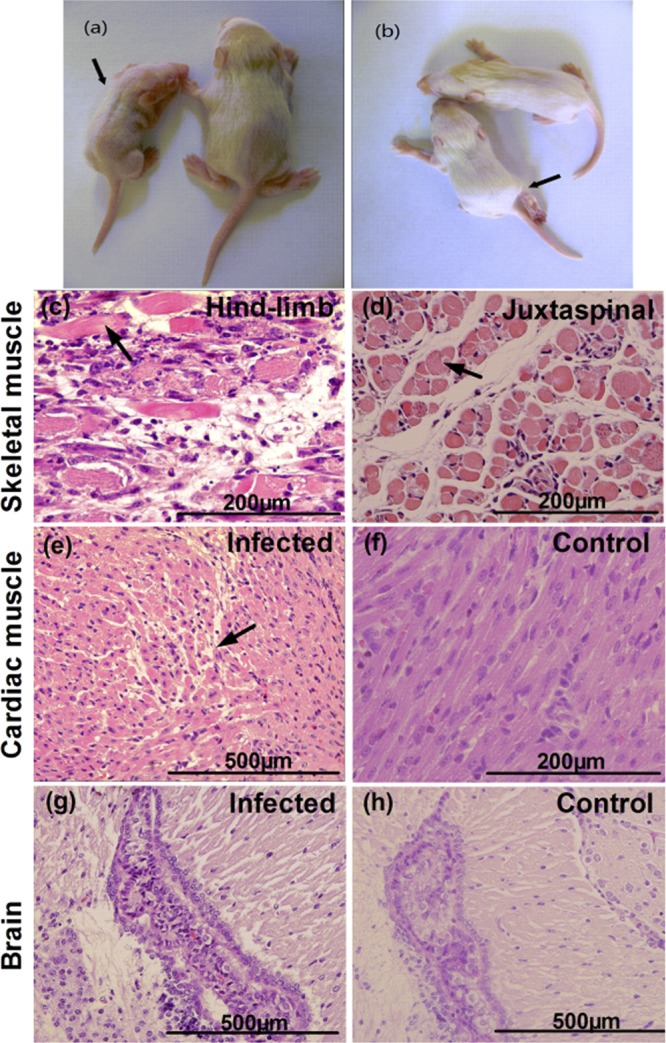
Histological examination of infected mice after intracerebral challenge with lethal doses of BJCA08/CA16. One-day-old ICR mice were intracerebrally inoculated with medium (mock control) or BJCA08 (260 LD50). (a) A representative picture of wasting caused by BJCA08 at day 8 of postinfection is shown (arrow). (b) A representative picture of right-hind-limb paralysis caused by BJCA08 at day 8 of postinfection is shown (arrow). Mice on the right side of panels a and b labeled in yellow were naive age-matched controls. Representative sections are shown. (c) Infected mice (grades 4 to 5) exhibited severe necrotizing myositis in hind-limb skeletal muscles (arrow). (d) Severe necrotizing myositis in skeletal muscles near the spine (arrow) was also detected in infected mice (grades 4 to 5). (e) A moribund mouse due to BJCA08 exhibited focal myocarditis (arrow). (f) In contrast, no histological change was observed in the heart of the mock-infected control mice. No histological change in the brain was observed in the brain of the infected mice (grades 3 to 5) (g) or the mock control mice (h). Samples in panels c to h were stained with hematoxylin and eosin stain; magnification, ×400.
Fig 4.
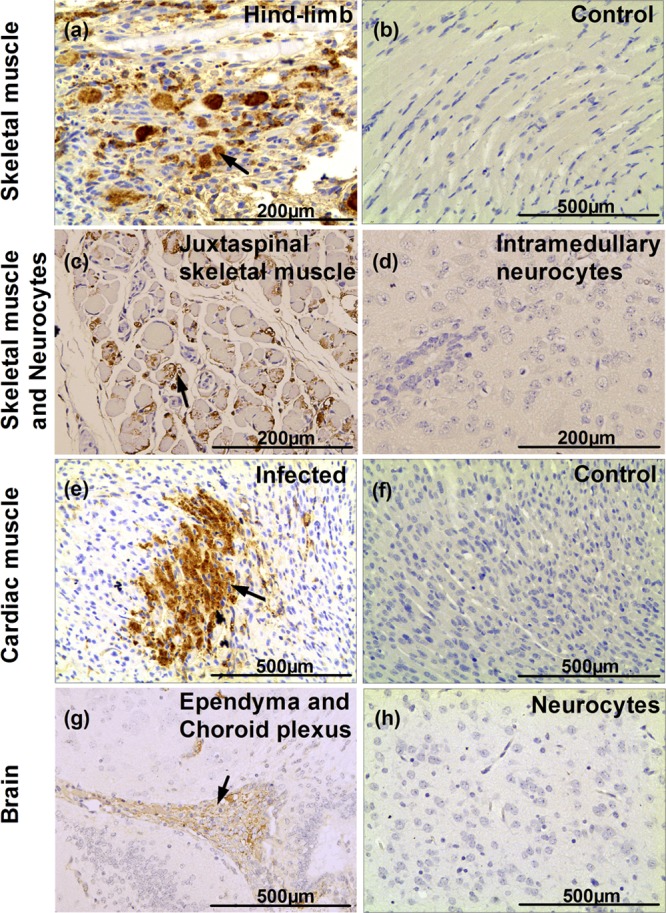
Immunohistochemical results for infected mice after intracerebral challenge with lethal doses of BJCA08/CA16. One-day-old ICR mice were intracerebrally inoculated with medium (mock control) or BJCA08 (260 LD50). Representative sections are shown. (a) Infected mice (grades 4 to 5) exhibited numerous viral antigen-positive fibers in hind-limb skeletal muscles (arrow). (b) In contrast, no viral antigen was observed in hind-limb skeletal muscles of the mock control mice. Numerous viral antigen-positive fibers (c, arrow) were also detected in skeletal muscles near the spine, but no viral antigen was observed in intramedullary nerve cells (d) of infected mice (grades 4 to 5). (e) A moribund mouse due to BJCA08 exhibited numerous viral antigen-positive fibers in the heart (arrow). (f) In contrast, no viral antigens were observed in the heart of the mock-infected control mice. Viral antigens were detected in ependymal cells and choroid plexus epithelium (g, arrow), but no viral antigen was found in other parts of the brain of the infected mice (grades 3 to 5) (h). Samples in panels a to h were subjected to IHC analysis with DAB (3,3′-diaminobenzidine) chromogen and hematoxylin counterstain; magnification, ×400.
Tissue viral loads in BJCA08/CA16-infected mice.
Detection of tissue viral loads may indicate the characteristics of virus distribution in infected mice at different time points after inoculation. Figure 5 shows that the virus was first detected in brain tissue (103.13 copies/mg) and blood (104.65 copies/ml) at 1 h after inoculation. At different time points postinfection (6, 12, 24, 48, 72, 96, and 120 h), the quantity of virus was at a stable level in the blood (105.47, 105.40, 106.47, 105.98, 105.80, 106.72, and 105.75 copies/ml, respectively). Twenty-four hours after inoculation, the virus was detected in the brain, hind-limb skeletal muscle, and cardiac tissues (106.33, 104.87, and 105.47 copies/mg, respectively). Seventy-two hours later, the virus was ubiquitous in all tissues, such as intestine (104.78 copies/mg), lung (104.71 copies/mg), liver (106.09 copies/mg), muscle (107.71 copies/mg), brain (104.56 copies/mg), spleen (105.45 copies/mg), kidney (104.37 copies/mg), and heart (105.81 copies/mg), suggesting that the virus had already spread throughout the body by means of the bloodstream. The viral loads in the hind-limb skeletal muscles steadily increased with time (from 104.56 copies/mg at 24 h to 109.68 copies/mg at 120 h). At 120 h, the viral load in the hind-limb skeletal muscles was 4 logs higher than that in the blood (105.75 copies/ml) and 2 to 3 logs higher than that in other tissues. For control mice, no viral loads were detected in the intestine, lung, liver, muscle, brain, spleen, kidney, heart, and blood. This observation was consistent with the results of pathological change and IHC staining, indicating BJCA08 has strong tropism to muscle tissues, which may be the major location for viral duplication.
Fig 5.

Mean tissue viral loads in BJCA08/CA16-infected mice. One-day-old ICR mice were intracerebrally inoculated with BJCA08 (260 LD50). Virus loads were assessed by real-time quantitative reverse transcriptase PCR in samples of the intestine, lung, liver, muscle, brain, spleen, kidney, heart, and blood from the infected mice. Samples were collected at the times indicated. Results represent the mean virus load (log10 copies) per milligram of tissue or per milliliter of blood ± SD (three mice per group).
Passive immunization with anti-G10/CA16 immune serum protected pups against CA16 challenge in vivo.
The anti-G10/CA16 serum (NTAb titer, 1,280) was serially diluted and was injected i.p. into neonatal mice. Within 1 h, the same mice were i.c. inoculated with BJCA08 at a dose of 260 LD50. The results (Fig. 6) showed that the control mice (i.p. injection of culture medium) got sick at 6 dpi (grade 3) and started to die at 7 dpi, and all were dead by 11 dpi. Mice i.p. inoculated with a 1:10 dilution of antiserum had 100% survival with no clinical symptoms. When i.p. inoculated with antiserum at 1:100 or 1:1,000 dilutions, the experimental mice began to die at the 9th or 6th day postinfection, respectively. Some mice in both groups suffered from mild symptoms and gradually recovered. The protection rates of the 1:100 and 1:1,000 dilution groups were 25 and 22%, respectively, at the end of the 21-day observation period. All mice i.p. inoculated with antiserum at a dilution of 1:10,000 were dead at 11 dpi. The Mantel-Cox log-rank tests suggested that the survival rates in mice treated with an antiserum of 1:10 or 1:100 dilution were statistically different from the control group (P < 0.0001). According to the survival rates for different serum-protective groups and the NTAb titer (1,280) of anti-G10 serum, ED50I was calculated using the Reed and Muench method (39). A significant dose-response effect was observed between the antibody concentration and the protection rates. Anti-G10 serum at the 85-times-diluted concentration may protect half of the experimental animals in the in vivo blocking test (i.e., ED50I = 15).
Fig 6.
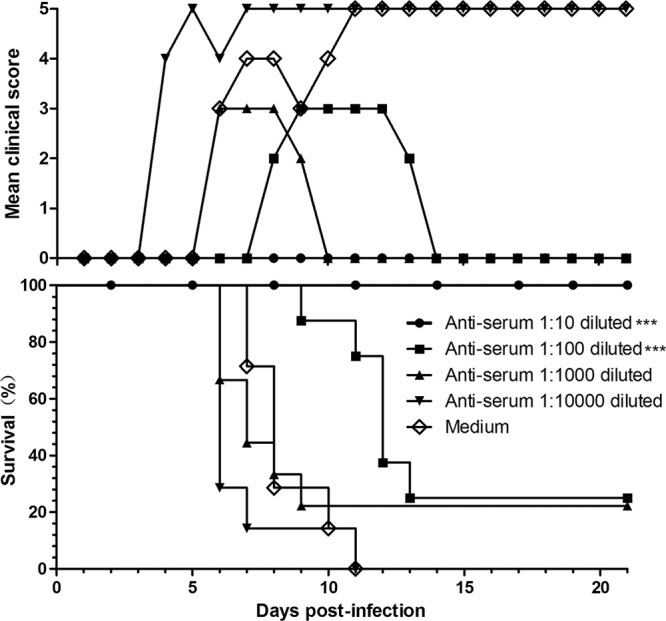
Passive immunization with anti-G10/CA16 serum protected pups against CA16 challenge in vivo. One-day-old ICR mice (n = 8 to 10 per group) were intraperitoneally inoculated with 50 μl of 10-fold serially diluted anti-G10 serum (NTAb titer, 1,280) or medium. Within 1 h after inoculation, each mouse was intracerebrally challenged with 260 LD50 of BJCA08. Mortality and clinical disease were monitored and recorded daily after infection. The Mantel-Cox log-rank test was used to compare the survival of pups between each antiserum group and the medium control group at 21 days postinfection. ***, P < 0.0001.
BJCA08/CA16 pretreated in vitro with anti-G10/CA16 serum could significantly lose its lethal effect in neonatal mice.
The in vivo neutralizing study showed that mice in the control group got sick at 3 dpi (grade 2), and all died by 9 dpi (Fig. 7). The survival rates for anti-G10 serum at dilutions of 1:10, 1:100, and 1:1,000 were 80, 50, and 40%, respectively, and the clinical grades for different dilutions also reduced with increasing quantities of antiserum. No protective role was found at a dilution of 1:10,000. At 9 dpi, all mice in the 1:10,000 group were dead. Compared to the culture medium control, the anti-G10 serum at dilutions of 1:10, 1:100, and 1:1,000 neutralized with BJCA08 in vitro at 37°C for an hour can significantly reduce the mortality rates of neonatal mice (P < 0.001). According to the survival rates for different serum dilution groups and the NTAb titer (1,280) of anti-G10 serum, ED50II was calculated using the Reed and Muench method (39). Anti-G10 serum at the 160-times-diluted concentration can neutralize virus and protect 50% of the experimental animals (ED50II = 8.3).
Fig 7.
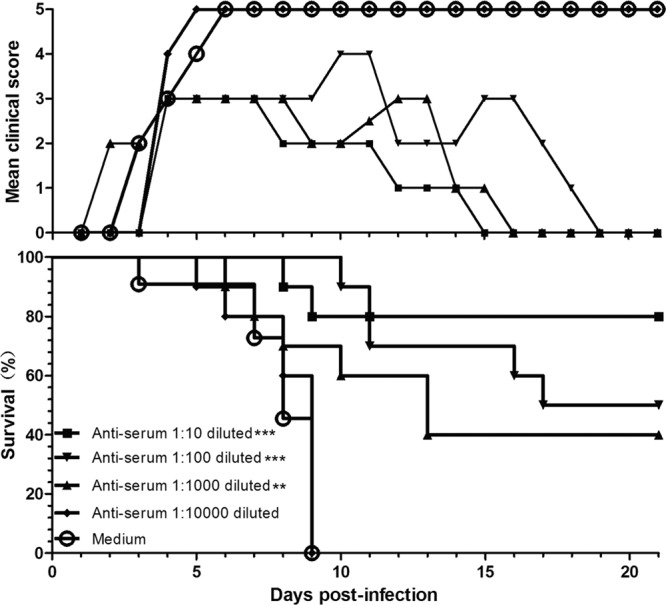
BJCA08/CA16 pretreated in vitro with anti-G10/CA16 serum could significantly lose its lethal effect in neonatal mice. Serially diluted anti-G10 serum (NTAb titer, 1,280) or medium was incubated with an equal volume of 300 PFU BJCA08 at 37°C for 1 h. One-day-old ICR mice (n = 8 to 10 per group) were intracerebrally inoculated with the mixture described above. Mortality and clinical disease were monitored and recorded daily after infection. The Mantel-Cox log-rank test was used to compare the survival of pups between each antiserum group and the medium control group at 21 days postinfection. ***, P < 0.0001; **, P < 0.001.
Protection role of maternal antibody.
In a previous study, we found that formaldehyde-inactivated G10/CA16 could induce a moderate humoral response in adult mice with neutralizing antibody titers from 64 to 512 1 week after the second inoculation (unpublished data). Whether the maternal antibody could neutralize BJCA08 and protect the neonatal mice from disease or death is still unknown. Inactivated BJCA08, inactivated G10, and culture medium were used to i.p. immunize female adult mice twice at 2-week intervals. They were allowed to mate 1 h after the first injection. The female mice delivered pups 5 to 10 days after the second inoculation. BJCA08 at a dose of 260 LD50 was used to i.c. inoculate the 1-day-old mice born to the immunized dams. Figure 8 shows that the neonatal mice in the control group started to die at 6 dpi, and all were dead at 10 dpi. One-day-old mice born to dams that were immunized by inactivated BJCA08 had a survival rate of 93% without severe clinical symptoms. One-day-old mice born to dams immunized by inactivated G10 exhibited disease and survived at a rate of 47%. These data suggest that using inactivated BJCA08 and G10 to immunize adult female mice significantly reduces mortality rates in the neonatal mice (P < 0.0001 by Mantel-Cox log-rank test). The weight gain for neonatal mice born to inactivated BJCA08-immunized dams was fastest, followed by the pups born to dams immunized with inactivated G10. Almost no weight gain was observed for the control mice, and all control mice were dead at 10 dpi (Fig. 8). Weight gain was significantly higher in the inactivated BJCA08 group and in the G10-immunized groups than in the control group (P < 0.0001).
Fig 8.
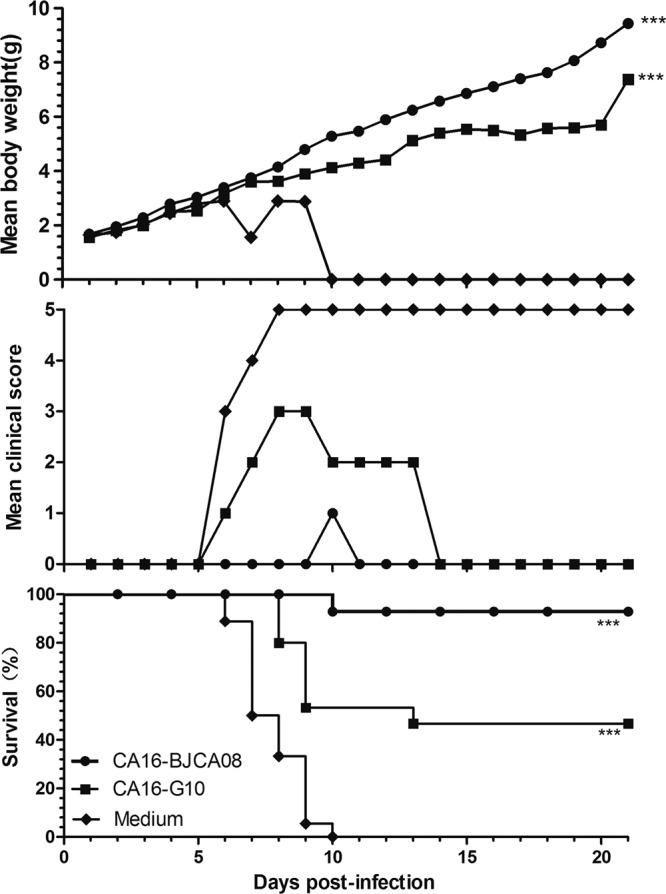
Maternal immunization with inactivated whole-virus CA16 antigen protected newborn mice against lethal challenge. Adult female ICR mice (n = 2 per group) were intraperitoneally injected with formaldehyde-inactivated G10 antigen (1.5 × 106 PFU/mouse), BJCA08 antigen (4.8 × 105 PFU/mouse), or medium twice at a 2-week interval and allowed to mate at 1 h after the first injection. After delivery, pups were challenged with BJCA08 (300 PFU/mouse) on postnatal day 1. Mortality and clinical disease were monitored and recorded daily after infection. The Mantel-Cox log-rank test was used to compare the survival of pups between each maternal immunization group and the medium control group at 21 days postinfection. Dunn's multiple-comparison test was used to compare the weight of pups between each maternal immunization group and the medium control group at 21 days postinfection. ***, P < 0.0001.
DISCUSSION
The animal model is very important in evaluating immunogenicity and the protective efficacy of a vaccine (29). To develop an EV71 vaccine, animal models for the neonatal mouse, the cynomolgus monkey, and the rhesus monkey have been established and improved. These animal models confirmed the protective role of vaccine in inducing neutralizing antibody and also promoted the development of the EV71 vaccine (1, 10, 33, 35). Until our study, no animal model had been developed to evaluate the protective efficacy of the CA16 vaccine. Sickles et al. discovered that coxsackievirus A generally had strong pathogenicity in newborn mice (43). However, the details of the infection are still unclear. In 1993, Hyypiä et al. detected the genomes of coxsackievirus in the tissues of newborn mice that were infected by five serotypes (A2, A9, A21, B3, and B4) using in situ hybridization. The results showed that coxsackievirus A could affect skeletal and cardiac muscles, while the coxsackievirus B subgroup infected a wide range of tissues (18).
To develop an animal model for evaluation of the CA16 vaccine's protective efficacy, the BJCA08 strain was used to establish the neonatal mouse model; it was the first CA16 strain isolated from a throat swab sample of a 3-year-old boy with HFMD in Beijing by our laboratory in 2008. The isolate was confirmed by serological and molecular biological detection without contamination of mycoplasma and adventitious viruses. The complete genome was sequenced (GenBank accession no. JX481738), which indicated that it belongs to the C subtype demonstrating homology in VP1 with other CA16 strains isolated from mainland China and Taiwan (Table 2). In the mouse model, newborn mice showed typical clinical symptoms and death following the infection of virus (see Fig. S2 in the supplemental material). Since subtype C was the major subtype of CA16 in mainland China in recent years, we selected BJCA08 to establish the neonatal mouse model to meet the need to evaluate CA16 vaccines in China, although the pathogenesis comparison to other CA16 strains was not carried out for newborn mice.
Table 2.
Nucleotide identity of BJCA08 with other CA16 virus strains in VP1 region
| Genotype (or subtype) | Isolate | GenBank accession no. | Location | Yr of isolation | % Nucleotide identity to BJCA08 in VP1 region |
|---|---|---|---|---|---|
| A | G10 | U05876 | South Africa | 1951 | 76.3 |
| B | SHZH00 | AY790926 | Shenzhen, China | 2000 | 95.5 |
| C1 | SHZH05 | EU262658 | Shenzhen, China | 2005 | 94.5 |
| C1 | GZ08 | FJ198212 | Guangdong, China | 2008 | 94.3 |
| C2 | TW01 | AF177911 | Taiwan | 2008 | 94.5 |
The appropriated route of challenge is one of the important considerations for animal model development. Because CA16 belongs to the enterovirus genus, in a previous study we first compared the sensitivity of oral inoculation (per os [p.o.]), i.p. inoculation, and i.c. inoculation in 1-day-old mice. The results showed that 1-day-old mice inoculated i.c. with 20 μl BJCA08 died earlier (4 to 6 days postinfection) than those inoculated i.p. with 50 μl BJCA08 (7 to 12 days postinfection), while 100 μl BJCA08 given orally to 1-day-old mice failed to induce 100% death (see Fig. S3 in the supplemental material). This result indicated that the i.c. route was the most sensitive for the neonatal mice. Furthermore, for the oral route, newborn mice should be fasted for hours, which may affect their normal growth. For the i.p. route, the abdominal capacity of 1-day-old mice was very limited, so leakage often occurred, which can make the challenge dose inaccurate. Given the aseptic meningitis and rhombencephalitis caused by CA16 in humans and the validity of the EV71 mouse model of i.c. inoculation (3, 5, 9), i.c. inoculation was finally selected to establish this CA16 mouse model. According to the results, we found that the effects of BJCA08 were significantly correlated with the age of mice and the dosage used. On the basis of experimental data, we chose a BJCA08 dosage of 260 LD50 as the challenge dose to i.c. inoculate the neonatal mice. The infected mice exhibited wasting, hind-limb paralysis, and even death. Under this challenge dose, the average onset time of disease was about 5 days postinfection, and all mice were dead between 6 and 10 days postinfection (100% mortality rate). These data indicated good feasibility and reproducibility (see Table S1 in the supplemental material).
Previous studies demonstrated that CA16 infection or immunization may induce neutralizing antibody generation (49). This is consistent with reports that the positive rate of neutralizing antibody was high in babies or infants living in epidemic areas (30, 56). Anti-CA16 serum was able to inhibit the cytopathic effect induced by CA16 in vitro (30). It is important for the development of a CA16 vaccine to verify whether anti-CA16 serum has a protective function in vivo similar to that of anti-poliovirus and anti-EV71 serum. In the current study, we intracerebrally inoculated neonatal mice with previously prepared hyperimmune anti-G10 serum to examine the serum's protective efficacy. We conducted the in vivo neutralizing experiment. Serially diluted anti-G10 sera were mixed with 260 LD50 of BJCA08 at the same volume. The neonatal mice were i.c. inoculated with the mixture after it had been neutralized at 37°C for 1 h. The results showed that anti-G10 serum could neutralize BJCA08 and reduce the severity of the diseases and the mortality rate, which displayed significant dose-response effects. A 10-μl anti-G10 serum with a neutralizing antibody titer of 8.3 may protect 50% of the neonatal mice from death. Second, a passive immunization protection test was performed to examine whether anti-G10 serum displayed a protective function similar to that of the in vivo neutralizing test. We intraperitoneally injected a series of diluted anti-G10 serum into 1-day-old mice. Within 1 h, the mice were i.c. inoculated by BJCA08 with a dosage of 260 LD50. The results showed that in a passive immunization protection test, the neutralizing antibody also had a protective efficacy with a significant dose-response effect but required a higher quantity to achieve the same effect as the in vivo neutralizing test. A 50-μl anti-G10 serum with a neutralizing antibody titer of 15 may protect 50% of the neonatal mice from death. These data suggest that sufficient anti-G10 serum was able to protect neonatal mice from lethal challenge.
To further explore the protective effect of vaccine in the neonatal mouse model, we chose maternal instead of neonatal immunization (36) because immunizing 1-day-old pups is not only technically difficult but also not suitable for the evaluation of CA16 vaccine, as the susceptibility of the animal only lasts a few days. Similarly, EV71, with very close gene homology and clinical symptoms to CA16, is also sensitive to suckling mice (1, 9). Thus, the maternal antibody protection method for the evaluation of EV71 vaccines was successfully used (1, 6–9, 47, 53), in which dams were immunized and specific maternal antibody vertically transferred from dams to pups demonstrated excellent protection against EV71 challenge in the pups. Moreover, the protection rates of newborn mice were dose dependent (1). In 2012, Li et al. first reported a phase I clinical trial of the EV71 vaccine (Clinicaltrials.gov no. NCT01273246 and NCT01273233), in which a clear dose-dependent correlation was observed (24). Referring to the EV71 maternal model, in our research, BJCA08/CA16 at a dosage of 260 LD50 was used to intracerebrally challenge the newborns of the dams immunized with inactivated CA16 (G10 and BJCA08). The clinical symptoms and survival rates of infected neonatal mice were analyzed to indirectly evaluate the protective effect of inactivated CA16. The results demonstrated that both inactivated G10 and BJCA08 may protect the infected neonatal mice to various degrees and also indicated that vertical immunity could be induced by the CA16 vaccine. The survival rates of neonatal mice born to dams immunized with inactivated G10 and BJCA08 were 47 and 93%, respectively. At the end of the observation period (21 days postinfection), the serum neutralizing antibody titer(s) of female mice immunized by G10 and BJCA08 were 28 and 14, respectively (see Table S2 in the supplemental material). These data indicated that immunization with inactivated CA16 virus can induce maternal antibody, which in turn prevents neonatal mice from lethal challenge with BJCA08. However, an unparalleled phenomenon was observed between the protection rates for neonatal mice and the antibody levels of immunized dams. The result was consistent with the observation by Wu et al. concerning the EV71 neonatal mouse model (47). It suggested that the higher protective level offered by the inactivated virus vaccine was not corroborated by a demonstrably higher neutralizing antibody titer(s). This may be explained by differences between in vivo and in vitro functions of pathogen-specific immunoglobulin. It may also be associated with the great differences in the genomic sequences between G10 and BJCA08. Comparing the nucleotide sequences of G10 and BJCA08, it can be seen that they belong to the same serotype but to a different genotype (A and C1). The homology of VP1 nucleotide sequences is only 76.3% (see Fig. S1) (51). Whether they have different neutralizing epitopes or cross-protective capacity requires further investigation.
Both EV71 and CA16 have similar viral structures, because they belong to the same Enterovirus genus. Studies concerning the EV71 neonatal mouse model showed that EV71 was neurotropic and might enter the brain through the nervous system and cause CNS complications (5, 35). To understand the pathogenic mechanism of CA16 infections, we conducted a pathological examination and IHC staining on the infected neonatal mice. No pathological change or positive CA16 antigen was found in neurocytes of the brain or the spinal cord, except for in the brain ependymal epithelium. This indicated that BJCA08/CA16, differing from EV71, had no significant neurotropism in the neonatal mouse. However, the hind-limb skeletal muscles showed severe necrosis. CA16 antigen was also observed in the corresponding areas of lesions. Moreover, severe necrosis in the juxtaspinal skeletal muscle fibers and positive CA16 antigen indicated that BJCA08 has a strong tropism to muscle tissues. These findings are consistent with the results reported by Sickles et al. (43). In addition, focal necrosis was also observed in the cardiac tissues of the moribund mice, and CA16 antigen was found in the corresponding IHC staining. These findings suggest that the lethal lesions occurring in the cardiac tissues at late stages of disease cause death in neonatal mice. The findings were also consistent with clinical observations that some deaths resulted from lethal myocarditis (12, 13, 45).
Using qRT-PCR, we found that CA16 was detected in brain tissue and blood at 1 h postinoculation, while the virus in the brain may be residual due to the i.c. inoculation at this time. The viral loads in the blood were steady and increased slightly from 1 to 120 h postinfection. Twenty-four hours after inoculation, the virus was first detectable in the brain, the hind-limb skeletal muscles, and the cardiac tissues. Thereafter, the virus gradually spread to all tissues (liver, lung, and intestine at 48 h; spleen and kidney at 72 h). These data revealed that after intracerebral injection, BJCA08 may enter the bloodstream by the capillary vessels of the brain, arrive and replicate in target tissues through blood circulation, and finally spread to the whole body. Among tissues having the virus at the earliest time, only the skeletal muscles had viral loads that steadily increased. It was speculated that CA16 was continuously replicated in the muscles and released into blood circulation, which then caused lesions and necrosis in cardiac muscles and finally led to death.
The population most vulnerable to severe disease from CA16 infection is children under 5 years of age. Clinical symptoms and disease caused by CA16 infection are usually mild, such as HFMD, fever, and herpangina. However, some reports indicate that CA16 infection also can cause severe complications, such as aseptic meningitis, rhombencephalitis, pulmonary complications, cardiac and pericardial disease, and even fatal myocarditis and pneumonitis (14, 21, 34, 40, 44–46, 50). The disease process caused by the CA16 infection progressed very quickly when severe complications occurred. However, the potential pathogenesis as well as the mechanism underlying lethal CA16 infection in humans is still unclear. Thus, in this study a newborn mouse model of CA16 infection was established by using i.c. inoculation with the clinical isolate strain BJCA08. Although the main cause of morbidity and mortality in our animal model is skeletal muscle myositis with no significant involvement of the CNS, a moribund mouse exhibited severe focal myocarditis that is similar to that of a human case of a 15-month-old boy who died of myocarditis following HFMD (45). Our data clearly indicated that this model is useful for the assessment of vaccine efficacy. A few cases of EV71 and CA16 coinfections have been reported in humans (17, 20, 52, 54, 55). Clearly, the incidence, pathogenicity, and pathogenesis of the coinfections need further investigation. It would be of interest to evaluate whether this mouse model could be applied to the study of such coinfection. Experiments are ongoing in our laboratories to address this potentially significant issue.
In summary, we used the clinically isolated BJCA08 viral strain to develop the first CA16 neonatal mouse model for the evaluation of the vaccine's protective efficacy. This animal model employed survival rate as an evaluation indicator with good reproducibility. Pathological observation, IHC staining, and quantitative real-time PCR revealed that BJCA08 had strong tropism for skeletal and cardiac muscles and caused necrosis. Using a maternal antibody protection study and an antiserum protection study, we demonstrated that the specific CA16 neutralizing antibody might block invasion of the virus and were able to evaluate the protective efficacy of the CA16 vaccine. In order to further improve this model, particularly to better understand virus transmission in vivo and the natural infection process in human beings, we are collecting CA16 virus strains of a more virulent nature to address the aforementioned issues.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the assistance of the Institute of Medical Biology, Chinese Academy of Medical Science, for the CA16/G10 and goat anti-G10 serum. We acknowledge the assistance of Beijing Wantai Biological Pharmacy Enterprise Co., Ltd., for the anti-CA16 MAb T26.
This work was supported by the National Science Project (no. 2008BAI69B01) and National 11th Five Major Special Projects Funding Program (no. 2009ZX10004-804).
Footnotes
Published ahead of print 5 September 2012
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1. Bek EJ, et al. 2011. Formaldehyde-inactivated vaccine provokes cross-protective immunity in a mouse model of human enterovirus 71 infection. Vaccine 29:4829–4838 [DOI] [PubMed] [Google Scholar]
- 2. Bendig JW, Fleming DM. 1996. Epidemiological, virological, and clinical features of an epidemic of hand, foot, and mouth disease in England and Wales. Commun. Dis. Rep. CDR Rev. 6:R81–R86 [PubMed] [Google Scholar]
- 3. Chan YF, AbuBakar S. 2005. Human enterovirus 71 subgenotype B3 lacks coxsackievirus A16-like neurovirulence in mice infection. Virol. J. 2:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chang LY, et al. 1999. Comparison of enterovirus 71 and coxsackievirus A16 clinical illnesses during the Taiwan enterovirus epidemic, 1998. Pediatr. Infect. Dis. J. 18:1092–1096 [DOI] [PubMed] [Google Scholar]
- 5. Chen CS, et al. 2007. Retrograde axonal transport: a major transmission route of enterovirus 71 in mice. J. Virol. 81(17):8996–9003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen CW, Lee YP, Wang YF, Yu CK. 2010. Formaldehyde-inactivated human enterovirus 71 vaccine is compatible for co-immunization with a commercial pentavalent vaccine. Vaccine 29:2772–2776 [DOI] [PubMed] [Google Scholar]
- 7. Chiu CH, Chu C, He CC, Lin TY. 2006. Protection of neonatal mice from lethal enterovirus 71 infection by maternal immunization with attenuated Salmonella enterica serovar Typhimurium expressing VP1 of enterovirus 71. Microbes Infect. 8:1671–1678 [DOI] [PubMed] [Google Scholar]
- 8. Chung YC, et al. 2008. Immunization with virus-like particles of enterovirus 71 elicits potent immune responses and protects mice against lethal challenge. Vaccine 26(15):1855–1862 [DOI] [PubMed] [Google Scholar]
- 9. Dong C, et al. 2011. Immunoprotection elicited by an enterovirus type 71 experimental inactivated vaccine in mice and rhesus monkeys. Vaccine 29(37):6269–6275 [DOI] [PubMed] [Google Scholar]
- 10. Dong C, et al. 2010. Optimized development of a candidate strain of inactivated EV71 vaccine and analysis of its immunogenicity in rhesus monkeys. Hum. Vaccine 6(12):1028–1037 [DOI] [PubMed] [Google Scholar]
- 11. Gilbert GL. 1988. Outbreak of enterovirus 71 in Victoria, Australia with a high incidence of neurologic involvement. Pediatr. Infect. Dis. J. 7:484–488 [DOI] [PubMed] [Google Scholar]
- 12. Gohd RS, Faigel HC. 1966. Hand, foot, and mouth disease resembling measles. A life-threatening disease: a case report. Pediatrics 37:644–648 [PubMed] [Google Scholar]
- 13. Goldberg MF, McAdams AJ. 1963. Myocarditis possibly due to coxsackie group A, type16, virus. J. Pediatr. 62:762–765 [DOI] [PubMed] [Google Scholar]
- 14. Goto K, et al. 2009. Rhombencephalitis and coxsackievirus A16. Emerg. Infect. Dis. 15(10):1689–1691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ho M, et al. 1999. An epidemic of enterovirus 71 infection in Taiwan. N. Engl. J. Med. 341:929–935 [DOI] [PubMed] [Google Scholar]
- 16. Hosoya M, et al. 2007. Genetic diversity of coxsackievirus A16 associated with hand, foot, and mouth disease epidemics in Japan from 1983 to 2003. J. Clin. Microbiol. 45(1):112–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huang WZ, et al. 2011. Genetic analysis and etiology study of hand-foot-mouth disease in surveillance hospitals of Hunan province during the period of 2008∼2010. Practical Preventive Medicine 18(5):779–784 [Google Scholar]
- 18. Hyypiä T, et al. 1993. Pathogenetic differences between coxsackie A and B virus infections in newborn mice. Virus Res. 27:71–78 [DOI] [PubMed] [Google Scholar]
- 19. Iwai M, et al. 2009. Genetic changes of coxsackievirus A16 and enterovirus 71 isolated from hand, foot, and mouth disease patients in Toyama, Japan between 1981 and 2007. Jpn. J. Infect. Dis. 62:254–259 [PubMed] [Google Scholar]
- 20. Jia L, et al. 2011. Comparisons of epidemiological and clinical characteristics in children with hand-foot-mouth disease caused by enterovirus 71 and coxsackievirus A16. Chin. J. Contemp. Pediatr. 13(8):635–637 [PubMed] [Google Scholar]
- 21. Legay F, et al. 2007. Fatal coxsackievirus A16 pneumonitis in adults. Emerg. Infect. Dis. 13:1084–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li LL, et al. 2005. Genetic characteristics of human enterovirus 71 and coxsackievirus A16 circulating from 1999 to 2004 in Shenzhen, People's Republic of China. J. Clin. Microbiol. 43:3835–3839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li WA, et al. 2009. Epidemiology and control of hand, foot and mouth disease in Singapore, 2001–2007. Ann. Acad. Med. Singapore 38:106–112 [PubMed] [Google Scholar]
- 24. Li YP, et al. 2012. Safety and immunogenicity of a novel human enterovirus 71 (EV71) vaccine: a randomized, placebo-controlled, double-blind, phase I clinical trial. Vaccine 30(22):3295–3303 [DOI] [PubMed] [Google Scholar]
- 25. Lin TY, Twu SJ, Ho MS, Chang LY, Lee CY. 2003. Enterovirus 71 outbreaks, Taiwan: occurrence and recognition. Emerg. Infect. Dis. 9:291–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu ML, et al. 2005. Type I interferons protect mice against enterovirus 71 infection. J. Gen. Virol. 86:3263–3269 [DOI] [PubMed] [Google Scholar]
- 27. Luo LF, et al. 2011. Severe hand foot and mouth disease (HFMD) suspected pathogen detection and epidemiological analysis. Chin. J. Health Lab. Technol. 21(4):971–972 [Google Scholar]
- 28. Luo XH, Zhang YM, Zhang JQ, He YY, Gao H. 2008. Etiological diagnosis and analysis of four outbreaks of hand-foot-mouth disease. Chin. Prev. Med. 9(12):1041–1043 [Google Scholar]
- 29. Mao QY, Yao X, Liang ZL. 2011. Animal infection models in the research of EV71 vaccine. Chin. J. Viral Dis. 1(3):222–226 [Google Scholar]
- 30. Mao QY, et al. 2010. Dynamic change of mother-source neutralizing antibodies against enterovirus 71 and coxsackievirus A16 in infants. Chin. Med. J. 123:1679–1684 [PubMed] [Google Scholar]
- 31. Martín J, Crossland G, Wood DJ, Minor PD. 2003. Characterization of formaldehyde-inactivated poliovirus preparations made from live attenuated strains. J. Gen. Virol. 84:1781–1788 [DOI] [PubMed] [Google Scholar]
- 32. McMinn PC. 2002. An overview of the evolution of enterovirus 71 and its clinical and public health significance. FEMS Microbiol. Rev. 26:91–107 [DOI] [PubMed] [Google Scholar]
- 33. Nagata N, et al. 2004. Differential localization of neurons susceptible to enterovirus 71 and poliovirus type 1 in the central nervous system of cynomolgus monkeys after intravenous inoculation. J. Gen. Virol. 85:2981–2989 [DOI] [PubMed] [Google Scholar]
- 34. Ogilvie MM, Tearne CF. 1980. Spontaneous abortion after hand, foot, and mouth disease caused by coxsackievirus A16. Br. Med. J. 281:1627–1628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ong KC, Devi S, Cardosa MJ, Wong KT. 2010. Formaldehyde-inactivated whole-virus vaccine protects a murine model of enterovirus 71 encephalomyelitis against disease. J. Virol. 84(1):661–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Paoletti LC, Pinel J, Kennedy RC, Kasper DL. 2000. Maternal antibody transfer in baboons and mice vaccinated with a group B streptococcal polysaccharide conjugate. J. Infect. Dis. 181(2):653–658 [DOI] [PubMed] [Google Scholar]
- 37. Podin Y, et al. 2006. Sentinel surveillance for human enterovirus 71 in Sarawak, Malaysia: lessons from the first 7 years. BMC Public Health 6:180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rabenau HF, Richter M, Doerr HW. 2010. Hand, foot and mouth disease: seroprevalence of coxsackie A16 and enterovirus 71 in Germany. Med. Microbiol. Immunol. 199(1):45–51 [DOI] [PubMed] [Google Scholar]
- 39. Reed LJ, Muench H. 1938. A simple method of estimating 50 percent end-points. Am. J. Hyg. 27:493–497 [Google Scholar]
- 40. Robinson CR, Doane FW, Rhodes AJ. 1958. Report of an outbreak of febrile illness with pharyngeal lesions and exanthem. Can. Med. Assoc. J. 79:615–621 [PMC free article] [PubMed] [Google Scholar]
- 41. Shekhar K, et al. 2005. Deaths in children during an outbreak of hand, foot and mouth disease in peninsular Malaysia–clinical and pathological characteristics. Med. J. Malaysia 60(3):297–304 [PubMed] [Google Scholar]
- 42. Shi WP, et al. 2010. Epidemiological survey of three outbreaks of CoxA16-induced hand, foot and mouth disease in kindergartens. Dis. Surveill. 25(1):22–24 [Google Scholar]
- 43. Sickles GM, Mutterer M, Feorino P, Plager H. 1955. Recently classified types of coxsackievirus, group A. Behavior in tissue culture. Proc. Soc. Exp. Biol. Med. 90:529–531 [DOI] [PubMed] [Google Scholar]
- 44. Urquhart GED. 1984. A survey of coxsackie A16 virus antibodies in human sera. J. Hyg. Camb. 93:205–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang CY, Li LF, Wu MH, Lee CY, Huang LM. 2004. Fatal coxsackievirus A16 infection. Pediatr. Infect. Dis. J. 23:275–276 [DOI] [PubMed] [Google Scholar]
- 46. Wright HT, Landing BH, Lennette EH, Mcallister RM. 1963. Fatal infection in an infant associated with coxsackie virus group A, type 16. N. Engl. J. Med. 9:1041–1044 [DOI] [PubMed] [Google Scholar]
- 47. Wu CN, et al. 2001. Protection against lethal enterovirus 71 infection in newborn mice by passive immunization with subunit VP1 vaccines and inactivated virus. Vaccine 20:895–904 [DOI] [PubMed] [Google Scholar]
- 48. Wu PC, et al. 2010. An outbreak of coxsackievirus A16 infection: comparison with other enteroviruses in a preschool in Taipei. J. Microbiol. Immunol. Infect. 43(4):271–277 [DOI] [PubMed] [Google Scholar]
- 49. Wu TC, et al. 2007. Immunity to avirulent enterovirus 71 and coxsackie A16 virus protects against enterovirus 71 infection in mice. J. Virol. 81(19):10310–12315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yang WS. 2011. Clinical characteristic analysis of 104 cases of children severe hand, foot and mouth disease. Chongqing Med. 40(17):1722–1727 [Google Scholar]
- 51. Yao X, et al. 2010. Genetic characteristics of coxsackievirus A16 complete genome isolated in Beijing, 2008. Chin. J. Epidemiol. 31(12):1437–1438 [Google Scholar]
- 52. Ye RX, Zhang YL, Pan KN, Tong YX. 2011. Significance of enterovirus 71 and coxsackievirus 16 pathogens detection in preventing epidemics of hand, foot and mouth disease. Chin. J. Nosocomiol. 21(7):1295–1296 [Google Scholar]
- 53. Yu CK, et al. 2000. Neutralizing antibody provided protection against enterovirus type 71 lethal challenge in neonatal mice. J. Biomed. Sci. 7:523–528 [DOI] [PubMed] [Google Scholar]
- 54. Zhang W, et al. 2011. Severe hand, foot, and mouth disease caused by mixed infection of enterovirus 71 and coxsackie A16: report of 6 cases. Chin. Gen. Practice 14(10):3341–3346 [Google Scholar]
- 55. Zhang YJ, Bai J, Huang XL. 2010. Clinical analysis of 54 cases of hand, foot and mouth disease with central nervous system damage. Chin. J. Crit. Care Med. 30(12):1090–1092 [Google Scholar]
- 56. Zhu Z, et al. 2010. Retrospective seroepidemiology indicated that human enterovirus 71 and coxsackievirus A16 circulated wildly in central and southern China before large-scale outbreaks from 2008. Virol. J. 7:300. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.