

Abstract
Herpes simplex virus 1 (HSV-1) virions, like those of all herpesviruses, contain a protein layer termed the tegument localized between the capsid and the envelope. VP22, encoded by the UL49 gene, is one of the most abundant tegument proteins in HSV-1 virions. Studies with a UL49-null mutant showed that the absence of VP22 resulted in decreased protein synthesis at late times in infection. VP22 is known to form a tripartite complex with VP16 and vhs through direct interactions with VP16. Given that UL49-null mutants have been shown to acquire spontaneous secondary mutations in the UL41 gene, which encodes vhs, we hypothesized that VP22 and vhs may play antagonistic roles during HSV-1 infections. In the present study, we show that the protein synthesis defect observed in UL49-null virus infections was rescued by a secondary, compensatory frameshift mutation in UL41. A double mutant bearing a deletion of UL49 and a point mutation in vhs previously shown to specifically abrogate vhs's RNase activity also resulted in a rescue of protein synthesis. To determine whether the UL49− protein synthesis defect, and the rescue by secondary mutations in vhs, occurred at the mRNA and/or translational levels, quantitative reverse transcriptase PCR (qRT-PCR) and polysome analyses were performed. We found that the absence of VP22 caused a small decrease in mRNA levels as well as a defect in polysome assembly that was independent of mRNA abundance. Both defects were complemented by the secondary mutations in vhs, indicating functional interplay between VP22 and vhs in both accumulation and translation of viral mRNAs.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) virions are composed of a double-stranded DNA (dsDNA) genome enclosed within an icosahedral capsid, a proteinaceous layer termed the tegument that surrounds the nucleocapsid, and a host-derived lipid membrane envelope that contains viral glycoproteins. The tegument layer is unique to the herpesviruses and is composed of at least 20 different viral proteins of various stoichiometries. Tegument proteins play a variety of roles in infection, including the regulation of viral and host gene expression and the promotion of virus assembly and egress (2, 14, 27, 31). Tegument proteins enter the cell upon fusion of the viral envelope with the host cell membrane and therefore can exert their activities prior to viral gene expression. As virion structural proteins, many of the tegument proteins are synthesized at late times in infection and may possess additional functions that are exerted at this time.
Viral protein 22 (VP22), encoded by the UL49 gene, is one of the most abundant tegument proteins, with an average of 2,000 copies present in each virion (10, 16, 20). Previous studies with a UL49-null virus showed that the absence of VP22 results in reduced plaque size and a nearly global shutoff protein synthesis at late times in infection (7, 8). Interestingly, passage of the UL49-null virus on noncomplementing cells led to a rescue of the small plaque phenotype thought to be the result of a secondary, compensatory mutation (7). Further work by other investigators showed that UL49 mutants propagated on noncomplementing cells frequently acquire secondary mutations in the UL41 gene. For example, Sciortino et al. isolated two UL49 deletion virus variants that had each acquired a spontaneous deletion of UL41 codons 22 to 75 and a third that had acquired a frameshift mutation at UL41 codon 286 (35).
The UL41 gene encodes the virion host shutoff protein (vhs), another tegument component of HSV-1 virions. vhs is an endoribonuclease that degrades both cellular and viral mRNAs (17, 24). Degradation of cellular mRNAs is thought to promote viral protein synthesis by increasing the availability of the cellular translation machinery, whereas degradation of viral mRNAs by vhs is thought to help regulate the sequential expression of different classes of viral genes (12, 17, 24, 25, 30). Although the RNase activity of vhs is nonspecific in vitro, during HSV-1 infections it is specific to mRNA, sparing other RNA species (11, 17, 24, 37, 39, 44). vhs associates with mRNAs via interactions with the translation initiation factors eIF4AII, eIF4H, and eIF4B, indicating that vhs is targeted to actively translating mRNAs (6, 12, 26). At late times in infection, the nuclease activity of vhs is downregulated by VP16, a tegument protein that also functions as the transactivator of immediate early (IE) gene expression (1, 2, 15, 18). In addition to its interactions with vhs, VP16 also directly interacts with VP22, allowing for the formation of a VP22-VP16-vhs tripartite complex (9, 38).
vhs can exert both positive and negative effects on viral protein translation independently of its RNase activity. In transient-transfection studies, vhs was shown to modulate reporter gene expression without greatly altering target mRNA levels (33). Specifically, Saffran et al. (33) used bicistronic reporter constructs bearing different internal ribosome entry sites (IRESs) located between two test cistrons. As expected, vhs inhibited the expression of the 5′ cistron in all constructs. Interestingly, the effect of vhs on expression of the 3′ cistron varied with the IRES: vhs strongly suppressed expression driven from the encephalomyocarditis virus (EMCV) IRES but activated expression from other IRESs. vhs-dependent IRES activation was observed in HeLa, HepG2, and 293 cells but not in Vero cells. In addition, several HSV-1 5′ untranslated region (UTR) mRNA sequences also served as positive vhs translational response elements (33). Further studies showed that the absence of vhs leads to the formation of stress granules, indicative of stalled translation initiation (5). Polysome analyses showed that true late viral transcripts were poorly translated in the absence of vhs. This effect was cell type specific in that the presence of vhs was necessary for robust translation of true late-gene products in HeLa cells, but not in Vero cells.
In the present study, we aimed to determine the mechanism by which VP22 promotes protein synthesis at late times in HSV-1 infections. The isolation of yet another UL49-null variant that had acquired a spontaneous, secondary frameshift mutation at vhs residue 284 pointed to a functional interplay between VP22 and vhs. However, it was possible that this spontaneous variant contained additional secondary mutations. Therefore, to examine possible antagonistic roles for VP22 and vhs in regulating protein synthesis, we generated a double mutant bearing a deletion of UL49 and the frameshift mutation at codon 284 of UL41 using a bacterial artificial chromosome (BAC) system. The frameshift mutation in vhs resulted in the rescue of protein synthesis late in infections of both Vero and HeLa cells. A double mutant bearing a deletion of UL49 and a point mutation in vhs previously shown to specifically abrogate vhs's RNase activity (13) also resulted in a rescue of protein synthesis. To determine whether the rescue in protein synthesis by secondary mutations in vhs occurred at the mRNA and/or the translational level, quantitative reverse transcription-PCR (qRT-PCR) and polysome analyses were performed. Relative to those of the wild type, viral mRNA levels were slightly decreased in UL49− infections and slightly increased in infections with the UL49/UL41 double mutants. Similarly, polysome levels were decreased in UL49− infections and increased with the double mutants relative to those of the wild type. The distributions of mRNAs across polysome gradients were quantified to determine whether the differences in polysome levels were caused solely by differences in mRNA levels or whether the various mutants displayed differences in translation efficiency. We found that the absence of VP22 caused a defect in translation that was independent of mRNA abundance, identifying a new role for this protein in HSV-1 replication. The secondary mutations in vhs compensated for this defect, pointing to an additional role for VP22 in modulating vhs's translational repression activity.
MATERIALS AND METHODS
Viruses and cells.
The generation of the wild-type HSV-1 strain F (WT) and UL49-null (UL49−) viruses using pYEbac102 was described previously (7). The generation of the vhsFS, UL49−/vhsFS, vhsD213N, and UL49−/vhsD213N viruses is described below. Vero and HeLa cells were purchased from the ATCC. V49 cells, a Vero-derived VP22-complementing cell line, were a gift from John Blaho (29). Vero and V49 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 4.0 mM l-glutamine, 4.5 g/liter glucose, 125 units/ml penicillin, 0.125 mg/ml streptomycin, and either 10% newborn calf serum (NBCS) or 15% fetal bovine serum (FBS), respectively. VP22 expression was maintained in V49 cells with the addition of Geneticin to 1 mg/ml. HeLa cells were maintained in DMEM–nutrient mixture–F-12 Ham medium supplemented with 2.5 mM l-glutamine without HEPES, 125 units/ml penicillin, 0.125 mg/ml streptomycin, and 10% FBS. All viruses were propagated on V49 cells; experiments were performed on Vero and HeLa cells. Because viruses with mutations in UL49 frequently acquire spontaneous secondary mutations in UL41 (35), all mutant virus stocks used in this study were tested prior to use. Testing was performed by PCR amplification and DNA sequencing of the UL41 gene from viral DNAs purified using phenol extraction and ethanol precipitation of virus stocks.
Generation of recombinant viruses.
Recombinant HSV-1(F) BACs were constructed as described previously by en passant mutagenesis, a two-step Red-mediated recombination system (7, 41, 42). The UL49−/vhsFS and UL49−/vhsD213N viruses were generated using the HSV-1(F) BAC pYEbac102:UL49−, which contains a deletion of the entire UL49 open reading frame (ORF) (7), and the Escherichia coli strain EL250 (42). The vhsFS and vhsD213N viruses were generated using the HSV-1(F) BAC pYEbac102 (40) and E. coli strain GS1783 (41). The primer pair used to generate the PCR amplicon for UL49−/vhsFS and vhsFS BAC construction via recombination was as follows: forward, 5′-CCGTGGAGGATGTGCTGCGCGAATGTCACTGGACCCCCCCCGAGTCGCTCTCAGACCCGGTAGGGATAACAGGGTAATCGATTTATTCAACAAA-3′; reverse, 5′-CCGTGGAGCGCGAGCTGGTGTGTTCCCGGCGGATGGCCCGCCGGGTCTGAGAGCGACTCGGCCAGTGGTACAACCAATTAACCAATTCT-3′. The primer pair used to generate the PCR amplicon for UL49−/vhsD213N and vhsD213N BAC construction via recombination was as follows: forward, 5′-TCCAACACAATATCACAGCCCATCAACGGAGGTCAGTGTTCGTGGTGTACACGTACGCGTAGGGATAACAGGGTAATCGATTT-3′; reverse, 5′-CTCTATCACACCAACACGGTCGCGTACGTGTACACCACGAACACTGACCTCCTGTTGATGGCCAGTGTTACAACCAATTAACC-3′. For the above primer sequences, nucleotides in roman type are homologous to HSV-1(F) BAC target sequences up- and downstream of the designed mutation, nucleotides in boldface are the intended sequence insertion or change, and nucleotides in italics are homologous to the SceI-Knr cassette of pLay2 (42). The expected mutations were confirmed by restriction fragment length polymorphism analysis and DNA sequencing. The resulting BACs were designated pYEbac102:vhsFS, pYEbac102:vhsD213N, pYEbac102:UL49−/vhsFS, and pYEbac102:UL49−/vhsD213N. Recombinant HSV-1(F) viruses were generated via cotransfection of V49 cells with purified BAC DNA and the Cre recombinase-expressing plasmid pCAGGS-nlsCre (a gift from Michael Kotlikoff, Cornell University). Plaques formed on V49 cells were purified and further propagated on V49 cells. Genotypes of the recombinant viruses were confirmed by PCR and DNA sequencing.
Protein synthesis time courses.
Vero or HeLa cells grown to 90 to 95% confluence in 6-well dishes were infected with WT, UL49−, vhsFS, UL49−/vhsFS, vhsD213N, or UL49−/vhsD213N in medium 199 supplemented with 1% NBCS (medium 199V) at a multiplicity of infection (MOI) of 10 PFU/cell for 1 h at 4°C to allow viral attachment. Virus-containing medium was replaced with fresh medium 199V, and plates were shifted to 37°C to synchronize infections. At various times after the 37°C shift, the overlying medium in each well was removed and replaced with DMEM (high glucose, with pyridoxine hydrochloride, no l-glutamine, no l-methionine, and no l-cysteine) supplemented with 10 μCi/ml Tran35S-label ([35S]methionine-cysteine; MP Biomedicals) for the time periods shown in Fig. 1 and 2. At the end of each labeling period, the overlying radioactive medium was removed, and cells were washed with phosphate-buffered saline (PBS), collected by scraping, resuspended in sodium dodecyl sulfate (SDS) Blue loading buffer (New England BioLabs), and boiled for 10 min. Radiolabeled lysates were separated by SDS-12% PAGE, transferred to a nitrocellulose membrane, and visualized by autoradiography on BioMax XAR film (Kodak). Radiolabeled lysates were also mixed with 2 ml of scintillation fluid (Ecoscint H; National Diagnostics), and incorporated radiolabeled methionine and cysteine were quantified using a Beckman scintillation counter.
Fig 1.

Analysis of global protein synthesis and accumulation in WT-, UL49−-, vhsFS-, and UL49−/vhsFS-infected cells. Vero (A and B) or HeLa (C and D) cells were synchronously infected at an MOI of 10 PFU/cell with the WT, UL49−, vhsFS, or UL49−/vhsFS virus and radiolabeled with [35S]methionine and [35S]cysteine for the time periods shown. Labeled proteins were detected by autoradiography (A and C) and scintillation counting (B and D). Data were compiled from two independent experiments, each performed in duplicate. Values in panels B and D are arithmetic means; error bars represent 1 standard deviation. Example IE proteins that are not shut off effectively in vhsFS- and UL49−/vhsFS-infected cells are indicated by < in panels A and C.
Fig 2.

Analysis of global protein synthesis and accumulation in WT-, UL49−-, vhsD213N-, and UL49−/vhsD213N-infected cells. Vero (A and B) or HeLa (C and D) cells were synchronously infected at an MOI of 10 PFU/cell with the WT, UL49−, vhsD213N, or UL49−/vhsD213N virus and radiolabeled with [35S]methionine and [35S]cysteine for the time periods shown. Labeled proteins were detected by autoradiography (A and C) and scintillation counting (B and D). Data were compiled from two independent experiments, each performed in duplicate. Values in panels B and D are arithmetic means; error bars represent 1 standard deviation. Example IE proteins that are not shut off effectively in vhsD213N- and UL49−/vhsD213N-infected cells are indicated by < in panels A and C.
Actinomycin D chase assay.
Vero cells grown in 100-mm dishes to 90 to 95% confluence were mock treated or infected with the WT, UL49−, vhsFS, UL49−/vhsFS, vhsD213N, and UL49−/vhsD213N viruses in medium 199V at an MOI of 10 PFU/cell in the presence of 10 μg/ml actinomycin D (Affymetrix, Inc.) for 1 h at 4°C to allow virus attachment. The overlying virus-containing media were then replaced with fresh medium 199V containing 10 μg/ml actinomycin D, and cells were placed at 37°C to synchronize the infections. At 6 h postinfection (hpi), total RNA was extracted from infected cells and prepared for quantitative real-time reverse transcription-PCR (RT-PCR) as described below. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA levels were quantified via qRT-PCR as described below. Because actinomycin D treatment affects 18S rRNA levels but not 5S rRNA levels (28), 5S rRNA was used as a control for template input. Primer sequences are listed in Table 1.
Table 1.
Primers used in qRT-PCR analyses
| cDNA | Forward primer | Reverse primer |
|---|---|---|
| 5S rRNA | 5′-GCCATACCACCCTGAACG-3′ | 5′-GGCGGTCTCCCATCCAAG-3′ |
| 18S rRNA | 5′-CCAGTAAGTGCGGGTCATAAGC-3′ | 5′-GCCTCACTAAACCATCCAATCGG-3′ |
| GAPDH | 5′-ACAGTCAGCCGCATCTTC-3′ | 5′-CTCCGACCTTCACCTTCC-3′ |
| Us8 (gE) | 5′-AGGTCTGCGGGGTTGGGATGG-3′ | 5′-GGTGCGACAGGCGGAACAGG-3′ |
| Us6 (gD) | 5′-AGGTCTGCGGGGTTGGGATGG-3′ | 5′-GGTGCGACAGGCGGAACAGG-3′ |
| UL29 (ICP8) | 5′-TGCGAGGGCGTCAGTTTCAG-3′ | 5′-GCGTGTCCGTCCGAAGGC-3′ |
Virion preparations and analysis.
Intracellular virions were prepared as described previously (7) from V49 cells infected with the WT, UL49−, vhsFS, UL49−/vhsFS, vhsD213N, and UL49−/vhsD213N viruses at an MOI of 0.005 PFU/cell at 37°C until 100% cytopathic effect (CPE) was reached. Three independent virion preparations were made for each virus. Purified virions were separated by SDS-PAGE and either stained with Coomassie brilliant blue or transferred to a nitrocellulose membrane. Transferred proteins were analyzed by immunoblotting using an antibody to VP5; the blot was then stripped and reprobed with an antibody specific to vhs. To determine the relative vhs abundances in virions produced by the different recombinant viruses, the vhs signal was normalized to the VP5 signal from the same virion preparation, and then each normalized vhs signal was calculated as a value relative to the WT normalized vhs signal.
Immunoblot analyses.
The 35S-labeled cell lysates and virion preparations described above were used in immunoblot analyses following separation via SDS-12% PAGE and transfer to nitrocellulose membranes. Free binding sites were blocked with 5% nonfat dry milk in PBS before incubation with either anti-gD (diluted 1:1,000; H170; Virusys), anti-gE (diluted 1:500; a kind gift from David Johnson [4, 21]), anti-ICP8 (diluted 1:2,000 [36]), anti-β-actin (diluted 1:1,000; SC-47778; Santa Cruz Biotechnology), anti-VP5 (diluted 1:1,000; HA018-100; Virusys), or anti-vhs (diluted 1:1,000; a kind gift from Duncan Wilson [19]) antibodies. After washing membranes extensively to remove unbound antibodies, bound primary antibodies were detected with anti-mouse (diluted 1:5,000; NA9310V; GE Healthcare Biosciences) or anti-rabbit (diluted 1:5,000; NA9340V; GE Healthcare Biosciences) immunoglobulin G conjugated to horseradish peroxidase. Membranes were again washed extensively, incubated in Pierce ELC Western blotting substrate (Thermo Scientific) for 1 min, and visualized via autoradiography on Amersham Hyperfilm ECL film (GE Healthcare Biosciences). For quantification, immunoblots were developed with the Amersham ECL Plus Western blotting detection system (GE Healthcare) and imaged on a Storm 860 phosphorimager (Molecular Dynamics, Inc.). Immunoblot signals were quantified using ImageQuant software (Amersham Biosciences, Inc.).
Quantitative real-time RT-PCR.
Vero cells grown to 95 to 100% confluence in 100-mm dishes were synchronously infected with the WT, UL49−, vhsFS, UL49−/vhsFS, vhsD213N, and UL49−/vhsD213N viruses in medium 199V at an MOI of 10 PFU/cell. At 18 hpi, total RNA was extracted from infected cells using the RNAqueous-4PCR kit (Ambion, Inc.) according to the manufacturer's instructions. Residual DNA was removed by treatment with DNase I, which was subsequently inactivated using DNase inactivation reagent (Ambion, Inc.). Before proceeding to the reverse transcription (RT) reactions, RNA samples were tested for contaminating DNA via quantitative PCR (qPCR) experiments using primers for both 18S rRNA and gE mRNA and Maxima SYBR green qPCR master mix (Fermentas, Inc.). All samples were found to be free of amplifiable contaminating DNA.
Samples (0.5 μg) of DNase I-treated total RNA were used as the templates for cDNA synthesis reactions primed with random decamers using the Retro-script kit (Ambion, Inc.). To determine template quality and obtain primer amplification efficiencies, serial dilutions of cDNAs were used as the templates in qRT-PCRs to construct standard curves. Amplification efficiencies were 92 to 110%, and the R2 value of all standard curves was ≥0.980.
Relative quantitative real-time RT-PCR experiments were performed to quantify gE, gD, and ICP8 mRNA levels using 18S rRNA internal controls as follows. Equal amounts of cDNAs were used in qRT-PCR mixtures containing primers to the mRNA of interest and, in separate reactions, primers to 18S rRNA as a control for normalizing template input. Primer pair sequences are listed in Table 1. All qRT-PCRs were performed in triplicate using Maxima SYBR green qPCR master mix (Fermentas, Inc.) in a MyiQ real-time PCR thermocycler and detection system (Bio-Rad Laboratories, Inc.). SYBR green fluorescence was measured over the course of 40 amplification cycles. This was immediately followed by a melt-curve analysis to confirm that nonspecific products did not contribute to the quantified signal. For each template quantified, the mean cycle number at which product accumulation entered the linear range (CT) was calculated. The 18S rRNA CT value for a given template was subtracted from the CT obtained for each mRNA of interest from the same template (same RT reaction) to obtain the normalized CT value (ΔCT) for each template. The change from that of the wild type was then calculated using the 2−ΔΔCT method.
Polysome analyses.
Methods outlined by Cheshenko et al. (3) and Dauber et al. (5) were employed as follows. Vero cells grown to 90 to 95% confluence in 100-mm dishes were either mock treated or infected with the WT, UL49−, UL49−/vhsFS, and UL49−/vhsD213N viruses at an MOI of 10 PFU/cell. Ten minutes prior to collection at 18 hpi, overlying medium was replaced with fresh medium 199V containing 100 μg/ml cycloheximide (CHX) to block translation elongation. Cells were collected via trypsin treatment, washed in PBS containing 100 μg/ml CHX, resuspended in gradient lysis buffer (50 mM Tris-HCl [pH 7.4], 250 mM sucrose, 25 mM KCl, 5 mM MgCl2, 0.5% Triton X-100, 100 μg/ml CHX, 2 mM dithiothreitol [DTT], and RNaseOUT), and incubated on ice for 10 min. Following centrifugation, supernatants were mixed with either MgCl2 (final concentration, 10 mM) or EDTA (final concentration, 25 mM), incubated on ice for 10 min, and layered atop a 10-ml 10% to 50% sucrose gradient containing 50 mM Tris-HCl (pH 7.4), 25 mM KCl, 1.5 mM MgCl2, 100 μg/ml CHX, and RNaseOUT. Samples were centrifuged in a Beckman SW41 rotor at 38,000 rpm for 105 min at 4°C. To examine the ribosome profile throughout each gradient, a density gradient fractionator (Labconco, Inc.) was connected to a UV-visible (UV-Vis) continuous flow cell (Shimadzu, Inc.). Absorbance was measured at 254 nm as each gradient was driven through the flow cell by the peristaltic pump on the fractionator. Each gradient was fractionated as it exited the flow cell. Gradient fractions were subsequently used in Northern blot analyses.
RNA was purified from gradient fractions via proteinase K digestion followed by phenol-chloroform extraction and ethanol precipitation. The RNA pellet from each fraction was resuspended in 10 μl, 4 μl of which was separated by 1.2% agarose-formaldehyde gel electrophoresis and transferred to an Immobilon charged nylon membrane (Millipore, Inc.). To generate the gE probe, a region of US8 spanning HSV-1 bp 141637 to 142159 was amplified via PCR from pRB123 (a gift from Bernard Roizman) using the primers 5′-AGGATGACAATGACGAGGGCGAGGG-3′ and 5′-GACGCGTCCCGGACTCCAGATTGA-3′. To generate the β-actin probe, a region spanning bp 398 to 1085 of the β-actin coding sequence was amplified from PlasmID clone HsCD00326994 purchased from the Dana-Farber/Harvard Cancer Center DNA Resource Core using the primers 5′-ACGTTGCTATCCAGGCTGTGCTAT-3′ and 5′-ACTCCTGCTTGCTGATCCACATCT-3′. The purified PCR products were used as the templates for probe generation via random priming using [α-32P]dCTP (MP Biomedicals) and the Prime-It II random primer labeling kit (Agilent Technologies). Hybridization was performed using ExpressHyb (Clontech) according to the manufacturer's instructions. Radioactive signals were analyzed with a Storm 860 phosphorimager (Molecular Dynamics, Inc.) and ImageQuant software (Amersham Biosciences, Inc.).
Statistical analyses.
Significant differences were analyzed using Student's t test and two-tailed distribution. Results were considered statistically significant if P was ≤0.05.
RESULTS
The vhsFS mutation compensates for the lack of VP22 in protein synthesis.
It was previously shown that UL49-null viruses can acquire secondary, compensatory mutations in UL41 (35). Upon sequencing PCR-amplified viral DNA isolated from plaques following several passages of the UL49− virus, we identified a UL49-null virus variant that contained a frameshift mutation at codon 284 of UL41. The above findings, combined with studies that showed that VP22, VP16, and vhs can form a tripartite complex (38), suggest a functional interplay between VP22 and vhs during HSV-1 infections. To examine whether VP22 and vhs play antagonistic roles in regulating viral protein synthesis, we first asked whether the secondary frameshift mutation at residue 284 of vhs could compensate for the lack of VP22 in protein synthesis late in infection. Because it was not known whether the UL49− isolate that had obtained the spontaneous, secondary frameshift mutation in UL41 contained additional mutations, we used a BAC recombination system to generate a recombinant virus containing a deletion of the UL49 ORF and the frameshift mutation at codon 284 of UL41. This virus was designated UL49−/vhsFS (Table 2). The frameshift mutation results in a protein that is 415 residues long, as opposed to 489 residues for wild-type vhs. As a control, we generated a virus containing only the frameshift mutation in UL41, designated vhsFS. Importantly, all virus stocks used throughout the following studies were propagated on a VP22-complementing cell line and were tested for the presence of additional mutations in vhs by sequencing the UL41 gene from PCR-amplified DNA purified from virus stocks.
Table 2.
HSV-1(F) recombinant viruses used
| Virus | Mutation(s) | Resulting coding change(s) | Source |
|---|---|---|---|
| WT | None | None | 7 |
| UL49− | Deletion of HSV-1 bp 105486 to 106391 | Deletion of VP22 (UL49 ORF deletion) | 7 |
| vhsFS | Guanine nucleotide insertion at HSV-1 bp 91789 | Frameshift at vhs residue 284 | This work |
| UL49−/vhsFS | Deletion of HSV-1 bp 105486 to 106391 and guanine nucleotide insertion at HSV-1 bp 91789 | Deletion of VP22 (UL49 ORF deletion) and frameshift at vhs residue 284 | This work |
| vhsD213N | C-to-T nucleotide change at HSV-1 bp 92001 | Aspartic acid (D)-to-asparagine (N) change at vhs residue 213 | This work |
| UL49−/vhsD213N | Deletion of HSV-1 bp 105486 to 106391 and C-to-T nucleotide change at HSV-1 bp 92001 | Deletion of VP22 (UL49 ORF deletion) and aspartate (D)-to-asparagine (N) change at vhs residue 213 | This work |
To examine the effect of the vhsFS mutation on protein synthesis at various times in infection, the WT, UL49−, vhsFS, and UL49−/vhsFS viruses were used in 35S label time courses. Vero cells infected with the above-mentioned viruses were radiolabeled with [35S]methionine and [35S]cysteine from 0 to 3 hpi, 3 to 6 hpi, 12 to 15 hpi, and 15 to 18 hpi. Labeled cell lysates were separated by SDS-PAGE, and proteins synthesized during the above time periods were visualized by autoradiography (Fig. 1A). Aliquots of labeled cell lysates were also analyzed by scintillation counting to determine total protein synthesis and accumulation during each labeling period (Fig. 1B). As shown previously (8), the absence of VP22 caused a decrease in protein synthesis at late times in infection (compare WT and UL49− at 12 to 18 hpi [Fig. 1A and B]). The data showed a trend toward complementation of the UL49 deletion by the vhsFS mutation inasmuch as mean protein synthesis from 15 to 18 hpi in UL49−- and UL49−/vhsFS-infected cells was 42% and 110%, respectively, of that in WT-infected cells. However, the difference in total protein expression at late times in UL49−- and UL49−/vhsFS-infected cells was not statistically significant. The vhsFS control virus produced protein levels similar to those produced by the WT and UL49−/vhsFS viruses during all time periods studied.
Dauber et al. showed that vhs is not required for efficient translation of late proteins in Vero cells but is required in HeLa cells and other restrictive cell lines (5). To determine whether UL49− protein synthesis complementation by the vhsFS mutation was cell line specific, we performed the above-described experiment in HeLa cells (Fig. 1C and D). We found that the vhsFS mutation was able to partially complement the UL49 deletion in HeLa cells, but not as efficiently as in Vero cells. Specifically, mean protein synthesis from 15 to 18 hpi in UL49−- and UL49−/vhsFS-infected HeLa cells was 49% and 78%, respectively, of that in WT-infected HeLa cells. Also, the difference in total protein expression in UL49−- and UL49−/vhsFS-infected HeLa cells was not statistically significant. Overall, however, our data show that the vhsFS mutation partially compensates for the lack of VP22 in protein synthesis late in infection in both a restrictive and a nonrestrictive cell line.
A number of different vhs mutants have been shown to possess defects in IE mRNA turnover and the shutoff of IE protein synthesis, exposing a role for vhs in regulating the sequential expression of different classes of viral genes (12, 17, 24, 25, 30). The vhsFS mutation also displayed a partial defect in IE protein synthesis shutoff inasmuch as synthesis of several IE proteins continued at late times postinfection in cells infected with the vhsFS virus, whereas those proteins were efficiently shut off at late times in wild-type-infected cells. This effect was more pronounced in HeLa cells than in Vero cells (Fig. 1A and C).
The vhsD213N mutation compensates for the lack of VP22 in protein synthesis.
The partial defect in IE protein synthesis shutoff displayed by the vhsFS mutation suggested that this mutant possesses a defect in vhs's RNase activity and that complementation of the UL49− defect in protein synthesis could occur via an increased abundance of viral mRNAs late in infection. However, we previously observed that viral mRNA levels were not decreased at late times in UL49− infections (8), making complementation by such a mechanism unlikely. To test whether a defect in vhs's RNase activity could compensate for the lack of VP22 in protein synthesis, we generated a recombinant virus containing a deletion of the UL49 ORF and an aspartate-to-asparagine change at codon 213 of UL41. The vhsD213N point mutation was previously shown to abrogate vhs's RNase activity in transfection experiments (11). The double mutant was designated UL49−/vhsD213N (Table 2). For a control, we generated a virus containing only the D213N point mutation in UL41, designated vhsD213N.
To determine whether the vhsD213N mutation could compensate for a lack of VP22 in protein synthesis, we again performed 35S label time courses. Vero cells infected with the WT, UL49−, vhsD213N, and UL49−/vhsD213N viruses were radiolabeled from 0 to 3 hpi, 3 to 6 hpi, 12 to 15 hpi, and 15 to 18 hpi. Labeled proteins synthesized during the above time periods were visualized by autoradiography following SDS-PAGE separation of cell lysates (Fig. 2A). Total protein synthesis and accumulation during each labeling period were quantified by scintillation counting of cell lysate aliquots (Fig. 2B). Interestingly, the vhsD213N mutation rescued the UL49− protein synthesis defect. Specifically, total protein synthesis in UL49−/vhsD213N-infected Vero cells was 180% and 144% of that in WT-infected cells from 12 to 15 hpi and 15 to 18 hpi, respectively, compared to 103% and 109% in UL49−/vhsFS-infected Vero cells for the same time periods. The vhsD213N control virus synthesized 186% and 177% of the protein levels produced by the WT virus in Vero cells from 12 to 15 hpi and 15 to 18 hpi, respectively. To ensure that this result was not cell line specific, the 35S label time courses were also performed in HeLa cells (Fig. 2C and D). The vhsD213N mutation compensated for a lack of VP22 in protein synthesis late in infection in both cell lines.
Similar to the vhsFS mutation, the vhsD213N mutation also displayed a partial defect in IE protein synthesis shutoff, in both the WT and UL49− genetic backgrounds, on both Vero and HeLa cells.
The vhsD213N mutation abrogates vhs's RNase activity during HSV-1 infections.
As mentioned above, the vhsD213N mutation has been previously shown to abrogate vhs's RNase activity in transfection experiments (11). To verify that this mutation possesses the same defect during HSV-1 infections, we performed actinomycin D chase assays. We included the UL49−/vhsFS and vhsFS mutant viruses in this assay to determine whether the frameshift at vhs residue 284 affects the protein's RNase activity. To examine mRNA turnover independently of transcription, Vero cells were mock treated or infected with the WT, UL49−, vhsFS, UL49−/vhsFS, vhsD213N, and UL49−/vhsD213N viruses for 6 h in the presence of actinomycin D. Total RNA was then collected, DNase I treated, reverse transcribed, and used in qRT-PCR experiments to quantify GAPDH mRNA levels. Because actinomycin D can affect 18S rRNA levels (28), 5S rRNA was used as a control to normalize template input.
We found that the vhsD213N mutation does possess a defect in mRNA degradation in the context of infection, as GAPDH mRNA levels were nearly 12-fold higher in vhsD213N-infected cells than in WT-infected cells (Fig. 3). However, the vhsD213N mutation did not completely abrogate vhs's RNase activity, as GAPDH mRNA levels in vhsD213N-infected cells were 47% of those in mock-treated cells. The vhsFS mutation also caused decreased mRNA degradation, although not as strongly as did the vhsD213N mutation. Specifically, cells infected with the vhsFS virus contained GAPDH levels 8-fold higher than those in WT-infected cells. However, further studies (see below) indicated that the decreased mRNA degradation observed with the vhsFS mutant may be due, at least in part, to decreased virion packaging of vhs rather than a specific defect in vhs's RNase activity.
Fig 3.
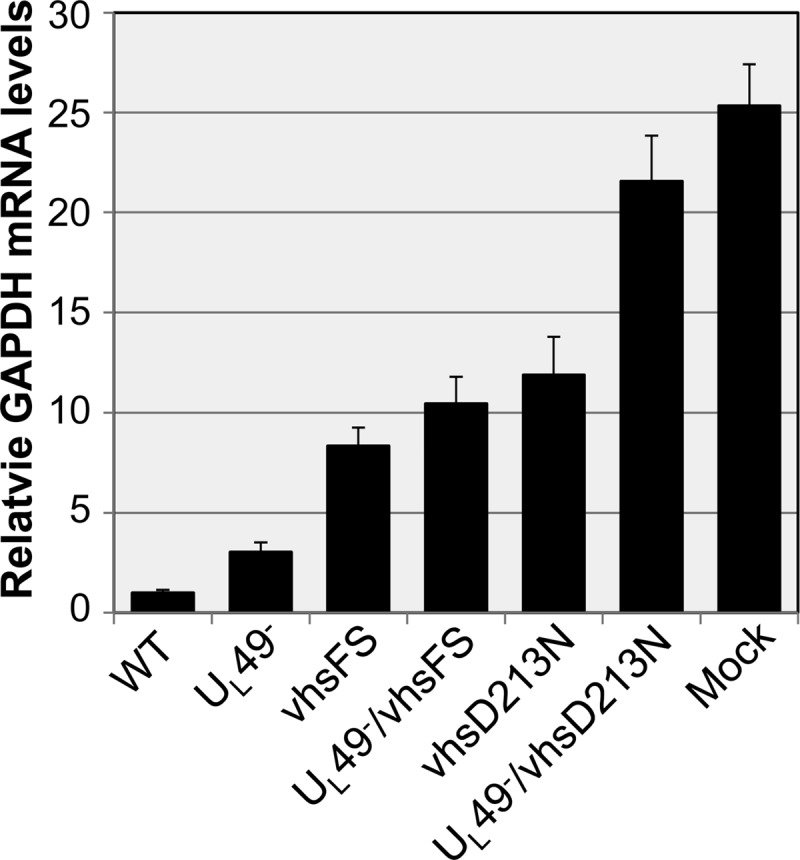
Analysis of mRNA stability. Data represent the relative GAPDH mRNA levels remaining in infected and mock-infected cells at 6 hpi in the absence of transcription. Vero cells were mock treated or infected at an MOI of 10 PFU/cell with the WT, UL49−, UL49−/vhsFS, or UL49−/vhsD213N virus for 6 h in the presence of actinomycin D. Total RNA was purified, reverse transcribed, and used as a template for qRT-PCR with primers specific to GAPDH mRNA. Separate reactions were performed using primers specific to 5S rRNA as a control to normalize for template input. GAPDH cDNA abundance, relative to that for WT, was determined using the 2−ΔΔCT method. Two independent experiments were performed, each in triplicate. Values given are arithmetic means; error bars represent 1 standard deviation.
UL49−/vhsD213N-infected cells contained significantly higher GAPDH mRNA levels than did vhsD213N-infected cells (Fig. 3). This was also observed, although to a lesser extent, in comparing UL49−- and UL49−/vhsFS-infected cells to WT- and vhsFS-infected cells, respectively. These results indicated that the absence of VP22 somehow decreased mRNA degradation in this experiment. However, all evidence to date points to a role for VP22 in negative, rather than positive, regulation of vhs's RNase activity. In the above-described assay, viral and cellular transcription were inhibited by actinomycin D. Therefore, the reductions in GAPDH mRNA levels in virus-infected cells relative to those in mock-treated cells were due to RNase activity delivered from infecting virions. The viral stocks used in these experiments were generated on V49 cells, which express a much lower level of VP22 than is expressed during WT infections (data not shown). We reasoned that the decreased levels of VP22 present in V49 cells could lead to decreased virion incorporation of vhs during stock production of viruses carrying a UL49 deletion. Consequently, this would result in decreased delivery of vhs upon experimental infection, leading to the reduced RNase activity observed in UL49−, UL49−/vhsFS, and UL49−/vhsD213N infections compared to that in WT, vhsFS, and vhsD213N infections, respectively. To test this hypothesis, we infected V49 cells with the WT, UL49−, vhsFS, UL49−/vhsFS, vhsD213N, and UL49−/vhsD213N viruses and purified virions from the infected cells (Fig. 4). Immunoblotting assays were performed on the purified virions using an antibody specific to VP5 as a loading control and an antibody specific to vhs to examine relative vhs levels (Fig. 4B and C). Consistent with our hypothesis, we found that virion incorporation of vhs was significantly reduced in UL49−/vhsD213N virions compared to that in vhsD213N virions (P = 0.05), which potentially explains the difference in GAPDH mRNA levels observed with these viruses in Fig. 3. Interestingly, vhsFS and UL49−/vhsFS virions did not differ from each other in vhs incorporation, but both contained significantly reduced levels of vhs relative to those in WT virions (P = 0.04 and 0.02, respectively). This finding suggests that the increased levels of GAPDH mRNA in vhsFS- and UL49−/vhsFS-infected cells relative to that in WT-infected cells may be due, at least in part, to decreased vhs delivery. It is likely that the vhsFS mutation causes misfolding of a large proportion of vhs expressed in vhsFS- and UL49−/vhsFS-infected cells, leading to decreased incorporation of vhs into vhsFS and UL49−/vhsFS virions and perhaps also causing a defect in vhs's RNase activity.
Fig 4.

Relative virion vhs levels. Virions were purified from V49 cells infected with the WT, UL49−, vhsFS, UL49−/vhsFS, vhsD213N, or UL49−/vhsD213N virus at an MOI of 0.005 PFU/cell and collected when CPE reached 100%. (A) Proteins present in purified virions were separated by SDS-PAGE and stained with Coomassie brilliant blue. Results are representative of three independent experiments. (B) Virion proteins were also examined by immunoblotting using an antibody specific to VP5 followed by blot stripping and reprobing with an antibody specific to vhs. The vhsFS mutation results in early translation termination and, thus, faster migration during SDS-PAGE. Results are representative of three independent experiments. (C) Immunoblot signals were quantified, and the vhs signal on each blot was normalized to the corresponding VP5 signal. Data were compiled from three independent experiments. Values shown are arithmetic mean VP5-normalized vhs levels present in virions relative to those in the WT; error bars represent 1 standard deviation.
Characterization of viral gene expression at late times in infection.
We previously observed that mRNA levels were unaffected at late times in UL49−-infected cells compared to those in WT-infected cells (8), leading us to hypothesize that the defect in protein synthesis seen in the absence of VP22 occurred at the translational level. However, given our present data with the UL49−/vhsD213N mutant showing that a defect in vhs's RNase activity can compensate for a lack of VP22 in protein synthesis, we wanted to reexamine mRNA levels in UL49−-infected cells at late times in infection. We first examined steady-state gE, gD, and ICP8 protein levels in order to identify target mRNAs to quantify. Vero cells were infected with the WT, UL49−, vhsFS, UL49−/vhsFS, vhsD213N, and UL49−/vhsD213N viruses for 3, 6, 15, or 18 h. Cell lysates were separated by SDS-PAGE and used in immunoblotting assays with antibodies specific to gE, gD, and ICP8 (Fig. 5A and B). An antibody specific to β-actin was used as a loading control. Steady-state gE and gD levels were greatly reduced in UL49−-infected cells compared to those in all other infections, whereas steady-state levels of ICP8 were only slightly reduced in UL49−-infected cells. This experiment was also performed on HeLa cells, with comparable results (data not shown).
Fig 5.

Relative viral gene expression. (A and B) Vero cells were synchronously infected at an MOI of 10 PFU/cell with the WT, UL49−, vhsFS (A), UL49−/vhsFS (A), vhsD213N (B), or UL49−/vhsD213N (B) virus for the time periods shown. Lysates from infected cells were analyzed for steady-state protein levels by immunoblotting using antibodies specific to gE, gD, and ICP8. An antibody specific to β-actin was used as a loading control. (C) Vero cells were synchronously infected at an MOI of 10 PFU/cell with the WT, UL49−, vhsFS, UL49−/vhsFS, vhsD213N, or UL49−/vhsD213N virus for 18 h. Total RNA was purified, reverse transcribed, and used as a template for qRT-PCR with primers specific to gE, gD, and ICP8 mRNAs. Separate reactions were performed using primers specific to 18S rRNA as a control to normalize for template input. Transcript abundance, relative to that for the WT, was determined using the 2−ΔΔCT method. Data are expressed as the means ± standard errors of the means of three independent experiments, each performed in triplicate.
We next examined gE, gD, and ICP8 mRNA levels via quantitative real-time RT-PCR. Total RNA was purified from Vero cells infected with the WT, UL49−, vhsFS, UL49−/vhsFS, vhsD213N, and UL49−/vhsD213N viruses for 18 h. Following DNase I treatment and reverse transcription, cDNAs were used in qRT-PCRs with primers specific to gE, gD, and ICP8 mRNAs (Table 1). Separate reactions were performed with primers specific to 18S rRNA to normalize for template input. The absence of VP22 resulted in a small reduction in steady-state mRNA levels at 18 hpi with gE, gD, and ICP8 mRNAs present in UL49−-infected cells at 57%, 25%, and 70%, respectively, of the levels present in WT-infected cells (Fig. 5C). The difference in gD mRNA levels present in WT- and UL49−-infected cells was statistically significant (P = 0.007), whereas the differences in gE and ICP8 mRNA levels were not significant. The decreased mRNA levels observed in UL49−-infected cells were complemented by the vhsFS and vhsD213N secondary mutations, with mean mRNA levels in UL49−/vhsFS- and UL49−/vhsD213N-infected cells being 1.8- to 4-fold higher than those in WT-infected cells.
VP22 is required for WT levels of protein translation.
Given the relatively small extent to which mRNA levels were reduced in the absence of VP22, we hypothesized that a reduction in translation efficiency also contributed to the protein shutoff observed at late times in UL49− infections. Vero cells were either mock infected or infected with the WT, UL49−, UL49−/vhsFS, and UL49−/vhsD213N viruses for 18 h. Postmitochondrial supernatants prepared in the presence of cycloheximide were separated by velocity sedimentation through sucrose density gradients. Gradients were driven through a UV-Vis continuous flow cell by the peristaltic pump on a gradient fractionator to measure absorbance at 254 nm across each gradient and then were fractionated as they exited the flow cell. The distribution of gE mRNAs across each gradient was also examined via Northern blotting assays performed on RNA purified from the gradient fractions.
The UV absorbance profiles across each gradient are shown in Fig. 6A. For controls to determine the gradient sedimentation positions of dissociated ribosomal subunits, 80S ribosomes, and polysomes, postmitochondrial supernatants from mock-treated cells were prepared in the presence and absence of EDTA. Mock-treated supernatants prepared in the presence of EDTA gave a large absorbance peak in fractions 4 and 5 and low absorbance in later fractions, where intact 80S ribosomes and polysomes sediment. In contrast, gradients of supernatants prepared from mock-treated cells in the absence of EDTA displayed low absorbance levels in fractions 4 and 5 and high absorbance in fractions 6 to 8, correlating with 80S ribosomes, and in fractions 11 to 18, correlating with polysome sedimentation. WT-infected cells contained both 80S ribosomes and polysomes as indicated by absorbance peaks in fractions 6 to 8 and 12 to 19, respectively. The WT-infected cell 80S peak was larger and the polysome peak was smaller than those in in mock-treated supernatants prepared in the absence of EDTA. This was expected, as translation is dampened in response to viral infection. Intriguingly, gradients from UL49−-infected cells gave a large peak at fractions 6 to 8, but absorbance was similar to that in mock-treated EDTA+ supernatant levels in later fractions, indicating that the absence of VP22 results in reduced polysome levels. Both the vhsFS and the vhsD213N mutations compensated for the absence of VP22, providing polysome levels similar to those in WT-infected cell or mock-treated EDTA− supernatants, respectively. Sharp spikes in the UV absorbance starting at fraction 16 were the result of air bubbles that formed in the peristaltic pump tubing in denser regions of the gradients.
Fig 6.

Analysis of translation efficiency. Postmitochondrial supernatants prepared from Vero cells mock treated or infected at an MOI of 10 PFU/cell for 18 h with the WT, UL49−, UL49−/vhsFS, or UL49−/vhsD213N virus were separated by 10% to 50% sucrose density centrifugation. Mock-treated supernatants were prepared in the presence and absence of EDTA. Gradients were driven, top to bottom, through a UV-Vis continuous flow cell by the peristaltic pump of a gradient fractionator. Gradients were fractionated as they exited the flow cell. (A) UV absorbance profiles at 254 nm of fractions 3 to 20 from each gradient. The top of the gradient is to the left, and the bottom is to the right. Sharp spikes in the UV absorbance toward the bottom of the gradient were caused by air bubbles that form in the peristaltic pump tubing in denser regions of the gradient. Similar results were observed in three independent experiments. (B and C) Distribution of mRNAs across gradient fractions collected following UV absorbance measurements. RNA was extracted from gradient fractions and analyzed by Northern blotting using probes specific to gE (B) or β-actin (C). Bands were quantified on a phosphorimager, and the signal in each fraction in a gradient was calculated as a percentage of the signal across all fractions in that gradient to determine the distribution of the mRNAs across the gradients. The graphs represent one of two independent, comparable experiments.
To determine whether the reduced polysomes present in UL49−-infected cell supernatants were due solely to the decreased mRNA levels observed above (Fig. 5C) or whether an additional defect in protein translation contributed, we quantified the distribution of specific mRNAs across each gradient. Northern blotting assays were performed on fractionated gradients from WT-, UL49−-, UL49−/vhsFS-, and UL49−/vhsD213N-infected cells using a probe to gE mRNA and on fractionated gradients from mock-treated cells using a probe to β-actin mRNA. The mRNA signal from each fraction was calculated as a percentage of the total signal across fractions 3 to 20 (Fig. 6B and C). Gradients of EDTA-treated and untreated mock-infected supernatants served as controls. β-Actin mRNA peaks corresponded to the 80S ribosome and polysome areas of the gradient (fractions 6 and 11 to 18, respectively) for untreated mock supernatants and to less-dense areas of the gradient (fractions 4 to 7) for EDTA-treated mock supernatants. In agreement with the absorbance measurements, gE mRNAs were found in the 80S and polysome peaks of WT gradients (fractions 6 to 8 and 12 to 19, respectively), with a larger 80S peak and a smaller polysome peak than those of mock supernatants prepared without EDTA. Importantly, UL49− gradients had a higher gE mRNA signal in the 80S region (fractions 6 to 8) and a lower signal in the polysome region (fractions 12 to 19) than did WT gradients, showing that the absence of VP22 causes a defect in translation that is independent of mRNA abundance. Both the UL49−/vhsFS and UL49−/vhsD213N gradients had large gE mRNA signals in the polysome areas (fractions 12 to 19), showing that the vhsFS and vhsD213N mutations compensated for the absence of VP22 by increasing polysome assembly, as well as by increasing mRNA levels.
DISCUSSION
Previous studies showed that the absence of VP22 results in a shutoff of protein synthesis at late times in infection (8). In the present study, we found that VP22 promotes both the accumulation of viral mRNAs and the efficiency of translation at late times in infection. Specifically, the absence of VP22 resulted in gE, gD, and ICP8 mRNA levels that were reduced by 43%, 75%, and 30%, respectively, compared to levels present in wild-type-infected cells. The effect of VP22 on protein translation, independent of mRNA abundance, was studied by quantifying the distribution of mRNAs across polysome gradients. Of the gE mRNA present in UL49−-infected cells, very little was associated with polysomes, indicating that the absence of VP22 also results in reduced translation efficiency. Both the mRNA and translation defects observed in UL49− infections were complemented by secondary mutations in UL41. These findings strongly suggest that functional interplay occurs between VP22 and vhs during wild-type HSV-1 infections. Both VP22 and vhs interact directly with VP16, allowing for the formation of a VP22-VP16-vhs tripartite complex (38). The functional interplay between VP22 and vhs may occur via structure/function changes upon complex formation. Alternatively, VP22 may act independently of complex formation to antagonize vhs's activities in mRNA degradation and/or translation suppression.
In addition to identifying roles for VP22 in regulating mRNA abundance and protein translation at late times in infection, the present study has also extended our understanding of vhs. Previous studies using transfected UL41 constructs and a lacZ reporter system indicated that the vhsD213N mutation completely abrogated vhs's RNase activity (11–13). Experiments performed here in the context of viral infections showed that the D213N mutation does not completely abolish vhs's RNase activity but does reduce mRNA degradation as well as compensate for the lack of VP22 in translation efficiency in a manner independent of mRNA abundance. The frameshift mutation at codon 284 of UL41 also compensated for the lack of VP22 in polysome assembly, indicating that the C-terminal region of vhs functions in translation suppression either directly or by contributing to correct vhs conformation and overall functionality. A large proportion of mutant vhs produced in vhsFS-infected cells is likely misfolded with reduced or abrogated functionality. A reduction in vhs activity caused by misfolding could result in the observed complementation of the UL49 deletion.
The vhs protein contains three domains predicted to be of high importance to its function (11). The first two domains, comprised of residues 1 to 103 and 165 to 265, share homology with mammalian, yeast, bacterial, and phage nucleases. The third domain is comprised of residues 365 to 489 and, along with domains 1 and 2, shares homology with UL41 homologues from other alphaherpesviruses. The frameshift mutation carried by the UL49−/vhsFS and the vhsFS viruses is located between domains 2 and 3, directly affecting the amino acid sequence of the third domain of vhs. We found that GAPDH mRNA in cells infected with the vhsFS virus in the presence of actinomycin D underwent less degradation than it did in wild-type-infected cells. This may be at least partially explained by our finding that the truncated protein produced by the vhsFS virus is incorporated into virions less efficiently than is full-length vhs, thereby decreasing the amount of vhs delivered upon infection. Mukhopadhyay et al. showed that the N-terminal 42 amino acids of vhs are sufficient for vhs membrane association and virion incorporation (22). Our data indicate that vhs residues 284 to 489 are also important for virion incorporation, perhaps by contributing to the conformational correctness of N-terminal regions of the protein.
VP22 could increase mRNA abundance at late times in infection by promoting transcription, inhibiting mRNA degradation, or both. For example, there is a growing body of evidence that HSV-1 replication is regulated in part by nucleosome-based mechanisms (reviewed in reference 23). VP22 may promote transcription at late times in infection, when it is present in infected cells in large amounts, via its interactions with the linker histone H1, the core histone H4, or the H2A-H2B-specific histone chaperone template-activating factor I (32, 43). Alternatively, VP22 may increase mRNA abundance by protecting it from degradation through its RNA-binding activity (34). Finally, as part of the VP22-VP16-vhs complex, VP22 may aid VP16 in modulating vhs's RNase activity at late times in infection.
There are also a number of mechanisms by which VP22 could promote protein translation at late times in infection. The data presented here show that the absence of VP22 resulted in reduced polysome levels and increased 80S ribosome levels, indicating that once a ribosome is loaded, further initiation events are abrogated. This effect was reversed by the vhsFS and vhsD213N secondary mutations and could occur by a number of different mechanisms. For example, a translation initiation-elongation transition defect would result in increased 80S ribosome levels and could be caused by a reduced abundance of translation elongation factors. In this scenario, VP22 would act to modulate vhs's RNase activity to maintain necessary levels of key cellular mRNAs and proteins, such as elongation factors. The absence of VP22 could then be complemented by secondary mutations that decrease vhs's RNase activity. Alternatively, VP22 may be necessary to protect the 5′ UTR from vhs cleavage following translation initiation. In this scenario, cleavage of 5′ UTRs in the absence of VP22 would abrogate further 5′ cap-dependent translation initiation events on the affected mRNAs, leading to an increase in 80S ribosomes and decreased protein synthesis. In our favored model, VP22 and VP16 interact with vhs during translation initiation and together serve to modulate vhs's RNase activity, thereby protecting 5′ UTRs from vhs cleavage. Future studies will focus on testing this model and identifying the mechanism(s) by which VP22 regulates mRNA abundance and protein translation at late times in infection.
ACKNOWLEDGMENTS
These studies were supported by an ASM Robert D. Watkins Graduate Research Fellowship to E.F.M., NIH R21 AI 083952-01 to C.D., institutional funds from the Saint Louis University School of Medicine to L.A.M., a Howard Hughes Medical Institute Undergraduate Science Education grant to The University of Alabama, and the Louis Stokes Alliance for Minority Participation Bridge to the Doctorate Program supported by NSF HRD-0602359.
We thank Duncan Wilson for the gift of the anti-vhs antibody and David Johnson for the gift of the anti-gE antibody. We thank Ian Mohr and Joel Baines for helpful discussions.
Footnotes
Published ahead of print 5 September 2012
REFERENCES
- 1. Batterson W, Roizman B. 1983. Characterization of the herpes simplex virion-associated factor responsible for the induction of alpha genes. J. Virol. 46:371–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Campbell ME, Palfreyman JW, Preston CM. 1984. Identification of herpes simplex virus DNA sequences which encode a trans-acting polypeptide responsible for stimulation of immediate early transcription. J. Mol. Biol. 180:1–19 [DOI] [PubMed] [Google Scholar]
- 3. Cheshenko N, Trepanier JB, Segarra TJ, Fuller AO, Herold BC. 2010. HSV usurps eukaryotic initiation factor 3 subunit M for viral protein translation: novel prevention target. PLoS One 5:e11829 doi:10.1371/journal.pone.0011829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cross AM, Hope RG, Marsden HS. 1987. Generation and properties of the glycoprotein E-related 32K/34K/35K and 55K/57K polypeptides encoded by herpes simplex virus type 1. J. Gen. Virol. 68:2093–2104 [DOI] [PubMed] [Google Scholar]
- 5. Dauber B, Pelletier J, Smiley JR. 2011. The herpes simplex virus 1 vhs protein enhances translation of viral true late mRNAs and virus production in a cell type-dependent manner. J. Virol. 85:5363–5373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Doepker RC, Hsu WL, Saffran HA, Smiley JR. 2004. Herpes simplex virus virion host shutoff protein is stimulated by translation initiation factors eIF4B and eIF4H. J. Virol. 78:4684–4699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Duffy C, et al. 2006. Characterization of a UL49-null mutant: VP22 of herpes simplex virus type 1 facilitates viral spread in cultured cells and the mouse cornea. J. Virol. 80:8664–8675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Duffy C, Mbong EF, Baines JD. 2009. VP22 of herpes simplex virus 1 promotes protein synthesis at late times in infection and accumulation of a subset of viral mRNAs at early times in infection. J. Virol. 83:1009–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Elliott G, Mouzakitis G, O'Hare P. 1995. VP16 interacts via its activation domain with VP22, a tegument protein of herpes simplex virus, and is relocated to a novel macromolecular assembly in coexpressing cells. J. Virol. 69:7932–7941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Elliott GD, Meredith DM. 1992. The herpes simplex virus type 1 tegument protein VP22 is encoded by gene UL49. J. Gen. Virol. 73:723–726 [DOI] [PubMed] [Google Scholar]
- 11. Everly DN, Jr, Feng P, Mian IS, Read GS. 2002. mRNA degradation by the virion host shutoff (Vhs) protein of herpes simplex virus: genetic and biochemical evidence that Vhs is a nuclease. J. Virol. 76:8560–8571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Feng P, Everly DN, Jr, Read GS. 2005. mRNA decay during herpes simplex virus (HSV) infections: protein-protein interactions involving the HSV virion host shutoff protein and translation factors eIF4H and eIF4A. J. Virol. 79:9651–9664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Feng P, Everly DN, Jr, Read GS. 2001. mRNA decay during herpesvirus infections: interaction between a putative viral nuclease and a cellular translation factor. J. Virol. 75:10272–10280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fuchs W, Granzow H, Klupp BG, Kopp M, Mettenleiter TC. 2002. The UL48 tegument protein of pseudorabies virus is critical for intracytoplasmic assembly of infectious virions. J. Virol. 76:6729–6742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Goding CR, O'Hare P. 1989. Herpes simplex virus Vmw65-octamer binding protein interaction: a paradigm for combinatorial control of transcription. Virology 173:363–367 [DOI] [PubMed] [Google Scholar]
- 16. Heine JW, Honess RW, Cassai E, Roizman B. 1974. Proteins specified by herpes simplex virus. XII. The virion polypeptides of type 1 strains. J. Virol. 14:640–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kwong AD, Frenkel N. 1987. Herpes simplex virus-infected cells contain a function(s) that destabilizes both host and viral mRNAs. Proc. Natl. Acad. Sci. U. S. A. 84:1926–1930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lam Q, et al. 1996. Herpes simplex virus VP16 rescues viral mRNA from destruction by the virion host shutoff function. EMBO J. 15:2575–2581 [PMC free article] [PubMed] [Google Scholar]
- 19. Lee GE, Church GA, Wilson DW. 2003. A subpopulation of tegument protein vhs localizes to detergent-insoluble lipid rafts in herpes simplex virus-infected cells. J. Virol. 77:2038–2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Leslie J, Rixon FJ, McLauchlan J. 1996. Overexpression of the herpes simplex virus type 1 tegument protein VP22 increases its incorporation into virus particles. Virology 220:60–68 [DOI] [PubMed] [Google Scholar]
- 21. McMillan TN, Johnson DC. 2001. Cytoplasmic domain of herpes simplex virus gE causes accumulation in the trans-Golgi network, a site of virus envelopment and sorting of virions to cell junctions. J. Virol. 75:1928–1940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mukhopadhyay A, Lee GE, Wilson DW. 2006. The amino terminus of the herpes simplex virus 1 protein Vhs mediates membrane association and tegument incorporation. J. Virol. 80:10117–10127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nevels M, Nitzsche A, Paulus C. 2011. How to control an infectious bead string: nucleosome-based regulation and targeting of herpesvirus chromatin. Rev. Med. Virol. 21:154–180 [DOI] [PubMed] [Google Scholar]
- 24. Oroskar AA, Read GS. 1989. Control of mRNA stability by the virion host shutoff function of herpes simplex virus. J. Virol. 63:1897–1906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Oroskar AA, Read GS. 1987. A mutant of herpes simplex virus type 1 exhibits increased stability of immediate-early (alpha) mRNAs. J. Virol. 61:604–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Page HG, Read GS. 2010. The virion host shutoff endonuclease (UL41) of herpes simplex virus interacts with the cellular cap-binding complex eIF4F. J. Virol. 84:6886–6890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pellett PE, McKnight JL, Jenkins FJ, Roizman B. 1985. Nucleotide sequence and predicted amino acid sequence of a protein encoded in a small herpes simplex virus DNA fragment capable of trans-inducing alpha genes. Proc. Natl. Acad. Sci. U. S. A. 82:5870–5874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Perry RP, Kelley DE. 1968. Persistent synthesis of 5S RNA when production of 28S and 18S ribosomal RNA is inhibited by low doses of actinomycin D. J. Cell. Physiol. 72:235–246 [DOI] [PubMed] [Google Scholar]
- 29. Pomeranz LE, Blaho JA. 2000. Assembly of infectious herpes simplex virus type 1 virions in the absence of full-length VP22. J. Virol. 74:10041–10054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Read GS, Frenkel N. 1983. Herpes simplex virus mutants defective in the virion-associated shutoff of host polypeptide synthesis and exhibiting abnormal synthesis of alpha (immediate early) viral polypeptides. J. Virol. 46:498–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Read GS, Karr BM, Knight K. 1993. Isolation of a herpes simplex virus type 1 mutant with a deletion in the virion host shutoff gene and identification of multiple forms of the vhs (UL41) polypeptide. J. Virol. 67:7149–7160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ren X, Harms JS, Splitter GA. 2001. Bovine herpesvirus 1 tegument protein VP22 interacts with histones, and the carboxyl terminus of VP22 is required for nuclear localization. J. Virol. 75:8251–8258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Saffran HA, Read GS, Smiley JR. 2010. Evidence for translational regulation by the herpes simplex virus virion host shutoff protein. J. Virol. 84:6041–6049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sciortino MT, Taddeo B, Poon AP, Mastino A, Roizman B. 2002. Of the three tegument proteins that package mRNA in herpes simplex virions, one (VP22) transports the mRNA to uninfected cells for expression prior to viral infection. Proc. Natl. Acad. Sci. U. S. A. 99:8318–8323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sciortino MT, et al. 2007. Replication-competent herpes simplex virus 1 isolates selected from cells transfected with a bacterial artificial chromosome DNA lacking only the UL49 gene vary with respect to the defect in the UL41 gene encoding host shutoff RNase. J. Virol. 81:10924–10932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shelton LS, Albright AG, Ruyechan WT, Jenkins FJ. 1994. Retention of the herpes simplex virus type 1 (HSV-1) UL37 protein on single-stranded DNA columns requires the HSV-1 ICP8 protein. J. Virol. 68:521–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Taddeo B, Roizman B. 2006. The virion host shutoff protein (UL41) of herpes simplex virus 1 is an endoribonuclease with a substrate specificity similar to that of RNase A. J. Virol. 80:9341–9345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Taddeo B, Sciortino MT, Zhang W, Roizman B. 2007. Interaction of herpes simplex virus RNase with VP16 and VP22 is required for the accumulation of the protein but not for accumulation of mRNA. Proc. Natl. Acad. Sci. U. S. A. 104:12163–12168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Taddeo B, Zhang W, Roizman B. 2006. The U(L)41 protein of herpes simplex virus 1 degrades RNA by endonucleolytic cleavage in absence of other cellular or viral proteins. Proc. Natl. Acad. Sci. U. S. A. 103:2827–2832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tanaka M, Kagawa H, Yamanashi Y, Sata T, Kawaguchi Y. 2003. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J. Virol. 77:1382–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tischer BK, Smith GA, Osterrieder N. 2010. En passant mutagenesis: a two step markerless red recombination system. Methods Mol. Biol. 634:421–430 [DOI] [PubMed] [Google Scholar]
- 42. Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step Red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197 [DOI] [PubMed] [Google Scholar]
- 43. van Leeuwen H, et al. 2003. Herpes simplex virus type 1 tegument protein VP22 interacts with TAF-I proteins and inhibits nucleosome assembly but not regulation of histone acetylation by INHAT. J. Gen. Virol. 84:2501–2510 [DOI] [PubMed] [Google Scholar]
- 44. Zelus BD, Stewart RS, Ross J. 1996. The virion host shutoff protein of herpes simplex virus type 1: messenger ribonucleolytic activity in vitro. J. Virol. 70:2411–2419 [DOI] [PMC free article] [PubMed] [Google Scholar]