

Abstract
Epstein-Barr virus (EBV) latent membrane protein 1 (LMP1) induces multiple signal transduction pathways during latent EBV infection via its C-terminal activating region 1 (CTAR1), CTAR2, and the less-studied CTAR3. One mechanism by which LMP1 regulates cellular activation is through the induction of protein posttranslational modifications, including phosphorylation and ubiquitination. We recently documented that LMP1 induces a third major protein modification by physically interacting with the SUMO-conjugating enzyme Ubc9 through CTAR3 and inducing the sumoylation of cellular proteins in latently infected cells. We have now identified a specific target of LMP1-induced sumoylation, interferon regulatory factor 7 (IRF7). We hypothesize that during EBV latency, LMP1 induces the sumoylation of IRF7, limiting its transcriptional activity and modulating the activation of innate immune responses. Our data show that endogenously sumoylated IRF7 is detected in latently infected EBV lymphoblastoid cell lines. LMP1 expression coincided with increased sumoylation of IRF7 in a CTAR3-dependent manner. Additional experiments show that LMP1 CTAR3-induced sumoylation regulates the expression and function of IRF7 by decreasing its turnover, increasing its nuclear retention, decreasing its DNA binding, and limiting its transcriptional activation. Finally, we identified that IRF7 is sumoylated at lysine 452. These data demonstrate that LMP1 CTAR3 does in fact function in intracellular signaling, leading to biologic effects. We propose that CTAR3 is an important signaling region of LMP1 that regulates protein function by sumoylation. We have shown specifically that LMP1 CTAR3, in cooperation with CTAR2, can limit the ability of IRF7 to induce innate immune responses by inducing the sumoylation of IRF7.
INTRODUCTION
Epstein-Barr virus (EBV) is a ubiquitous gammaherpesvirus that infects more than 90% of the world's population and establishes a lifelong infection within the host. Latent EBV infection is associated with several lymphoid tumors, including posttransplant lymphoproliferative disorder (PTLD), and AIDS-associated central nervous system (CNS) lymphomas (32, 33), which are associated with type III EBV latency. This form of latency is observed in lymphoblastoid cell lines (LCLs), which are established following EBV infection of primary B cells and exhibit sustained cellular proliferation and survival due to the constitutive activation of cellular signaling pathways. The main viral protein important in regulating these signal transduction events is latent membrane protein 1 (LMP1), an integral membrane signaling protein that mimics the tumor necrosis factor receptor family members, such as CD40, with the exception that its activation is ligand independent and it is constitutively active (22). LMP1 activates multiple signal transduction events via its C-terminal region (3, 9, 22; A. Kieser, presented at the 13th Biennial Conference of the International Association for Research on Epstein-Barr Virus and Associated Diseases, Guangzhou, China, 2008). One signaling pathway of continuing interest in our laboratory is the regulation of interferon regulatory factor 7 (IRF7) by LMP1.
IRFs are a family of transcription factors that regulate interferon responses, and IRFs have also been implicated in the regulation of cell growth and differentiation, apoptosis, and oncogenesis (31, 34). The N-terminal regions of IRFs contain the conserved DNA-binding domain (DBD), allowing the IRFs to bind to consensus motifs found in interferon (IFN)-stimulated response elements (ISREs), including promoters of type I IFNs. The C-terminal regions of the IRFs are much more variable, resulting in distinct IRF-protein interactions, and guide the various functions of IRFs and the specificity of their interactions.
Of all the IRFs, IRF7 and IRF3 are key regulators of the expression of type I IFNs, which include alpha IFN (IFN-α) and beta IFN (IFN-β) (11–13, 34, 36, 46). While IRF3 is considered to be responsible for the early phase of type I IFN induction, IRF7 is now understood to be the master regulator of all type I IFN-dependent immune responses (13). Our laboratory identified IRF7 in a search for a regulator of the EBV nuclear antigen 1 (EBNA-1) promoter used in type I latency: Qp, to which IRF7 binds (31, 45). We also found that IRF7 binds to and activates the LMP1 promoter (28, 29). Since then, IRF7 has been recognized as having a major role in the control of the host immune response, including the induction of IFN-γ through cooperation with STAT1 and IRF1 (8, 12, 13). In lymphoid cells, IRF7 is constitutively expressed at low levels. Increased expression can be induced by various stimuli, including EBV LMP1 (31, 46). In addition to inducing IRF7 expression, LMP1 regulates the function of IRF7 by inducing posttranslational modifications of this protein (14, 27, 42, 43).
Two major posttranslational modifications of IRF7 that we have examined are phosphorylation and ubiquitination. LMP1 induces the phosphorylation and K63-linked ubiquitination of IRF7, resulting in its nuclear translocation and increased transcriptional activity (14, 27, 42). The interactions of LMP1 CTAR2 with RIP and TRAF6, which serves as an E3 ligase responsible for the ubiquitination of IRF7 (14, 27), are required for LMP1-induced activation of IRF7. Recently, we also found that LMP1 can induce A20-mediated deubiquitination of IRF7, leading to the transcriptional repression of IRF7 (30). Additional work suggests that LMP1 may also mediate K48-linked ubiquitination of IRF7, resulting in its proteasome-mediated degradation (unpublished data). Together these findings suggest that LMP1 has several mechanisms by which it modifies IRF7 and regulates its function, which results in the regulation of cellular immune responses. Following our report that LMP1 induces the sumoylation of cellular proteins, we initiated studies to determine if IRF7 is a target of LMP1-induced sumoylation that provides an additional mechanism by which LMP1 can modulate the function of IRF7.
Protein sumoylation is a dynamic and reversible process that can regulate protein function by altering a protein's intracellular location, turnover, ability to interact with other proteins, or ability to interact with DNA (16, 17, 19). Only 5 to 10% of a protein is in a sumoylated form at any given time. Sumoylation typically occurs at lysine residues found within the conserved ΨKxE motif, where Ψ represents a hydrophobic residue (17). Analysis of the IRF7 protein sequence revealed a putative sumoylation site at position K452. Recent reports showed that murine IRF7 is sumoylated at K405, which corresponds to K452 of human IRF7, during vesicular stomatitis virus (VSV) and Ebola Zaire virus infections (6, 20). In addition, human IRF7 is reported to be sumoylated at K446 during Sendai virus infection (23). These reports focused on the sumoylation of IRF7 during RNA virus infections. Here we explored whether EBV can induce the sumoylation of IRF7 during latent infection. We propose that LMP1 limits the transcriptional activity of IRF7 and inhibits induction of innate immune responses through sumoylation. We have shown that IRF7 is sumoylated endogenously in type III EBV latency. In addition, LMP1-induced sumoylation of IRF7 decreases the turnover of IRF7, resulting in its nuclear accumulation, and limits its transcriptional activity due to the decreased interaction of IRF7 with the chromatin.
MATERIALS AND METHODS
Cells.
Human embryonic kidney (HEK) 293T cells were maintained in Dulbecco's modified Eagle's medium (DMEM) plus 10% fetal bovine serum (FBS). KR4 cells (a lymphoblastoid cell line representing type III EBV latency), BL41 EBV wild-type (WT) cells, and BL41 EBV mutant cells (P3HR1) (4, 5, 7) were maintained in RPMI plus 10% FBS.
Plasmids.
Flag-IRF7 and the IFN-β promoter construct were gifts from John Hiscott and have been described previously (24). FLAG-LMP1, FLAG-LMP1 PQAA, FLAG-LMP1 YIID, and FLAG-LMP1 DM were gifts from Nancy Raab-Traub (25). FLAG-LMP1Δ33bpr (FLAG-LMP1 dCTAR3) was a gift from Wolfgang Hammerschmidt (9).
Luciferase assays.
293T cells were grown in 6-well plates and transfected with 0.6 μg of plasmids using polyethylenimine (PEI) (VWR). Empty vector was used to equalize the total amounts of DNA in the transfections. β-Galactosidase served as a transfection control. Luciferase and β-galactosidase assays were performed 24 h posttransfection.
Immunoprecipitation.
293T cells were grown in 100-mm dishes and transfected with 8 μg of DNA using PEI. Forty-eight hours posttransfection, cells were lysed in NP-40 lysis buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1% NP-40, 5 mM dithiothreitol [DTT], 50 μM Na3VO4, 100 mM NaF, 1 mM phenylmethylsulfonyl fluoride [PMSF], 20 μM N-ethylmaleimide [NEM], and complete protease inhibitors; Sigma) or denaturing lysis buffer (10 mM NaCl, 1.5 mM MgCl2, 10 mM Tris, pH 7.5, 1% SDS, 20 μM NEM, benzonase, and complete protease inhibitors). For denaturing immunoprecipitations, cells were incubated at 95°C for 10 min to denature all proteins. Following lysis, supernatant fluids were collected; 10% was used to examine protein expression and labeled whole-cell lysates (WCL). The remaining 90% was used for immunoprecipitations where the lysates were incubated with 1 μg of antibody for 1 h at 4°C. Washed protein A/G agarose beads (Santa Cruz) were added to the samples, which were then incubated overnight at 4°C. Beads were washed four times with NP-40 lysis buffer. Immunoprecipitations of KR4 cells were performed in a similar manner with IRF7, SUMO-1, or isotype control antibodies.
Protein turnover.
293T cells were grown in 6-well plates and transfected using PEI. Cells were treated with cycloheximide (75 μg/ml; Sigma), MG132 (50 μM; Sigma), or dimethyl sulfoxide (DMSO) (Sigma) for 18 h prior to harvesting. Forty-eight hours after transfection, WCLs were harvested.
Cell compartmentalization.
293T cells were grown and transfected in 100-mm dishes. Forty-eight hours after transfection, cells were harvested, and cell pellets were processed using the Qproteome cell compartment kit (Qiagen).
Immunofluorescence microscopy.
293T cells were grown and transfected on glass coverslips. Twenty-four hours after transfection, cells were fixed with 4% paraformaldehyde. Expression of GFP-IRF7 and that of DAPI were examined at ×100 magnification.
Western blot analysis.
WCLs and immunoprecipitants were denatured in sodium dodecyl sulfate (SDS) (Sigma) loading buffer and boiled for 5 min. The samples were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride (PVDF) membranes (GE). Membranes were blocked with 5% milk in Tris-buffered saline–Tween 20 (TBST) and incubated overnight at 4°C with primary antibodies. The membranes were washed and incubated with appropriate horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. Membranes were washed again, and bands were visualized with enhanced chemiluminescence reagent from GE.
Collecting chromatin fractions.
293T cells were grown and transfected in 100-mm dishes. Cells were harvested 48 h after transfection, and WCLs and chromatin fractions were collected. As described previously (15), cells were washed and resuspended in 200 μl of CSK buffer (10 mM PIPES, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 1 mM EGTA, 1 mM DTT, 0.1 mM ATP, 0.1% Triton X-100, and complete protease inhibitor), and pellets were collected following centrifugation at 4,000 rpm for 5 min. Pellets were washed twice with 1 ml CSK buffer, and insoluble pellets were resuspended in 50 μl of 4× SDS sample buffer and boiled for 10 min.
Mutagenesis.
GFP-IRF7 k452R was constructed with wild-type GFP-IRF7 and use of a site-directed mutagenesis kit (Stratagene). Using a forward primer [K452R(F)] (5′-GAG CCT GGT CCT GGT GCG GCT GGA ACC CTG GCT G-3′) and reverse primer [K452R(R)] (5′ CAG CCA GGG TTC CAG CCG CAC CAG GAC CAG GCT C-3′), PCR was performed. DNA was denatured at 95°C for 30 s, followed by 20 cycles of 95°C for 30 s, 55°C for 60 s, and 68°C for 9 min. A final elongation step was performed at 68°C for 10 min. The PCR product was subjected to DpnI (Stratagene) digestion, transformed into XL-1 supercompetent cells (Stratagene), and grown on LB-Kan plates. Colonies were selected and sequenced to verify the mutation of K452. Protein expression was verified by Western blot analyses.
Semiquantitative RT-PCR.
293T cells were grown and transfected in 100-mm dishes. Cells were collected, and RNA was harvested 24 h after transfection using the Qiagen RNeasy Plus kit. Total RNA was reverse transcribed using the ABI Prism RT kit. With the use of the Promega GoTaq PCR kit and primers for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (5′-AGGTGAAGGTCGGAGTCAACG-3′ and 5′-AGGGGTCATTGATGGCAACA-3′) and IFN-β (5′-GATTCATCTAGCACTGGCTGG-3′ and 5′-CTTCAGGTAATGCAGAATCC-3′), the cDNA was amplified by PCR. The cDNA was denatured at 95°C for 30 s, followed by 30 cycles of 95°C for 30 s, 55°C for 60 s, and 68°C for 60 s. PCR products were fractionated on 2.0% agarose gels and stained with ethidium bromide, and bands were detected by UV. Relative RNA expression was determined by densitometry.
Antibodies.
Anti-FLAG (M2) and antiactin (AG-15) antibodies were purchased from Sigma. Anti-SUMO-1 (D-11), anti-hemagglutinin (HA) (F-7), anti-IRF7 (H-246 and G-8), anti-Myc (9E10), anti-histone H1 (AE-4), anti-EBV Ea-R p85 (6G7), and anti-GAPDH (FL-335) antibodies were purchased from Santa Cruz. Anti-LMP1 antibodies (CS 1-4) were purchased from Dako. Anti-poly(ADP-ribose) polymerase (PARP) antibodies were from Cell Signaling.
RESULTS
IRF7 is sumoylated endogenously.
We recently documented that LMP1 expression coincides with increased sumoylation of cellular proteins (2). In this new work, we investigate whether IRF7 could be specifically targeted for sumoylation by LMP1. Endogenous sumoylation of IRF7 was examined in EBV-transformed LCLs. Results from nondenaturing immunoprecipitations showed that IRF7 and a higher-molecular-mass form of IRF7 (∼70 kDa, the predicted size of sumoylated IRF7) were detected when immunoprecipitations were performed with SUMO-1- or IRF7-specific antibodies (Fig. 1A). While control antibodies did pull down unmodified IRF7, the higher-molecular-mass form of IRF7 was not detected, indicating that this band is specific for SUMO-1 and IRF7. Immunoprecipitations performed for all SUMO-1-associated proteins confirmed that higher-molecular-mass forms of IRF7 could be pulled down with SUMO-1-specific antibodies (Fig. 1B). These data suggest that IRF7 forms a complex with SUMO-1 and may be sumoylated during latent EBV infection.
Fig 1.

IRF7 is sumoylated endogenously in type III EBV LCLs. (A to C) KR4 cells were grown, and cell lysates were collected. (A) Nondenaturing immunoprecipitations were performed with SUMO-1, IRF7, or isotype control (anti-EBV EA-R p85) antibodies. Additional nondenaturing (B) or denaturing (C) immunoprecipitations were performed with SUMO-1 or isotype control antibodies. Western blot analyses were used to detect IRF7 expression. Duplicate experiments are shown. (D and E) BL41 EBV WT and BL41 EBV mut cells were grown, and cell lysates were collected. Denaturing immunoprecipitations were performed with 1 mg of cell lysates with SUMO-1, IRF7, or isotype control antibodies. Western blot analyses were used to detect IRF7 or SUMO-1 expression. Duplicate experiments are represented.
To elucidate whether IRF7 is covalently modified by SUMO-1, denaturing immunoprecipitations were used to collect all sumoylated proteins. Results revealed a 70-kDa form of IRF7 as well as higher-molecular-mass forms of IRF7 among the sumoylated proteins (Fig. 1C). These data demonstrate that IRF7 is covalently modified endogenously by SUMO-1 during type III EBV latency.
Because we hypothesized that LMP1 induces the sumoylation of IRF7, we examined the sumoylation of IRF7 in a set of paired cell lines (BL41 EBV WT, which expresses LMP1, and BL41 EBV mut [P3HR1], which lacks EBNA2, resulting in no detectable LMP1 expression) (5, 7, 28). IRF7 expression was not substantial in either cell line (data not shown); however, endogenous sumoylated IRF7 was consistently detected by immunoprecipitations (Fig. 1D and E). When all sumoylated proteins were collected, results showed that increased levels of sumoylated IRF7 were detected in BL41 EBV WT cells compared with levels in BL41 EBV mut cells (Fig. 1D). Immunoprecipitations performed with IRF7-specific antibodies and probed for SUMO-1 (Fig. 1E) supported these findings. Taken together, these data suggest that LMP1 induces the covalent modification of IRF7 by SUMO-1 during latent EBV infection.
LMP1 induces the sumoylation of IRF7.
Next, we further investigated if LMP1 expression could induce the sumoylation of IRF7. Denaturing immunoprecipitations to pull down all sumoylated proteins showed that sumoylated IRF7 was detected only when IRF7, SUMO-1, and LMP1 were coexpressed (Fig. 2A). Analyses of whole-cell lysates (WCLs) demonstrated that LMP1 mediated the sumoylation of IRF7 with endogenous SUMO-1 and with FLAG-SUMO-1 (Fig. 2A, lanes 4 and 6). However, only IRF7 sumoylated with FLAG-SUMO-1 was detected by immunoprecipitation (lane 6).
Fig 2.

LMP1 expression increases the sumoylation of IRF7. (A) 293T cells were transfected as indicated and cultured for 48 h. Cell lysates were collected, and denaturing immunoprecipitations were performed with FLAG antibodies. Western blot analyses of the immunoprecipitates and whole-cell lysates (WCL) (2% of total lysates) were performed for detection of IRF7, sumoylated IRF7, and LMP1. (B) 293T cells were transfected as indicated and cultured for 48 h. Cell lysates were collected, followed by denaturing immunoprecipitations with c-Myc antibodies. (C and D) 293T cells were transfected as indicated, and WCLs were collected 48 h posttransfection. Western blot analyses of the immunoprecipitates and WCLs were performed to detect IRF7, sumoylated IRF7, LMP1, SENP1, and SENP2.
To confirm that the observed LMP1-induced modification of IRF7 was in fact by SUMO-1, we used the SUMO protease SENP1, which differentially cleaves SUMO-1 over SUMO-2 (Fig. 2B) (37, 40). Following denaturing immunoprecipitations to collect all proteins modified by Myc-SUMO-1, the findings again showed that sumoylated IRF7 was detected only when LMP1 was coexpressed (lane 4). Expression of SENP1 significantly reduced the levels of sumoylated IRF7 (lane 5), demonstrating that the detected LMP1-induced modification of IRF7 was by SUMO-1. Additionally, as the expression of SENP1 increased, the levels of sumoylated IRF7 decreased (Fig. 2C, lanes 2, 3, and 4), indicating that IRF7 is desumoylated in a dose-dependent manner. The ability of additional SUMO proteases to desumoylate IRF7 was also tested. While SENP1 efficiently cleaves SUMO-1, SENP2 processes SUMO-1 less efficiently than SUMO-2 (35). Results showed that expression of both of these SENPs was able to significantly reduce LMP1-induced sumoylation of IRF7 (Fig. 2D, lanes 2 and 3). Together, these data demonstrate that LMP1 expression is sufficient to induce the sumoylation of IRF7 and that SENP1 and SENP2 can serve as SUMO proteases for IRF7.
LMP1-induced sumoylation of IRF7 is dependent on LMP1 CTAR3.
Because we previously showed that most of the protein sumoylation induced by LMP1 can be attributed to LMP1 CTAR3 (2), we examined if CTAR3 was necessary for LMP1-induced sumoylation of IRF7. We used wild-type LMP1 and LMP1dCTAR3 (LMP1Δ33bpr [2, 9]), which lacks LMP1 amino acids 245 to 307, is unable to interact with the SUMO-conjugating enzyme Ubc9, and exhibits decreased ability to induce the sumoylation of cellular proteins (2). Denaturing immunoprecipitations were performed to detect sumoylated IRF7 in the presence of LMP1 and LMP1dCTAR3. Sumoylated IRF7 was detected with expression of intact LMP1; however, significantly less sumoylated IRF7 was detected when LMP1dCTAR3 was expressed (Fig. 3A, lane 2 versus lane 3). Densitometry to quantitate levels of sumoylated IRF7 showed a reduction of sumoylated IRF7 (approximately 80%, as described previously [2]) when LMP1dCTAR3 was expressed in place of LMP1. The magnitude of the decrease is similar to the reduction of total protein sumoylation produced by LMP1dCTAR3 (2).
Fig 3.
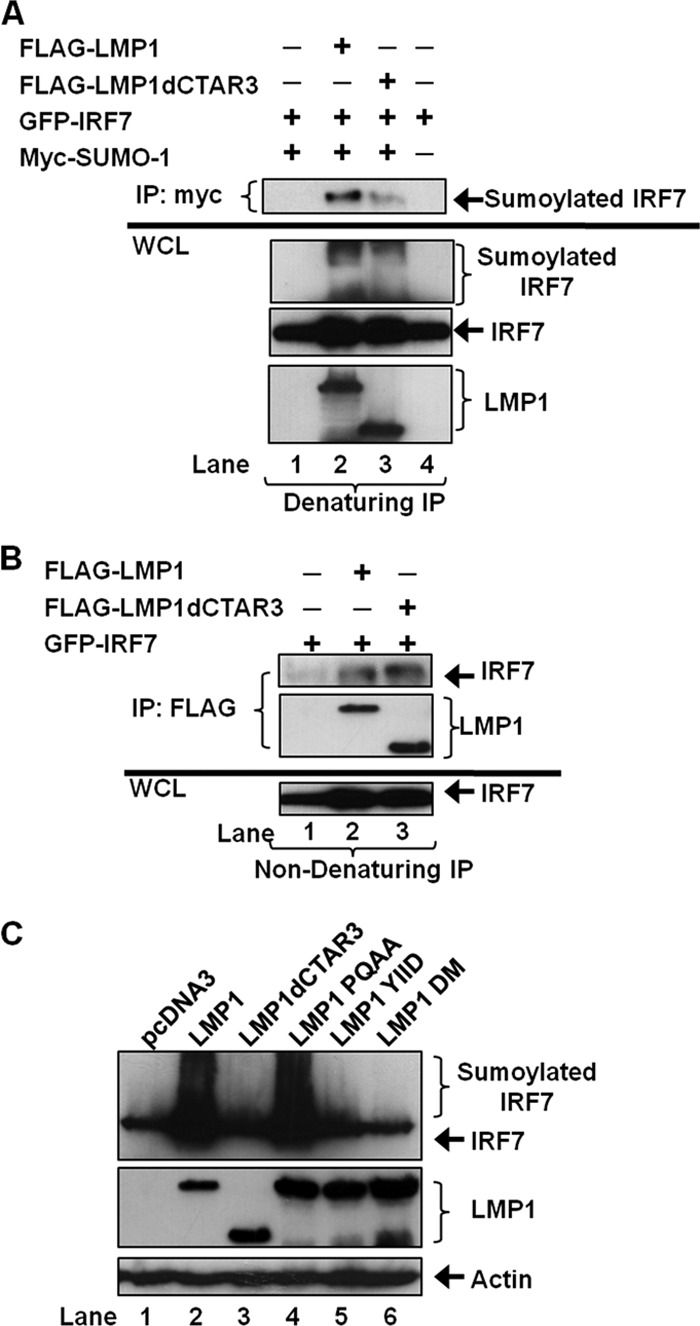
LMP1-induced sumoylation of IRF7 is dependent on CTAR3. (A) 293T cells were transfected as indicated and cultured for 48 h. Cell lysates were collected, and denaturing immunoprecipitations were performed with c-Myc antibodies. Western blot analyses of the immunoprecipitates and WCLs were used to detect IRF7, sumoylated IRF7, and LMP1. (B) 293T cells were transfected as indicated and cultured for 48 h. Cell lysates were collected, and nondenaturing immunoprecipitations were performed with FLAG antibodies, followed by Western blot analyses as described above. (C) 293T cells were transfected with GFP-IRF7, Myc–SUMO-1, and the indicated FLAG-LMP1 constructs. Cell lysates were collected 48 h posttransfection, and Western blot analyses were used to detect IRF7, sumoylated IRF7, and LMP1. Actin served as a loading control.
Because IRF7 has been shown to bind indirectly to LMP1 CTAR2 through its interaction with RIP and TRAF6 (14, 27, 39), we next verified whether LMP1dCTAR3 still interacted with IRF7. Nondenaturing immunoprecipitations (Fig. 3B) showed that IRF7 interacted with LMP1dCTAR3 and LMP1. Because LMP1 CTAR2 is intact in LMP1dCTAR3, these data support our previous observations that CTAR2 is important for the LMP1-IRF7 interaction (14, 27, 39) and demonstrate that LMP1dCTAR3 has a functional CTAR2 domain.
To investigate whether there is possible cooperation between LMP1 CTAR3 and CTAR2 in the regulation of IRF7, we analyzed the sumoylation of IRF7 using several LMP1 mutants. LMP1 PQAA has a nonfunctional CTAR1 domain (Fig. 3C, lane 4), LMP1 YIID has a nonfunctional CTAR2 domain (lane 5), and LMP1 DM has nonfunctional CTAR1 and CTAR2 domains (lane 6). We previously documented that these three mutants still interacted with the SUMO-conjugating enzyme Ubc9 (2), so we determined their effect on the sumoylation of IRF7 (Fig. 3C). The data showed that, as expected, LMP1 expression coincided with increased sumoylation of IRF7 and that LMP1dCTAR3 abrogated this effect. Additionally, LMP1 PQAA seemed unimpaired in its ability to sumoylate IRF7, whereas neither LMP1 YIDD nor LMP1 DM induced the sumoylation of IRF7. These results suggest that while CTAR3, the binding site for Ubc9, is necessary for LMP1-induced sumoylation of IRF7, CTAR2 also contributes to this process.
Sumoylated IRF7 accumulates in the nucleus.
Next, we explored the effect of sumoylation on IRF7 function. We have shown that LMP1-mediated activation of IRF7 coincides with the translocation of IRF7 to the nucleus (14, 27, 28, 42, 43), so we investigated the localization of sumoylated IRF7. Findings, both by cell fractionation and by microscopy, showed that LMP1 expression increased the nuclear accumulation of IRF7 (Fig. 4A and B). Deletion of CTAR3 did not affect this response, confirming our findings that LMP1 CTAR2 induces the nuclear accumulation of IRF7 (27, 42). Sumoylated IRF7 also localized to the nucleus (Fig. 4A), and expression of LMP1dCTAR3 significantly reduced amounts of modified IRF7 found in the nucleus. Thus, our data demonstrate that CTAR3 contributes to but is not alone required for the nuclear accumulation and retention of IRF7. Additionally, these data demonstrate that sumoylated IRF7 localizes to the nucleus.
Fig 4.

LMP1-induced sumoylated IRF7 localizes to the nucleus. (A) 293T cells were transfected with FLAG-LMP1 or FLAG-LMP1dCTAR3 along with GFP-IRF7, Myc–SUMO-1, or the vector control and cultured for 48 h. Cytoplasmic (CE) and nuclear (NE) extracts were collected, and Western blot analyses were performed for detection of IRF7, sumoylated IRF7, and LMP1. PARP and GAPDH were used as loading controls. (B) 293T cells were transfected with FLAG-LMP1 or FLAG-LMP1dCTAR3 along with GFP-IRF7 or the vector control and cultured for 24 h. Cells were fixed and examined by immunofluorescence microscopy (magnification, ×100).
Sumoylation promotes the stability of IRF7.
Western blot analyses consistently showed increased amounts of total IRF7 when it was sumoylated compared with nonsumoylated IRF7. Because sumoylation and ubiquitination both occur at lysine residues and sumoylation has been shown to protect specific proteins from proteasomal degradation (16, 19), we investigated if LMP1-induced sumoylation of IRF7 increased its stability. 293T cells were transfected and treated with DMSO, cycloheximide (CHX), or the proteasome inhibitor MG132 for 18 h before harvesting, and the relative expression of IRF7 was determined (Fig. 5). Western blot analyses showed that CHX treatment resulted in a significant loss of IRF7 expression (Fig. 5, lane 2; approximately a 75% decrease) while MG132 treatment greatly increased IRF7 levels (lane 3; approximate a 250% increase). These data suggest that the turnover of IRF7 is readily detectable (approximate half-life of 9 h) and that IRF7 is ubiquitinated and undergoes proteasome-mediated degradation.
Fig 5.

LMP1-induced sumoylation of IRF7 inhibits IRF7 turnover. 293T cells were transfected as indicated, and 18 h before harvesting, cells were treated with DMSO, cycloheximide (CHX) (75 μg/ml), or MG132 (50 μM). Cell lysates were harvested 48 h posttransfection and immunoblotted to determine IRF7 and LMP1 levels. Actin was used as a loading control. Densitometry was used to determine relative protein levels (normalized to actin). Data are shown as means ± SD.
Compared with expression of the vector control, LMP1 expression coincided with a smaller decrease in IRF7 expression following CHX treatment (Lane 8; approximately a 30% decrease) and a lesser increase in IRF7 levels following MG132 treatment (lane 9). Coexpression of SUMO-1 with LMP1 abrogated the loss of IRF7 expression produced by CHX. Instead, there was an increase in IRF7 expression levels (lane 11; approximately 50%), which suggests that sumoylation stabilizes IRF7 expression. Deletion of CTAR3 yielded results similar to those observed in the absence of LMP1 expression. CHX treatment resulted in the loss of IRF7 expression, and MG132 treatment increased IRF7 expression regardless of whether SUMO-1 was overexpressed.
Together, these data demonstrate that sumoylation of IRF7 promotes the stability of IRF7, decreasing its turnover rate. Additionally, the findings suggest that IRF7 is targeted for proteasomal degradation. Finally, sumoylation decreases its proteasomal degradation, which suggests that there is competition between the ubiquitination and sumoylation of IRF7.
LMP1-induced IRF7 sumoylation limits its transcriptional activity.
Next we investigated effects of sumoylation on the transcriptional activity of IRF7. IRF7 binds to and activates ISREs (14, 27, 42, 44, 45), and we studied the activation of ISRE-luciferase constructs when IRF7 was sumoylated. Luciferase and β-gal assays were performed 24 h after transfection. Luciferase expression was normalized to β-gal expression, and fold changes in promoter activity were determined. LMP1 expression significantly (P < 0.05) increased the ability of IRF7 to bind to and activate ISREs (Fig. 6A), confirming our previous data (42). Deletion of CTAR3 significantly (P < 0.05) increased the transcriptional activity of IRF7 compared to that when wild-type LMP1 was expressed. When SUMO-1 was coexpressed with LMP1 to increase the sumoylation of IRF7, a significant (P < 0.05) decrease in IRF7 transcriptional activity was observed. However, no effects on activity were observed when SUMO-1 was coexpressed with LMP1dCTAR3.
Fig 6.
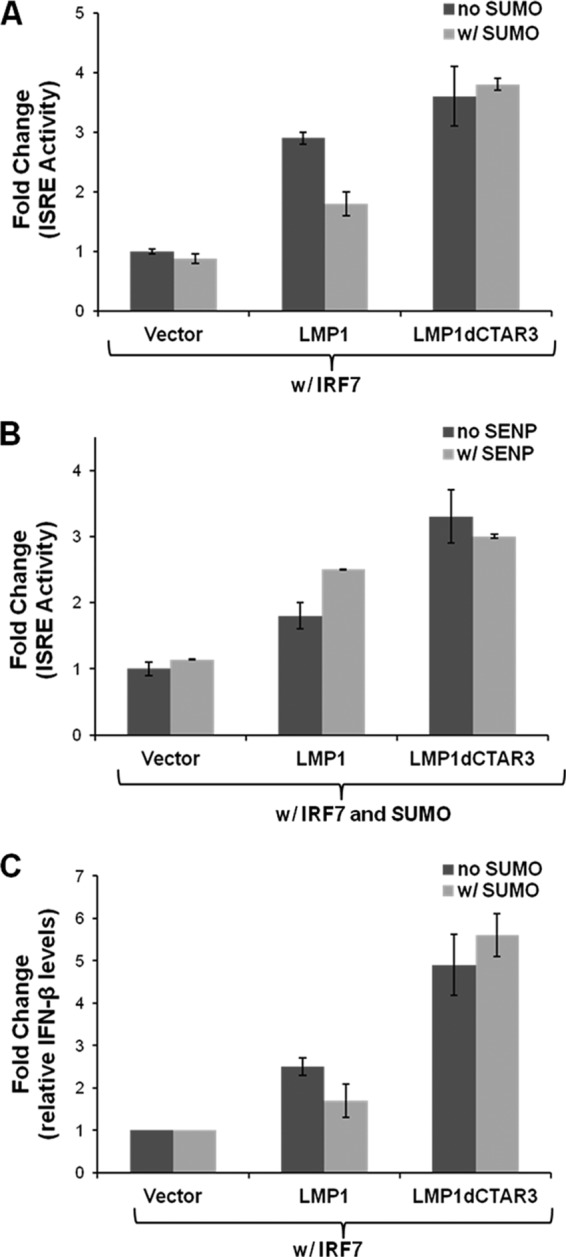
LMP1-induced sumoylation of IRF7 limits its transcriptional activity. (A and B) 293T cells were transfected as indicated along with ISRE-luciferase and beta-galactosidase and assayed 24 h posttransfection. Firefly luciferase activity was normalized to beta-galactosidase expression, and fold changes in ISRE activity were calculated. Data are shown as means ± SD. (C) 293T cells were transfected as indicated, and total RNA was harvested 24 h posttransfection. RT-PCR was carried out to determine IFN-β and GAPDH mRNA levels, and fold changes in IFN-β expression (relative to GADPH expression) were determined. Data are shown as means ± SD.
To confirm that the changes in IRF7 transcriptional activity were due to effects of SUMO-1, we used the SUMO protease SENP1 in parallel experiments (Fig. 6B). Confirming our earlier data, we found that when SUMO-1 was overexpressed, LMP1dCTAR3 significantly (P < 0.05) increased the transcriptional activity of IRF7 compared with that with LMP1. Overexpression of SENP1 significantly (P < 0.05) increased the ability of LMP1 to induce the transcriptional activity of IRF7 but did not affect the activation of IRF7 by LMP1dCTAR3.
Additional support for the hypothesis that LMP1-induced sumoylation limits the activation of IRF7 was obtained by examining the levels of IFN-β mRNA in transiently transfected 293T cells. Relative IFN-β mRNA expression was normalized to relative GAPDH mRNA expression, and fold changes in IFN-β mRNA levels were calculated (Fig. 6C). The findings were similar to results of reporter assays in that LMP1 expression significantly (P < 0.05) increased IFN-β mRNA levels, and deletion of CTAR3 produced even higher levels of IFN-β mRNA. Overexpression of SUMO-1 significantly (P < 0.05) decreased the ability of LMP1 to induce IFN-β mRNA expression in the presence of IRF7 but had no such effect with LMP1 dCTAR3. Taken together, these data demonstrate that LMP1-induced sumoylation of IRF7 limits or modulates the transcriptional activity of IRF7. These findings suggest that CTAR3 may serve as a negative regulatory domain for LMP1-induced activation of IRF7.
LMP1-induced sumoylation of IRF7 inhibits its ability to associate with chromatin.
Because we observed that sumoylation limits the transcriptional activity of IRF7, we next investigated whether this regulation was due to the loss in the ability of sumoylated IRF7 to associate with chromatin. Chromatin fractions were collected from transfected cells (15), and fold changes in relative IRF7 expression in the fractions (normalized to total IRF7 expression) were determined. Results showed that LMP1 expression significantly (P < 0.05) increased the association of IRF7 with chromatin, but coexpression of SUMO-1, which increased the sumoylation of IRF7, significantly (P < 0.05) decreased the association of IRF7 with the chromatin fractions (Fig. 7A). The association of IRF7 with the chromatin fractions remained significantly (P < 0.05) increased when LMP1dCTAR3 was expressed, regardless of SUMO-1 overexpression. SENP1 expression abrogated the effects of SUMO on LMP1-induced IRF7 association with the chromatin (Fig. 7B), but SENP1 did not affect the association of IRF7 with the chromatin induced by LMP1dCTAR3. Together, these data suggest that LMP1 CTAR3-induced sumoylation of IRF7 inhibits its ability to interact with DNA.
Fig 7.

LMP1-induced sumoylation of IRF7 inhibits its association with chromatin. (A and B) 293T cells were transfected as indicated. WCLs and chromatin fractions were collected 48 h posttransfection (15), and Western blot analyses were used to detect IRF7 expression. Densitometry was performed, and IRF7 expression in the chromatin fractions was normalized to IRF7 expression in the WCLs. Fold changes in IRF7 chromatin association were determined. Data are shown as means ± SD.
LMP1 modifies IRF7 by SUMO-1 at lysine 452.
Finally, we investigated the residue of IRF7 modified by SUMO-1 with expression of LMP1. Analysis of the IRF7 sequence using the SUMOplot software program (Abgent) revealed three putative sumoylation sites: K452, which has a high probability of being sumoylated, and K179 and K45, which have low probabilities of being sumoylated. We mutated K452 to abolish the SUMO motif (ΨKxE → ΨRxE) and examined its ability to be sumoylated by LMP1 (Fig. 8). Denaturing immunoprecipitations were performed for all sumoylated proteins. Results revealed that while LMP1 could induce the sumoylation of wild-type IRF7, IRF7 K452R was not detected in the sumoylated proteins.
Fig 8.

LMP1-induced sumoylation of IRF7 occurs at IRF7 lysine 452. 293T cells were transfected as indicated. (A) Cell lysates were collected 48 h posttransfection. Denaturing immunoprecipitations were performed with c-Myc antibodies. Western blot analyses were performed on the immunoprecipitants and whole-cell lysates to determine IRF7, sumoylated IRF7, IRF7 K452R, sumoylated IRF7 K452R, and LMP1 expression. (B) Cytoplasmic (CE) and nuclear (NE) extracts were collected, and Western blot analyses were used to detect IRF7 K452R and LMP1. Histone H1 and GAPDH were used as loading controls. (C) Eighteen hours before harvesting, cells were treated with DMSO or CHX (75 μg/ml). Cell lysates were harvested 48 h posttransfection and immunoblotted to determine IRF7/IRF7 K452R and actin levels. Densitometry was used to determine relative protein levels, and the fold change in relative IRF7/IRF7 K452R expression was determined. (D) Reporter assays were performed as described for Fig. 6. (E) WCLs and chromatin fractions were collected, and fold changes in IRF7 chromatin association were determined as described for Fig. 7. Data are shown as the means ± SD.
To verify that LMP1-induced sumoylation of IRF7 limits its function, we examined the localization, stability, transcriptional activity, and chromatin association of the sumoylation-defective IRF7 K452R protein. As expected, LMP1 expression could still induce the nuclear accumulation of IRF7 K452R (Fig. 8B), demonstrating that sumoylation does not affect the localization of IRF7. Experiments with cells treated with cycloheximide or DMSO revealed that mutation of IRF7 K452 abrogated LMP1- and SUMO-induced stability of IRF7 (Fig. 8C), supporting our suggestion that LMP1-mediated sumoylation of IRF7 reduces the turnover of IRF7. Reporter assays confirmed that LMP1-mediated sumoylation of IRF7 limits its transcriptional activity (Fig. 8D). Specifically, while LMP1-mediated sumoylation of wild-type IRF7 significantly (P < 0.05) limited the ability of IRF7 to activate ISREs, it had no effect on the SUMO-deficient IRF7 K452R. The data showed that in addition to abrogating the repressive effect on the transcriptional activity, mutation of IRF7 K452 also reversed the inhibitory effect that sumoylation had on LMP1-induced association of IRF7 with chromatin (Fig. 8E). LMP1 expression increased the association of both wild-type IRF7 and IRF7 K452R with the chromatin, confirming our earlier findings. However, while LMP1-induced sumoylation inhibited the association of wild-type IRF7 with the chromatin, it had no effect on IRF7 K452R. Taken together, these data suggest that LMP1 induces the sumoylation of IRF7 at K452, which inhibits IRF7 turnover and limits the ability of IRF7 to bind to DNA and to act as a transcription factor during latent EBV infection.
DISCUSSION
We recently documented that LMP1 interacts with the SUMO-conjugating enzyme Ubc9 and induces the sumoylation of cellular proteins (2). In addition, we have shown that LMP1 can activate IRF7 by both phosphorylation and ubiquitination (14, 27, 42). We now identify IRF7 as a primary target for LMP1-induced sumoylation. Reports on the sumoylation of IRFs are limited, and to date, they all show that sumoylation of IRF family members represses their transcriptional activity (18, 20, 21, 26). Of the three reports of IRF7 sumoylation during RNA viral infections (6, 20, 23), two document that murine IRF7 is sumoylated at K405, which corresponds to human K452. These reports show that during vesicular stomatitis virus (VSV) and Ebola virus infections, IRF7 is sumoylated, resulting in the inhibition of a type I IFN response (20). The third report showed that IRF7 is sumoylated at K446 by TRIM28 during Sendai virus infection (23). We have documented that in addition to K452, K444 and K446 are critical for LMP1-mediated K63-linked ubiquitination of IRF7 (6, 20, 23). Accordingly, whether LMP1 could also sumoylate IRF7 at one of these residues and limit its activity was of considerable interest. Our data show that LMP1 can in fact induce the sumoylation of IRF7 at K452.
In addition to the overexpression studies, we documented by both denaturing and nondenaturing immunoprecipitations that IRF7 was sumoylated endogenously. Denaturing immunoprecipitations revealed that IRF7 is covalently modified by SUMO-1. Nondenaturing immunoprecipitations were performed in the presence of the SUMO protease inhibitor N-ethylmaleimide, which covalently modified the catalytic cysteine residue of the SUMO proteases. This modification enabled examination of both the sumoylation of IRF7 and the interaction of IRF7 with SUMO-1. The results suggested that IRF7 both interacts with and is modified by SUMO-1. A consensus SUMO-interacting motif has been identified (V/I/L-x-V/I/L-V/I/L or V/I/L-V/I/L-x-V/I/L, where x is any amino acid) (10, 38). Analysis of the IRF7 sequence revealed that it contains a SUMO-interacting motif between amino acids 448 and 451, adjacent to the residue covalently modified by SUMO-1 (K452). Because we previously reported that LMP1 dysregulates sumoylation processes within the cell, increasing overall levels of sumoylated proteins (2), it is possible that LMP1 has two mechanisms by which it negatively regulates the function of IRF7: (i) by the covalent modification of IRF7 by SUMO-1 and (ii) by increasing the interaction of IRF7 with SUMO-1. Here we have shown how the covalent modification of IRF7 by SUMO-1 affects its function.
LMP1-induced sumoylation of IRF7 was dependent on LMP1 CTAR3, and we have shown, for the first time, that there is cooperation between LMP1 CTAR2 and LMP1 CTAR3 in the regulation of a target protein, specifically IRF7. Previous studies have focused on the role of CTAR2 in inducing the phosphorylation, K63-linked ubiquitination, and A20-mediated deubiquitination of IRF7 (27, 30). Here, we identified a novel role for LMP1 CTAR3 in the regulation of IRF7, specifically in the sumoylation of IRF7, which is mediated by the binding of Ubc9 to CTAR3. Our findings show that while a functional CTAR2 domain is still required for CTAR3-induced sumoylation of IRF7, these findings could result from the loss of the LMP1-IRF7 interaction upon mutation of CTAR2. The importance of CTAR2 for the LMP1-IRF7 interaction is compounded by the observations that CTAR3 did not abrogate the LMP1-IRF7 interaction. Together, these findings expose a new area of IRF7 regulation that warrants further examination (i.e., cooperation or competition between CTAR2 and CTAR3 in the regulation of IRF7).
The expression of the cellular SUMO protease SENP1 (as well as SENP2 and SENP5; data not shown) could reverse the effects of LMP1 CTAR3-induced sumoylation of IRF7. SENP1 is involved in SUMO maturation, or the initial processing of immature SUMO to generate a C-terminal diglycine motif (37, 40). Additionally, SENP1 can deconjugate SUMO-1, SUMO-2, and SUMO-3 from the target protein, resulting in protein desumoylation. SENP1 can itself be covalently modified by SUMO-1 (1); however, the functional consequence of SENP1 sumoylation remains unknown. We found that in the presence of LMP1, sumoylated SENP1 could be detected by immunoprecipitation and Western blot analyses of whole-cell lysates. Whether LMP1-induced sumoylation of SENP1 has a role in LMP1 CTAR3-induced sumoylation of cellular proteins remains unexamined. However, it is possible that sumoylation of SENP1 by LMP1 is a second mechanism through which the viral oncoprotein hijacks the cellular SUMO machinery during latent EBV infection.
In addition to identifying the IRF7 residue modified by SUMO1 and the LMP1 requirements for sumoylation of IRF7, we also document the effects of sumoylation on IRF7 function. While sumoylation did not alter LMP1-induced nuclear translocation of IRF7, sumoylated IRF7 was detected in the nuclear fractions and not the cytoplasmic extracts, suggesting that sumoylation may act to retain IRF7 in the nucleus. Because the putative nuclear export signal of IRF7 is located within amino acids 416 to 467 (24) and we identified K452 as the primary site of sumoylation, LMP1-induced sumoylation of IRF7 possibly blocks the nuclear export of IRF7, thus helping retain it within the nucleus.
Regardless of its localization, our data show that turnover of IRF7 is inhibited by sumoylation. IRF7 is targeted by K48-linked ubiquitination for proteasome-mediated degradation (41), and both ubiquitination, K48-linked and K63-linked, and sumoylation occur at lysine residues, which results in competition between different posttranslational modifications. Our previous results suggest that LMP1 induces the K63-linked ubiquitination of IRF7 (14, 27, 30). Here our data suggest that LMP1 also targets IRF7 for K48-linked ubiquitination, which leads to its proteasomal degradation during latent EBV infection. We now show that sumoylation may compete with ubiquitination at K452 in regulating IRF7 function and expression. This competition would result in increased accumulation of IRF7 within the cell, which would add to the increased expression of IRF7 induced by LMP1 (42). Thus, increases in transcription of the irf7 gene along with decreases in protein degradation together result in the increased levels of IRF7 observed in latently infected cells.
Although sumoylation of IRF7 resulted in increased nuclear retention and stability of the protein, it had a negative effect on the ability of IRF7 to interact with the chromatin and activate promoters. While the activation of ISRE reporters and the association of IRF7 with the chromatin were significantly higher when LMP1 was coexpressed than with the vector control, these levels were even greater with expression of SENP1 or LMP1dCTAR3 due to the loss of IRF7 sumoylation. These findings confirm and explain the previous reports of the negative effect of sumoylation on IRF7 transcriptional activity (29, 30) and suggest that sumoylation of IRF7 inhibits its ability to interact with DNA, thus repressing its transcriptional activity.
In summary, our studies of the regulation of IRF7 by LMP1 document that LMP1, in addition to phosphorylating IRF7, can induce the following modifications of IRF7: (i) the K63-linked ubiquitination of IRF7 (27), resulting in its transcriptional activation, (ii) the A20-mediated deubiquitination of IRF7 (30), resulting in its transcriptional repression, and (iii) the sumoylation of IRF7, limiting its activity. Thus, LMP1 tightly modulates the induction of innate immune responses during latent EBV infection. We propose that during latent EBV infections there is a balance between the ubiquitination, deubiquitination, and sumoylation status of IRF7. This balance regulates both the activation and repression of IRF7 function and transcriptional activity. Therefore, if an innate immune response is involved, LMP1 can signal the K63-linked ubiquitination of IRF7, leading to the activation of ISREs. When the need for an immune response wanes, LMP1, acting through A20, induces the deubiquitination of IRF7, and through its interaction with Ubc9 via CTAR3, LMP1 can then induce the sumoylation of IRF7, limiting the activation of ISREs yet protecting IRF7 from degradation in case the need for an immune response arises. This delicate balance, between the activation and repression of IRF7, may be beneficial for the maintenance of viral latency, and disruption of this balance may amplify the oncogenic potential of LMP1. Therefore, deciphering mechanisms by which LMP1 both activates and represses the functions of IRF7 may help elucidate ways by which this viral oncoprotein may be manipulated to yield the required outcome.
ACKNOWLEDGMENTS
We thank Cary A. Moody for providing the PARP antibody.
This work was supported by the National Institutes of Health, grants CA19014-26 (to J.S.P.) and CA142132 (to G.L.B.). This research was also funded in part by supplemental funding for HIV-associated malignancy research to the UNC Lineberger Cancer Center and the UNC CFAR.
Footnotes
Published ahead of print 5 September 2012
REFERENCES
- 1. Bailey D, O'Hare P. 2004. Characterization of the localization and proteolytic activity of the SUMO-specific protease, SENP1. J. Biol. Chem. 279:692–703 [DOI] [PubMed] [Google Scholar]
- 2. Bentz GL, Whitehurst CB, Pagano JS. 2011. Epstein-Barr virus latent membrane protein 1 (LMP1) C-terminal-activating region 3 contributes to LMP1-mediated cellular migration via its interaction with Ubc9. J. Virol. 85:10144–10153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brennan P, Floettmann JE, Mehl A, Jones M, Rowe M. 2001. Mechanism of action of a novel latent membrane protein-1 dominant negative. J. Biol. Chem. 276:1195–1203 [DOI] [PubMed] [Google Scholar]
- 4. Calender A, et al. 1987. Epstein-Barr virus (EBV) induces expression of B-cell activation markers on in vitro infection of EBV-negative B-lymphoma cells. Proc. Natl. Acad. Sci. U. S. A. 84:8060–8064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Calender A, Cordier M, Billaud M, Lenoir GM. 1990. Modulation of cellular gene expression in B lymphoma cells following in vitro infection by Epstein-Barr virus (EBV). Int. J. Cancer 46:658–663 [DOI] [PubMed] [Google Scholar]
- 6. Chang TH, et al. 2009. Ebola Zaire virus blocks type I interferon production by exploiting the host SUMO modification machinery. PLoS Pathog. 5:e1000493 doi:10.1371/journal.ppat.1000493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cherney BW, Sgadari C, Kanegane C, Wang F, Tosato G. 1998. Expression of the Epstein-Barr virus protein LMP1 mediates tumor regression in vivo. Blood 91:2491–2500 [PubMed] [Google Scholar]
- 8. Farlik M, et al. 2012. Contribution of a TANK-binding kinase 1-interferon (IFN) regulatory factor 7 pathway to IFN-gamma-induced gene expression. Mol. Cell. Biol. 32:1032–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gires O, et al. 1999. Latent membrane protein 1 of Epstein-Barr virus interacts with JAK3 and activates STAT proteins. EMBO J. 18:3064–3073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hecker CM, Rabiller M, Haglund K, Bayer P, Dikic I. 2006. Specification of SUMO1- and SUMO2-interacting motifs. J. Biol. Chem. 281:16117–16127 [DOI] [PubMed] [Google Scholar]
- 11. Honda K, Takaoka A, Taniguchi T. 2006. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 25:349–360 [DOI] [PubMed] [Google Scholar]
- 12. Honda K, Taniguchi T. 2006. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 6:644–658 [DOI] [PubMed] [Google Scholar]
- 13. Honda K, et al. 2005. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434:772–777 [DOI] [PubMed] [Google Scholar]
- 14. Huye LE, Ning S, Kelliher M, Pagano JS. 2007. Interferon regulatory factor 7 is activated by a viral oncoprotein through RIP-dependent ubiquitination. Mol. Cell. Biol. 27:2910–2918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Izumi M, Yatagai F, Hanaoka F. 2001. Cell cycle-dependent proteolysis and phosphorylation of human Mcm10. J. Biol. Chem. 276:48526–48531 [DOI] [PubMed] [Google Scholar]
- 16. Kerscher O. 2007. SUMO junction—what's your function? New insights through SUMO-interacting motifs. EMBO Rep. 8:550–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kerscher O, Felberbaum R, Hochstrasser M. 2006. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu. Rev. Cell Dev. Biol. 22:159–180 [DOI] [PubMed] [Google Scholar]
- 18. Kim EJ, Park JS, Um SJ. 2008. Ubc9-mediated sumoylation leads to transcriptional repression of IRF-1. Biochem. Biophys. Res. Commun. 377:952–956 [DOI] [PubMed] [Google Scholar]
- 19. Kroetz MB. 2005. SUMO: a ubiquitin-like protein modifier. Yale J. Biol. Med. 78:197–201 [PMC free article] [PubMed] [Google Scholar]
- 20. Kubota T, et al. 2008. Virus infection triggers SUMOylation of IRF3 and IRF7, leading to the negative regulation of type I interferon gene expression. J. Biol. Chem. 283:25660–25670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lefort S, Soucy-Faulkner A, Grandvaux N, Flamand L. 2007. Binding of Kaposi's sarcoma-associated herpesvirus K-bZIP to interferon-responsive factor 3 elements modulates antiviral gene expression. J. Virol. 81:10950–10960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li HP, Chang YS. 2003. Epstein-Barr virus latent membrane protein 1: structure and functions. J. Biomed.Sci. 10:490–504 [DOI] [PubMed] [Google Scholar]
- 23. Liang Q, et al. 2011. Tripartite motif-containing protein 28 is a small ubiquitin-related modifier E3 ligase and negative regulator of IFN regulatory factor 7. J. Immunol. 187:4754–4763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lin R, Mamane Y, Hiscott J. 2000. Multiple regulatory domains control IRF-7 activity in response to virus infection. J. Biol. Chem. 275:34320–34327 [DOI] [PubMed] [Google Scholar]
- 25. Miller WE, Mosialos G, Kieff E, Raab-Traub N. 1997. Epstein-Barr virus LMP1 induction of the epidermal growth factor receptor is mediated through a TRAF signaling pathway distinct from NF-kappaB activation. J. Virol. 71:586–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nakagawa K, Yokosawa H. 2002. PIAS3 induces SUMO-1 modification and transcriptional repression of IRF-1. FEBS Lett. 530:204–208 [DOI] [PubMed] [Google Scholar]
- 27. Ning S, Campos AD, Darnay BG, Bentz GL, Pagano JS. 2008. TRAF6 and the three C-terminal lysine sites on IRF7 are required for its ubiquitination-mediated activation by the tumor necrosis factor receptor family member latent membrane protein 1. Mol. Cell. Biol. 28:6536–6546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ning S, Hahn AM, Huye LE, Pagano JS. 2003. Interferon regulatory factor 7 regulates expression of Epstein-Barr virus latent membrane protein 1: a regulatory circuit. J. Virol. 77:9359–9368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ning S, Huye LE, Pagano JS. 2005. Interferon regulatory factor 5 represses expression of the Epstein-Barr virus oncoprotein LMP1: braking of the IRF7/LMP1 regulatory circuit. J. Virol. 79:11671–11676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ning S, Pagano JS. 2010. The A20 deubiquitinase activity negatively regulates LMP1 activation of IRF7. J. Virol. 84:6130–6138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ning S, Pagano JS, Barber GN. IRF7: activation, regulation, modification and function. Genes Immun. 12:399–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pagano JS. 2009. EBV diseases, p 217–240 In Damania B, Pipas JM. (ed), DNA tumor viruses. Springer Science + Business Media, New York, NY [Google Scholar]
- 33. Pagano JS, et al. 2004. Infectious agents and cancer: criteria for a causal relation. Semin. Cancer Biol. 14:453–471 [DOI] [PubMed] [Google Scholar]
- 34. Paun A, Pitha PM. 2007. The IRF family, revisited. Biochimie 89:744–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Reverter D, Lima CD. 2004. A basis for SUMO protease specificity provided by analysis of human Senp2 and a Senp2-SUMO complex. Structure 12:1519–1531 [DOI] [PubMed] [Google Scholar]
- 36. Sakaguchi S, et al. 2003. Essential role of IRF-3 in lipopolysaccharide-induced interferon-beta gene expression and endotoxin shock. Biochem. Biophys. Res. Commun. 306:860–866 [DOI] [PubMed] [Google Scholar]
- 37. Shen LN, Dong C, Liu H, Naismith JH, Hay RT. 2006. The structure of SENP1-SUMO-2 complex suggests a structural basis for discrimination between SUMO paralogues during processing. Biochem. J. 397:279–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Song J, Zhang Z, Hu W, Chen Y. 2005. Small ubiquitin-like modifier (SUMO) recognition of a SUMO binding motif: a reversal of the bound orientation. J. Biol. Chem. 280:40122–40129 [DOI] [PubMed] [Google Scholar]
- 39. Song YJ, Izumi KM, Shinners NP, Gewurz BE, Kieff E. 2008. IRF7 activation by Epstein-Barr virus latent membrane protein 1 requires localization at activation sites and TRAF6, but not TRAF2 or TRAF3. Proc. Natl. Acad. Sci. U. S. A. 105:18448–18453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xu Z, Au SW. 2005. Mapping residues of SUMO precursors essential in differential maturation by SUMO-specific protease, SENP1. Biochem. J. 386:325–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yu Y, Wang SE, Hayward GS. 2005. The KSHV immediate-early transcription factor RTA encodes ubiquitin E3 ligase activity that targets IRF7 for proteosome-mediated degradation. Immunity 22:59–70 [DOI] [PubMed] [Google Scholar]
- 42. Zhang L, Pagano JS. 2000. Interferon regulatory factor 7 is induced by Epstein-Barr virus latent membrane protein 1. J. Virol. 74:1061–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang L, Pagano JS. 2001. Interferon regulatory factor 7 mediates activation of Tap-2 by Epstein-Barr virus latent membrane protein 1. J. Virol. 75:341–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang L, Pagano JS. 2001. Interferon regulatory factor 7: a key cellular mediator of LMP-1 in EBV latency and transformation. Semin. Cancer Biol. 11:445–453 [DOI] [PubMed] [Google Scholar]
- 45. Zhang L, Pagano JS. 1997. IRF-7, a new interferon regulatory factor associated with Epstein-Barr virus latency. Mol. Cell. Biol. 17:5748–5757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang L, Pagano JS. 2002. Structure and function of IRF-7. J. Interferon Cytokine Res. 22:95–101 [DOI] [PubMed] [Google Scholar]