

Abstract
Epstein-Barr virus (EBV) BGLF4 is a member of the conserved herpesvirus kinases that regulate multiple cellular and viral substrates and play an important role in the viral lytic cycles. BGLF4 has been found to phosphorylate several cellular and viral transcription factors, modulate their activities, and regulate downstream events. In this study, we identify an NF-κB coactivator, UXT, as a substrate of BGLF4. BGLF4 downregulates not only NF-κB transactivation in reporter assays in response to tumor necrosis factor alpha (TNF-α) and poly(I·C) stimulation, but also NF-κB-regulated cellular gene expression. Furthermore, BGLF4 attenuates NF-κB-mediated repression of the EBV lytic transactivators, Zta and Rta. In EBV-positive NA cells, knockdown of BGLF4 during lytic progression elevates NF-κB activity and downregulates the activity of the EBV oriLyt BHLF1 promoter, which is the first promoter activated upon lytic switch. We show that BGLF4 phosphorylates UXT at the Thr3 residue. This modification interferes with the interaction between UXT and NF-κB. The data also indicate that BGLF4 reduces the interaction between UXT and NF-κB and attenuates NF-κB enhanceosome activity. Upon infection with short hairpin RNA (shRNA) lentivirus to knock down UXT, a spontaneous lytic cycle was observed in NA cells, suggesting UXT is required for maintenance of EBV latency. Overexpression of wild-type, but not phosphorylation-deficient, UXT enhances the expression of lytic proteins both in control and UXT knockdown cells. Taking the data together, transcription involving UXT may also be important for EBV lytic protein expression, whereas BGLF4-mediated phosphorylation of UXT at Thr3 plays a critical role in promoting the lytic cycle.
INTRODUCTION
The outcome of viral infection is a battle between the host antiviral signaling pathways and the blocking effects of viral factors. Upon infection, multiple cellular transactivators are activated through signal transduction pathways to turn on the expression of various cellular genes that trigger the innate immune response and the expression of cytokines to prevent viral replication and spread. Therefore, viruses have evolved counteracting mechanisms to escape the host immune system and to modify cellular factors to enable virus replication. Nuclear factor κB (NF-κB) is an important cellular factor that can be activated by viral infection and plays important roles in many cellular events. NF-κB, a widely expressed transcription factor, regulates a series of genes involved in inflammation, immune responses, and cell survival, differentiation, and proliferation (12, 18). The NF-κB family is composed of various homo- or heterodimers, including 105/p50 (NF-κB1), p100/p52 (NF-κB2), p65 (RelA), RelB, and c-Rel. All members of the NF-κB family are characterized by the presence of a Rel homology domain involved in DNA binding and dimerization (42). In unstimulated cells, NF-κB is predominantly localized in the cytoplasm through binding to the inhibitory IκB proteins, which inhibit the nuclear translocation signal of NF-κB (19). NF-κB signaling can be activated by various stimuli, including growth factors and cytokines that activate the IκB kinase complex (IKK). Once activated, IKK phosphorylates IκB proteins, which results in polyubiquitination and degradation of IκBs by the 26S proteasome (8, 17). As a result, the NF-κB complex translocates into the nucleus and binds to NF-κB-responsive elements to modulate the expression of target genes (11, 29).
NF-κB activation is one of the hallmarks of viral infection (37). Activation of NF-κB, inducing inflammatory responses during viral infection, is believed to protect hosts from viral pathogens. Hence, many viruses counteract NF-κB activity with various strategies to evade the immune responses. Furthermore, viruses may modulate NF-κB signaling to enhance viral replication and prevent virus-induced apoptosis (20, 48).
Epstein-Barr virus (EBV) is a human gammaherpesvirus that infects mainly B lymphocytes and epithelial cells and persists in the majority of the world's population (1). As a successful virus, EBV has evolved several mechanisms to antagonize the host immune response (58). Like other herpesviruses, EBV has two distinct life cycles, and the switch between latency and the lytic cycle is well tuned. Following initial infection, EBV persists in host cells preferentially in a latent state, with no virus production, and only a few viral proteins are expressed. EBV displays various forms of latency in host cells and expresses distinct patterns of latent proteins, which contribute to the latent replication and modulate cellular signaling pathways and the host immune system to maintain EBV latency. For instance, the EBV nuclear antigen 1 (EBNA1) protein binds to viral DNA and allows the EBV genome to be maintained as a circular DNA episome in the host cells. LMP1, which is critical for B cell transformation, functions as a constitutively activated member of the tumor necrosis factor receptor (TNFR) superfamily and activates several signaling pathways in a ligand-independent manner (58). Following certain stimulations, such as chemical treatment or IgG cross-linking, the latent EBV is reactivated into the lytic cycle and replicates productively. The immediate-early transactivators, Zta and Rta, are expressed first and transactivate the expression of early and late genes. Early proteins usually take part in or help viral DNA replication, and most late proteins are structural proteins that form viral particles. To ensure successful lytic reactivation, EBV modulates various cellular events to facilitate virus production. Some lytic proteins modulate cell cycle progression, such as Rta, which induces G1 arrest primarily originating at the transcriptional level (6). Some suppress the immune response to facilitate the progression of the viral lytic cycle. For example, the EBV transactivator Zta inhibits the expression of interferon regulatory factor 7 (IRF7) to block the antiviral response (16) and disrupts the gamma interferon (IFN-γ) signaling pathway to prevent IFN-γ-induced HLA class II surface expression (39).
In addition, EBV BGLF4 kinase also plays an important role in facilitating virus replication. BGLF4 is the only known proline-dependent Ser/Thr protein kinase of EBV and is expressed at the early stage of the lytic cycle (5, 56). It is known that BGLF4 phosphorylates the EBV latent transactivator EBNA2 to suppress LMP1 expression, contributing to the promotion of lytic progression (59). Moreover, BGLF4 also phosphorylates the EBV processivity factor BMRF1 to downregulate its transactivation and synergistically enhances the transactivation of the EBV BHLF1 lytic promoter by Zta to facilitate lytic replication (57). BGLF4 also modifies cellular proteins to facilitate virus replication. Phosphorylation of MCM4 in the MCM complex by BGLF4 may block cellular chromosomal DNA replication for efficient virus replication in lytic cells (25). BGLF4 induces premature chromosome condensation to provide more space and resources for viral DNA replication through activating condensin and topoisomerase II (26). BGLF4 phosphorylates lamin A/C to promote the reorganization of nuclear lamina and facilitate the nuclear egress of the nucleocapsid (27). On the other hand, BGLF4 phosphorylates IRF3 and suppresses the IRF3 signaling pathway to inhibit the host innate immune response (55). Knockdown of BGLF4 attenuates viral DNA replication and inhibits the nuclear egress of the nuclear capsid of EBV (10, 40), indicating that BGLF4 is crucial for EBV lytic replication. Recent studies of protein arrays identified more cellular and viral factors that can be phosphorylated by BGLF4, such as the EBV single-stranded DNA binding protein BALF2 (61) and cellular proteins involved in the regulation of the cell cycle and the DNA damage response (30), suggesting that BGLF4 may regulate multiple signaling pathways and contribute to effective lytic progression.
To identify the cellular events in which BGLF4 may participate, we searched for cellular proteins interacting with BGLF4 using a yeast two-hybrid system. One of the candidates is a protein named ubiquitously expressed transcript (UXT). UXT, also named androgen receptor trapped clone 27 (ART-27), is an 18-kDa protein predominantly distributed in the nucleus (35). Computer modeling predicts that UXT is an α-class prefoldin (PFD) family protein (13). Most members of the PFD family are small proteins with molecular masses around 14 to 23 kDa and composed of coiled-coil structures, and they function as molecular chaperones in protein folding (50, 54). Velocity gradient sedimentation analysis of nuclear extracts suggests that native UXT is part of a multiprotein complex. The components of the complex, identified by mass spectrometric analysis, include subunits shared by RNA polymerases I, II, and III and other factors involved in transcription, suggesting that UXT is a component of a large transcription complex and is associated with proteins involved in the regulation of transcription (13, 31, 32). In addition, UXT has been identified as a coactivator that binds to the N terminus of the androgen receptor to regulate androgen receptor-responsive genes (35, 52). UXT is also a coactivator of the NF-κB enhanceosome and is important for the nuclear function of NF-κB (51).
In this study, we show that BGLF4 interacts with and phosphorylates UXT. Because UXT is important for NF-κB nuclear function, we investigated whether BGLF4 modulates NF-κB transactivation to affect cellular and viral gene expression. The results indicate that BGLF4 decreases the interaction between UXT and NF-κB through phosphorylation of UXT, leading to attenuated NF-κB transactivation.
MATERIALS AND METHODS
Yeast two-hybrid screening.
pJTW2, which encodes GAL4DBD-BGLF4(1-293), and pJTW3, which encodes GAL4DBD-BGLF4(201-429), were constructed on the basis of pGBDUC1 and used as the bait to screen a human HeLa cell cDNA library as described previously (24). Briefly, the prey HeLa cell cDNA library and the bait were cotransformed into Saccharomyces cerevisiae strain PJ69-4A. Positive clones were selected on plates of synthetic complete medium lacking Ura, Leu, and Ade and confirmed for interaction on plates of synthetic complete medium lacking Ura, Leu, and His.
Plasmid construction.
pYPW17 and pYPW20 are plasmids expressing wild-type BGLF4 kinase and the K102I kinase-dead mutant, respectively (56). pcDNA3-HA-p65 was kindly provided by W. W. Lin (Graduate Institute of Pharmacology, National Taiwan University College of Medicine). The cDNA sequence of UXT (GenBank accession no. AF092737) was isolated from the human HeLa cDNA library constructed in pGADGH/X and cloned into the BglII/XhoI site of hemagglutinin (HA)-containing pSG5 (pHY25) to generate pYSW1 (pSG5-HA-XUT). To generate pLSC5 (pcDNA3-HA-UXT) containing two HA tags at the N terminus, the HA-UXT fragment was isolated from pYSW1 and then cloned into the EcoRI/XhoI sites of the pcDNA3-HA vector, which contains an HA tag inserted into the KpnI/BamHI sites of the pcDNA3 vector (Invitrogen). For pLSC6 (pGEX-4T-1-UXT) construction, the UXT cDNA fragment was generated by PCR amplification and cloned into the BamHI/EcoRI sites of the pGEX-4T-1 vector (GE Healthcare). pLSC7 [pGEX-4T-1-UXT(T3V)], pLSC8 [pcDNA3-HA-UXT(T3V)], and pLSC10 [pcDNA3-HA-UXT(T3E)] were generated by a single-primer-based site-directed mutagenesis strategy (34). The primer for UXT(T3V) was 5′-GAGCCCATCATGGCGGCGCCCCCTAAGCGGCG-3′, and the primer for UXT(T3E) was 5′-ATTACGCTGGCATGGCGGAGCCCCCTAAGCGGCGGG-3′. Underlining indicates the mutation site of the UXT cDNA sequence. The reporter plasmid pNF-κB-Luc contains five copies of the NF-κB enhancer elements in front of a TATA box and a firefly luciferase gene (Stratagene). pBHLF1-Luc was generated as described previously (57). The pGL2-basic (Promega) Zp luciferase reporter containing nucleotides −221 to +12 relative to the transcription initiation site of the Zta promoter was kindly provided by C. H. Tsai (7). The Zta expression plasmid pRC/CMV-Zta was described previously (33). The Rta expression plasmid RTS15 was a gift from Diane Hayward (48a). The pPRDII2-Luc reporter plasmid was kindly provided by K. Fitzgerald (9).
Cell culture, transfection, double-stranded RNA stimulation, and phosphatase treatment.
The 293-TLR3 stable cell line was a gift from K. A. Fitzgerald (9). NA is a recombinant Akata EBV-converted NPC-TW01 cell line (3). 293T, 293-TLR3, and NA cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 8% fetal calf serum and cultured in the presence of penicillin (100 U/ml) and streptomycin (100 μg/ml) at 37°C with 5% CO2. Plasmid DNA was transfected into 293T or 293-TLR3 cells using the calcium phosphate-N,N-bis(2-hydroxyethyl)-2-aminoethanesulfonic acid method (4) or transfected into NA cells with Lipofectamine 2000 (Invitrogen). The BGLF4-targeted small interfering RNAs (siRNAs), siBGLF4-1 (5′-CCCUCUAUGUAAAGCUGCCGGAGAA-3′) and siBGLF4-2 (5′-UGGGUAGGCUGGUCCUGACUGAUUA-3′), and a control siRNA (siCtrl; 5′-CCCGUAUAAAUGUCGGGCCACUGAA-3′) with comparable GC content were purchased from Invitrogen and transfected into NA cells using Lipofectamine 2000. For double-stranded RNA treatment, 293-TLR3 cells were treated with poly(I·C), with a final concentration of 25 μg/ml in the complete medium.
For phosphatase treatment, 15 μg of protein lysate was incubated with phosphatase buffer in the presence or absence of 10 U calf intestinal alkaline phosphatase (CIP) (New England BioLabs) at 37°C for 1.5 h.
Immunoblotting.
Cells were washed once with ice-cold phosphate-buffered saline (PBS) and then resuspended in lysis buffer (50 mM Tris/HCl, pH 7.5, 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM NaF, and 1× protease inhibitor cocktail [Roche]). Protein concentrations were determined by the Bradford protein assay. Immunoblotting was performed as described previously (5). The antibodies used were BGLF4 monoclonal antibodies (MAbs) 2224 and 2616 (56), Rta MAb 467 (23), p65 antibody (Santa Cruz), HA MAb (Covance), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Biodesign), and α-tubulin (Calbiochem). UXT antibody was kindly provided by W. Krek (ETH-Hönggerberg, Institute of Cell Biology, Switzerland) (13).
Coimmunoprecipitation.
After transfection, 293T cells were lysed in NP-40 lysis buffer (1% NP-40, 50 mM Tris, pH 8.0, 150 mM NaCl, 2 mM EDTA, and 1 mM Na3VO4), and 300 μg of cell lysates was precleared with 100 μl of 20% protein A-Sepharose beads (Pharmacia) at 4°C for 1 h and incubated with specific antibodies at 4°C for 16 h. The mixtures were incubated with 125 μl of 20% protein A-Sepharose beads at 4°C for 2 h. The beads were washed three times with NP-40 lysis buffer and resuspended in 2× sodium dodecyl sulfate (SDS) sample buffer. The immunoprecipitated complexes were separated by SDS-PAGE and analyzed by immunoblotting.
Luciferase assay.
293T or 293-TLR3 cells were seeded into 12-well plates (1 × 105 cells/well) and transfected with the indicated reporter and effecter plasmids. A Renilla luciferase (Rluc) reporter plasmid (pCMV-Rluc) or pEGFP-C1 (BD Bioscience) expression plasmid was cotransfected as a control for transfection efficiency. At 48 h posttransfection, cells were harvested and assayed for firefly luciferase and Renilla luciferase activities using the Dual-Glo Luciferase Assay system (Promega) or for firefly luciferase activity and green fluorescent protein (GFP) intensity using the Bright-Glo Luciferase Assay system (Promega). Relative promoter activities were determined by the firefly luciferase activity normalized to Renilla luciferase activity or GFP intensity in each reaction. For NF-κB transactivation assays, cells were stimulated at 48 h posttransfection with tumor necrosis factor alpha (TNF-α) (10 ng/ml) or poly(I·C) (25 μg/ml) for the indicated time periods and harvested for luciferase assays.
RNA purification and quantitative real-time PCR.
293T cells were starved in DMEM containing 0.5% serum at 24 h posttransfection. After 24 h of starvation, the cells were stimulated for 90 min with TNF-α (10 ng/ml), and total cellular RNA was isolated with TRIzol (Invitrogen) according to the manufacturer's instructions. Reverse transcription of purified RNA was performed using random primers. The quantification of gene transcripts was detected by real-time PCR using SYBR green I dye (Invitrogen) and the iCycler iQ Detection System (Bio-Rad). The reverse-transcribed cDNAs were denatured at 95°C for 1 min and amplified with 40 cycles of denaturation at 95°C for 10 s and annealing at 56°C for 25 s, followed by extension at 72°C for 20 s and 80°C for 10 s. Expression values of target genes were normalized with the values of control GAPDH and analyzed using iQ5 Optical system software. The relative fold expression was the ratio of the expression value in each reaction to the value of vector-transfected cells. The primers used were as follows: interleukin 8 (IL-8), sense (AGGTGCAGTTTTGCCAAGGA) and antisense (TTTCTGTGTTGGCGCAGTGT); IκBα, sense (CTGAGCTCCGAGACTTTCGAGG) and antisense (CACGTGTGGCCATTGTAGTTGG); A20, sense (GCGTTCAGGACACAGACTTG) and antisense (GCAAAGCCCCGTTTCAACAA) (51); and GAPDH, sense (CATCATCCCTGCCTCTACTG) and antisense (GCCTGCTTCACCACCTTC).
Protein expression in a recombinant baculovirus system.
Baculoviruses expressing His-BGLF4 or His-K102I protein were generated with the Bac-to-Bac system (Invitrogen). Briefly, pFastbac-HTB-BGLF4 or pFastbac-HTB-K102I was transformed into DH10Bac competent cells to generate a recombinant bacmid, and the clone was isolated with blue/white selection. Recombinant bacmid DNA was confirmed by PCR and then transfected into Sf9 insect cells for the production of recombinant viruses. His-BGLF4 or His-K102I protein expressed from recombinant-virus-infected Sf9 cells was further purified using a BGLF4 2224 antibody affinity column (56).
Expression, purification, and thrombin cleavage of GST-UXT and GST-UXT(T3V).
Bacterially expressed UXT and UXT(T3V) proteins were generated from glutathione S-transferase (GST)-UXT and GST-UXT(T3V) proteins purified according to the Amersham handbook with slight modifications. Competent Escherichia coli BL21(DE3) bacteria were transformed with GST-UXT or GST-UXT(T3V) expression plasmid, and the bacteria were then induced in LB medium at exponential phase with 0.1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) and grown at 30°C for 3 h. The bacteria were lysed in cold PBS containing 1% Triton X-100 and incubated with lysozyme (1 mg/ml) once for 1 h. Then, the bacterial lysates were frozen at 80°C overnight. After being quickly thawed, the protein sample was harvested by sonication and incubated with glutathione-Sepharose 4B (Sigma) at 4°C overnight. After three washes with cold PBS containing 1% Triton X-100, the protein was eluted with glutathione elution buffer (50 mM Tris, pH 8.0, and 5 mM reduced glutathione). To avoid possible hindering effects of GST on the N terminus of UXT or UXT(T3V) protein, eluted GST-UXT or GST-UXT(T3V) was cleaved with thrombin (Sigma) at 22°C for 16 h (1 cleavage unit/100 μg protein). The cleavage reaction was stopped with 1 mM phenylmethylsulfonyl fluoride (PMSF), and proteins were stored at −80°C.
In vitro kinase assay.
Bacterially expressed UXT and UXT(T3V) proteins were used as the substrates for the in vitro kinase assay. Purified His-BGLF4 or His-K102I from baculovirus was incubated with 0.5 μg UXT or UXT(T3V) protein in the presence of 10 μCi [γ-32P]ATP and kinase buffer A (20 mM Tris HCl, pH 7.5, 1 mM EDTA, 1 mM dithiothreitol [DTT], 10 mM MgCl2, 0.2 mM Na3VO4, 100 μM ATP) at 37°C for 1 h (25). To stop the reaction, 2× SDS sample buffer was added, and the reaction mixtures were boiled for 5 min. After the proteins were resolved in 15% SDS-PAGE, the gels were dried, stained with Coomassie blue to detect the substrate input, and subjected to autoradiography or transferred onto Hybond-C Extra membranes (Amersham) and detected with anti-BGLF4 2616 antibody.
Preparation and infection of UXT shRNA (shUXT) lentivirus.
pCMV-dR891, pMD2.G, pLKO-shLuc (encoding luciferase shRNA), and pLKO-shUXT expression plasmids were from the National RNAi Core Facility of Academia Sinica. The knockdown efficiencies of pLKO-shUXT1 (clone identifier [ID], TRCN0000154852; GCTGTAACTTCTTCGTTGACA), pLKO-shUXT2 (clone ID, TRCN0000151893; CCTTCAACTGAGAAATGTCAT), and pLKO-shUXT3 (clone ID, TRCN0000155295; GCTAAGCACTCGGAGTTATAT) were examined, and pLKO-shUXT1 was chosen. The preparation and infection of shUXT expression lentivirus were modified from the standard protocol suggested by the RNAi Core. Briefly, 293T cells were seeded in a 10-cm plate (5 × 106 cells/plate) and then transfected with 9 μg of pCMV-dR8.91, 1 μg of pMD2.G, and 10 μg of pLKO-shLuc or pLKO-shUXT using Lipofectamine 2000 (Invitrogen) for the lentivirus package. The supernatant containing pLKO-shLuc or pLKO-shUXT virus was harvested at 40 h and 64 h posttransfection and filtered through the 0.22-μm filter. For lentivirus infection, NA cells (3 × 106 cells) were infected with pLKO-Luc or pLKO-shUXT lentivirus at a multiplicity of infection (MOI) of 1 and selected in DMEM containing 2 μg/ml puromycin at 24 h postinfection.
RESULTS
BGLF4 interacts with UXT.
To identify potential BGLF4-interacting cellular proteins, a yeast two-hybrid screening was performed with a HeLa cDNA library, using GAL4DBD-BGLF4(1-293) or GAL4DBD-BGLF4(201-429) as the bait. One clone, which contained a cDNA copy of UXT, was found to interact with GAL4DBD-BGLF4(1-293). To confirm the interaction between BGLF4 and UXT, BGLF4, or the kinase-dead mutant (K102I), and HA-UXT expression plasmids were transfected into 293T cells for coimmunoprecipitation (Fig. 1). The size difference between BGLF4 and K102I is caused by the autophosphorylation of BGLF4 itself. Without kinase activity, the band of K102I is lower than that of wild-type BGLF4. Using anti-BGLF4 2224 MAb, HA-UXT could be coimmunoprecipitated with BGLF4 and also K102I (Fig. 1B). Reciprocally, BGLF4 or K102I was coimmunoprecipitated with HA-UXT by anti-HA antibody. Although a very light signal was seen in the lane transfected with only K102I, the signal of K102I coimmunoprecipitated with HA-UXT was much stronger, indicating that K102I could also bind to HA-UXT (Fig. 1C). The results show that HA-UXT interacts with BGLF4 and that the interaction is independent of BGLF4 kinase activity.
Fig 1.
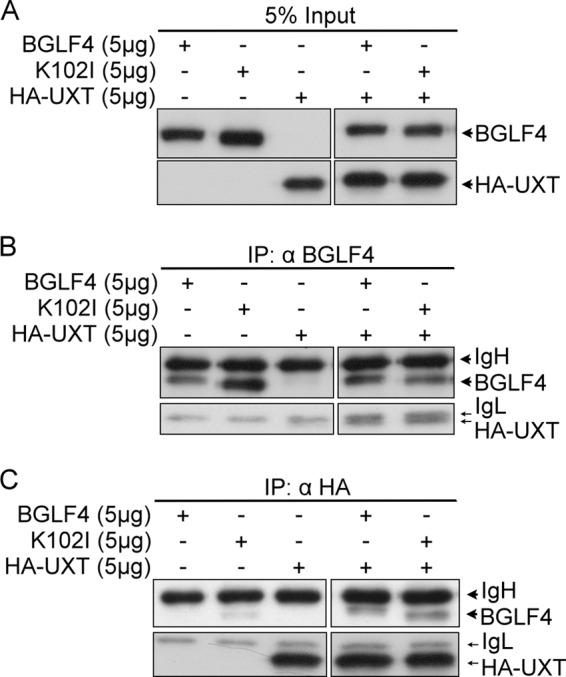
BGLF4 interacts with UXT. 293T cells were transiently transfected with BGLF4, K102I, or HA-UXT expression plasmid. At 48 h posttransfection, 300 μg of cell lysates was precleared with protein A-Sepharose beads. (A) The input represented 5% of cell lysates used for each immunoprecipitation (IP). (B and C) The precleared lysates were immunoprecipitated with BGLF4 (α-BGLF4 2224) (B) or HA-specific antibody (C). The immunocomplexes were resolved by SDS-PAGE and immunoblotted with BGLF4 (α-BGLF4 2616) and HA-specific antibodies.
BGLF4 downregulates NF-κB transactivation.
UXT has been reported to interact with several transcription factors participating in transcriptional regulation (35, 36, 51). Among these interactions, NF-κB was found to downregulate EBV reactivation (2, 43) and contribute to multiple antiviral effects. Therefore, we used reporter assays to determine whether BGLF4 affects the NF-κB activity through its interaction with UXT. To this end, BGLF4 or K102I expression plasmid was cotransfected with pNF-κB-Luc and control pCMV-Rluc plasmids into 293T cells, and NF-κB activity was induced by TNF-α treatment. With the expression of BGLF4, but not K102I, there was an approximately 2- to 5-fold decrease of NF-κB transactivation in a dose-dependent manner (Fig. 2A). Many genes involved in inflammation and the immune response, such as IFN-β, are regulated by NF-κB. 293-TLR3 cells, which overexpress Toll-like receptor 3 (TLR3) to recognize double-stranded RNA, can be used as a model system to monitor the double-stranded RNA-mediated NF-κB signaling (9). To investigate whether BGLF4 also modulates NF-κB-mediated gene expression, pNF-κB-Luc or pPRDII2-Luc containing two repeats of the NF-κB-responsive PRDII region on the IFN-β promoter was cotransfected with BGLF4 or K102I and pCMV-Rluc into 293-TLR3 cells. With poly(I·C) stimulation, BGLF4, but not K102I, suppressed poly(I·C)-induced NF-κB activity on pNF-κB-Luc with a 2- to 5-fold decrease (Fig. 2B) and on pPRDII2-Luc with a 2- to 2.5-fold decrease after normalization (Fig. 2C). Taking the data together, BGLF4 downregulates both TNF-α- and double-stranded RNA-induced NF-κB transactivation in a dose- and kinase activity-dependent manner.
Fig 2.
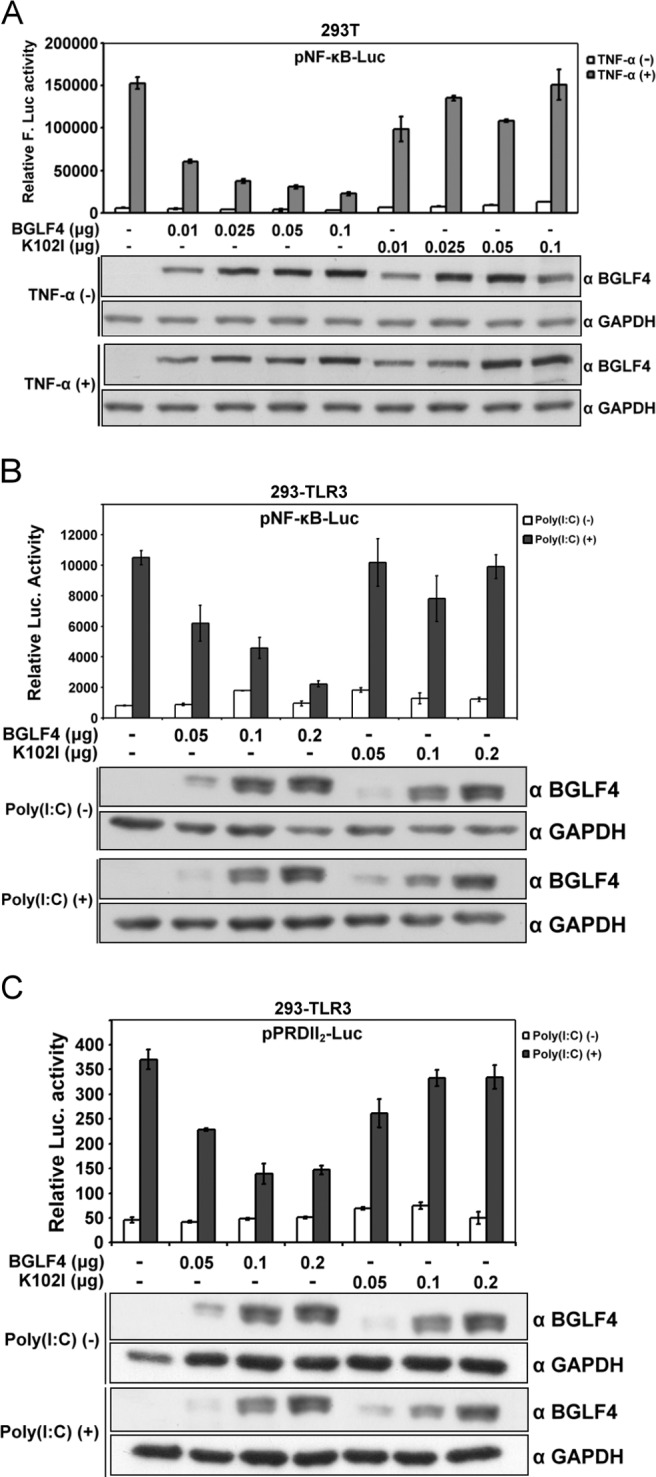
BGLF4 downregulates NF-κB transactivation in a kinase activity-dependent manner. (A) The amounts of BGLF4 or K102I expression plasmid indicated were cotransfected with pNF-κB-Luc and pCMV-Rluc reporter plasmids (as transfection controls) into 293T cells. At 48 h posttransfection, the cells were stimulated with TNF-α (10 ng/ml) for 7 h, and the luciferase activity was determined and normalized to Rluc activity. The expression of BGLF4 was detected with specific antibody by Western blotting. The error bars indicate standard deviations of duplicated wells. F-Luc, firefly luciferase. (B and C) BGLF4 or K102I expression plasmid was transfected into 293-TLR3 cells with pNF-κB-Luc reporter plasmid or pPRDII2-Luc reporter plasmid. Equal amounts of pCMV-Rluc reporter plasmid were also transfected into 293-TLR3 cells in each reaction. At 24 h posttransfection, the cells were stimulated by poly(I·C) (25 μg/ml), and the transactivation activity of NF-κB was analyzed by the luciferase assay and normalized to Rluc activity.
BGLF4 rescues NF-κB-mediated suppression of Zta and Rta transactivation.
It is known that NF-κB inhibits Zta transactivation (14). In addition, a previous study reported that NF-κB inhibits the activation of lytic promoters and impedes lytic cycle progression of gammaherpesviruses, including EBV, Kaposi's sarcoma-associated herpesvirus (KSHV), and murine herpesvirus 68 (MHV68) (2). The EBV lytic promoter containing 1 kb of the BHLF1 promoter is activated by the EBV transactivators, Zta and Rta, during EBV lytic replication. The transcription activity of the BHLF1 promoter is considered important for the initiation of EBV lytic replication (45, 49). Because we showed that BGLF4 inhibited NF-κB transactivation activity, we sought to determine whether BGLF4 can rescue NF-κB-mediated inhibition of the EBV lytic promoter. Therefore, BGLF4 or K102I was coexpressed with HA-p65, Zta expression plasmids, and the pBHLF1-Luc reporter plasmid in 293T cells. The pEGFP-C1 expression plasmid was also transfected as a control in each reaction. With the expression of HA-p65, there was a 1.5- to 4-fold decrease of Zta-induced pBHLF1 promoter activity after normalization. Coexpression of BGLF4 rescued the NF-κB-mediated suppression of Zta transactivation. The expression of K102I at a higher dose (0.5 μg) also partially rescued the NF-κB-mediated inhibition of Zta activity (Fig. 3A). A similar effect on the reporter was observed, driven by the Zta promoter (pZp-Luc) (Fig. 3B). The effect of NF-κB on Rta transactivation was also investigated on pBHLF1-Luc, and the result showed that HA-p65 inhibited Rta transactivaton, while BGLF4 could also rescue the inhibition caused by NF-κB on the BHLF1 promoter (Fig. 3C). Overall, BGLF4 inhibits NF-κB transactivation and reverses the NF-κB-mediated suppression of Zta or Rta transactivation.
Fig 3.
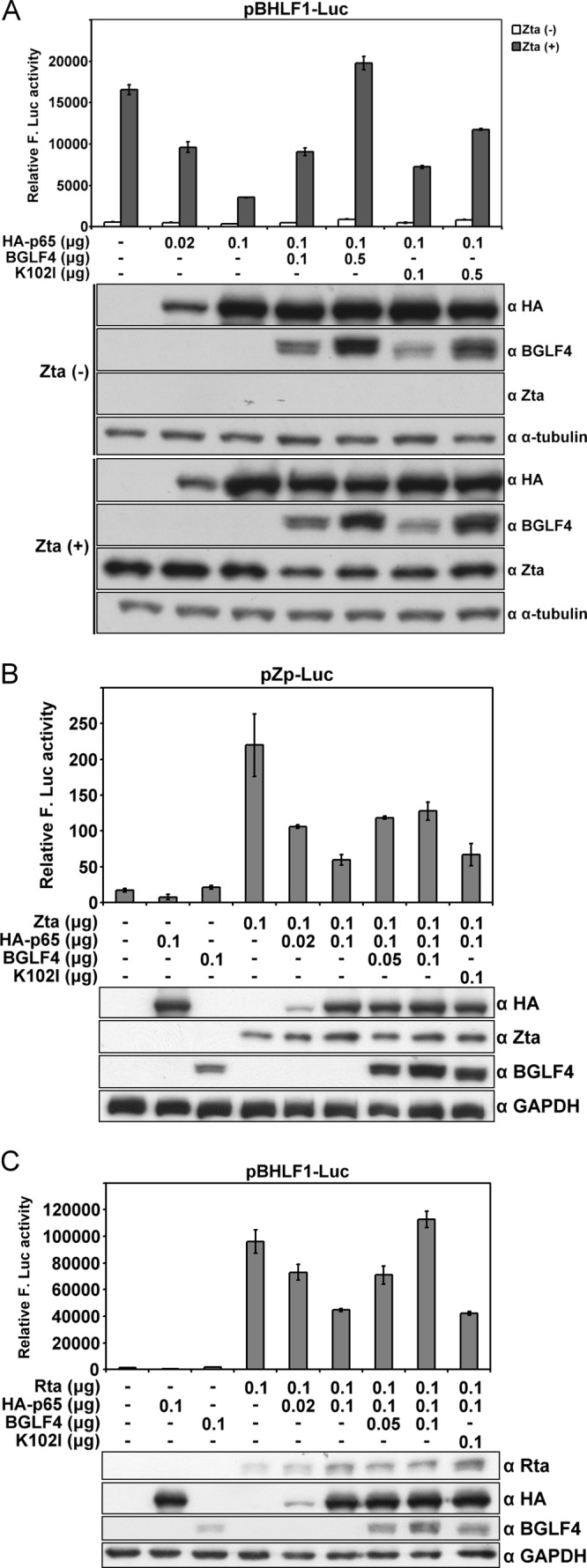
BGLF4 rescues p65-mediated inhibition of Zta and Rta transactivation. (A) The indicated expression plasmids were cotransfected with pBHLF1-Luc and pEGFP-C1 (as a transfection control) into 293T cells. At 48 h posttransfection, cells were harvested and the luciferase activity was measured and normalized to GFP intensity. The error bars indicate standard deviations. (B) The indicated amounts of Zta, HA-p65, BGLF4, and K102I expression plasmids were cotransfected with pZp-Luc and pEGFP-C1 into 293T cells. At 48 h posttransfection, cells were harvested and Zta transactivation was examined using a luciferase assay. (C) The indicated amounts of Rta, HA-p65, BGLF4, and K102I were cotransfected with pBHLF1-Luc and pEGFP-C1 into 293T cells for 48 h. Rta transactivation was analyzed using the luciferase assay.
Knockdown of BGLF4 during EBV lytic replication elevates NF-κB activity and downregulates the reporter activity of the EBV oriLyt BHLF1 promoter.
To investigate whether BGLF4 represses NF-κB transactivation during EBV lytic replication, EBV-positive NA cells were transfected with pNF-κB-Luc reporter and Rta-expressing plasmids to induce the lytic cycle. The dynamics of NF-κB activity during the EBV lytic cycle were monitored in the presence or absence of siBGLF4 oligonucleotides. Compared to the siControl at 18, 24, and 48 h posttransfection, NF-κB transactivation activity was elevated while BGLF4 was knocked down during EBV lytic replication (Fig. 4A). The activity of NF-κB transactivation was increased in both sets of siBGLF4 cells when BGLF4 was diminished. At 48 h posttransfection, however, NF-κB activity in siBGLF4-2 cells was decreased along with the increase of BGLF4 expression. The result implies that BGLF4 plays a role in downregulating NF-κB activity during EBV lytic progression. Because the oriLyt BHLF1 promoter is recognized as a hallmark for the initiation of viral replication (45, 49), we demonstrated the role of BGLF4 in regulating BHLF1 promoter activity upon EBV lytic replication. To this end, the pBHLF1-Luc reporter plasmid was cotransfected with Rta expression plasmid, siBGLF4, or siControl into NA cells. The activity of the EBV oriLyt pBHLF1 promoter was examined at 24 and 48 h posttransfection. Compared with the control, there was no obvious difference in BHLF1 promoter activity at 24 h posttransfection in BGLF4 knockdown cells, whereas knockdown of BGLF4 decreased BHLF1 promoter activity at 48 h posttransfection (Fig. 4B), indicating BGLF4 plays an important role in lytic cycle progression. The downregulation of BHLF1 promoter activity was in concordance with the elevation of NF-κB activity, suggesting that the suppression of NF-κB activity by BGLF4 benefits EBV lytic replication.
Fig 4.

Knockdown of BGLF4 during EBV lytic replication increases NF-κB transactivation. (A) BGLF4-targeted siRNA or control siRNA was cotransfected with Rta expression plasmid, pEGFP-C1, and pNF-κB-Luc into NA cells. At 18, 24, and 48 h posttransfection, luciferase activities were determined and normalized with GFP intensities. The error bars indicate standard deviations. (B) BGLF4-targeted siRNA or siCtrl was cotranfected with pSG5-Rta or pSG5, pEGFP-C1, and pBHLF1-Luc into NA cells. The luciferase activity was measured at 24 and 48 h posttransfection and normalized with GFP intensity. *, P < 0.05; **, P < 0.01 (Student's t test); VC, vector control.
BGLF4 downregulates TNF-α-stimulated NF-κB-dependent gene expression in 293T cells.
NF-κB is an important signal transducer that can be activated by multiple signaling pathways and regulates many key cellular processes by binding to various target sequences. Thus, by inhibiting NF-κB transactivation, BGLF4 may modulate cellular responses and promote viral lytic progression. To determine whether BGLF4 attenuates the expression of NF-κB-regulated cellular genes in a natural context, 293T cells were transfected with BGLF4 or K102I expression plasmid for 48 h and stimulated with TNF-α. Total RNA was isolated after stimulation, and the NF-κB-regulated gene transcripts, A20, IκBα, and IL-8, were quantitated by quantitative real-time PCR (qRT-PCR). In accordance with the data from reporter assays, the expression of A20, IκBα, and IL-8 was downregulated 4- to 8-fold in the presence of BGLF4, while K102I also had a slight inhibitory effect on these genes (Fig. 5A, B, and C). Collectively, these data suggest that BGLF4 attenuates NF-κB-mediated gene expression, and K102I also has a partial inhibitory effect on NF-κB-regulated genes in a cellular context.
Fig 5.
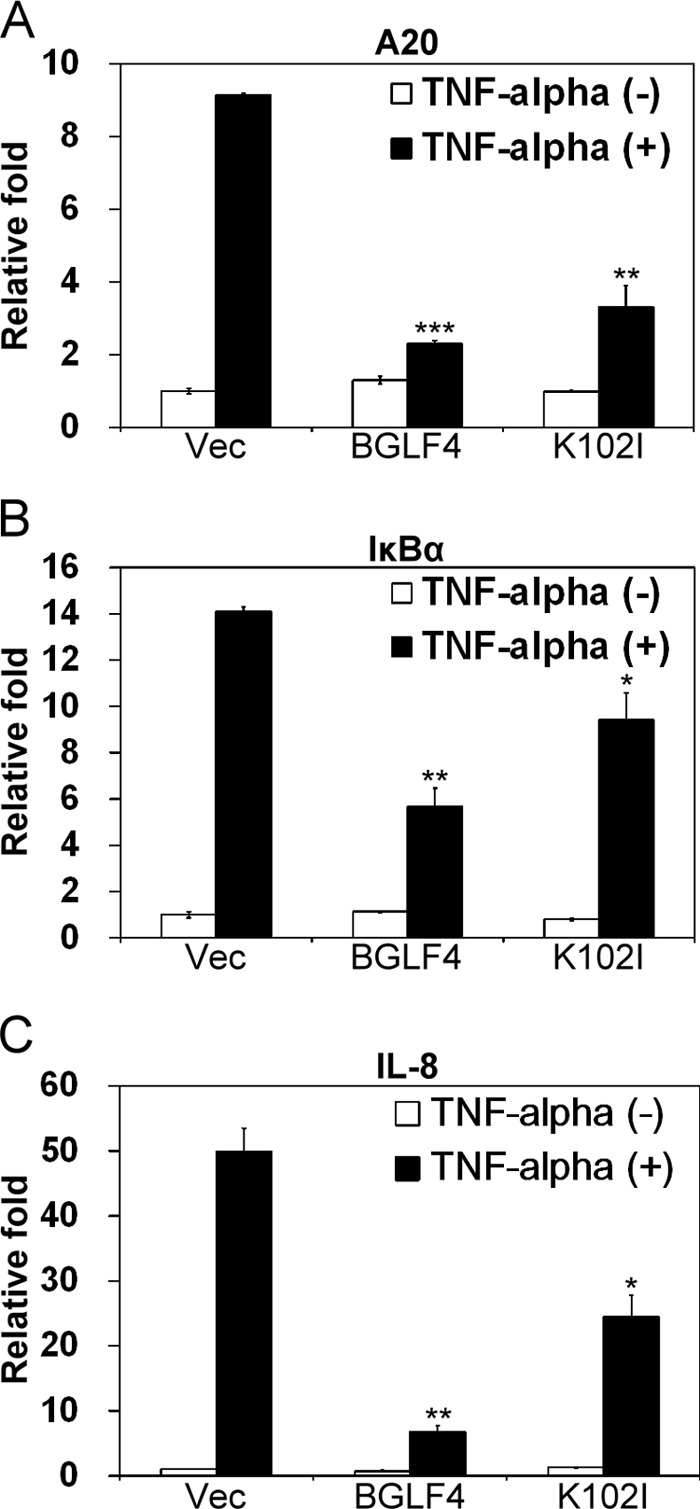
BGLF4 attenuates the expression of NF-κB-regulated genes. 293T cells were transfected with BGLF4, K102I, or pSG5 expression plasmid. At 48 h posttransfection, the cells were stimulated with TNF-α (10 ng/ml) for 90 min. Total RNA was extracted and subjected to quantitative real-time RT-PCR with primers for A20 (A), IκBα (B), and IL-8 (C). The expression levels of target genes were normalized with that of control GAPDH. The “relative fold” is the ratio of the expression value in each reaction to the value of vector (Vec)-transfected cells. The error bars indicate standard deviations. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (Student's t test).
To elucidate the mechanism whereby BGLF4 downregulates NF-κB transactivation, we first determined whether the TNF-α-mediated NF-κB signaling pathway was interfered with by BGLF4. From the subcellular fractionation and indirect immunofluorescence assay (IFA) data, neither BGLF4 nor K102I affected IκBα degradation or p65 nuclear translocation (data not shown). We investigated further whether BGLF4 reduces the DNA-binding ability of NF-κB on the A20, IκBα, or IL-8 promoter by chromatin immunoprecipitation (ChIP) assay. With the expression of BGLF4, only a moderate reduction of p65 binding to each promoter was found (data not shown). This suggests that, instead of interfering with the DNA-binding ability of p65, other mechanisms may be involved in BGLF4-mediated NF-κB downregulation.
UXT phosphorylation attenuates its interaction with p65 and modulates NF-κB activity.
UXT is known to be a coactivator of the NF-κB enhanceosome, and its assembly into the complex is important for the integrity of that enhanceosome (51). Because BGLF4 interacts with UXT (Fig. 1), we speculated that, by interacting with and modifying UXT, BGLF4 might disrupt the stability of the NF-κB enhanceosome complex to downregulate NF-κB activity. To first investigate whether BGLF4 phosphorylates UXT, HA-UXT was cotransfected with BGLF4 or K102I into 293T cells, and the cell lysates were treated with alkaline phosphatase or left untreated. Compared with the presence of vector or K102I, the mobility of HA-UXT became a little lower in the presence of BGLF4, while the mobility change of HA-UXT disappeared after phosphatase treatment (Fig. 6A). However, the BGLF4-induced mobility change of HA-UXT was very subtle, probably because there is only one putative BGLF4 phosphorylation site (Thr/Pro) at the third amino acid of UXT. To further reveal whether BGLF4 phosphorylates UXT at the Thr3 site, we generated an UXT phosphorylation site-deficient mutant, UXT(T3V), with Thr3 mutated to valine, and an in vitro kinase assay was performed with purified UXT and UXT(T3V) proteins. To avoid a possible masking effect of the N-terminal GST tag on the putative phosphorylation site on UXT, the GST tag was removed by thrombin cleavage to generate UXT and UXT(T3V) proteins as substrates for the in vitro kinase assay. GST-lamin A tail 1, containing the amino acids 369 to 519 of lamin A, served as a positive control (27), and the purified GST protein served as a negative control. As shown in Fig. 6B, UXT, but not UXT(T3V), was phosphorylated by BGLF4 in vitro, suggesting that BGLF4 phosphorylates UXT at Thr3.
Fig 6.

BGLF4 phosphorylates UXT, and phosphorylation of UXT modulates NF-κB transactivation. (A) HA-UXT expression plasmid was cotransfected with BGLF4 or K102I expression plasmid into 293T cells. At 48 h posttransfection, cell lysates were harvested and treated with CIP or left untreated for 1.5 h. The reaction mixture was resolved by immunoblotting (IB) with specific antibodies. (B) His-BGLF4 and His-K102I purified from recombinant baculovirus-infected Sf9 cells were used as the kinase sources. GST-UXT and GST-UXT(T3V) purified from BL21(DE3) were digested with thrombin to remove the GST tag and used as the kinase substrates. Lamin tail 1, which was identified as a BGLF4 substrate (27), served as a positive control. GST served as a negative control. The arrowhead indicates a nonspecific signal in the reaction. (C) HA-UXT expression plasmid was cotransfected with BGLF4 or K102I expression plasmid into 293T cells. At 48 h posttransfection, cell lysates were immunoprecipitated with p65-specific antibody. The immunocomplexes were analyzed by SDS-PAGE and detected by p65-, BGLF4-, and HA-specific antibodies. Ctrl., control. (D) 293T cells were transfected with HA-UXT, HA-UXT(T3V), or HA-UXT(T3E) expression plasmid. At 48 h posttransfection, cell lysates were immunoprecipitated with p65-specific antibody. The immunocomplexes were resolved by SDS-PAGE and immunoblotted with p65- and HA-specific antibodies. (E) The indicated amounts of HA-UXT and HA-UXT(T3V) expression plasmids and pNF-κB-Luc and pCMV-Rluc reporter plasmids were cotransfected with BGLF4 or K102I expression plasmid into 293T cells. At 48 h posttransfection, the cells were stimulated with TNF-α (10 ng/ml). At 7 h poststimulation, the luciferase activity was measured and normalized by Rluc activity. Protein expression was confirmed by immunoblotting using the antibodies indicated. GAPDH served as a loading control. The error bars indicate standard deviations. F-Luc. firefly luciferase.
To investigate whether BGLF4 interferes with the interaction between p65 and UXT, HA-UXT was coexpressed with BGLF4 or K102I in 293T cells, and the interaction between p65 and HA-UXT was determined by the coimmunoprecipitation assay (Fig. 6C). In the presence of BGLF4, but not K102I, the amount of UXT coimmunoprecipitated with p65 was dramatically reduced, indicating that BGLF4 impedes the interaction between p65 and UXT, and kinase activity plays a major role in this interference.
Moreover, to verify the influence of Thr3 phosphorylation on UXT function, an UXT phosphorylation site-deficient mutant, HA-UXT(T3V), and a phosphorylation-mimicking mutant with Thr3 mutated to glutamate, HA-UXT(T3E), were generated. The interaction between p65 and HA-UXT, HA-UXT(T3V), or HA-UXT(T3E) was explored using the coimmunoprecipitation assay. With anti-p65 antibody, HA-UXT was coimmunoprecipitated with endogenous p65 protein (Fig. 6D). Compared to HA-UXT, HA-UXT(T3V) showed a stronger interaction with p65, while HA-UXT(T3E) showed a much weaker interaction with p65, suggesting that phosphorylation of UXT on Thr3 attenuates the interaction between UXT and p65.
Because the phosphorylation-defective HA-UXT(T3V) bound more strongly to p65, we then wondered whether UXT(T3V) could rescue BGLF4-mediated downregulation of NF-κB transactivation. To this end, HA-UXT, HA-UXT(T3V), BGLF4, and K102I expression plasmids were cotransfected with pNF-κB-Luc reporter plasmid into 293T cells. After stimulation with TNF-α for 7 h, NF-κB activity was detected using the luciferase assay. The result showed that HA-UXT(T3V) was more able than wild-type HA-UXT to restore BGLF4-mediated inhibition of NF-κB activity (Fig. 6E), suggesting that BGLF4-mediated phosphorylation of UXT plays a negative regulatory role in NF-κB transactivation.
BGLF4-mediated phosphorylation of UXT helps EBV lytic cycle progression.
To elucidate the role of UXT in EBV lytic replication, EBV-positive NA cells were infected with shUXT lentivirus to knock down UXT. Among three different clones, shUXT1 and shUXT3 appeared to have good knockdown efficiency, coupling with significant numbers of floating cells in the first week after lentivirus infection (data not shown). On day 5 postinfection, the UXT knockdown cells were transfected with Rta expression plasmid to induce the lytic cycle, and we noticed that the expression of Zta and BGLF4 proteins was obvious without Rta transfection. However, the induction was not further enhanced by Rta, suggesting that UXT is required for both the maintenance of EBV latency and efficient replication after the expression of Rta (Fig. 7A). After 14 days of puromycin selection, spontaneous lytic protein expression was not seen, which could be due to the fact that the residual UXT is able to support the NF-κB activity to inhibit spontaneous EBV reactivation. Of note, shUXT cells expressed less Zta and BGLF4 after Rta induction (Fig. 7A, lanes 1 to 4 and 5 to 8, B, and C). Overexpression of HA-UXT, but not HA-UXT(T3V), in shLuc and shUXT NA cells further enhanced the expression of lytic proteins at 24 and 48 h after Rta transfection (Fig. 7B and C, lanes 5 and 8), suggesting that UXT may also participate in the transcription of viral lytic genes and that phosphorylation of UXT at the Thr3 site is important for lytic replication. The data here indicate that transcription involving UXT may be important for EBV lytic protein expression, whereas BGLF4-mediated phosphorylation of UXT at Thr3 plays a critical role in promoting the lytic cycle.
Fig 7.
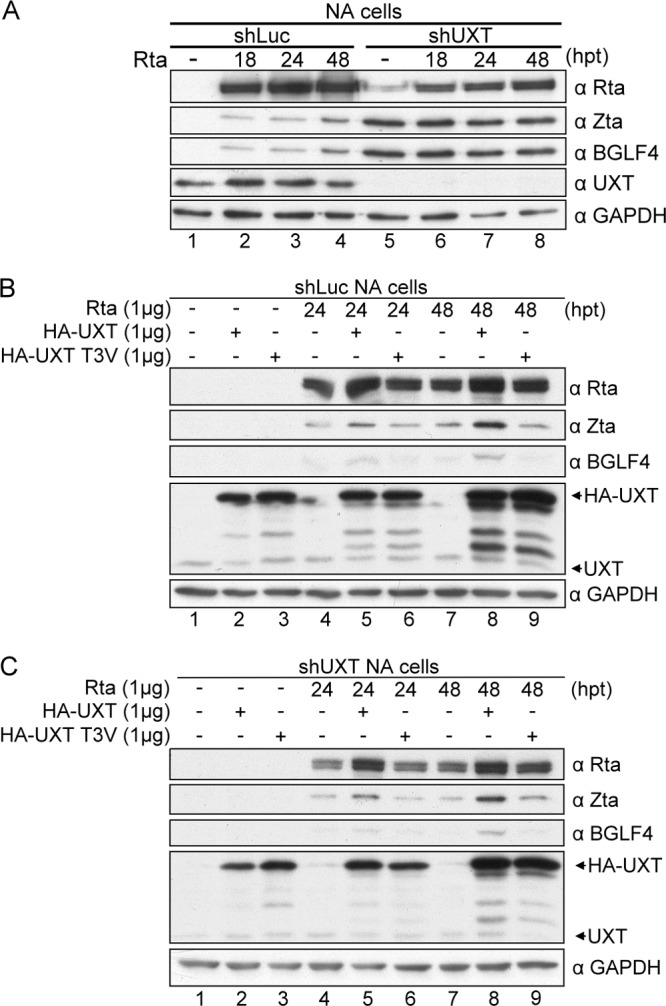
Complementation of HA-UXT, but not HA-UXT(T3V), promotes EBV lytic protein expression in shUXT NA cells. (A) NA cells were infected with shLuc or shUXT lentivirus at an MOI of 1 and then selected with pyromycin (2 μg/ml) at 24 h postinfection. After 5 days of infection, cells were transfected with pSG5-Rta and harvested at 0, 24, and 48 h posttransfection (hpt). Protein expression was detected by immunoblotting using the indicated antibodies. (B and C) On day 15 after shRNA lentivirus infection, shLuc NA cells (B) and shUXT NA cells (C) were transfected with Rta, HA-UXT, or HA-UXT(T3V). Cells were harvested at 0, 24, and 48 h posttransfection, and protein expression was demonstrated by immunoblotting using the indicated antibodies. GAPDH served as a loading control.
DISCUSSION
Upon virus infection, NF-κB and interferon regulatory factors (IRFs) are activated to induce the expression of cytokines and IFNs and to defend against the virus. We have reported previously that BGLF4 suppresses IRF3 transactivation through direct phosphorylation at its linker region, subverting antiviral mechanisms in host cells and enhancing viral lytic progression (55). In this study, we show further that BGLF4 downregulates NF-κB transactivation activity, suggesting that BGLF4 can effectively attenuate the antiviral response through the combined effects of IRF3 and NF-κB suppression. Both TNF-α- and poly(I·C)-induced NF-κB activities were inhibited by BGLF4, suggesting that BGLF4 may suppress immune responses activated by both intra- and intercellular signaling. Knockdown of BGLF4 during lytic replication increases NF-κB transactivation and downregulates EBV oriLyt BHLF1 promoter activity in Rta-transduced EBV-positive NA cells, which indicates that BGLF4 has a negative regulatory effect on NF-κB activity during the lytic cycle to benefit EBV lytic progression. Moreover, we found that BGLF4 phosphorylates UXT at the Thr3 residue to diminish the interaction between UXT and NF-κB, attenuating NF-κB transactivation. Furthermore, depletion of UXT leads to spontaneous EBV lytic replication. Overexpression of HA-UXT, but not HA-UXT(T3V), in UXT knockdown NA cells enhances EBV lytic protein expression. We propose that BGLF4 can inhibit the host immune response activated during the lytic cycle and facilitate viral replication through this mechanism.
NF-κB regulates many genes involved in the immune response and is an attractive target for viruses to escape immune attack. For example, human cytomegalovirus (HCMV) immediate-early protein IE86 blocks the DNA-binding activity of NF-κB and attenuates NF-κB-dependent cytokine and chemokine gene expression (53). Molluscum contagiosum virus (MCV) MC160 protein prevents IKK complex formation to inhibit NF-κB activation, suppressing the production of antiviral molecules (41). Here, we show that BGLF4 downregulates NF-κB activity, which indicates that BGLF4 may suppress the expression of inflammatory cytokines and inhibit the immune response mediated by NF-κB during EBV lytic replication. In addition, as a tegument protein in virions, BGLF4 could also suppress the immune response activated by EBV upon primary infection.
EBV BGLF4 downregulates NF-κB transactivation by disrupting the interaction between NF-κB and the coactivator UXT. UXT has been reported to be a component of a multiprotein transcriptional complex (13). Furthermore, UXT acts as a cofactor to modulate the activities of transcription factors (35, 36, 51). As a result, it is not surprising that knockdown of UXT affects overall viral gene transcription in the EBV lytic cycle (Fig. 7B and C). UXT, but not UXT(T3V), was phosphorylated by BGLF4 (Fig. 6B), indicating that Thr3 of UXT is the only site targeted by BGLF4. Because HA-UXT(T3V) can restore NF-κB transactivation activity in the presence of BGLF4 (Fig. 6E), it would be interesting to determine whether Thr3 of UXT is also a target site of a cellular kinase(s) and involved in the regulation of NF-κB in the absence of virus infection. In addition, the luciferase assay (Fig, 3A) and qRT-PCR (Fig. 5) data show that K102I in a high dose also partially inhibits NF-κB transactivation activity, which may result from the interaction between K102I and UXT. Large amounts of K102I would hinder the interaction of UXT and other transcription factors to affect the stability of the NF-κB enhanceosome. Similar results were also observed in our previous study of the suppression of the IRF3 signaling pathway by BGLF4 (55). BGLF4 inhibits endogenous poly(I·C)-triggered IRF3 activation, downregulating the mRNA level of IFN-β, whereas K102I also partially decreases IRF3-regulated transcription, which may also be attributed to the interference of K102I with the enhanceosome assembly.
Interestingly, the shRNA experiment demonstrates that UXT may play multiple roles in EBV-positive cells. We found that UXT depletion induces a spontaneous EBV lytic cycle within the first week of shUXT lentivirus infection in NA cells (Fig. 7A). After 2 weeks of selection, however, the leaking of lytic protein expression was not observed, and shUXT cells behaved similarly to shLuc cells in complementation experiments (Fig. 7B and C), which may be attributed to the fact that cells with spontaneous EBV lytic replication were dead and live cells were able to maintain a level of UXT sufficient to sustain NF-κB activity for the maintenance of EBV latency. To sum up, it is possible that UXT functions as a key player to maintain EBV latency, and phosphorylation of UXT at the Thr3 site by BGLF4 can disrupt the NF-κB-mediated repression of virus replication. On the other hand, UXT is also required for the general transcription of both cellular and viral promoters. In addition, UXT was reported to be essential for cell viability (60). Thus, the floating cells during the first week of shUXT lentivirus infection could be caused by either the decrease of UXT-mediated survival signaling or the activation of the EBV lytic cycle.
Persistence of EBV within the host is achieved by downregulation of viral protein expression. To inhibit lytic activation, EBV appears to use NF-κB to help in latency maintenance. It was shown that NF-κB physically interacts with the Zta dimerization domain and inhibits Zta transactivation (14). Inhibition of NF-κB induces EBV from latency into lytic replication (2). As a result, NF-κB plays an important role in the EBV latent-lytic switch and must be downregulated during lytic replication. Nevertheless, the mechanism by which NF-κB inhibits lytic promoter transactivation remains unclear. The inhibition may result from competition or steric hindrance by NF-κB, preventing interaction between the viral transactivators and other transcription factors. Alternatively, an NF-κB downstream cellular factor(s) may inhibit the functions of viral transactivators. On the other hand, it was reported that NF-κB and the EBV immediate-early transactivator Zta restrain each other's transcriptional functions (14, 38). Therefore, the balance between Zta and NF-κB activities determines the fate of EBV in its host cells. It is known that sumoylation inhibits Zta transactivation of lytic gene expression, promoting viral latency. However, BGLF4 reduces Zta sumoylation, enhancing the function of Zta as a transactivator (15). Here, we show that BGLF4 can also inhibit NF-κB transactivation activity at the early stage of lytic replication and promote lytic progression. We have reported previously that BGLF4 enhances Zta transactivation of the EBV lytic promoter (57). Thus, BGLF4 may function through the simultaneous inhibition of Zta sumoylation and NF-κB activation to help EBV lytic replication. Moreover, BGLF4 can also restore NF-κB-mediated downregulation of Rta transactivation of the oriLyt promoter (Fig. 3C), suggesting the blockage of NF-κB activity benefits virus replication through multiple mechanisms. We have also observed that knockdown of BGLF4 in EBV-positive NA cells during EBV lytic replication causes a decrease of EBV BHLF1 oriLyt promoter activity (Fig. 4B). The downregulation may be attributed to the decrease of Zta and Rta transactivation activity or to the activation of NF-κB and its downstream signaling pathways to inhibit EBV lytic replication in the absence of BGLF4. This result correlates with the previous observation that BGLF4 knockdown partially inhibits viral DNA replication and the expression of some late genes (10).
EBV is a very successful virus that has evolved multiple mechanisms to escape immune recognition. EBV gp42 is modified posttranslationally to produce soluble gp42, which mediates HLA class II immune evasion (46). Furthermore, Zta binds to the CIITA promoter to inhibit CIITA transcription, downregulating major histocompatibility complex (MHC) class II expression to escape T cell immunity (28). Zta also decreases the expression of the IFN-γ receptor to counteract the antiviral response (39). EBV exonuclease BGLF5 imposes a shutoff of host protein synthesis that leads to a decrease of surface HLA molecules and may contribute to immune evasion (47). EBV BNLF2a blocks the binding of peptides and ATP to the TAP complex, impairs peptide loading of HLA class I molecules, and downregulates surface HLA class I molecules (21, 22). Together with the fact that BGLF4 suppresses the activation of NF-κB and IRF3, EBV is able to counteract the antiviral response during the lytic cycle.
Taking the data together, we have shown that BGLF4 phosphorylates the NF-κB coactivator UXT at the Thr3 site, hindering the interaction between UXT and p65, and downregulates NF-κB transactivation. In this way, BGLF4 may inhibit the immune response activated by NF-κB during lytic replication and benefit the progression of virus replication.
ACKNOWLEDGMENTS
We thank Katherine A. Fitzgerald (Division of Infectious Diseases and Immunology, University of Massachusetts Medical School, Worcester, MA) for providing the pPRDII2-Luc reporter plasmid and 293-TLR3 cells. We also thank W. W. Lin (Graduate Institute of Pharmacology, National Taiwan University College of Medicine) for providing the pcDNA3-HA-p65 expression plasmid. We are very grateful to W. Krek (ETH-Hönggerberg, Institute of Cell Biology, Switzerland) for providing UXT antibody, and we are grateful to Tim J. Harrison of University College London for critical reading and modification of the manuscript.
This study was supported by the National Science Council (NSC98-2320-B-002-054-MY3), the National Health Research Institutes (NHRI-EX-99-9928BI and NHRI-EX100-9928BI), and National Taiwan University (98R0302 and 99R71423).
Footnotes
Published ahead of print 29 August 2012
REFERENCES
- 1. Baumforth KR, Young LS, Flavell KJ, Constandinou C, Murray PG. 1999. The Epstein-Barr virus and its association with human cancers. Mol. Pathol. 52:307–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brown HJ, et al. 2003. NF-kappaB inhibits gammaherpesvirus lytic replication. J. Virol. 77:8532–8540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chang Y, et al. 1999. Requirement for cell-to-cell contact in Epstein-Barr virus infection of nasopharyngeal carcinoma cells and keratinocytes. J. Virol. 73:8857–8866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chen C, Okayama H. 1987. High-efficiency transformation of mammalian cells by plasmid DNA. Mol. Cell. Biol. 7:2745–2752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen MR, Chang SJ, Huang H, Chen JY. 2000. A protein kinase activity associated with Epstein-Barr virus BGLF4 phosphorylates the viral early antigen EA-D in vitro. J. Virol. 74:3093–3104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen YJ, et al. 2011. Epstein-Barr virus (EBV) Rta-mediated EBV and Kaposi's sarcoma-associated herpesvirus lytic reactivations in 293 cells. PLoS One 6:e17809 doi:10.1371/journal.pone.0017809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chua HH, et al. 2007. Role of the TSG101 gene in Epstein-Barr virus late gene transcription. J. Virol. 81:2459–2471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. 1997. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature 388:548–554 [DOI] [PubMed] [Google Scholar]
- 9. Fitzgerald KA, et al. 2003. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4:491–496 [DOI] [PubMed] [Google Scholar]
- 10. Gershburg E, Raffa S, Torrisi MR, Pagano JS. 2007. Epstein-Barr virus-encoded protein kinase (BGLF4) is involved in production of infectious virus. J. Virol. 81:5407–5412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ghosh S, Karin M. 2002. Missing pieces in the NF-kappaB puzzle. Cell 109(Suppl.):S81–S96 [DOI] [PubMed] [Google Scholar]
- 12. Ghosh S, May MJ, Kopp EB. 1998. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 16:225–260 [DOI] [PubMed] [Google Scholar]
- 13. Gstaiger M, et al. 2003. Control of nutrient-sensitive transcription programs by the unconventional prefoldin URI. Science 302:1208–1212 [DOI] [PubMed] [Google Scholar]
- 14. Gutsch DE, et al. 1994. The bZIP transactivator of Epstein-Barr virus, BZLF1, functionally and physically interacts with the p65 subunit of NF-kappa B. Mol. Cell. Biol. 14:1939–1948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hagemeier SR, et al. 2010. Sumoylation of the Epstein-Barr virus BZLF1 protein inhibits its transcriptional activity and is regulated by the virus-encoded protein kinase. J. Virol. 84:4383–4394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hahn AM, Huye LE, Ning S, Webster-Cyriaque J, Pagano JS. 2005. Interferon regulatory factor 7 is negatively regulated by the Epstein-Barr virus immediate-early gene, BZLF-1. J. Virol. 79:10040–10052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hatakeyama S, et al. 1999. Ubiquitin-dependent degradation of IkappaBalpha is mediated by a ubiquitin ligase Skp1/Cul 1/F-box protein FWD1. Proc. Natl. Acad. Sci. U. S. A. 96:3859–3863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hayden MS, Ghosh S. 2008. Shared principles in NF-kappaB signaling. Cell 132:344–362 [DOI] [PubMed] [Google Scholar]
- 19. Hayden MS, Ghosh S. 2004. Signaling to NF-kappaB. Genes Dev. 18:2195–2224 [DOI] [PubMed] [Google Scholar]
- 20. Hiscott J, Kwon H, Genin P. 2001. Hostile takeovers: viral appropriation of the NF-kappaB pathway. J. Clin. Invest. 107:143–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hislop AD, et al. 2007. A CD8+ T cell immune evasion protein specific to Epstein-Barr virus and its close relatives in Old World primates. J. Exp. Med. 204:1863–1873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Horst D, et al. 2009. Specific targeting of the EBV lytic phase protein BNLF2a to the transporter associated with antigen processing results in impairment of HLA class I-restricted antigen presentation. J. Immunol. 182:2313–2324 [DOI] [PubMed] [Google Scholar]
- 23. Hsu TY, et al. 2005. Reactivation of Epstein-Barr virus can be triggered by an Rta protein mutated at the nuclear localization signal. J. Gen. Virol. 86:317–322 [DOI] [PubMed] [Google Scholar]
- 24. James P, Halladay J, Craig EA. 1996. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics 144:1425–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kudoh A, et al. 2006. Phosphorylation of MCM4 at sites inactivating DNA helicase activity of the MCM4-MCM6-MCM7 complex during Epstein-Barr virus productive replication. J. Virol. 80:10064–10072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee CP, et al. 2007. Epstein-Barr virus BGLF4 kinase induces premature chromosome condensation through activation of condensin and topoisomerase II. J. Virol. 81:5166–5180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee CP, et al. 2008. Epstein-Barr virus BGLF4 kinase induces disassembly of the nuclear lamina to facilitate virion production. J. Virol. 82:11913–11926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li D, et al. 2009. Down-regulation of MHC class II expression through inhibition of CIITA transcription by lytic transactivator Zta during Epstein-Barr virus reactivation. J. Immunol. 182:1799–1809 [DOI] [PubMed] [Google Scholar]
- 29. Li Q, Verma IM. 2002. NF-kappaB regulation in the immune system. Nat. Rev. Immunol. 2:725–734 [DOI] [PubMed] [Google Scholar]
- 30. Li R, et al. 2011. Conserved herpesvirus kinases target the DNA damage response pathway and TIP60 histone acetyltransferase to promote virus replication. Cell Host Microbe 10:390–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu L, Amy V, Liu G, McKeehan WL. 2002. Novel complex integrating mitochondria and the microtubular cytoskeleton with chromosome remodeling and tumor suppressor RASSF1 deduced by in silico homology analysis, interaction cloning in yeast, and colocalization in cultured cells. In Vitro Cell. Dev. Biol. Anim. 38:582–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu L, McKeehan WL. 2002. Sequence analysis of LRPPRC and its SEC1 domain interaction partners suggests roles in cytoskeletal organization, vesicular trafficking, nucleocytosolic shuttling, and chromosome activity. Genomics 79:124–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lu J, et al. 2000. Upregulation of tyrosine kinase TKT by the Epstein-Barr virus transactivator Zta. J. Virol. 74:7391–7399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Makarova O, Kamberov E, Margolis B. 2000. Generation of deletion and point mutations with one primer in a single cloning step. Biotechniques 29:970–972 [DOI] [PubMed] [Google Scholar]
- 35. Markus SM, et al. 2002. Identification and characterization of ART-27, a novel coactivator for the androgen receptor N terminus. Mol. Biol. Cell 13:670–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McGilvray R, Walker M, Bartholomew C. 2007. UXT interacts with the transcriptional repressor protein EVI1 and suppresses cell transformation. FEBS J. 274:3960–3971 [DOI] [PubMed] [Google Scholar]
- 37. Mogensen TH, Paludan SR. 2001. Molecular pathways in virus-induced cytokine production. Microbiol. Mol. Biol. Rev. 65:131–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Morrison TE, Kenney SC. 2004. BZLF1, an Epstein-Barr virus immediate-early protein, induces p65 nuclear translocation while inhibiting p65 transcriptional function. Virology 328:219–232 [DOI] [PubMed] [Google Scholar]
- 39. Morrison TE, Mauser A, Wong A, Ting JP, Kenney SC. 2001. Inhibition of IFN-gamma signaling by an Epstein-Barr virus immediate-early protein. Immunity 15:787–799 [DOI] [PubMed] [Google Scholar]
- 40. Murata T, et al. 2009. Efficient production of infectious viruses requires enzymatic activity of Epstein-Barr virus protein kinase. Virology 389:75–81 [DOI] [PubMed] [Google Scholar]
- 41. Nichols DB, Shisler JL. 2006. The MC160 protein expressed by the dermatotropic poxvirus molluscum contagiosum virus prevents tumor necrosis factor alpha-induced NF-kappaB activation via inhibition of I kappa kinase complex formation. J. Virol. 80:578–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Perkins ND. 2007. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat. Rev. Mol. Cell Biol. 8:49–62 [DOI] [PubMed] [Google Scholar]
- 43. Prince S, et al. 2003. Latent membrane protein 1 inhibits Epstein-Barr virus lytic cycle induction and progress via different mechanisms. J. Virol. 77:5000–5007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Reference deleted.
- 45. Rennekamp AJ, Lieberman PM. 2011. Initiation of Epstein-Barr virus lytic replication requires transcription and the formation of a stable RNA-DNA hybrid molecule at OriLyt. J. Virol. 85:2837–2850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ressing ME, et al. 2005. Epstein-Barr virus gp42 is posttranslationally modified to produce soluble gp42 that mediates HLA class II immune evasion. J. Virol. 79:841–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rowe M, et al. 2007. Host shutoff during productive Epstein-Barr virus infection is mediated by BGLF5 and may contribute to immune evasion. Proc. Natl. Acad. Sci. U. S. A. 104:3366–3371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Santoro MG, Rossi A, Amici C. 2003. NF-kappaB and virus infection: who controls whom. EMBO J. 22:2552–2560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48a. Sarisky RT, et al. 1996. A replication function associated with the activation domain of the Epstein-Barr virus Zta transactivator. J. Virol. 70:8340–8347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schepers A, Pich D, Mankertz J, Hammerschmidt W. 1993. cis-acting elements in the lytic origin of DNA replication of Epstein-Barr virus. J. Virol. 67:4237–4245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Siegert R, Leroux MR, Scheufler C, Hartl FU, Moarefi I. 2000. Structure of the molecular chaperone prefoldin: unique interaction of multiple coiled coil tentacles with unfolded proteins. Cell 103:621–632 [DOI] [PubMed] [Google Scholar]
- 51. Sun S, et al. 2007. UXT is a novel and essential cofactor in the NF-kappaB transcriptional enhanceosome. J. Cell Biol. 178:231–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Taneja SS, et al. 2004. ART-27, an androgen receptor coactivator regulated in prostate development and cancer. J. Biol. Chem. 279:13944–13952 [DOI] [PubMed] [Google Scholar]
- 53. Taylor RT, Bresnahan WA. 2006. Human cytomegalovirus IE86 attenuates virus- and tumor necrosis factor alpha-induced NFkappaB-dependent gene expression. J. Virol. 80:10763–10771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Vainberg IE, et al. 1998. Prefoldin, a chaperone that delivers unfolded proteins to cytosolic chaperonin. Cell 93:863–873 [DOI] [PubMed] [Google Scholar]
- 55. Wang JT, et al. 2009. Epstein-Barr virus BGLF4 kinase suppresses the interferon regulatory factor 3 signaling pathway. J. Virol. 83:1856–1869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang JT, et al. 2005. Detection of Epstein-Barr virus BGLF4 protein kinase in virus replication compartments and virus particles. J. Gen. Virol. 86:3215–3225 [DOI] [PubMed] [Google Scholar]
- 57. Yang PW, Chang SS, Tsai CH, Chao YH, Chen MR. 2008. Effect of phosphorylation on the transactivation activity of Epstein-Barr virus BMRF1, a major target of the viral BGLF4 kinase. J. Gen. Virol. 89:884–895 [DOI] [PubMed] [Google Scholar]
- 58. Young LS, Rickinson AB. 2004. Epstein-Barr virus: 40 years on. Nat. Rev. Cancer 4:757–768 [DOI] [PubMed] [Google Scholar]
- 59. Yue W, Gershburg E, Pagano JS. 2005. Hyperphosphorylation of EBNA2 by Epstein-Barr virus protein kinase suppresses transactivation of the LMP1 promoter. J. Virol. 79:5880–5885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhao H, et al. 2005. UXT is a novel centrosomal protein essential for cell viability. Mol. Biol. Cell 16:5857–5865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhu J, et al. 2009. Protein array identification of substrates of the Epstein-Barr virus protein kinase BGLF4. J. Virol. 83:5219–5231 [DOI] [PMC free article] [PubMed] [Google Scholar]