

Abstract
Herpes simplex virus (HSV) is a large DNA virus which is characterized by its ability to form latent infections in neurons of the peripheral nervous system. Although histones are found in the capsids of small DNA viruses (papovaviruses), none are found in the capsids of large HSV. However, after entry into the infected cell nucleus, the HSV genome begins to associate with nucleosomes during the earliest stages of infection. In contrast, late during infection, newly replicated viral DNA does not appear to associate with nucleosomes, suggesting that histones are deposited specifically on input viral DNA. The mechanisms of deposition and removing histones from the viral genome are unclear. Recently, histone chaperones, involved in the assembly and disassembly of nucleosomes, have been identified. Human antisilencing factor 1 (Asf1) is one such factor which is involved in both the assembly and disassembly of nucleosomes in cellular systems. In this study, we have examined the effect of small interfering RNA (siRNA) knockdown of Asf1a on HSV infections in HeLa cells. Both viral replication and growth were found to be decreased. Also, viral DNA was significantly less protected from micrococcal nuclease (MNase) digestion up to 6 h postinfection (hpi). However, transcription of the immediate early (IE) genes ICP0 and ICP4 was significantly upregulated at 3 h postinfection. Also, these genes were found to be less protected from MNase digestion and, therefore, less associated with nucleosomes. These results suggest that Asf1a plays a role in regulating IE genes by assembling chromatin onto histone-free viral DNA by 3 h postinfection.
INTRODUCTION
Herpes simplex virus (HSV) is a large DNA virus (152-kb double-stranded DNA [dsDNA]) that infects humans. Initially, it infects epithelial cells at the periphery, and it eventually establishes latent infections in neurons of the innervating peripheral nervous system. Reactivation of this latent infection leads to recurrent disease and characterizes HSV infections.
Unlike small DNA viruses, which have nucleosomes in their virion capsids neutralizing the charge on the DNA (10, 44), the charge on HSV DNA in virion capsids is assumed to be neutralized by polyamines (9). After docking of the HSV capsid at the nuclear pore and entry of the viral DNA into the nucleus, the viral DNA appears to start to be assembled into chromatin. However, it is not chromatinized in a 200-bp, evenly spaced nucleosomal pattern during lytic infection, and no ladder-like MNase digestion pattern is detected on partial digestion during a productive lytic infection (28, 29, 36).
Chromatin immunoprecipitation (ChIP) experiments suggest that nucleosomes are present in smaller amounts on HSV than cellular DNA (18, 20, 21, 24, 39). Furthermore, during lytic infection, it has been suggested that the HSV DNA nucleosomes are less stable than normal (27). If they are in an unstable dynamic state, they may be distributed evenly along the viral genome unless their positioning is sequence specific. Nevertheless, an evenly spaced nucleosomal pattern that is similar to that seen in partial MNase digestion of cellular DNA is detected during latent infection (5). Little is known of the mechanism of chromatin assembly on the viral genome other than that it occurs at early times before DNA replication. ICP0 has been shown to dissociate the histone deacetylase 1 (HDAC1) from the REST/CoREST/HDAC repressor complex, resulting in an increase in H3 present on the viral genome (3, 12, 13). VP22 has also been implicated in blocking nucleosome assembly on HSV DNA (51).
Chromatin can be assembled on cellular DNA at the time of DNA replication or during DNA repair or transcription (1). The basic unit of chromatin is the nucleosome, consisting of 2 copies each of the H3, H4, H2a, and H2b histone proteins, which protects 147 bp of DNA from MNase digestion. Histones are assembled into nucleosomes by histone chaperone proteins, such as antisilencing factor 1 (Asf1), histone regulator A (HIRA), and chromatin assembly factor 1 (CAF-1) (16). Although Asf-1 is involved with both replication-dependent and replication-independent chromatin assembly, CAF-1 is involved with replication-dependent chromatin assembly and HIRA is involved with replication-independent chromatin assembly (48). Little is known of the role of these histone chaperones in the formation of nucleosomes on HSV-1 DNA during infection.
Asf1 participates in both nucleosome assembly and disassembly (23, 40). In cellular DNA replication, Asf1 acts as a histone H3/H4 chaperone and, with CAF-1 and PCNA, assembles nucleosomes on replicated DNA (8, 11, 33, 45). The absence of the Asf1 protein results in a delay in the advancing replication fork, leading to inefficient DNA replication (46).
The chromatin structures on HSV DNA clearly play a role in viral gene expression during HSV infection. Evidence for the role of modifications (epigenetic markers) on histones comprising viral nucleosomes in gene expression from the viral genome during both lytic and latent infection of HSV is growing (22, 26, 30, 42), as is evidence on their role in gene expression of other herpesviruses (34).
In this study, we have examined the role of Asf1a in HSV infection. Specifically, viral DNA synthesis, viral replication, and chromatin structure were examined during the knockdown of the Asf1a protein. Viral replication and viral growth were found to be decreased. Also, viral DNA was found to be significantly less protected from MNase digestion up to 6 h postinfection (hpi) in Asf1a knockdown cells. However, transcription of the immediate early (IE) genes ICP0 and ICP4 was dramatically upregulated during infection in these cells, and the ICP0 and ICP4 genes were less associated with histones (less protected from MNase digestion). These results suggest that Asf1a is playing a significant role in regulating IE genes by assembling chromatin into histone-free viral DNA during early times of infection.
MATERIALS AND METHODS
Cells and viruses.
HeLa cells and Vero cells (African green monkey kidney cell line) were grown in Dulbecco's modified Eagle medium (DMEM) supplemented with 5% calf serum and antibiotics. The F strain of HSV-1 was used to infect cells at a multiplicity of infection (MOI) of 5.
Transfection.
A total of 2 pmol of Asf1a siGENOME SMART pool small interfering RNA (siRNA) or control negative siRNA (Dharmacon) was transfected with 2.5 μl of Lipofectamine RNAiMAX (Invitrogen) into one well of a 6-well plate and incubated for 2 days before infection.
Quantitative real-time PCR.
Purified immunoprecipitated DNAs were amplified by previously described methods (39).
Plaque assay.
Infected cells were harvested and freeze-thawed 3 times, and total virus was determined by standard plaque assays on Vero cells (15). Briefly, serially diluted samples of freeze-thawed infected cells were transferred to monolayer cultures of Vero cells in 48-well plates. After 16 h incubation, the monolayers were fixed with methanol-acetone (2:1) for 20 min two times. The plaques were counted after immunostaining the fixed infected monolayers.
MNase digestion.
Micrococcal nuclease (MNase) digestion was performed by previously described methods (21). Briefly, cells were harvested by scraping, resuspended in 0.5 ml RSB (10 mM Tris, pH 7.5, 10 mM 5 M NaCl, and 3 mM MgCl2), and incubated for 5 min on ice. Following addition of 0.5 ml 1% NP-40 (in RSB), the cells were mixed, incubated for 2 min on ice, overlaid with 0.5 ml of 0.33 M sucrose (in RSB), and centrifuged at 1,700 rpm for 10 min at 4°C. The pelleted nuclei were resuspended in 200 μl of MNase buffer (20 mM PIPES [piperazine-N,N′-bis(2-ethanesulfonic acid)], pH 7, 1 mM MgCl2, 10 mM NaCl, 1 mM CaCl2, 5 mM 2-mercaptoethanol, 0.25 M sucrose, and 0.1 mM phenylmethylsulfonyl fluoride [PMSF]) and digested with S7 micrococcal nuclease (MNase) (Roche) at 5 Kunitz units/μl for 30 min at room temperature. EDTA was added (12 mM; on ice) to stop the reaction before treating the mixture with 100 g/ml of proteinase K overnight at 37°C. DNA was purified using a Wizard DNA cleanup kit (Promega) according to the manufacturer's instructions.
Southern blot.
A total of 5 μg of total DNA prepared from MNase digestion was used to load in 1.5% low-melting-point (LMP) agarose gel electrophoresis. The gel was denatured in 1.5 M NaCl and 0.5 M NaOH for 20 min, neutralized in 1.5 M NaCl and 0.5 M Tris buffer (pH 7) for 20 min two times, and transferred into a Hybond-N+ membrane (Amersham) overnight. Then the membrane was UV cross-linked in UV Stratalinker 2400 and preincubated in the Rapid-Hyb buffer (Amersham) for 1.5 h. The labeled probes were prepared by using 5 cosmids covering the entire HSV-1 genome (4) with the nick translation kit (Invitrogen). The membrane was hybridized with the radioactive probes at 62°C for 1.5 h. After washing, it was incubated in a phosphorus screen overnight and scanned by a Typhoon imager (Amersham).
Reverse transcription-quantitative PCR (RT-qPCR).
Total RNA was extracted from cells using the All Prep DNA/RNA minikit (Qiagen) in accordance with the manufacturer's instructions. The reverse transcription (RT) part was performed using SuperScript First Strand (Invitrogen) in accordance with the manufacturer's instructions. cDNA was amplified by previously described methods (39).
SDS-PAGE and Western blots.
F strain-infected and mock-infected HeLa cells were processed for Western blots by previously described methods (39). The following antibodies were used: rabbit monoclonal antibody to Asf1a (Cell Signaling), rabbit polyclonal antibody to tubulin (Santa Cruz), ICP4 (Virusys), and VP16 (Sigma), and mouse monoclonal antibody to CAF-1 (Abcam) and ICP0 (Santa Cruz).
Chromatin immunoprecipitation assays.
F strain-infected and mock-infected HeLa cells were processed for a ChIP assay by previously described methods (39).
RESULTS
HSV-1 DNA replication and viral replication are delayed in cells treated with Asf1a siRNA.
To determine the impact of Asf1a on viral DNA replication and viral replication, HeLa cells in 6-well plates were transfected with Asf1a-specific siRNA or control siRNA for 2 days and then infected with HSV-1 strain F (MOI of 5). As shown in Fig. 1A and B, Asf1a mRNA and protein were knocked down while Asf1b mRNA was not changed during the time course of the experiment (0 to 16 hpi). The amount of Asf1b mRNA and CAF-1 protein were not significantly changed in Asf1a siRNA- and control siRNA-treated cells, suggesting that the knockdown of Asf1a is specific (Fig. 1A and B). Interestingly, the Asf1a protein level increased during late HSV-1 infection while CAF-1 protein levels were not changed (Fig. 1A and B). However, Asf1a mRNA levels did not change. In cytomegalovirus (CMV) infection, the Asf1a protein has also been noted to accumulate, as early as 8 hpi (38). Before viral infection, total and live cell numbers of Asf1a knockdown cells were determined with a TC10 automated cell counter (Bio-Rad). Cell doubling time and cell viability were not changed in Asf1a siRNA-treated cells compared to negative siRNA-treated cells during the 2 days prior to infection, suggesting that knockdown of Asf1a had no major effect on cellular physiology over the time of our experiments (data not shown). In our experiments, viral DNA was reduced and delayed from 6 hpi through 16 hpi in Asf1a siRNA compared to that in control siRNA-treated cells, while DNA at 3 hpi in Asf1a siRNA was similar to that in control siRNA-treated cells (Fig. 1C). Viral yield from Asf1a knockdown cells was significantly reduced at 8 hpi (18-fold), 10 hpi (7-fold), and 16 hpi (4-fold) (Fig. 1D). Taken together, these data indicate that Asf1a plays an important role in HSV-1 DNA replication and viral replication from the earliest stages of viral infection.
Fig 1.

Effect of Asf1a knockdown on HSV-1 growth. HeLa cells were transfected with Asf1a siRNA for 2 days, infected with HSV, and harvested at the indicated time p.i. (A) Asf1a siRNA knocks down Asf1a protein levels. Cells were lysed and immunoblotted with rabbit anti-Asf1a or β-tubulin or CAF-1 antibody. The reduction in the Asf1a protein is 93%. Interestingly, HSV infection induces Asf1a expression. (B) Asf1a siRNA specifically knocks down Asf1a mRNA, not Asf1b. Total mRNA was isolated and reverse transcribed. Asf1a and Asf1b mRNA levels (relative to a mock-infected sample) were determined at various times p.i. by qPCR and normalized to 18S rRNA levels. (C) Asf1a siRNA delayed viral DNA replication. Accumulated viral DNA was quantified by qPCR with TK promoter primers and normalized to GAPDH (glyceraldehyde-3-phosphate dehydrogenase) levels. The relative viral DNA amount was calculated based on a 3-hpi sample in negative (neg) siRNA-treated cells. (D) Asf1a siRNA delayed virus replication. Viral yield from Asf1a knockdown cells was significantly reduced at 8 hpi (18-fold), 10 hpi (7-fold), and 16 hpi (4-fold). Viral yields were determined by a plaque assay as described in Materials and Methods. All data are the averages of three independent experiments, and error bars represent the standard deviation. Student t test values are displayed with asterisks (*, P < 0.01; **, P < 0.001; ***, P < 0.0001).
HSV DNA is significantly less protected from MNase digestion at early times postinfection (p.i.) after Asf1a siRNA treatment.
Because the Asf1a protein is a histone H3/H4 chaperone, the chromatin status of HSV DNA was determined after Asf1a siRNA treatment. MNase digestion was used to determine the protection of viral DNA assembled into nucleosomes. Cells were transfected with Asf1a siRNA or control siRNA, incubated for 2 days, infected with HSV-1 strain F (MOI = 5), harvested at various times p.i., and digested with MNase as described in Materials and Methods. The nucleosome-protected viral DNA was purified, separated by agarose gel electrophoresis, blotted onto a membrane, and visualized by hybridization with probes from 5 cosmids covering the entire HSV-1 genome. Figure 2A shows less 150-bp nucleosome-protected viral DNA in the Asf1a siRNA-treated samples. Figure 2B shows the quantitated data from Fig. 2A, corrected for the total amount of viral DNA (before MNase digestion) in each sample. The 150-bp viral DNA fragment was measured by a Typhoon phosphor imager using ImageQuant software (GE Healthcare) and normalized to the total viral DNA at each time course (Fig. 1C). Viral DNA was found to be significantly less protected in the Asf1a siRNA-treated samples at 3 hpi and 6 hpi (Fig. 2B) but not at later times in infection. Figure 2C shows the fraction of 150-bp fragment in total viral DNA. The fraction of viral 150-bp fragment in total viral DNA at 3 hpi (1%) was found to be significantly less than that at other times. Viral DNA was most covered with nucleosome-like particles (7%) at 6 hpi in control siRNA-treated cells. These results suggest that Asf1a is important in assembling nucleosomes on viral DNA, especially at the early stage of infection. The fact that nucleosomes are deposited in the absence of Asf1a (Asf1a siRNA-treated cells) but at later times (peaking at 8 hpi) suggests that there may be a second (Asf1a-independent) mechanism for nucleosome deposition on HSV DNA, operating at later times in the infectious cycle.
Fig 2.

The HSV genome is less protected from MNase digestion in Asf1a siRNA-treated cells at early times of infection. (A) Asf1a siRNA decreased the protection of viral DNA from MNase digestion. Nuclei were isolated and digested with MNase. DNA was purified, electrophoresed, transferred into membrane, and hybridized with 32P-labeled nick-translated probes prepared from 5 cosmids covering the entire HSV-1 genome. These data represent one of three independent experiments. (B) The amount of viral genome protected from MNase digestion was significantly decreased even after correction for the delayed accumulation of viral DNA in the Asf1a siRNA-treated cells. The 150-bp viral DNA fragment was measured by a phosphor imager and normalized to the total viral DNA at each time course. The relative amount of protected viral DNA was calculated based on the 3-hpi sample in Asf1a siRNA-treated cells. (C) The 150-bp fragment in total viral DNA at 3 hpi was lower than at other times (1%). The signal from 150-bp fragments and total viral DNA from Fig. 2A Southern data were measured by a phosphor imager, and the value of the 150-bp fragments was divided by the value of the total viral DNA in the lane to give the percentage of HSV DNA in nucleosome-like particles. All data are the averages of three independent experiments, and error bars represent the standard deviation.
Transcription of viral immediate early genes but not early or late genes is upregulated at early times in Asf1a siRNA-treated cells.
To investigate the impact of the reduction of nucleosomes on viral DNA (caused by suppression of Asf1a) on viral gene expression, representative genes from each class of viral genes (IE, early [E], and late [L]) were examined. HeLa cells were treated with siRNA to Asf1a and infected with HSV-1 (F) at a MOI of 5. At various times p.i., cells were harvested and RNA was extracted and quantitated by RT-qPCR as described in Materials and Methods. Relative mRNA levels for ICP0, ICP4, TK, and VP16 were determined by qPCR and normalized to 18S rRNA levels. The relative mRNA amount was calculated based on a 3-hpi sample in negative siRNA-treated cells.
As shown in Fig. 3A and B, the accumulation of both ICP0 and ICP4 (IE) transcripts was dramatically increased in Asf1a siRNA-treated cells. However, there was no significant change in early (TK) and late (VP16) gene transcription (Fig. 3C and D). Together, these results suggest that the knockdown of Asf1a results in less-chromatized viral DNA, which leads to more transcription of IE genes (ICP0 and ICP4) but not E or L genes (TK, VP16).
Fig 3.

Asf1a knockdown increased IE viral transcription. HeLa cells were transfected with Asf1a siRNA for 2 days, infected, and harvested at the indicated time. Asf1a siRNA treatment dramatically increased mRNA levels of ICP0 (A) and ICP4 (B), while it did not affect mRNA levels of TK (C) and VP16 (D). Total mRNA was isolated and reverse transcribed. Relative mRNA levels for ICP0, ICP4, TK, and VP16 were determined by qPCR and normalized to 18S rRNA levels. The relative mRNA amount was calculated based on a 3-hpi sample in negative siRNA-treated cells. Three independent experiments were performed, and the error bars represent the standard deviation.
Viral protein expression is altered after suppression of nucleosome deposition.
In order to determine if the increase in ICP0 and ICP4 transcripts in Asf1a siRNA-treated cells results in an increase in protein expression, we performed a Western blot analysis of ICP0, ICP4, and VP16 protein at various times p.i. in the presence and absence of Asf1a siRNA. Interestingly, only ICP0 protein expression was shown to be increased after knockdown of Asf1a, while the level of ICP4 and VP16 protein was not changed (Fig. 4; 3 independent experiments).
Fig 4.
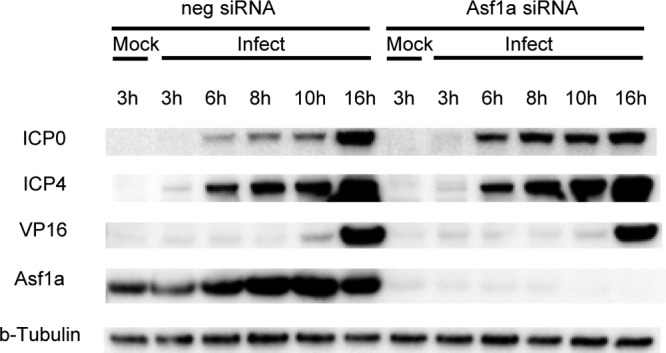
Asf1a knockdown only increased ICP0 protein expression. Total lysates from siRNA-treated and infected cells at various times postinfection were Western blotted and probed with ICP0, ICP4, and VP16 antibodies. β-Tubulin was used as a loading control.
The promoter regions of ICP0 and ICP4 were less associated with nucleosomes in Asf1a siRNA-treated, HSV-1 infected cells.
To confirm the reduction in association of nucleosomes with viral genes in Asf1a siRNA-treated cells, HeLa cells were transfected with Asf1a siRNA or control siRNA for 2 days, infected with HSV-1, and harvested at various times p.i., as previously described. A chromatin immunoprecipitation assay (ChIP) was performed to determine the amount of viral DNA associated with histone H3 (as described in Materials and Methods).
The promoter regions of ICP0, ICP4, and TK were shown to be significantly less associated with histone H3 at 3 hpi but associated at a similar level at 6 hpi and up to 16 hpi in the Asf1a-treated and untreated cells (Fig. 5A, B, and C). However, the association of the promoter region of VP16 with histone H3 was not changed. Interestingly, although the transcribed region of ICP0 was a little less associated with histone H3 at 3 hpi after Asf1a siRNA treatment (P = 0.01), the transcribed regions of ICP4, TK, and VP16 showed no change in association with histone H3 (Fig. 5B, C, and D).
Fig 5.

Asf1a knockdown lowers the deposition of histone H3 on viral promoter regions at 3 hpi. The levels of histone H3 on promoter and transcribed regions of the ICP0 (A), ICP4 (B), TK (C), and VP16 (D) genes were investigated by ChIP. Values are calculated as percentages of input. Student t test values are displayed with asterisks (*, P < 0.01; **, P < 0.001; ***, P < 0.0001). All data are the averages of three independent experiments, and error bars represent the standard deviation.
To confirm these data, we used the alternative method of MNase digestion and qPCR to determine the association of promoter and transcribed regions with nucleosomes in Asf1a siRNA-treated HSV-1 infected cells (Fig. 6). MNase digestion was performed, and purified 150-bp DNA was amplified by qPCR as described in Materials and Methods. The promoter regions of all genes tested, IE (ICP0 and ICP4), E (TK), and L (VP16), were significantly less protected from MNase digestion at 3 hpi but maintained at a similar level between 6 hpi and 16 hpi (Fig. 6). There was one exception, the promoter region of VP16 at 16 hpi (P < 0.001). The transcribed regions of ICP4, TK, and VP16, but not ICP0, were significantly different at 3 hpi (P < 0.01). ICP0 had marginal significance, with a P value of 0.02. There was no difference at later times (6 to 16 hpi).
Fig 6.

Asf1a knockdown lowers the protection of the viral genome from MNase digestion at 3 hpi. The levels of histone H3 on promoter and transcribed regions of the ICP0 (A), ICP4 (B), TK (C), and VP16 (D) genes were examined by qPCR with 150-bp fragments purified after MNase digestion. Values are calculated as percentages of input. Student t test values are displayed with asterisks (*, P < 0.01; **, P < 0.001; ***, P < 0.0001). All data are the averages of three independent experiments, and error bars represent the standard deviation.
There is a 10- to 50-fold difference of percent input values between Fig. 5 and Fig. 6. Overall, ChIP samples have a higher percent input than MNase samples. This may be due to the cross-linking treatment for ChIP samples. ChIP samples have tighter binding, leading to the higher percent input retained compared to that for MNase samples. Alternatively, the difference may be due in part to the difference in size of the DNA fragments. MNase digestion and selection of 150-bp-sized DNA fragments result in only nucleosome-sized particles being detected, whereas the ChIP technique relies on sonication of DNA to approximately 500-bp fragments for its technique, resulting in less specificity for nucleosome-sized particles. The fact that the difference between the results for the 2 techniques is so marked for ICP0 compared to ICP4 suggests that there may be a lot of nonnucleosomal protein material on the ICP0 promoter but not many nucleosomes at 3 hpi.
Taken together, these data broadly support each other. Clearly, the effect of Asf1a on nucleosome deposition on HSV DNA occurs early in the infectious cycle, at a time before DNA replication is occurring.
DISCUSSION
Asf1 is a histone H3/H4 chaperone that plays critical roles in various cellular functions, including DNA replication, gene transcription, and DNA repair (35). Asf1 has two isoforms in humans, Asf1a and Asf1b, which are functionally different (6, 47, 50). Asf1a differs in its serine/threonine-rich C-terminal region and is more phosphorylated than Asf1b (32, 37, 47). However, there are similarities between the two isoforms (49). Human Asf1a (204 amino acids [aa]) and Asf1b (202 aa) show 71% identity in protein sequences, and both of them interact with the p60 subunit of CAF-1 (2, 49). The expression of Asf1a is ubiquitous in various human cells, while Asf1b is specifically expressed in tissue (50). Asf1b has been shown to be recruited by HCF-1 into viral replication forks during viral DNA replication (41). In HSV-1-infected cells, Asf1b has been shown to be required for efficient viral DNA replication (41). In varicella-zoster virus (VZV)-infected cells, IE63 gene expression was shown to increase the association of Asf1a with H3.1 and H3.3 (2). VZV replication was not changed following the knockdown of the Asf1 protein. In cytomegalovirus (CMV) infections, Asf1a was localized to the region of the replication compartment (38).
Asf1 is known to interact with other chromatin-associated proteins, such as histone cell cycle regulation-defective homolog A (HIRA) and chromatin assembly factor (CAF-1) (8, 49). HIRA has been shown to be required for efficient histone variant H3.3 deposition on the HSV-1 genome, viral gene expression, and DNA replication (42, 43). Asf1a interacts more preferentially with HIRA than does Asf1b (48, 49, 52). Asf1 can also form complexes with the DNA damage checkpoint kinase Rad53 (7, 19).
In contrast to the role that Asf1b plays in viral DNA replication and late (but not immediate early or early) viral gene expression (41), it appears that Asf1a plays an important role in viral growth and gene expression at the early stages of viral infection. Viral replication and viral growth were decreased in Asf1a knockdown (siRNA-treated) cells, supporting a role for Asf1a in these processes (Fig. 1). Reduction in these processes, rather than complete blocking, suggests that other chaperones, like Asf1b, may also play a role in this process.
Interestingly, overall protection of the viral genome from MNase digestion was highest at 6 hpi (suggesting that the nucleosome content of the viral DNA is highest at this time p.i.) (Fig. 2), while promoter regions and transcribed regions of representative genes from IE, E, and L classes (ICP0, ICP4, TK, VP16) were most associated and protected at 3 hpi (Fig. 5 and 6), indicating that the nucleosome content of these genes can vary from that of the whole genome.
Although only about 1% of the viral DNA is protected from nuclease digestion at 1 hpi, this percentage rises to 7% of total viral DNA at 6 hpi before dropping off at later times (Fig. 2C). This number may be more or less due to the digestion conditions, which were optimized to give 150-bp fragments. Overdigestion with MNase will digest these 150-bp fragments to smaller sizes. However, our data show that at least some HSV DNA is protected from MNase digestion in 150-bp fragments, indicative of the presence of nucleosomes. However, because we have no direct evidence for nucleosomes (histone octamers consisting of dimers of H2A and B, H3, and H4 present on HSV DNA), perhaps it is more correct to call them nucleosome-like particles.
The highly upregulated ICP0 and ICP4 transcription seen in Asf1a siRNA-treated cells (Fig. 3) suggests that the addition of nucleosomes to these genes by an Asf1A-mediated pathway leads to decreased, or suppressed, transcription. A decrease in H3/H4 deposition (possibly with heterochromatin marks) by Asf1a knockdown may contribute to the derepression of IE promoters. The dramatic rise in levels of transcripts from the IE genes ICP0 and ICP4 is in contrast to that of the early gene TK and the late gene VP16 (Fig. 3). This rise may be the result of the VP16 transcriptional coactivator complex's ability to dislodge nucleosomes from the IE genes (25) or the entire HSV-1 genome (14), the rate-limiting step in IE gene expression. The failure to deposit nucleosomes in the absence of Asf1a would remove this bottleneck, allowing increased IE gene expression.
The overexpression of the IE transcripts did lead to a detectable overexpression of ICP0, but not ICP4, protein (Fig. 4). The IE transcripts are exported into the cytoplasm for translation. However, with the limited resources of a cellular translation system under the stress of a viral infection, they may not be processed quickly. Nevertheless, the spliced ICP0 mRNA can be exported and translated through the altered nucleosome positioning. In the cellular system, splicing regulators can be recruited by modulation of the positioning of nucleosomes along exons (31). We suggest that an increase in ICP0 protein expression was observed because the splicing of ICP0 was helpful for the export and translation of ICP0 transcripts during the knockdown of Asf1a. This supports a role of chromatinization of the viral genome by Asf1a in reduction of viral transcription and, hence, limited protein expression.
In conclusion, Asf1 is considered to play a role in both cellular nucleosome assembly and disassembly (17), and our data indicate that it also plays a role in viral chromatin assembly. Overall chromatin assembly on HSV-1 DNA up to 6 hpi was dependent on a mechanism involving Asf1a, and in its absence, nucleosomes were not deposited and transcription was increased in IE genes. At later times, some nucleosomes appear to accumulate on the viral genome by an Asf1a-independent mechanism.
ACKNOWLEDGMENTS
We gratefully acknowledge Wenpei Liu and Mark Boyer for supporting these studies. We thank Shelley L. Berger for the use of a Bioruptor sonicator for the ChIP method.
This work was supported by a grant from the NIH, NS-33768.
Footnotes
Published ahead of print 5 September 2012
REFERENCES
- 1. Akey CW, Luger K. 2003. Histone chaperones and nucleosome assembly. Curr. Opin. Struct. Biol. 13:6–14 [DOI] [PubMed] [Google Scholar]
- 2. Ambagala AP, et al. 2009. Varicella-zoster virus immediate-early 63 protein interacts with human antisilencing function 1 protein and alters its ability to bind histones h3.1 and h3.3. J. Virol. 83:200–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cliffe AR, Knipe DM. 2008. Herpes simplex virus ICP0 promotes both histone removal and acetylation on viral DNA during lytic infection. J. Virol. 82:12030–12038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cunningham C, Davison AJ. 1993. A cosmid-based system for constructing mutants of herpes simplex virus type 1. Virology 197:116–124 [DOI] [PubMed] [Google Scholar]
- 5. Deshmane S, Fraser NW. 1989. During latency, herpes simplex virus type 1 DNA is associated with nucleosomes in a chromatin structure. J. Virol. 63:943–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Eitoku M, Sato L, Senda T, Horikoshi M. 2008. Histone chaperones: 30 years from isolation to elucidation of the mechanisms of nucleosome assembly and disassembly. Cell. Mol. Life Sci. 65:414–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Emili A, Schieltz DM, Yates JR, III, Hartwell LH. 2001. Dynamic interaction of DNA damage checkpoint protein Rad53 with chromatin assembly factor Asf1. Mol. Cell 7:13–20 [DOI] [PubMed] [Google Scholar]
- 8. Franco AA, Lam WM, Burgers PM, Kaufman PD. 2005. Histone deposition protein Asf1 maintains DNA replisome integrity and interacts with replication factor C. Genes Dev. 19:1365–1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gibson W, Roizman B. 1971. Compartmentalization of spermine and spermidine in the herpes simplex virion. Proc. Natl. Acad. Sci. U. S. A. 68:2818–2821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Griffith JP, Griffith DL, Rayment I, Murakami WT, Caspar DL. 1992. Inside polyomavirus at 25-A resolution. Nature 355:652–654 [DOI] [PubMed] [Google Scholar]
- 11. Groth A, et al. 2007. Regulation of replication fork progression through histone supply and demand. Science 318:1928–1931 [DOI] [PubMed] [Google Scholar]
- 12. Gu H, Liang Y, Mandel G, Roizman B. 2005. Components of the REST/CoREST/histone deacetylase repressor complex are disrupted, modified, and translocated in HSV-1-infected cells. Proc. Natl. Acad. Sci. U. S. A. 102:7571–7576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gu H, Roizman B. 2007. Herpes simplex virus-infected cell protein 0 blocks the silencing of viral DNA by dissociating histone deacetylases from the CoREST-REST complex. Proc. Natl. Acad. Sci. U. S. A. 104:17134–17139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hancock MH, Cliffe AR, Knipe DM, Smiley JR. 2010. Herpes simplex virus VP16, but not ICP0, is required to reduce histone occupancy and enhance histone acetylation on viral genomes in U2OS osteosarcoma cells. J. Virol. 84:1366–1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Harland J, Brown SM. 1998. HSV growth, preparation, and assay. Methods Mol. Med. 10:1–8 [DOI] [PubMed] [Google Scholar]
- 16. Haushalter KA, Kadonaga JT. 2003. Chromatin assembly by DNA-translocating motors. Nat. Rev. Mol. Cell Biol. 4:613–620 [DOI] [PubMed] [Google Scholar]
- 17. Henikoff S. 2008. Nucleosome destabilization in the epigenetic regulation of gene expression. Nat. Rev. Genet. 9:15–26 [DOI] [PubMed] [Google Scholar]
- 18. Herrera FJ, Triezenberg SJ. 2004. VP16-dependent association of chromatin-modifying coactivators and underrepresentation of histones at immediate-early gene promoters during herpes simplex virus infection. J. Virol. 78:9689–9696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hu F, Alcasabas AA, Elledge SJ. 2001. Asf1 links Rad53 to control of chromatin assembly. Genes Dev. 15:1061–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huang J, et al. 2006. Trimethylation of histone H3 lysine 4 by Set1 in the lytic infection of human herpes simplex virus 1. J. Virol. 80:5740–5746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kent JR, et al. 2004. During lytic infection herpes simplex virus type 1 is associated with histones bearing modifications that correlate with active transcription. J. Virol. 78:10178–10186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Knipe DM, Cliffe A. 2008. Chromatin control of herpes simplex virus lytic and latent infection. Nat. Rev. Microbiol. 6:211–221 [DOI] [PubMed] [Google Scholar]
- 23. Krebs A, Tora L. 2009. Keys to open chromatin for transcription activation: FACT and Asf1. Mol. Cell 34:397–399 [DOI] [PubMed] [Google Scholar]
- 24. Kutluay SB, Doroghazi J, Roemer ME, Triezenberg SJ. 2008. Curcumin inhibits herpes simplex virus immediate-early gene expression by a mechanism independent of p300/CBP histone acetyltransferase activity. Virology 373:239–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kutluay SB, Triezenberg SJ. 2009. Regulation of histone deposition on the herpes simplex virus type 1 genome during lytic infection. J. Virol. 83:5835–5845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kutluay SB, Triezenberg SJ. 2009. Role of chromatin during herpesvirus infections. Biochim. Biophys. Acta 1790:456–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lacasse JJ, Schang LM. 2010. During lytic infections, herpes simplex virus type 1 DNA is in complexes with the properties of unstable nucleosomes. J. Virol. 84:1920–1933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Leinbach SS, Summers WC. 1980. The structure of herpes simplex virus type 1 DNA as probed by micrococcal nuclease digestion. J. Gen. Virol. 51:45–59 [DOI] [PubMed] [Google Scholar]
- 29. Lentine AF, Bachenheimer SL. 1990. Intracellular organization of herpes simplex virus type 1 DNA assayed by staphylococcal nuclease sensitivity. Virus Res. 16:275–292 [DOI] [PubMed] [Google Scholar]
- 30. Lu X, Triezenberg SJ. 2010. Chromatin assembly on herpes simplex virus genomes during lytic infection. Biochim. Biophys. Acta 1799:217–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Luco RF, Allo M, Schor IE, Kornblihtt AR, Misteli T. 2011. Epigenetics in alternative pre-mRNA splicing. Cell 144:16–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mello JA, et al. 2002. Human Asf1 and CAF-1 interact and synergize in a repair-coupled nucleosome assembly pathway. EMBO Rep. 3:329–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Miller A, Yang B, Foster T, Kirchmaier AL. 2008. Proliferating cell nuclear antigen and ASF1 modulate silent chromatin in Saccharomyces cerevisiae via lysine 56 on histone H3. Genetics 179:793–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mounce BC, Tsan FC, Kohler S, Cirillo LA, Tarakanova VL. 2011. Dynamic association of gammaherpesvirus DNA with core histone during de novo lytic infection of primary cells. Virology 421:167–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mousson F, Ochsenbein F, Mann C. 2007. The histone chaperone Asf1 at the crossroads of chromatin and DNA checkpoint pathways. Chromosoma 116:79–93 [DOI] [PubMed] [Google Scholar]
- 36. Muggeridge MI, Fraser NW. 1986. Chromosomal organization of the herpes simplex virus genome during acute infection of the mouse central nervous system. J. Virol. 59:764–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Munakata T, Adachi N, Yokoyama N, Kuzuhara T, Horikoshi M. 2000. A human homologue of yeast anti-silencing factor has histone chaperone activity. Genes Cells 5:221–233 [DOI] [PubMed] [Google Scholar]
- 38. Nitzsche A, Paulus C, Nevels M. 2008. Temporal dynamics of cytomegalovirus chromatin assembly in productively infected human cells. J. Virol. 82:11167–11180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Oh J, Fraser NW. 2008. Temporal association of the herpes simplex virus genome with histone proteins during a lytic infection. J. Virol. 82:3530–3537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Park YJ, Luger K. 2008. Histone chaperones in nucleosome eviction and histone exchange. Curr. Opin. Struct. Biol. 18:282–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Peng H, Nogueira ML, Vogel JL, Kristie TM. 2010. Transcriptional coactivator HCF-1 couples the histone chaperone Asf1b to HSV-1 DNA replication components. Proc. Natl. Acad. Sci. U. S. A. 107:2461–2466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Placek BJ, Berger SL. 2010. Chromatin dynamics during herpes simplex virus-1 lytic infection. Biochim. Biophys. Acta 1799:223–227 [DOI] [PubMed] [Google Scholar]
- 43. Placek BJ, et al. 2009. The histone variant H3.3 regulates gene expression during lytic infection with herpes simplex virus type 1. J. Virol. 83:1416–1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Roitman-Shemer V, Stokrova J, Forstova J, Oppenheim A. 2007. Assemblages of simian virus 40 capsid proteins and viral DNA visualized by electron microscopy. Biochem. Biophys. Res. Commun. 353:424–430 [DOI] [PubMed] [Google Scholar]
- 45. Sanematsu F, et al. 2006. Asf1 is required for viability and chromatin assembly during DNA replication in vertebrate cells. J. Biol. Chem. 281:13817–13827 [DOI] [PubMed] [Google Scholar]
- 46. Schulz LL, Tyler JK. 2006. The histone chaperone ASF1 localizes to active DNA replication forks to mediate efficient DNA replication. FASEB J. 20:488–490 [DOI] [PubMed] [Google Scholar]
- 47. Sillje HH, Nigg EA. 2001. Identification of human Asf1 chromatin assembly factors as substrates of Tousled-like kinases. Curr. Biol. 11:1068–1073 [DOI] [PubMed] [Google Scholar]
- 48. Tagami H, Ray-Gallet D, Almouzni G, Nakatani Y. 2004. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell 116:51–61 [DOI] [PubMed] [Google Scholar]
- 49. Tang Y, et al. 2006. Structure of a human ASF1a-HIRA complex and insights into specificity of histone chaperone complex assembly. Nat. Struct. Mol. Biol. 13:921–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Umehara T, Horikoshi M. 2003. Transcription initiation factor IID-interactive histone chaperone CIA-II implicated in mammalian spermatogenesis. J. Biol. Chem. 278:35660–35667 [DOI] [PubMed] [Google Scholar]
- 51. van Leeuwen H, et al. 2003. Herpes simplex virus type 1 tegument protein VP22 interacts with TAF-I proteins and inhibits nucleosome assembly but not regulation of histone acetylation by INHAT. J. Gen. Virol. 84:2501–2510 [DOI] [PubMed] [Google Scholar]
- 52. Zhang R, et al. 2005. Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev. Cell 8:19–30 [DOI] [PubMed] [Google Scholar]