

Abstract
Paramyxoviruses use a specialized fusion protein to merge the viral envelope with cell membranes and initiate infection. Most paramyxoviruses require the interaction of two viral proteins to enter cells; an attachment protein binds cell surface receptors, leading to the activation of a fusion (F) protein that fuses the viral envelope and host cell plasma membrane. In contrast, human metapneumovirus (HMPV) expressing only the F protein is replication competent, suggesting a primary role for HMPV F in attachment and fusion. We previously identified an invariant arginine-glycine-aspartate (RGD) motif in the HMPV F protein and showed that the RGD-binding integrin αVβ1-promoted HMPV infection. Here we show that both HMPV F-mediated binding and virus entry depend upon multiple RGD-binding integrins and that HMPV F can mediate binding and fusion in the absence of the viral attachment (G) protein. The invariant F-RGD motif is critical for infection, as an F-RAE virus was profoundly impaired. Further, F-integrin binding is required for productive viral RNA transcription, indicating that RGD-binding integrins serve as receptors for the HMPV fusion protein. Thus, HMPV F is triggered to induce virus-cell fusion by interactions with cellular receptors in a manner that is independent of the viral G protein. These results suggest a stepwise mechanism of HMPV entry mediated by the F protein through its interactions with cellular receptors, including RGD-binding integrins.
INTRODUCTION
Enveloped virus surface proteins attach to cell surface receptors and fuse viral membranes with cell membranes during entry. Several unrelated enveloped viruses, including influenza virus, human immunodeficiency virus (HIV), and paramyxoviruses, use class I viral fusion proteins to induce membrane fusion. Class I fusion proteins initiate fusion by springing open to insert a hydrophobic fusion peptide into the cell membrane, creating a molecular bridge between the viral and cellular membranes, which are merged by fusion protein refolding (8, 19). Although all class I fusion proteins appear to use this spring-loaded mechanism, each virus family has adapted different strategies for triggering fusion. Paramyxoviruses encode two viral proteins, an attachment protein and a fusion protein, both of which are typically necessary for fusion. Paramyxovirus attachment and fusion are tightly connected events, such that attachment protein binding to cell surface receptors activates the fusion protein to induce fusion at the cell membrane (1, 9, 18, 20, 23, 25, 28–30, 36). This does not appear to be the mechanism used by the members of the Pneumovirinae subfamily of paramyxoviruses, which includes two important human respiratory viruses: human metapneumovirus (HMPV) and human respiratory syncytial virus (hRSV) (34). HMPV and hRSV encode a separate attachment (G) protein; however, viruses with only the fusion protein on the surface are replication competent in vitro and in vivo, although HMPV lacking G is attenuated in primates (4, 5, 17). Thus, the HMPV fusion (F) protein can mediate both attachment and membrane fusion, suggesting that F binds to specific cell surface receptors during attachment, drives fusion, and mediates virus entry in a manner that supports productive infection. In contrast to the current model of paramyxovirus fusion, where attachment protein binding is coupled to fusion activity of a separate fusion protein, the mechanism by which Pneumovirinae fusion proteins, including the HMPV F protein, bind to receptors and induce fusion is a mystery.
We previously identified an invariant arginine-glycine-aspartate (RGD) motif that was unique to HMPV F among human paramyxoviruses. This discovery led us to hypothesize that integrins may serve as receptors for HMPV F. Integrins are cell surface adhesion receptors composed of one α subunit and one β subunit; 18 α subunits and 8 β subunits combine to form 24 distinct heterodimers. A subset of integrins, αVβ1, αVβ3, αVβ5, αVβ6, αVβ8, α5β1, α8β1, and αIIbβ3, bind proteins with RGD motifs (16), and several other viruses with conserved RGD motifs bind integrins to mediate entry (reviewed in reference 35). We previously demonstrated that HMPV infection depends upon RGD-binding integrins and suggested that HMPV F utilizes αVβ1 integrin as a receptor during entry (11). However, whether an F-RGD interaction was sufficient for HMPV binding or whether HMPV F attachment to RGD-binding integrins was linked to fusion activity remained unclear.
We hypothesized that HMPV F binding to RGD-binding integrins was necessary for virus entry and that integrin binding triggered fusion. To test this hypothesis, we developed assays to measure HMPV binding and fusion. Here we show that HMPV binds to RGD-binding integrins and that this interaction is necessary for virus attachment, viral RNA transcription, and subsequent infection. Multiple RGD-binding integrins are capable of mediating HMPV attachment, and the F protein RGD motif is required for productive infection. While HMPV F-integrin binding is required for efficient virus entry, F binding to RGD-binding integrins is not sufficient to initiate virus-cell membrane fusion. HMPV hemifusion proceeds efficiently both during RGD-binding integrin blockade and in the absence of G protein. We propose that HMPV entry is a stepwise process whereby HMPV F mediates entry through its interactions with RGD-binding integrins and other unidentified cell surface receptors, eliminating the absolute requirement for an additional viral attachment protein.
MATERIALS AND METHODS
Cells.
BEAS-2B (ATCC CRL-9609) and LLC-MK2 (ATCC CCL-7) cells were maintained in Opti-MEM I (Invitrogen) containing 2% fetal bovine serum (FBS), 2 mM l-glutamine, 50 μg/ml gentamicin, and 2.5 μg/ml amphotericin B. Suspension 293-F cells were maintained as recommended by the manufacturer (293 Freestyle expression system; Invitrogen). BSR T7/5 cells that constitutively express T7 RNA polymerase (6) were kindly provided by Ursula Buchholz and maintained in Dulbecco modified Eagle medium (DMEM) supplemented with 10% FBS, 2 mM l-glutamine, antimicrobials as described above, and 1 mg/ml Geneticin.
Antibodies.
Anti-human integrin monoclonal antibodies (MAbs) MAb2021Z (αv; clone AV1), MAb1957 (β3; clone 25E11), MAb1976Z (αVβ3; clone LM609), and MAb1950 (α2; clone P1E6) were purchased from Millipore. Anti-human integrin MAb AIIB2 (β1) and BIIG2 (α5) developed by Caroline H. Damsky were obtained from the Developmental Studies Hybridoma Bank under the auspices of the NICHD and maintained by the University of Iowa, Department of Biology, Iowa City, IA. A heat-inactivated polyclonal HMPV-immune human serum, which neutralizes HMPV infection in an in vitro plaque neutralization assay (50% inhibitory concentration [IC50], 1:1,500), was used in fusion experiments. The HMPV F-specific MAb, DS7, used for immunoprecipitation experiments has been previously described (39). HMPV F-specific MAb 1017 (38) and a rabbit polyclonal antiserum generated against a synthetic peptide representing residues 131 to 145 of the HMPV TN/94-49 M protein were used for Western blot analyses.
Viruses.
HMPV strain TN/94-49 (virus genotype A2) was used for all virus binding and fusion experiments. HMPV TN/94-49 is a clinical isolate passaged 5 to 7 times and thrice plaque purified in LLC-MK2 cells. Stock virus was propagated using LLC-MK2 cells in serum-free growth medium supplemented with 5 μg/ml trypsin (Invitrogen) as described previously (40). Working virus was propagated in suspension 293-F cells in 293 Freestyle expression medium supplemented with 5 μg/ml trypsin-EDTA (both from Invitrogen). Briefly, 293-F cells were inoculated at 0.1 PFU/cell. At 4 days postinoculation, supernatant was collected and infected cells were freeze-thawed thrice, clarified by centrifugation at 300 × g for 5 min, and added to the supernatant fraction. This crude virus preparation was further purified through a 20% sucrose cushion via ultracentrifugation at 100,000 × g for 90 min at 4°C. Octadecyl rhodamine B chloride (R18)-labeled HMPV (R18-MPV) was prepared by incubating 10 mM R18 dissolved in ethanol with sucrose-purified HMPV at 20 nmol R18 per mg total virus protein for 1 h at room temperature. The reaction mixture was pelleted through a discontinuous sucrose gradient by ultracentrifugation at 100,000 × g for 90 min at 4°C. Labeled virus (pink by visible light) was collected at the 20/60% sucrose interface, snap-frozen in dry ice-alcohol, and stored at −80°C. All virus preparations were titrated on LLC-MK2 and BEAS-2B cells as previously described (40). Heat-inactivated virus was prepared by heating R18-MPV at 70°C for 15 min.
Recovery of recombinant HMPV with F-RAE mutation.
The HMPV reverse genetics system designed and kindly provided by Ron Fouchier was used (13). Nucleotides 4054 to 4059 in the viral genome plasmid FLG NL00-1 were changed from GGAGAC to GCAGAG by site-directed mutagenesis using QuikChange II (Stratagene) and confirmed by DNA sequencing. This genetic mutation changes the invariant F-RGD motif to F-RAE in the translated fusion protein. We chose the RAE mutation based upon observations from other viruses that utilize RGD binding motifs during virus entry, where often only RGD revertants can be recovered. Thus, we decided to make conservative mutations to two amino acids in the motif, since no comparable amino acids substitute well for the Arg unique structure. Wild-type (F-wt; unmodified FLG NL00-1 genome) and F-RAE viruses were rescued in BSR T7/5 cells using the previously described method (13), except LLC-MK2 cells were used in lieu of Vero cells and viruses were harvested at 13 to 15 days postinfection of LLC-MK2 monolayers. Because the reverse genetics system relies on plasmid-carried genomes, individual viral clones were recovered in replicate wells from three independent experiments. We rescued four F-RAE virus clones and four F-wt clones as a positive control. The viral genome was extracted from centrifuge-clarified virus preparations with the RNeasy minikit (Qiagen) according to the manufacturer's protocol. A nested PCR amplification strategy was used to amplify the F protein open reading frame, with flanking intragenic genomic sequences. Primers for the nested PCR amplification of F have been previously described (41). Reverse transcription-PCR (RT-PCR) bands were gel purified, subcloned into pGEM (Promega), and sequenced to confirm that the desired genetic mutation was maintained and no other mutations had been introduced during virus propagation.
VLPs.
HMPV virus-like particles (VLPs) were generated in suspension 293-F cells by transient expression of the HMPV matrix (M), fusion (F), and glycoprotein (G) proteins. Cloning and sequence optimization for mammalian expression of the full-length F sequence from a pathogenic clinical HMPV isolate, TN/92-4 (virus genotype A2), have been previously described (12). The full-length M and G sequences from TN/94-49 (virus genotype A2) were also sequence optimized (GeneArt) and subcloned into pcDNA3.1. Suspension 293-F cells were transfected with pcDNA3.1-M (40 μg), pcDNA3.1-F (10 μg), and either empty vector (pcDNA3.1) (10 μg) for F-VLPs or pcDNA3.1-G (10 μg) for F+G-VLPs using 293fectin transfection reagent (60 μl) as recommended by the manufacturer (Invitrogen). G-VLP producer cells were transfected with pcDNA3.1-M (40 μg), empty vector (pcDNA3.1) (10 μg), and pcDNA3.1-G (10 μg). At 18 hours posttransfection, growth medium was changed and 5 μg/ml trypsin was added. At 3 days posttransfection, cells and debris were separated from the supernatant by centrifugation at 300 × g for 5 min. Clarified supernatant was pelleted through 20% sucrose as described above for virus. White VLP pellets were resuspended in MHN solution (0.1 M MgSO4, 50 mM HEPES, 150 mM NaCl), R18 labeled as described for virus, and stored at −80°C.
Western blotting.
For the analysis presented in Fig. 5C, sucrose-purified HMPV or VLPs were diluted in phosphate-buffered saline (PBS), immunoprecipitated with 2 μg of an F-specific MAb (DS7) (39) for 4 h, and captured on protein G-agarose beads (Sigma). Beads were washed thrice with PBS and heated to 70°C for 10 min in NuPAGE LDS sample buffer (Invitrogen) containing 5% β-mercaptoethanol (Sigma). Proteins were separated on 10% NuPAGE bis-Tris gels (Invitrogen) and transferred to polyvinylidene fluoride (PVDF) membranes. Membranes were blocked for 1 h with 5% milk in PBS plus 0.1% Tween (blocking buffer). Polyclonal guinea pig antiserum, which was generated against sucrose-purified HMPV (TN/94-49) and recognizes both HMPV F and G proteins, and rabbit polyclonal anti-M serum were diluted in blocking buffer and incubated with membranes for 12 to 14 h at 4°C. Li-Cor IRDye 800CW anti-guinea pig and IRDye 680CW anti-rabbit secondary antibodies were diluted in blocking buffer and incubated with membranes for 1 h at room temperature. Membranes were washed three times in PBS plus 0.1% Tween and twice in PBS to remove residual detergent and dried. Bands were imaged and quantified using an Odyssey infrared imaging system (Li-Cor). For the analysis shown in Fig. 8G, producer cell lysates were prepared in lysis buffer containing 20 mM Tris (pH 7.4), 150 mM NaCl, 1 mM EDTA, and 1% NP-40 supplemented with a complete protease inhibitor cocktail (Roche). Total protein concentrations for cell lysates and sucrose-purified, R18-labeled VLPs were determined with the Bio-Rad DC protein assay (Bio-Rad). Proteins were separated by SDS-PAGE, transferred to PVDF membranes, and incubated with HMPV F-specific MAb 1017 and Cy5-conjugated anti-Armenian hamster secondary antibody (Jackson ImmunoResearch) as described above.
Fig 5.

HMPV F binds to RGD-binding integrins in the absence of G. (A) Electron micrographs of HMPV and F-only and F+G virus-like particles (VLPs). Sucrose-purified particles were stained with uranyl formate and imaged on an FEI Morgagni electron microscope (magnification, ×28,000). Protein spikes are visible projecting from the surfaces of both virus and VLPs. (B) Ten to 30 particles were chosen for morphology analysis; diameters and glycoprotein spike lengths were measured using AMT Image Capture Engine software. (C) Viral matrix (M), fusion (F), and attachment (G) protein incorporation into VLPs was confirmed by Western blotting. Virus and VLPs were immunoprecipitated with an anti-F monoclonal antibody (2 μg) and analyzed by Western blotting. Viral proteins were detected with HMPV M-specific or HMPV polyclonal A2 virus-specific (F and G) antibodies and fluorescent secondary antibodies using the Li-Cor Odyssey infrared imaging system. Lanes: 1, mock; 2, HMPV; 3, F-VLP; 4, F+G-VLP. Uncleaved protein (F0) and the large subunit from the cleaved form (F1) of the fusion protein were detected. M bands were detected in a different channel than F and G bands on the same blot and thus appear on a different image. (D) R18-MPV or R18-VLP binding was measured in the absence or presence of integrin function-blocking antibodies (α2 or a combination of αV, α5, and β1 to block all available RGD-binding integrins). Results from three independent experiments are expressed as mean percent inhibition relative to an untreated control (no MAb). Error bars indicate SEM; *, P ≤ 0.05.
Fig 8.

HMPV fusion is not triggered by HMPV G or RGD-binding integrins. (A) R18 F-VLP fusion was measured in the absence (circles) or presence (triangles) of neutralizing HMPV antiserum or for heat-inactivated particles (squares). R18-G-VLP (diamonds) dequenching was monitored as a background control. Percent R18 dequenching was calculated as described in Materials and Methods, and curves represent means for three independent experiments monitored for triplicate wells. Error bars indicate SEM. (B and C) HMPV G does not alter HMPV F-mediated fusion kinetics or the extent of fusion at 4 h. R18-labeled HMPV (black circles), F-VLPs (gray squares), or F+G-VLPs (open squares) were bound to the surface of BEAS-2B cells, and R18 fluorescence was monitored for 4 h. Triton X-100 was added after 4 h to determine the extent of virus or VLP fusion. Curves in panel B represent mean percent R18 dequenching for three independent experiments, monitored for duplicate wells. Bars in panel C represent the extent of fusion observed after 4 h for three independent experiments; error bars indicate SEM. *, P < 0.05; N.S., P > 0.05. (D to F) HMPV F binding to RGD-binding integrins does not alter HMPV fusion kinetics. R18-labeled HMPV (D), F-VLP (E), or F+G-VLP (F) fusion was assessed in the absence of antibodies (black lines) or in the presence of integrin function-blocking antibodies against α2 integrins (gray lines) or all RGD-binding integrins (αV plus α5 plus β1) (dotted lines). Curves represent mean percent R18 dequenching for three independent experiments monitored for duplicate wells. Error bars are not shown for figure clarity. Dequenching rates were not significantly altered in the presence of integrin antibodies; however, significantly less HMPV and VLPs bound during RGD-binding integrin blockade (data not shown). (G) F-RGD (wt) VLPs (10 μg), F-RAE VLPs (10 μg), and producer cell lysates (50 μg) were analyzed by Western blotting. Uncleaved (F0) and cleaved (F1) HMPV F were detected with an F-specific MAb and fluorescent secondary antibody using the Li-Cor Odyssey infrared imaging system. C, untransfected 293-F cell lysate. (H) The RGD integrin-binding motif is not required for efficient F-mediated hemifusion. R18 VLP fusion was measured for F-RGD (circles), F-RAE (triangles), or G-only (squares) particles as described for panel A.
Flow cytometry.
BEAS-2B cells were harvested and incubated with either isotype control or anti-human integrin antibodies, followed by Alexa Fluor 647-conjugated secondary antibodies (Invitrogen). Expression levels of different integrins were detected with a BD LSRII flow cytometer.
Quantifying HMPV infection.
At 24 hours postinoculation, BEAS-2B cell monolayers were fixed with buffered formalin (3.3% in growth medium) and immunostained for HMPV infection with a precipitating peroxidase substrate (True Blue; KPL) as previously described (11). Individual wells were imaged on a light box with a macro zoom lens (Navitar, 18 to 108 mm), and infected cells, which appear blue, were enumerated using ImageJ. Cells in entire wells were counted. The image contrast threshold was held constant during counting for all wells in a single plate.
Visualizing HMPV plaques by microscopy.
Serial dilutions of F-wt or F-RAE viruses were adsorbed to LLC-MK2 cell monolayers for 1 h at room temperature. Cells were overlaid with Opti-MEM I medium containing 0.75% methylcellulose and 5 μg/ml trypsin and incubated at 37°C with 5% CO2 for 4 days. Cell monolayers were fixed and immunostained, and plaques were counted. Cell images were captured with a Zeiss Axiovert 200 microscope using a 2.5× or 10× objective.
Electron microscopy.
Sucrose-purified HMPV and VLPs were stained with uranyl formate and prepared for transmission electron microscopy (TEM) as described previously (26). Briefly, 2.5 μl of particles was adsorbed to a glow-discharged 200-mesh copper grid covered with carbon-coated collodion film. The grid was washed with water and stained with uranyl formate (0.75%). Samples were imaged on a FEI Morgagni electron microscope operated at an acceleration voltage of 100 kV. Images were recorded at a magnification of 28,000 and collected using a 1,000 by 1,000 charge-coupled device (CCD) camera (AMT). Ten to 30 representative HMPV, F-VLP, or F+G-VLPs were chosen for morphology analysis. Measurements of particle diameters and glycoprotein spike length were made using AMT Image Capture Engine software.
Fluorescence microscopy.
Viral fusion was monitored by fluorescence microscopy as R18-MPV fused with live cells. BEAS-2B cells, grown to ∼90% confluence on a thin layer of Matrigel (BD Biosciences) in 6-well plates, were incubated with 25 μg/ml 4′,6-diamidino-2-phenylindole (DAPI) in culture medium for 1 h at 37°C. Excess DAPI was washed away, and cells were incubated with R18-MPV (0.2 PFU/cell) for 1 h on ice. Unbound virus was washed away, and Opti-MEM I medium (without phenol red) supplemented with 2% FBS was added to cells. The zero time point was imaged with cells at 4°C. Virus fusion was initiated by incubating cells at 37°C. Live cell images were captured with a Zeiss Axiovert 200 microscope using a 40× objective with 359/461-nm (DAPI) and 556/573-nm (R18) filters at the indicated time points.
R18-MPV binding assay.
To study HMPV binding, we developed a fluorescence-based assay to quantify binding independently of fusion and infection, allowing us to discriminate the first critical step in the entry process. We loaded HMPV particles with self-quenching concentrations of octadecyl rhodamine B chloride (R18-MPV) (15). To measure binding, R18-MPV was bound to cells and subsequently solubilized with detergent, resulting in fluorescence proportional to the amount of bound virus. Specifically, R18-MPV (0.02 PFU/cell) was incubated with BEAS-2B cells grown to 90 to 95% confluence on a thin layer of Matrigel (BD Biosciences) in 48-well plates for 1 h on ice. Cells were washed with ice-cold PBS to remove unbound virus. PBS plus 1% Triton X-100 was added to each well, and fluorescence (excitation, 544 nm; emission, 590 nm) was measured using a SpectraMax M5 (Molecular Devices) plate reader. For dose-response experiments (results in Fig. 1), serial 2-fold dilutions of 2 × 103 PFU were incubated with cell monolayers prior to detecting virus binding (reported as fluorescence intensity) or infectivity. For function-blocking antibody experiments, integrin-specific antibodies were diluted in Opti-MEM I plus 5% FBS and incubated with BEAS-2B cell monolayers for 1 h at 37°C, followed by 30 min on ice, before addition of ice-cold virus inoculum. Binding was expressed as mean percent inhibition relative to an untreated control. For infectivity experiments, instead of detergent, growth medium was added and cells were incubated at 37°C with 5% CO2. At 24 hours postinoculation, cell monolayers were fixed with buffered formalin and immunostained for HMPV infection as previously described (11). Infected cells were enumerated as described above, and infectivity was expressed as mean percent inhibition relative to an untreated control. R18-VLP experiments were conducted as described for R18-MPV.
Fig 1.

A fluorescence-based assay to quantify HMPV binding. (A) Schematic of HMPV binding assay. R18-MPV was bound to cells on ice to prevent virus fusion. Adding detergent chemically dequenched the R18 dye associated with virus membranes, resulting in measurable fluorescence proportional to the amount of virus bound to cells. (B) R18-MPV binding to human bronchial epithelial (BEAS-2B) cells results in a linear increase in fluorescence (slope = 144.1 ± 8.8; r2 = 0.9817). Binding was measured as fluorescence of cell-bound virus after addition of 1% Triton X-100. Results are means ± standard errors of the means (SEM) for three independent experiments performed in triplicate. (C) R18-MPV binding correlates with infectivity (slope = 0.009 ± 0.0004; r2 = 0.9910). R18-MPV binding was assessed as for panel B; identical wells were incubated at 37°C in culture medium, and HMPV-infected cells were enumerated at 24 h postbinding as described in Materials and Methods. Results are means ± SEM for three independent experiments performed in triplicate.
R18-MPV fusion assay.
To measure fusion, we incubated cells with R18-MPV at 4°C, washed away unbound virus, added fresh cell culture medium, and incubated cells at 37°C, monitoring R18 fluorescence in real time. During virus-cell fusion, R18 dilutes from the virus membrane into unlabeled cell membranes, resulting in a measureable increase in fluorescence that reflects the extent of virus fusion. This assay allowed us to monitor HMPV fusion while virus and cell membranes were in the process of merging. While developing the assay, we tested various plate reader settings and found that top-read and bottom-read fluorescence readings generated equivalent R18 dequenching curves during R18-MPV fusion with BEAS-2B cells. HMPV fusion kinetics were similar when monitored in either clear 48-well plates or black (opaque) transparent-bottom 96-well plates, indicating that cross talk between adjacent wells did not significantly affect fluorescence readings for this assay. Background fluorescence during experiments measured with top-read mode (excitation, 544 nm; emission, 590 nm) was typically 20-fold lower than the initial R18 fluorescence reading (see Fig. 7B). Moreover, R18-MPV dequenching kinetics were equivalent over a 30-fold range of virus binding, despite declining fluorescence signals for lower multiplicities of infection (MOIs).
Fig 7.

HMPV fusion occurs slowly over the course of several hours. (A) R18-MPV fusion with live, adherent human bronchial epithelial (BEAS-2B) cells. R18 fluorescence increases over time as R18 dye self-quenched in the virus membrane dilutes into cellular membranes during virus-mediated membrane fusion. The zero time point was imaged before incubating cells at 37°C to initiate virus fusion. Live cell images were captured with a Zeiss Axiovert 200 microscope using a 40× objective with 359/461-nm (DAPI) and 556/573-nm (R18) filters at the indicated time points. (B) R18-MPV fusion was measured with a plate reader. Curves represent fluorescence of cells (gray diamonds), cells plus R18-MPV at 4°C (blue squares), or cells plus R18-MPV at 37°C (red circles). Results for triplicate wells (mean ± standard deviation [SD]) from a representative experiment are shown. (C) R18-MPV fusion was measured in the absence (red circles) or presence (green triangles) of neutralizing HMPV antiserum (dilution, 1:40) or for heat-inactivated virus (black squares). Percent R18 dequenching was calculated as described in Materials and Methods. Curves represent mean percent R18 dequenching for three independent experiments, monitored for triplicate wells. Error bars indicate SEM.
For the experiments described here, R18-MPV (0.1 PFU/cell) was bound to BEAS-2B cells grown to 90 to 95% confluence on a thin layer of Matrigel (BD Biosciences) in 48-well plates for 1 h on ice. Unbound virus was removed by washing with ice-cold PBS before Opti-MEM I (without phenol red) supplemented with 2%FBS was added to cells. Ice-cold plates were transferred to a preheated (37°C) SpectraMax M5 plate reader. Fluorescence (excitation, 544 nm; emission, 590 nm; top-read mode) was measured in real time for 2 h with readings collected every 2 min or for 4 h with readings in 5-min intervals. At the end of the time course, Triton X-100 (final concentration of 1%) was added to each well, and final fluorescence readings were acquired. R18-VLP experiments were conducted as described for R18-MPV. Function-blocking integrin antibody experiments were performed as described above. The ability of bound R18-MPV or R18-VLPs to subsequently undergo fusion was defined as R18 dequenching and calculated according to the following equation: % R18 dequenching = 100 × (F − F0)/(Fd − F0), where F is fluorescence, F0 is fluorescence at t = 0 min, and Fd is fluorescence after the addition of Triton X-100 (at the end of the experiment).
Real-time RT-PCR.
Function-blocking integrin-specific antibodies (anti-α2 [10 μg/ml] or a combination of anti-αV [40 μg/ml], anti-α5 [6 μg/ml], and anti-β1 [7.5 μg/ml]) were diluted in Opti-MEM I and incubated with 90% confluent BEAS-2B cells grown on a thin layer of Matrigel in 48-well plates for 1 h at 37°C and then for 30 min on ice. Cells incubated with Opti-MEM I only (no MAb) were used as the negative control. HMPV (0.25 PFU/cell) was added to cells and allowed to bind for 1 h on ice with occasional rocking. Cells were washed twice with ice-cold PBS to remove unbound virus. Half of the wells were used to determine the input viral genome (lysed immediately [t = 0]), and half of the wells were incubated in Opti-MEM I supplemented with 2% FBS for 8 h at 37°C to allow virus entry and transcription. At t = 0 or 8 h, cells were washed with PBS to remove medium, and lysates were prepared in 350 μl of MagNA Pure LC total nucleic acid lysis/binding buffer (Roche Applied Sciences) and stored at 4°C. RNA was extracted using the MagNA Pure LC total nucleic acid isolation kit (Roche Applied Sciences) on a MagNA Pure LC using the total NA external lysis protocol and stored at −80°C until further use. Real-time RT-PCR was performed in 25-μl reaction mixtures containing 5 μl of extracted RNA on an ABI StepOnePlus real-time PCR system (Life Technologies/Applied Biosystems) using the AgPath-ID one-step RT-PCR kit (Applied Biosystems/Ambion). Primers and probe targeting the HMPV N gene have been previously published (2) and were used to detect HMPV genome and transcripts. Cycling conditions were 50°C for 30 min, followed by an activation step at 95°C for 10 min and then 45 cycles of 15 s at 95°C and 30 s at 60°C. All sample cycle threshold (CT) values were less than 26 and were considered positive; the HMPV N CT values for mock-infected cells were undetectable at >45. To determine the amount of HMPV N transcript for each treatment condition, values were normalized to the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) housekeeping gene and compared to input genome levels using the 2−ΔΔCT method, where ΔΔCT = (HMPV N CT8 − GAPDH CT8) − (HMPV N CT0 − GAPDH CT0) (3, 22, 27). The effect of integrin inhibitors was determined by comparing the fold change in HMPV N over time relative to the treatment control (no MAb).
Statistical analysis.
For binding and infectivity experiments, data are expressed as mean percent inhibition, relative to an untreated control, for at least three independent experiments performed in triplicate. Student's t test was used to determine whether the level of inhibition observed in the presence of integrin-blocking antibodies was significantly greater than that for the untreated control (no MAb). For the combination blockade experiments, a t test was used to determine whether the level of inhibition observed for one MAb differed from the level of inhibition observed when two MAbs were used to target different integrins during HMPV binding or infection. Virus and VLP fusion extents were compared using a one-way analysis of variance (ANOVA) and Tukey's multiple-comparison test. For all analyses, a P value of ≤0.05 was considered statistically significant.
RESULTS
HMPV binding and subsequent infection depend upon RGD-binding integrins.
We previously showed that RGD-binding integrins promote HMPV infection (11), but we did not determine whether integrin engagement was required for HMPV entry. Investigating HMPV entry presented methodological challenges because both attachment and fusion are necessary for virus entry; therefore, a complete understanding of virus entry required assays that quantify binding and measure fusion. As virus receptors, RGD-binding integrins could be required for HMPV F binding, fusion, or both steps in virus entry. First, we developed a fluorescence-based assay to quantify binding independently from fusion and infection, allowing us to discern virus attachment (see Materials and Methods). HMPV binding resulted in a dose-dependent increase in fluorescence that directly correlated with infectivity (Fig. 1), confirming that the assay measured productive virus binding to cells.
Next, we sought to determine whether HMPV binds RGD-binding integrins during virus attachment to human bronchial epithelial (BEAS-2B) cells. BEAS-2B cells express several RGD-binding integrin subunits, i.e., αV, α5, β1, β3, and β5, and the collagen-binding integrin subunit α2 (Fig. 2). Therefore, all the heterodimeric RGD-binding integrins on BEAS-2B cells contain αV, α5, and/or β1 integrin subunits. We blocked each individual integrin subunit with function-blocking antibodies and examined the effect on HMPV attachment and infectivity. We found that HMPV binding and infection were inhibited in a dose-dependent manner as a result of RGD-binding integrin blockade (Fig. 3). HMPV bound in an α5, β1, and αV integrin-dependent manner (Fig. 3C to E), but β3 MAb had no effect (Fig. 3B). Based upon the low expression level of β3 that we observed in our flow cytometry experiments despite high expression levels of the αVβ3 heterodimer (compare Fig. 2E and F), we were concerned that the β3 MAb might not efficiently bind αVβ3 integrin on the surface of BEAS-2B cells. Therefore, we tested whether αVβ3 function-blocking MAb (40 μg/ml) inhibited HMPV binding or infectivity and found that blocking αVβ3 had no effect (data not shown). HMPV binding and infection did not depend upon the collagen-binding integrin α2, as expected (Fig. 3A). These results indicate that HMPV can bind to different RGD-binding integrins, i.e., α5β1 and either αVβ1, αVβ5, αVβ6, or αVβ8, on BEAS-2B cells.
Fig 2.

Integrin expression on human bronchial epithelial (BEAS-2B) cells. BEAS-2B cells were stained for surface expression of specific integrins and analyzed by flow cytometry. Expression of α2 (A), αV (B), α5 (C), β1 (D), and β3 (E) integrin subunits or the αVβ3 heterodimer (F) is shown in shaded histograms, relative to an isotype control antibody (open histograms). The number in the upper right corner of each panel represents mean fluorescence intensity of integrin expression.
Fig 3.

HMPV binding and subsequent infection depend upon RGD-binding integrins. (A to E) HMPV binding (black circles) or infection at 24 h (gray squares) was determined in the absence or presence of α2 (A), β3 (B), α5 (C), β1 (D), or αV (E) integrin function-blocking antibodies. Results from at least three independent experiments performed in triplicate are expressed as mean percent inhibition relative to untreated control; error bars indicate SEM. *, P ≤ 0.05 from Student's t test, comparing antibody treatment to no treatment. (F) The highest concentrations of αV, β1, or α5 function-blocking antibodies were used alone or in combination to target multiple integrins, and HMPV binding (black bars) or infectivity (gray bars) was determined. Asterisks indicate an additive effect of combination blockade on HMPV infectivity (gray bars); Student's t test comparing two treatments, P ≤ 0.05.
To further explore whether HMPV engaged multiple RGD-binding integrins during entry, we measured HMPV binding while all RGD-binding integrins were blocked using combinations of MAbs. Blocking all RGD-binding integrins resulted in ∼40% reduced virus binding, comparable to the inhibition observed when individual integrins were blocked (Fig. 3F, αV + β1 and αV + α5). This suggests to us that HMPV is capable of engaging different RGD-binding integrins at the cell surface but that integrin-mediated attachment is saturable. Residual virus binding is mediated by other cell surface molecules that participate in initial HMPV attachment, such as heparan sulfate, which has recently been identified as critical for HMPV F attachment (7). Further, the HMPV G protein has been shown to bind cellular glycosaminoglycans (37) and likely contributes to HMPV binding, in an integrin-independent manner.
Blocking all RGD-binding integrins resulted in 90% reduction of HMPV infection, significantly more than when individual integrin subunits were blocked (Fig. 3F, αV + β1 and αV + α5). Thus, we observed an additive inhibitory effect on infectivity when all RGD-binding integrins were blocked during entry. The additive effect on HMPV infectivity was also observed when lower, nonsaturating concentrations of α5 and αV MAbs were used to block integrin receptors during HMPV entry (Fig. 4). These results suggest that HMPV is capable of engaging multiple RGD-binding integrins rather than a single, specific integrin during the entry process. Further, infection was inhibited to a greater degree than binding for all RGD-binding integrins (Fig. 3C to F), suggesting that RGD-binding integrins serve a postbinding role during HMPV infection. We confirmed that the integrin MAbs did not exert an indirect effect on endocytosis by measuring the uptake of fluorescent dextran, which was not inhibited in the presence of integrin MAbs (data not shown). Further, we performed integrin antibody binding at 4°C with similar results (not shown), showing that MAb binding at 37°C did not induce an indirect effect on HMPV entry. Thus, while ∼50% of HMPV particles are capable of binding in an integrin-independent manner, these attachment events do not lead to productive entry in the presence of RGD-binding integrin-specific antibodies. These data indicate that HMPV engages RGD-binding integrins during virus attachment and that integrin-mediated binding is necessary for infection.
Fig 4.

HMPV infection depends upon α5β1 and αV integrins. HMPV infection at 24 h was determined in the absence or presence of αV, α5, or a combination of the two integrin function-blocking antibodies. Infected cells were enumerated as described in Materials and Methods. Results from at least three independent experiments performed in triplicate are expressed as mean percent inhibition relative to untreated control (no MAb); error bars indicate SEM. *, additive effect of combination blockade on HMPV infectivity; Student's t test comparing two treatments, P ≤ 0.05.
HMPV F binds to RGD-binding integrins in the absence of G.
To test the hypothesis that HMPV F directly engages RGD-binding integrins during attachment, we established a system to investigate F-mediated binding in the absence or presence of the viral attachment (G) protein. We generated virus-like particles (VLPs) with only HMPV F (F-VLP) or both the F and G proteins (F+G-VLP) (see Materials and Methods). F-VLPs and F+G-VLPs were similar to virus in morphology, and F glycoprotein spikes of 13.3 nm were visible on the surfaces of virus and VLPs (Fig. 5A and B). The majority of the F spikes appear to resemble the “ball-and-stem” conformation observed for prefusion soluble paramyxovirus F protein (10). Rarely, we observed F spikes that appeared elongated with a wider base and compacted head domain, which may be F trimers that have adopted the postfusion conformation, as the shape is consistent with previous EM images of paramyxovirus postfusion soluble F protein (10). Spikes with a stem and a branched head domain were occasionally visible in the virus and F+G-VLP electron micrographs (Fig. 5A and C). These “tree-shaped” spikes may be the HMPV G protein, but further characterization would be required to confirm this speculation. We confirmed the incorporation of HMPV M, F, and G by Western blot analysis, following immunoprecipitation of virus and VLPs with a MAb recognizing the F protein. HMPV M and G coimmunoprecipitated with the F protein (Fig. 5C), indicating that these viral proteins were contained in VLPs with F protein. Two forms of the fusion protein were detected: the uncleaved precursor (F0) and a large subunit from the cleaved form (F1). Although we observed differences in F cleavage (conversion of F0 to F1) in the virus preparation, the F-VLP and F+G-VLP preparations contained similar levels of HMPV F in both forms. HMPV G bands migrate as a hazy smear at 70 to 90 kDa due to heavy glycosylation of the G protein (5, 21, 31). Levels of G were similar for virus and F+G-VLPs.
HMPV, F-VLP, and F+G-VLP binding were significantly impaired by αV, α5, and β1 integrin blockade, while blocking α2 integrin had no effect (Fig. 5D). Both HMPV and VLP binding were inhibited by ∼50% in the presence of RGD-binding integrin-specific antibodies. Importantly, there were no differences in binding or susceptibility to MAb blockade between F-VLPs and F+G-VLPs. Thus, HMPV F binding occurs in an RGD-binding integrin-dependent manner, independently of any contribution that HMPV G-receptor binding may add to virus binding. These results indicate that F engages RGD-binding integrins during attachment and the viral G protein does not contribute to integrin engagement during virus attachment.
The HMPV F RGD motif is required for HMPV infection.
To further define the importance of HMPV F binding to RGD-binding integrins, we investigated whether the conserved RGD motif was necessary for HMPV infection. We used reverse genetics to replace the F protein RGD motif with an RAE mutation in the viral genome. F-RAE virus growth was delayed and attenuated, resulting in >2-log10-lower peak titers in the supernatant (Fig. 6A). The total F-RAE virus yield obtained from pooling supernatant and cell-associated virus fractions, harvested 13 to 15 days postinoculation when F-wt virus infected cells exhibited maximal cytopathic effect, had >3-log10-lower titers than F-wt viruses (data not shown), indicating that lower supernatant titers were not due to a defect in virus budding. F-RAE plaques were tiny and could not be enumerated without microscope magnification, while F-wt plaques could be counted by eye (Fig. 6B). F-RAE plaques also lacked characteristic syncytia visible in F-wt plaques (Fig. 6C), suggesting that budding F-RAE virus cannot infect neighboring cells and that cell-cell F-mediated fusion was also impaired when the conserved RGD motif was changed to RAE. To confirm that the RAE mutation did not alter F expression, we expressed HMPV F-RAE from a plasmid and determined that F-RAE was expressed on the surface of transfected cells at wild-type levels and was cleaved from the immature (F0) form to the functional (F1/F2) form by the addition of trypsin at the same efficiency as for F-wt (data not shown). Therefore, the defect in F-RAE virus replication is unlikely to be due to a defect in fusion protein expression or cleavage. These results suggest that F-RGD-mediated binding is required for HMPV infection and support a mechanism where F interacts directly with RGD-binding integrins during attachment.
Fig 6.

The HMPV F RGD motif is required for HMPV infection. (A) Titers from reverse-engineered viruses recovered with either the native RGD motif (F-wt) or F-RAE are shown as mean titer for two independently recovered viruses; error bars indicate SEM. The limit of detection is shown as a dotted line. (B and C) Light microscopy images depicting typical plaque size and morphology for HMPV F-wt and F-RAE. LLC-MK2 cells were infected with either HMPV F-wt or F-RAE and incubated for 4 days under a semisolid medium, permitting only cell-to-cell virus spread. HMPV-infected cells are stained black. Magnifications, 2.5× (B) and 10× (C).
HMPV fusion is not triggered by HMPV G or RGD-binding integrins.
The finding that HMPV infection was inhibited more potently than attachment by RGD-binding integrin blockade suggested that not only binding but also subsequent fusion depend upon RGD-binding integrin engagement. To investigate whether HMPV F fusion activity was linked to RGD-integrin binding, we sought to determine HMPV virus-cell fusion kinetics.
We developed an R18-dequenching assay to monitor virus-cell membrane fusion in real time (see Materials and Methods). During virus-mediated fusion, R18 dilutes from a quenched state in the viral membrane into unlabeled cell membranes, resulting in fluorescence (R18 dequenching) that reflects the extent of virus fusion. R18-MPV attached to the cell surface resulted in minimal R18 fluorescence (Fig. 7A, time = 0 min). However, during virus-cell fusion, we observed a steady increase in R18 fluorescence over time (Fig. 7A). HMPV fusion appeared to progress from punctuate foci to fluorescent cell membranes. R18-MPV fusion resulted in a linear increase in R18 fluorescence over 2 h, which reached a plateau after 160 min (Fig. 7B, red circles). As expected, fusion was temperature dependent, because cells maintained at 4°C did not increase in fluorescence intensity over time (Fig. 7B, blue squares).
HMPV fusion appeared to be a slow process, but we sought to confirm that the increased R18 signal was due to virus fusion and that known inhibitors of HMPV fusion could inhibit R18 dequenching in our assay. To monitor HMPV fusion kinetics but normalize for any differences in particle binding, we converted the R18 fluorescence intensity to percent R18 dequenching over time (as described in Materials Methods) for the positive inhibition controls (Fig. 7C) and all conditions tested and reported in Fig. 8. Paramyxovirus fusion proteins can be artificially triggered to refold into an inactive postfusion structure by heat (10). Heat-inactivated R18-MPV could bind but not fuse with cells (Fig. 7C, black squares), confirming that the R18 fusion signal was due to active virus fusion rather than passive transfer of R18 dye. R18-MPV fusion was significantly inhibited in the presence of neutralizing HMPV antiserum (Fig. 7C, green triangles). The polyclonal antiserum reduced R18-MPV binding by ∼50% but completely neutralized HMPV infection (data not shown). Thus, HMPV F-specific antibodies in the serum blocked binding but also prevented bound virus from fusing with cells. Importantly, this inhibition of virus-cell fusion resulted in significantly less R18 dequenching in the fusion assay. Thus, our findings indicate that HMPV fusion is a slow process that occurs over several hours.
Next, we investigated whether the HMPV attachment (G) protein was required for virus-cell fusion. Schowalter et al. previously reported that HMPV G was not required for HMPV F-mediated cell-cell fusion, although the authors suggested that the G protein might enhance fusion in certain cell types (33). HMPVΔG is attenuated in vivo, although it is currently not clear whether lower viral titers are due to a defect in virus binding and/or entry (4, 5). Further, no previous studies have assessed whether virus-cell fusion requires the presence of the HMPV G protein. To determine whether HMPV G was required to trigger F-mediated fusion, we analyzed VLP fusion kinetics. Results for R18-F-VLP fusion controls are shown in Fig. 8A. As expected from our virus-cell fusion experiments, heat inactivation abolished R18 dequenching. G-VLP dequenching (Fig. 8A, diamonds) provided a measure of background R18 transfer in our VLP fusion assay, as these particles are expected to bind but not fuse because they do not have HMPV F. Neutralizing antiserum significantly reduced HMPV F-mediated VLP fusion, nearly to the background levels observed for G-only VLPs. These fusion controls indicate that there is some nonspecific R18 dye transfer in the VLP fusion assay but that HMPV F-mediated fusion kinetics can be quantified because F-VLP dequenching was significantly over background. Next, F-only and F+G VLP fusion kinetics were monitored to define whether the HMPV G protein contributed to HMPV fusion. We found that F-only VLP and F+G-VLP fusion proceeded with the same kinetics (Fig. 8B) and to the same extent (Fig. 8C). Thus, G does not contribute to F-mediated VLP fusion in the in vitro fusion assay. We noted that VLP fusion was not as efficient as virus fusion (Fig. 8B and C); however, despite differences in fusion extent, the early kinetics of virus and VLP fusion were identical. Fusion began after a discernible delay, the initial fusion rate became lower after 2 h, and fusion reached a plateau at around 4 h. These similarities suggest that VLP fusion represents an appropriate model for early virus fusion kinetics. Thus, these results indicate that the HMPV F protein is necessary and sufficient for virus-cell fusion and can regulate fusion in the absence of G. Our findings suggest that HMPV F-receptor interactions are sufficient to trigger fusion, and G interactions with F do not appear to be essential for HMPV virus-cell fusion in vitro.
We hypothesized that HMPV F binding to RGD-binding integrins may serve to trigger fusion. To explore this possibility, we assessed HMPV and VLP fusion kinetics in the presence of integrin function-blocking antibodies. As expected, blockade of RGD-binding integrins significantly decreased the amount of virus and VLP binding (inhibiting binding by ∼50%), while blocking the collagen-binding integrin subunit α2 had no effect (data not shown). However, we found that HMPV fusion was not significantly impaired by RGD-binding integrin blockade (Fig. 8D to F). Blocking RGD-binding integrins during virus or VLP binding altered neither fusion kinetics nor fusion extent after 4 h. In other words, even though ∼50% less virus or VLPs bound to the surface of cells during RGD-binding integrin blockade, the particles that did bind were still capable of mediating fusion.
In order to examine the importance of the integrin-binding motif for F-mediated particle-cell fusion, we generated F-RAE VLPs. F-RAE VLPs budded with slightly less efficiency than F-wt VLPs, although the fusion protein was efficiently cleaved into the fusogenic form (indicated by F0-to-F1 conversion) (Fig. 8G). We found that F-RAE VLPs fused with kinetics identical to those of wild-type F-VLPs (Fig. 8H), which would be expected if integrin binding was not the primary trigger for the initiation of HMPV fusion. We tested F-RAE versus F-RGD VLP fusion at different concentrations of bound particles (a range of 30-fold) to confirm that the number of particles bound did not affect fusion kinetics, and we found that R18-dequenching kinetics were unaltered (data not shown). These results strongly suggest that although efficient HMPV binding and productive infection require RGD-binding integrin engagement, integrin binding alone does not trigger fusion.
Productive HMPV transcription depends upon RGD-binding integrin-mediated virus entry.
R18 dequenching measures virus hemifusion, mixing of the outer lipid leaflets of virus and cellular membranes as fusion begins. After hemifusion, HMPV fusion and virus entry require merging of the inner leaflets of the virus and cell membranes, formation of a fusion pore, fusion pore expansion, and, finally, delivery of the viral genome into the cytoplasm for viral transcription. Thus, productive virus transcription can be used as a measure of the entire virus entry process. Because RGD-binding integrins were not required for HMPV hemifusion, we next investigated whether integrins were required for full fusion and virus genome entry, as measured by virus transcription at 8 h postinfection. We sought to determine whether RGD-binding integrins mediated virus entry postbinding and designed the experiment to elucidate whether HMPV bound in the presence of RGD-binding integrin function-blocking antibodies was capable of entering cells and producing virus transcripts. We used real-time RT-PCR to quantify the level of HMPV nucleoprotein (N) transcribed during the first 8 h of infection (see Materials and Methods) and found that blocking the interaction between HMPV F and all RGD-binding integrins (αV plus α5 plus β1) resulted in 50% fewer viral transcripts than for an untreated control (Fig. 9, hatched bar versus black bar). As expected, blocking the collagen-binding α2 integrin subunit had no effect on HMPV transcription (Fig. 9, gray bar). These results suggest that RGD-binding integrins mediate postbinding events required for productive virus entry and necessary for efficient virus transcription early in HMPV infection.
Fig 9.
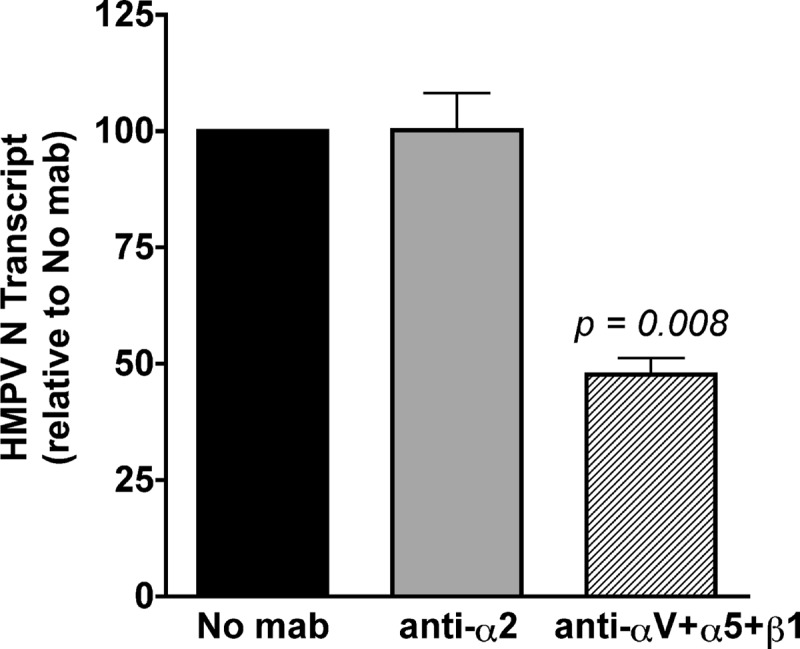
Productive HMPV transcription depends upon RGD-binding integrin-mediated virus entry. HMPV (MOI = 0.25 PFU/cell) was bound to the surface of BEAS-2B cells in the absence or presence of α2 integrin or RGD-binding integrin (αV plus α5 plus β1) function-blocking antibodies. Levels of HMPV N transcript were determined at 8 h postinfection by real-time RT-PCR, relative to the GAPDH cellular gene. The 2−ΔΔCT method was used to correct for input genome and compare transcript levels in treated and untreated samples. Data are presented as the average level of HMPV N transcript detected relative to an untreated (no-MAb) control; error bars indicate SEM for six biological replicates from two independent experiments.
DISCUSSION
HMPV gains entry into cells in a manner that can be mediated solely by one surface glycoprotein, the fusion (F) protein. How HMPV F mediates both attachment and fusion has not been clearly defined. Here, we present evidence that F mediates HMPV entry through an interaction with RGD-binding integrins. HMPV F binds to RGD-binding integrins during virus attachment, and this binding event is necessary for virus entry and subsequent productive infection. In lieu of an interaction between the viral attachment (G) protein and cellular receptors, F is capable of initiating virus entry by binding directly to cellular receptors, including RGD-binding integrins. Interestingly, while an F-integrin interaction is necessary for virus entry, integrin binding does not appear to trigger F-mediated hemifusion. RGD-binding integrins are important for postbinding events, which occur after virus hemifusion but before HMPV transcription. Thus, we propose a model of HMPV entry as a stepwise process where (i) F binds to cell surface receptors, including RGD-binding integrins, (ii) unidentified events trigger virus hemifusion, and (iii) RGD-binding integrins facilitate postbinding events that promote virus entry and lead to efficient virus transcription and productive infection.
We previously demonstrated that HMPV infection depends upon RGD-binding integrins and suggested that αVβ1 was a receptor for HMPV (11). Here, we show that multiple RGD-binding integrins can be used for infection, and integrins are required for HMPV attachment and entry. HMPV F-mediated attachment depends, in part, upon RGD-binding integrin engagement on the surface of human respiratory epithelial cells. RGD-binding integrin-specific binding accounted for about 40% of total R18-MPV binding in our experiments, indicating that other cell surface receptors also bind HMPV. When all RGD-binding integrins were blocked, we observed a 40% decrease in HMPV attachment that resulted in a 90% reduction in HMPV infectivity. This discrepancy led us to explore whether RGD-binding integrins were required for virus entry after initial virus binding, and we found that RGD-binding integrins promote virus entry after HMPV hemifusion begins. HMPV transcription was reduced by 50% when HMPV F-integrin binding was blocked. This suggests that although HMPV binds to cell surface receptors other than integrins, most of these particles are not capable of delivering the genome into target cells for virus transcription. Although it appears that HMPV attaches to more than one receptor on the cell surface, HMPV F must engage RGD-binding integrins during attachment for efficient virus entry and productive infection.
Chang et al. recently reported that HMPV binding and infection are mediated by interactions between the F protein and heparan sulfate and suggested that heparan sulfate is the first binding partner for HMPV F (7). In experiments using β1 integrin-deficient murine fibroblasts, β1 integrin was critical for HMPV infection, but the authors concluded that β1 integrin is involved after initial binding and is not a direct cellular receptor for HMPV because virus still bound β1 integrin-deficient cells (7). In light of our findings that HMPV F binds multiple integrins during attachment, HMPV F would be expected to bind to other RGD-binding integrins such as αVβ5, αVβ6, and/or αVβ8. HMPV F binding to αV integrins could explain some of the residual F-mediated virus-cell binding and low levels of infection that Chang et al. (7) observed in β1 integrin-deficient cells at a higher multiplicity of infection. Moreover, when β1-null fibroblasts were complemented with human β1 integrin, HMPV F-only virus bound more readily and infection was significantly enhanced (7). These findings are consistent with our results implicating multiple RGD-binding integrins (including α5β1) in regulating HMPV entry following initial virus attachment. This suggests that heparan sulfate may be an important adhesion factor for HMPV F but that integrin engagement is critical for infection. Our results do not exclude the possibility that F also interacts with other adhesion factors during cell attachment. Indeed, our results strongly suggest that HMPV F binds to multiple cellular receptors, and further investigation is required to clarify which F-receptor interactions are necessary to trigger F refolding and initiate virus hemifusion. Taken together, the data suggest that HMPV F interacts with multiple binding partners during attachment, perhaps first attaching to heparan sulfate before engaging specific proteinaceous receptors such as RGD-binding integrins, which mediate postbinding events that lead to productive HMPV infection.
Paramyxoviruses are believed to enter cells in a pH-independent manner at the cell surface, a mechanism that requires receptor binding to trigger virus-cell fusion. However, others have hypothesized that HMPV fusion may be triggered by exposure to low pH following virus internalization into cellular endosomes (32, 33). HMPV F-mediated cell-cell fusion of some lineage A strains is enhanced by exposure to low pH; however, lineage B strains fuse in a pH-independent manner (14, 24, 32, 33). Mas et al. identified a tetrad of residues at positions 294, 296, 396, and 404 in the F protein and found that the presence of residues GKRN conferred a low-pH-dependent cell-cell fusion phenotype to HMPV F proteins from all lineages (24). Here, we present virus-cell fusion experiments with A2 F proteins that have tetrad residues EKRN, typical of most A2 HMPV strains. Of note, A2 F proteins with the tetrad residues EKRN poorly induced cell-cell membrane fusion in a pH-independent manner, but low-pH-induced fusion was enhanced with an E294G mutation according to Mas et al. (24). Thus, our A2 F protein sequence would be predicted to support pH-independent cell-cell fusion, although we have not tested this directly. In our virus-cell fusion assay, we found that low-pH pulses did not significantly enhance fusion of our A2 lineage virus strain (R. G. Cox and J. V. Williams, unpublished data). Whether the low-pH cell-cell fusion phenotype is indicative of a requirement for low-pH exposure within endosomes during HMPV entry is not clear.
The HMPV F protein alone is necessary and sufficient for virus attachment, and VLPs with only F protein require RGD-binding integrins for binding. Moreover, mutating the RGD to RAE resulted in a 3-log10-PFU defect in HMPV replication, showing that the invariant RGD motif in the F protein is required for HMPV infectivity. This strongly suggests that integrin binding is mediated largely by the RGD motif and that HMPV F-integrin engagement is essential for receptor-binding activity. The F RAE mutation did not completely abolish HMPV infection, although F-RAE viruses did not replicate efficiently and exhibited clear defects in cell-to-cell spread. It is possible that simply mutating the 3-amino-acid motif is not sufficient to completely abolish an F-integrin interaction during virus binding; there are likely other residues in both proteins that contribute to the interaction. The reproducible recovery of low levels of an F-RAE virus and the detection of F-RAE protein on the surface of infected cells suggest that the F-RAE protein is expressed and folded into a fusion-competent form that mediates low levels of virus entry. We confirmed that the F-RAE mutation did not alter cell surface F expression or cleavage levels when the protein was expressed from a plasmid in transfected cells (data not shown). Unfortunately, due to the extremely low virus titer, it was not feasible to measure F-RAE virus binding and fusion. However, we were able to compare F-RGD and F-RAE VLP fusion kinetics, and we found that the integrin-binding motif was not required for efficient HMPV hemifusion. Collectively, these results provide evidence that supports a mechanism where F engages RGD-binding integrin receptors during virus entry. Our VLP experiments suggest that HMPV F-mediated attachment and virus-cell fusion are regulated independently of HMPV G. Because HMPV ΔG is not defective for replication in vitro (5) and HMPV F-RAE is quite debilitated, our findings are consistent with a mechanism whereby HMPV F interacts with cell surface receptors in a manner that triggers fusion and facilitates virus entry in a G-independent manner. It is important to note that HMPV ΔG is attenuated in hamsters and nonhuman primates (4, 5), and G likely plays an important role in infection in vivo.
By definition, virus receptors mediate attachment and facilitate entry. In principle, cellular receptors for HMPV F could facilitate entry by inducing conformational changes in F, recruiting coreceptors to sites of virus attachment, recruiting cellular factors that facilitate fusion pore expansion, or promoting internalization of virus particles. Although HMPV F attachment was inhibited, virus entry leading to efficient viral replication was further impaired in the presence of RGD-binding integrin antibodies. These results explain why the inhibition of attachment is much less potent than the inhibition of infectivity (Fig. 3). If HMPV binding and postbinding events during entry depend upon RGD-binding integrins, then infectivity should be affected to a greater degree than attachment during integrin blockade. Our experiments suggest that integrin engagement does not trigger HMPV hemifusion but that RGD-binding integrins do promote HMPV entry. The mechanism by which RGD-binding integrins regulate HMPV entry requires further investigation.
ACKNOWLEDGMENTS
This work was supported by Public Health Service grants AI-73697 and AI-85062 (J.V.W.) and T32 AI-7611 (R.G.C.) from the National Institute of Allergy and Infectious Diseases and T32 CA-9682 (S.B.L.) from the National Cancer Institute. The VMC Flow Cytometry Shared Resource is supported by the Vanderbilt Ingram Cancer Center (NIH grant P30 CA-68485) and the Vanderbilt Digestive Disease Research Center (NIH grant DK-58404). This work was supported in part by Vanderbilt CTSA grant UL1 RR-24975-01 from the NCRR/NIH.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. J.V.W. serves on the Scientific Advisory Board of Quidel.
We thank current and past members of the Williams laboratory for helpful discussions. We are grateful to H. Earl Ruley and Roy Zent for manuscript reviews. We thank members of the James Crowe laboratory, especially Fyza Shaikh, for helpful suggestions, and James Crowe, Christopher Aiken, and Terence Dermody for use of equipment.
Footnotes
Published ahead of print 29 August 2012
REFERENCES
- 1. Aguilar HC, et al. 2009. A novel receptor-induced activation site in the Nipah virus attachment glycoprotein (G) involved in triggering the fusion glycoprotein (F). J. Biol. Chem. 284:1628–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ali SA, et al. 2011. Real-world comparison of two molecular methods for detection of respiratory viruses. Virol. J. 8:332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Applied Biosystems 2001. User bulletin 2: ABI PRISM 7700 sequence detection system. Applied Biosystems, Foster City, CA [Google Scholar]
- 4. Biacchesi S, et al. 2005. Infection of nonhuman primates with recombinant human metapneumovirus lacking the SH, G, or M2-2 protein categorizes each as a nonessential accessory protein and identifies vaccine candidates. J. Virol. 79:12608–12613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Biacchesi S, et al. 2004. Recombinant human metapneumovirus lacking the small hydrophobic SH and/or attachment G glycoprotein: deletion of G yields a promising vaccine candidate. J. Virol. 78:12877–12887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Buchholz UJ, Finke S, Conzelmann KK. 1999. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J. Virol. 73:251–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chang A, Masante C, Buchholz UJ, Dutch RE. 2012. Human metapneumovirus (HMPV) binding and infection are mediated by interactions between the HMPV fusion protein and heparan sulfate. J. Virol. 86:3230–3243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Colman PM, Lawrence MC. 2003. The structural biology of type I viral membrane fusion. Nat. Rev. Mol. Cell Biol. 4:309–319 [DOI] [PubMed] [Google Scholar]
- 9. Connolly SA, Leser GP, Jardetzky TS, Lamb RA. 2009. Bimolecular complementation of paramyxovirus fusion and hemagglutinin-neuraminidase proteins enhances fusion: implications for the mechanism of fusion triggering. J. Virol. 83:10857–10868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Connolly SA, Leser GP, Yin HS, Jardetzky TS, Lamb RA. 2006. Refolding of a paramyxovirus F protein from prefusion to postfusion conformations observed by liposome binding and electron microscopy. Proc. Natl. Acad. Sci. U. S. A. 103:17903–17908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cseke G, et al. 2009. Integrin alphavbeta1 promotes infection by human metapneumovirus. Proc. Natl. Acad. Sci. U. S. A. 106:1566–1571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cseke G, et al. 2007. Human metapneumovirus fusion protein vaccines that are immunogenic and protective in cotton rats. J. Virol. 81:698–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Herfst S, et al. 2004. Recovery of human metapneumovirus genetic lineages A and B from cloned cDNA. J. Virol. 78:8264–8270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Herfst S, et al. 2008. Low-pH-induced membrane fusion mediated by human metapneumovirus F protein is a rare, strain-dependent phenomenon. J. Virol. 82:8891–8895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hoekstra D, de Boer T, Klappe K, Wilschut J. 1984. Fluorescence method for measuring the kinetics of fusion between biological membranes. Biochemistry 23:5675–5681 [DOI] [PubMed] [Google Scholar]
- 16. Humphries JD, Byron A, Humphries MJ. 2006. Integrin ligands at a glance. J. Cell Sci. 119:3901–3903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Karron RA, et al. 1997. Respiratory syncytial virus (RSV) SH and G proteins are not essential for viral replication in vitro: clinical evaluation and molecular characterization of a cold-passaged, attenuated RSV subgroup B mutant. Proc. Natl. Acad. Sci. U. S. A. 94:13961–13966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Krishnan A, et al. 2009. A histidine switch in hemagglutinin-neuraminidase triggers paramyxovirus-cell membrane fusion. J. Virol. 83:1727–1741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lamb RA, Jardetzky TS. 2007. Structural basis of viral invasion: lessons from paramyxovirus F. Curr. Opin. Struct. Biol. 17:427–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee JK, et al. 2008. Functional interaction between paramyxovirus fusion and attachment proteins. J. Biol. Chem. 283:16561–16572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu L, Bastien N, Li Y. 2007. Intracellular processing, glycosylation, and cell surface expression of human metapneumovirus attachment glycoprotein. J. Virol. 81:13435–13443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- 23. Mahon PJ, Mirza AM, Iorio RM. 2011. Role of the two sialic acid binding sites on the Newcastle disease virus HN protein in triggering the interaction with the F protein required for the promotion of fusion. J. Virol. 85:12079–12082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mas V, Herfst S, Osterhaus AD, Fouchier RA, Melero JA. 2011. Residues of the human metapneumovirus fusion (F) protein critical for its strain-related fusion phenotype: implications for the virus replication cycle. J. Virol. 85:12650–12661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Melanson VR, Iorio RM. 2006. Addition of N-glycans in the stalk of the Newcastle disease virus HN protein blocks its interaction with the F protein and prevents fusion. J. Virol. 80:623–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ohi M, Li Y, Cheng Y, Walz T. 2004. Negative staining and image classification—powerful tools in modern electron microscopy. Biol. Proc. Online 6:23–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45 doi:10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Plemper RK, Hammond AL, Gerlier D, Fielding AK, Cattaneo R. 2002. Strength of envelope protein interaction modulates cytopathicity of measles virus. J. Virol. 76:5051–5061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Porotto M, Murrell M, Greengard O, Moscona A. 2003. Triggering of human parainfluenza virus 3 fusion protein (F) by the hemagglutinin-neuraminidase (HN) protein: an HN mutation diminishes the rate of F activation and fusion. J. Virol. 77:3647–3654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Russell CJ, Kantor KL, Jardetzky TS, Lamb RA. 2003. A dual-functional paramyxovirus F protein regulatory switch segment: activation and membrane fusion. J. Cell Biol. 163:363–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ryder AB, Tollefson SJ, Podsiad AB, Johnson JE, Williams JV. 2010. Soluble recombinant human metapneumovirus G protein is immunogenic but not protective. Vaccine 28:4145–4152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schowalter RM, Chang A, Robach JG, Buchholz UJ, Dutch RE. 2009. Low-pH triggering of human metapneumovirus fusion: essential residues and importance in entry. J. Virol. 83:1511–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schowalter RM, Smith SE, Dutch RE. 2006. Characterization of human metapneumovirus F protein-promoted membrane fusion: critical roles for proteolytic processing and low pH. J. Virol. 80:10931–10941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Smith EC, Popa A, Chang A, Masante C, Dutch RE. 2009. Viral entry mechanisms: the increasing diversity of paramyxovirus entry. FEBS J. 276:7217–7227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stewart PL, Nemerow GR. 2007. Cell integrins: commonly used receptors for diverse viral pathogens. Trends Microbiol. 15:500–507 [DOI] [PubMed] [Google Scholar]
- 36. Tanabayashi K, Compans RW. 1996. Functional interaction of paramyxovirus glycoproteins: identification of a domain in Sendai virus HN which promotes cell fusion. J. Virol. 70:6112–6118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Thammawat S, Sadlon TA, Hallsworth PG, Gordon DL. 2008. Role of cellular glycosaminoglycans and charged regions of viral G protein in human metapneumovirus infection. J. Virol. 82:11767–11774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ulbrandt ND, et al. 2006. Isolation and characterization of monoclonal antibodies which neutralize human metapneumovirus in vitro and in vivo. J. Virol. 80:7799–7806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Williams JV, et al. 2007. A recombinant human monoclonal antibody to human metapneumovirus fusion protein that neutralizes virus in vitro and is effective therapeutically in vivo. J. Virol. 81:8315–8324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Williams JV, Tollefson SJ, Johnson JE, Crowe JE., Jr 2005. The cotton rat (Sigmodon hispidus) is a permissive small animal model of human metapneumovirus infection, pathogenesis, and protective immunity. J. Virol. 79:10944–10951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yang CF, et al. 2009. Genetic diversity and evolution of human metapneumovirus fusion protein over twenty years. Virol. J. 6:138. [DOI] [PMC free article] [PubMed] [Google Scholar]