

Abstract
Human noroviruses are one of the major causes of acute gastroenteritis in the developed world, yet our understanding of their molecular mechanisms of genome translation and replication lags behind that for many RNA viruses. Due to the nonculturable nature of human noroviruses, many related members of the Caliciviridae family of small RNA viruses are often used as model systems to dissect the finer details of the norovirus life cycle. Murine norovirus (MNV) has provided one such system with which to study the basic mechanisms of norovirus translation and replication in cell culture. In this report we describe the use of riboproteomics to identify host factors that interact with the extremities of the MNV genome. This network of RNA-protein interactions contains many well-characterized host factors, including PTB, La, and DDX3, which have been shown to play a role in the life cycle of other RNA viruses. By using RNA coimmunoprecipitation, we confirmed that a number of the factors identified using riboproteomics are associated with the viral RNA during virus replication in cell culture. We further demonstrated that RNA inhibition-mediated knockdown of the intracellular levels of a number of these factors inhibits or slows norovirus replication in cell culture, allowing identification of new intracellular targets for this important group of pathogens.
INTRODUCTION
The genomes of small positive-strand RNA viruses must not only encode the viral proteins required for viral genome replication and infectious virus production, but they must also contain cis-acting RNA sequences and structures that play critical roles in the virus life cycle (40). Host cell factors that interact with these RNA elements play important roles in all aspects of the virus life cycle (49); consequently, the manipulation of these interactions via the introduction of mutations into the viral genome is a viable approach to rational attenuation (24, 26, 57, 78). In addition, the modification of these RNA-protein interactions by using small-molecule or peptide inhibitors has been shown to provide antiviral activity, at least in cell culture (8, 19).
Noroviruses, first identified in 1972 by Albert Kapikian (33), are now accepted as the major cause of viral gastroenteritis in the developed world (21). Reports indicate that in excess of 23 million cases occur each year in the United States, resulting in a significant economic impact. Now over 20 years since the first full genome sequence of a norovirus became available (77), investigators are still unable to efficiently propagate human noroviruses in cell culture (15, 37). Recent data have indicated that the viral RNA purified from the feces of individuals infected with Norwalk virus, the prototype human norovirus, can replicate when transfected into immortalized cells in culture (23). These data clearly demonstrate that one of the major blocks in the ability of human noroviruses to replicate in cell culture is the ability of the virus to enter and spread from cell to cell. It is also clear that the intracellular environment, in particular the innate immune response and cholesterol levels, may also modulate norovirus replication in cell culture (6, 7, 54) and may in some way contribute to the nonculturable nature of human noroviruses. Until the missing factor or factors required for the efficient replication and spread of human norovirus in cell culture are identified, other members of the Caliciviridae family of small RNA viruses provide excellent models to dissect various aspects of the calicivirus life cycle (68).
Murine norovirus (MNV), the only cultivatable norovirus identified to date, was isolated in 2003 as the causative agent of a lethal infection in mice deficient in STAT-1 and RAG (36), factors involved in the innate and adaptive immune responses, respectively. With the development of an appropriate cell culture system (74) and reverse genetics systems (9, 72, 80), MNV has now become a widely used model for human noroviruses. While the MNV pathogenesis model lacks some features of an ideal model for human noroviruses (i.e., MNV does not routinely cause diarrhea), the genetic similarity makes it a valid model that allows the molecular characterization of viral genome translation and replication.
To date, our understanding of host cell factors that are involved in the norovirus life cycle is limited. Previous reports have illustrated that one of the nonstructural proteins from the prototype human norovirus Norwalk virus interacts with components of the vacuolar sorting pathway and alters host cell secretion (16, 63). Studies have also identified a number of host cell nucleic acid binding proteins that interact with the 5′ and 3′ extremities of the human norovirus genomic RNA (25, 27). While a definitive function of these interactions has yet to be confirmed due to lack of suitable experimental systems, it is likely they contribute to genome circularization (60). We previously reported the bioinformatic and functional identification of RNA structures involved in the MNV life cycle (3, 65). This finding highlighted that, as has been well established for other RNA viruses, the termini of the viral genome contain RNA structures that play functional roles in the norovirus life cycle. In the current report we use RNA affinity chromatography followed by mass spectrometry analysis to identify host cell factors that interact with these RNA structures. We subsequently demonstrate using confocal microscopy that some of these host cell factors can be found associated with the viral replication complex during virus replication, and based on small interfering RNA-mediated knockdown studies, that they also play a functional role in the norovirus life cycle. This work identifies a number of host cell factors that may provide additional targets for antiviral intervention or the generation of attenuated noroviruses.
MATERIALS AND METHODS
Cells and antibodies.
Murine macrophage RAW264.7 cells were maintained at 37°C with 10% CO2 in Dulbecco's modified Eagle's medium (DMEM; Gibco) containing 10% fetal calf serum (FCS; Biosera) and an antibiotic cocktail of penicillin (100 SI units/ml) and streptomycin (100 μg/ml). The murine microglial BV-2 cell line (5) was maintained in DMEM supplemented with 10% FCS, 2 mM l-glutamine, 0.075% sodium bicarbonate, and antibiotics (100 units/ml penicillin and 100 μg/ml streptomycin). Rabbit polyclonal antisera against the MNV NS7 polymerase protein and poly r(C)-binding proteins 1 and 2 (PCBP1/2) were developed in the laboratory. Primary antibodies against the following host factors were purchased from the indicated suppliers: glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Ambion), PTB (Santa Cruz Biotechnology), La (Santa Cruz Biotechnology), heat shock protein 90 (HSP90; Santa Cruz Biotechnology), HuR (Santa Cruz Biotechnology), YBX-1 (Sigma), Talin-1 (Sigma). Anti-DDX3 antiserum was generously provided by Arvind Patel, University of Glasgow. The secondary antibodies were purchased from Invitrogen and used at the dilutions recommended.
Preparation and purification of RNA transcripts.
The regions of interest, namely, nucleotides (nt) 1 to 250 and 7141 to 7397 of the MNV-1 genome, were PCR amplified from the MNV cDNA clone pT7:MNV 3′Rz already described (9). The forward primers contained T7 promoter sequence at the 5′ end of viral RNA-specific sequence. The 5′ extremity of the viral RNA was amplified using the primers IGIC16 (TAATACGACTCACTATAGGGGTGAAATGAGGATGGCAACGC) and IGIC17 (AGCACGCGTGCATCACTACGCG). The 3′ extremity of the MNV genome was amplified using the primers IGIC127 (TAATACGACTCACTATAGGGACATCCCCTCTACCGATCTCGG) and IGIC128 (TTTTTTTTTTTTTTTAAAATGC). A control RNA was generated by PCR amplification of part of the ampicillin resistance gene by using the primers IGIC479 (GCGTAATACGACTCACTATAGGGTCGTATTAAATTCGTAATC) and IGIC480 (GGATCCGACCGTGGTGCCCTTGCGGGCAGAAGTCCAAATGCGATCCTATATACGGAAGAGCGCCCAATAC) to produce a PCR product that would generate a transcript of 250 nt in length. The amplified PCR products were gel purified and subjected to in vitro transcription using T7 RNA polymerase. The in vitro transcription reactions (reaction mixtures containing 40 mM Tris [pH 8.0], 40 mM dithiothreitol [DTT], 6 mM MgCl2, 2 mM spermidine, 0.5 mM ATP, CTP, GTP, and UTP, 600 ng of PCR product, 2.5 μg T7 polymerase, and 100 units of RNasin in a 100-μl volume) were carried out at 37°C for 4 h. Afterwards, the reaction mixtures were incubated with 10 units of DNase at 37°C for 30 min. The RNA was then purified by excision from the gel, following denaturing acrylamide gel electrophoresis (urea-PAGE), ethanol precipitated, and resuspended in RNA storage solution (Ambion). The RNA was quantified using spectrophotometry and stored at −20°C until further use. The integrity of the RNA was checked by denaturing PAGE prior to use.
RNase sensitivity mapping.
RNA transcripts generated as described above were subjected to limited RNase T2, T1, A, and V1 digestion in vitro and subsequent primer extension as previously described (3).
Preparation of cell extracts and fractionation of rabbit reticulocyte lysated.
Rabbit reticulocyte lysates (RRLs; Green Hectares) and RAW264.7 cells were used to make ribosomal salt wash and S-100 extracts, respectively. Aliquots (35 ml) of untreated RRLs were pooled and mixed with 350 μl protease inhibitor (Perbio). RRLs were then ultracentrifuged at 100,000 × g for 2 h at 4°C, and the supernatant was removed. The pellet containing ribosomes was dissolved in 10 ml of buffer C (30 mM Tris [pH 7.5], 50 mM KCl, 6 mM MgCl2, 2 mM DTT, 0.25 M sucrose, and protease inhibitors), KCl was added to a final concentration of 0.5 M, and the mixture was ultracentrifuged as described above. The supernatant was harvested as ribosomal salt wash (RSW) and dialyzed at 4°C overnight against buffer D (44 mM KCI, 1.2 mM magnesium acetate, 1 mM DTT, 0.1 mM EDTA, 10% glycerol, 10 mM HEPES; pH 7.6). RAW264.7 cells were washed in phosphate-buffered saline (PBS), centrifuged, and resuspended in a 5× packed cell volume in buffer A (10 mM HEPES [pH 7.9], 1.5 mM MgCl2, 10 mM KCl, 1 mM DTT). The cells were allowed to swell on ice from 30 to 60 min, following which the swollen cells were centrifuged and resuspended again in 15 ml of buffer A. The swollen cells were homogenized using a Dounce homogenizer and centrifuged at 2,000 × g for 20 min. The supernatant was mixed with a 1/10 volume of buffer B (300 mM HEPES [pH 7.9], 30 mM MgCl2, 1.4 M KCl) and centrifuged at 100,000 × g for 1 h. The supernatant was harvested as the S-100 cytoplasmic extract, mixed with protease inhibitors, aliquoted, and stored at −80°C.
Preparation of RNA affinity columns.
Approximately 100 μg of in vitro-transcribed RNA was covalently coupled to cyanogen bromide (CNBR)-activated Sepharose beads as described previously (31). Briefly, 125 μl of packed preswollen CNBR-activated Sepharose beads (Sigma) was equilibrated in 200 mM MES (pH 6.0) and mixed with 100 μg of RNA. This mixture was kept at 4°C overnight with rotation. The beads were later washed three times with 100 mM Tris (pH 8.0) and incubated in 100 mM Tris (pH 8.0) for 1 h at 4°C. The prepared RNA-linked Sepharose beads were washed three times in RNA-binding buffer (50 mM HEPES [pH 7.6], 50 mM KCl, 5 mM MgO-acetate, 125 mM NaCl, 2 mM DTT, 10% glycerol) and used for affinity purification.
Enrichment of RNA-binding protein by using RNA affinity purification.
The RNA-linked Sepharose beads were incubated with approximately 7.5 mg of RRL RSW or RAW264.7 S-100 lysates in the presence of 100 μg yeast RNA, 1 mM ATP, 1 mM GTP, 100 U RNasin RNase inhibitor (Promega) at 4°C for 2 h with continuous mixing. RNA-protein complexes bound to the beads were washed three times with binding buffer at 4°C, and the bound proteins were eluted by boiling in presence reducing SDS-PAGE loading buffer. The eluted proteins were run on NuPAGE Novex 4-to-12% bis-Tris gels (Invitrogen) and stained with Coomassie colloidal blue stain (Invitrogen). Protein bands of interest were excised and submitted to McGill University and the Genome Quebec Innovation Centre, Canada, for mass spectrometry.
Mass spectrometry.
The McGill University and Genome Quebec Innovation Centre, Canada, performed all mass spectrometry (MS). Proteins present in bands excised from SDS-PAGE gels were identified using a QTof micro mass spectrometer (Waters Micromass) at McGill University and the Genome Quebec Innovation Centre, Canada. Briefly, proteins from gel bands were subjected to reduction, cysteine alkylation, and in-gel tryptic digestion as described previously (73). Extracted peptides were injected onto a desalting column and subsequently chromatographically separated on a Biobasic C18 capillary column, by using a Nano high-performance liquid chromatography system. Eluted peptides from the in-gel digestion were analyzed on a QTof micro mass spectrometer. Individual sample tandem mass spectrometry spectra were listed by peak, and a search against a copy of the Uniprot database by using Mascot (version 2.1.4.04) was performed. Mascot was set up to search the Mus musculus (taxonomy ID10090) database (17 September 2010 release; 61,462 sequenced protein entries), assuming the digestion enzyme trypsin, with a fragment ion mass tolerance of 0.80 Da and a parent ion tolerance of 1.5 Da. The iodoacetamide derivative of cysteine was specified in both search engines as a fixed modification. Oxidation of methionine residues was specified in Mascot as a variable modification. Scaffold (Proteome Software Inc.) was used to validate tandem MS (MS/MS)-based peptide and protein identifications.
Western blot analysis.
Samples were boiled for 10 min in the presence of SDS loading buffer and electrophoresed on 12% SDS-polyacrylamide gels. Proteins were then transferred to polyvinylidene difluoride membranes for Western blotting. The membranes were blocked for 2 h at 4°C with 5% milk in PBS–Tween 20 (0.1%) and incubated for 1 h with the appropriate primary antibody (Ab). The excess antibody was washed three times in PBS–Tween 20 (0.1%) and incubated for 1 h with corresponding secondary Ab conjugated to horseradish peroxidase. After washing away excess antibody, the bands were detected by using enhanced chemiluminescence reagent (GE Healthcare).
RNA coimmunoprecipitation.
The interaction of various host proteins with MNV RNA during infection was investigated using RNA-coimmunoprecipitation (co-IP). Briefly, 2 × 106 RAW264.7 cells per assay were infected for 15 h with MNV-1 (multiplicity of infection [MOI] of 1 50% tissue culture infective dose [TCID50] per cell). In parallel, protein A/G beads (20 μl, packed volume), preequilibrated with NT2 buffer (50 mM Tris [pH 7.4], 150 mM NaCl, 1 mM MgCl2, and 0.05% Nonidet P-40) were incubated with 2 μg of the corresponding antibodies at 4°C. After 15 h, cells were washed twice with ice-cold PBS and lysed in polysome lysis buffer (10 mM HEPES [pH 8.0], 100 mM KCl, 5 mM MgCl2, 0.5% NP-40) containing 2 mM DTT, 100 units/ml RNasin (Promega), 1 mM ribonucleoside vanadyl complex, and protease inhibitor (Perbio); lysis was aided by passing the mixture through a 25-gauge needle. The lysate was centrifuged at 16,000 × g for 15 min at 4°C, and supernatant was diluted 10 times in NT2 buffer for co-IP. The labeled protein A/G beads (washed three times using NT2 buffer), along with RNAsin and protease inhibitors, were added to the lysate and incubated at 4°C for 4 h with rotation. Finally, beads were collected by centrifugation at 2,000 × g for 2 min and washed 3 times in NT2 buffer. The beads were used to extract RNA with the GenElute mammalian total RNA miniprep kit (Sigma). For reverse transcription-PCR (RT-PCR) analysis, 1/10 of the RNA was reverse transcribed to cDNA by using a primer that bound to nt 1779 to 1760 of the MNV-1 genome and Moloney murine leukemia virus reverse transcriptase (Promega) as per the manufacturer's protocol. A 1/10 aliquot of the cDNA was then subjected to PCR using the primers IGIC200 (nt 1439 to 1416) and IGIC464 (nt 1779 to 1760) to produce a 364-bp product encompassing nucleotides 1416 to 1779 of the MNV genome, and the PCR products were analyzed on a 2% agarose gel.
Immunofluorescence assays.
BV-2 cells were plated on glass coverslips and infected with MNV (MOI, 5 TCID50 per cell) at 37°C. After 12 h, the coverslips were washed with cold PBS, fixed in 4% paraformaldehyde solution containing 250 mM HEPES (pH 7.4) for 10 min on ice, and permeabilized with 0.2% Triton X-100 in 4% paraformaldehyde for an additional 10 min at room temperature, and aldehyde groups were quenched with 0.2 M glycine in PBS. The cells were then stained for various host or viral proteins and double-stranded RNA (dsRNA) by using the corresponding primary and secondary antibodies in block buffer (1% bovine serum albumin in PBS) for 45 min. The glass coverslips were washed three times for 5 min each with ice-cold PBS after each step. Finally, the cells were washed in water and mounted on glass slides in mounting medium (MOWIOL containing 4′,6-diamidino-2-phenylindole [DAPI]) overnight before visualization on a Zeiss LSM 510 META confocal microscope. The images were processed with Zeiss Meta 510 software (Carl Zeiss) and later analyzed by using Image J application version 1.43.
siRNA-mediated inhibition of cellular gene expression.
Actively growing MNV-permissive BV-2 cells were used for analyzing the effects of protein knockdown on virus replication. Briefly, 6 × 106 BV-2 cells were electroporated with 100 pmol of siRNA duplexes using the Neon transfection system (Invitrogen) at 1,700 V for 10 ms and 3 pulses. siRNAs were purchased from Eurogentec, and the target sequences were as follows: nonspecific nontargeting siRNA, GCGCGCUUUGUAGGAUUCG (67); PTB, AACUUCCAUCAUUCCAGAG; La, UUUGCCACGGGACAAGUUU. For DDX3, a combination of two siRNAs was used: UCAUAUCCACUAGACGUCC and CCUGAACUCUUCAGAUAAU. The cells were plated in antibiotic-free medium to allow recovery and incubated for 12 h. The cells were then infected with MNV-1 (MOI, 0.01 TCID50/cell), and samples were harvested for RNA isolation, virus titer determinations, and Western blotting at various time points postinfection. The virus titer was determined based on the TCID50, and the viral RNA copy number was quantified using RT-quantitative PCR as described previously (3).
Computations and RNA structure analysis.
Computer analysis of the RNA secondary structures was performed using the Mfold2 software through the Web interface at http://frontend.bioinfo.rpi.edu/applications/mfold.
RESULTS
The 5′ and 3′ extremities of the murine norovirus genome contain an extensive RNA structure.
Our previous bioinformatics analysis, which examined synonymous codon suppression and the folding energy within various regions of calicivirus genomes, clearly indicated that the majority of the RNA structure lays at the 5′ and 3′ termini of genomic and subgenomic viral RNAs (65). More-detailed characterization of the 3′ end of the MNV genome, including biochemical structure probing and mutational analysis, confirmed that the 3′ end contains at least 3 stem-loop (SL) structures (Fig. 1), the integrity of which are required for efficient virus replication in tissue culture (3, 65). To extend this analysis to the 5′ end of the viral genome, we used bioinformatics to predict the structures adopted by this region of the viral genome (Fig. 1). Computational structure predictions suggested the presence of a number of RNA structures, in addition to the stem-loop structures present at positions 8 and 29, previously referred to as SL8 and SL29, respectively (65). RNase sensitivity mapping was then used to biochemically probe the structure adopted by the 5′ 250 nucleotides, as this was the region previously shown to contain the evolutionarily conserved RNA structures (65). In the majority of instances, the RNase sensitivity mapping correlated with the predicted RNA secondary structure (Fig. 1). However, a number of RNase T1 cleavage-sensitive residues, single-stranded G specific, were observed at positions predicted to be in double-stranded regions, namely, nucleotide positions 53, 58, 205, and 206 (Fig. 1). In addition, nucleotide positions 139 and 138, predicted to lie within a single-stranded region of RNA, were found to be sensitive to RNase V1, suggestive of the presence of double-stranded base pairing and/or RNA in a helical conformation (42). This heterogeneity may be a reflection of an alternative RNA conformation present within the population, or that particular regions of the RNA structure are more promiscuous in their base pairing, in agreement with the relatively high P-num values (data not shown). Overall, however, the biochemical mapping data confirmed that extensive RNA structures lay within the coding region of the MNV genome.
Fig 1.

Murine norovirus genome organization and positions of conserved RNA structures. The NS1 to -7 nomenclature, as proposed by Sosnovtsev et al. (66), is presented in italics below the various open reading frames. Note that the RNA structures are not drawn to scale. (A) Predicted RNA structures adopted by the 5′ end of the MNV genome, highlighting the biochemically determined cleavage sites for single-stranded RNA-specific RNases T1, T2, and A, along with the double-stranded RNA-specific RNase V1 cleavage sites. (B) Primary data for RNase sensitivity mapping of the 5′ end of the MNV genome. In vitro-transcribed RNA encompassing the region was subjected to limited RNase digestion and subsequent primer extension analysis as described previously. A sequencing ladder prepared using the same primer was also performed to allow the identification of the RNase cleavage sites. Analysis was performed a minimum of 3 times, and 1 representative gel shown. Nucleotide positions are numbered according to their positions in the murine norovirus genome. Note that data are shown for regions containing RNase cleavage sites only. (C) Schematic illustration of the RNA structures adopted by the 3′ 237 nucleotides of the MNV genome. For clarity, the sequence of this region is not shown. The structure shown is as previously computationally predicted and biochemically confirmed (3).
The 5′ and 3′ extremities of the norovirus genome interact with host cell factors.
Our previous work, based on mutational analysis of the RNA structures present at the extremities of the MNV genome, illustrated that the RNA structures present in these regions play a role in the norovirus life cycle; destabilization of SL8 or SL29 resulted in reduced virus yield, and in the case of SL8 there was also a reduction in plaque size (65). The biochemical data described above also illustrate that additional stem-loop structures are formed within the first 250 nucleotides of the viral genome, in agreement with our previous bioinformatic data. Mutational analysis of the RNA structures at the 3′ extremity has been limited to the noncoding region only, and has indicated that the integrity of the two terminal stem-loop structures are essential for virus viability in cell culture and that the polypyrimidine tract contributes to virulence in the natural host (3). To begin to further understand the role of these RNA structures in the norovirus life cycle, RNA affinity chromatography followed by mass spectrometry was used to identify host factors that interact with these structures in vitro. This type of riboproteomics approach has been widely used to identify host factors involved in internal ribosome entry site (IRES)-mediated translation initiation (52, 53). Regions encompassing the most highly conserved RNA structures of the MNV genome, namely, nt 1 to 250 and 7141 to 7397 of the 5′ and 3′ extremities of the MNV-1 genome, respectively, were PCR amplified with the inclusion of a T7 RNA polymerase promoter at their 5′ end. In the case of the 3′ extremity, the product generated included a 3′ poly(A) tail of 15 nt to produce an RNA target that would best represent the authentic 3′ end of the polyadenylated viral RNA genome. In vitro transcription was then used to generate RNA targets, which were subsequently coupled to CNBR-activated Sepharose. As controls, an irrelevant RNA or Sepharose beads alone were also prepared in the same manner. Cytoplasmic extracts from RAW264.7 cells, a murine macrophage cell line previously shown to be highly permissive for MNV replication (74), or RSW from RRL, likely to be highly enriched in factors involved primarily in viral translation initiation, were then incubated with the RNA-coupled Sepharose beads After extensive washing, the bound cellular proteins were resolved by SDS-PAGE and visualized by colloidal Coomassie staining (Fig. 2). Visual inspection and densitometry were used to identify areas of the lanes that contained proteins within the test samples that were either absent or appeared reduced in the alternate viral RNA target or the nonrelated RNA and bead-alone controls. These areas were subsequently excised from the gel, along with the same region in the nonrelated RNA control, and the identities of proteins within these bands were then determined using mass spectrometry. By combining data from 4 independent experiments, we identified 99 proteins from RAW264.7 cell S-100 extracts with 2 or more unique peptides in the sample prepared using the 5′ extremity of the genomic RNA (Table 1; see also Table S1 in the supplemental material). Using the same criteria, we identified 33 proteins in the sample prepared using the 3′ extremity of the MNV genome to purify proteins from RAW264.7 S-100 extracts (Table 2; see also Table S2 in the supplemental material). In contrast, many fewer proteins (17 and 7 for the 5′ and 3′ extremities, respectively) were identified from the RRL RSW (Tables 3 and 4; see also Tables S3 and S4 in the supplemental material).
Fig 2.

Identification of host factors that interact with the extremities of the murine norovirus genome. Shown are results SDS-PAGE analysis of RNA affinity chromatography-purified host factors that interact with the 5′ or 3′ extremities of the murine norovirus genome, 5′G and 3′Ex, respectively. N/R and beads refer to controls generated using either a nonrelated RNA coupled to cyanogen bromide-activated Sepharose or Sepharose beads alone. (A) Samples purified using S-100 extracts from RAW264.7cells; (B) samples purified using a ribosomal salt wash from rabbit reticulocyte lysates. The positions of proteins enriched in the viral RNA samples compared to the control are highlighted with arrowheads. Note that for clarity only a single gel from each analysis is shown. The protein identities from the data shown are listed in Tables 1 and 2 for the RAW264.7 cells and RRL, respectively.
Table 1.
Host factors from RAW264.7 cell S-100 extracts that interacted with the 5′ extremity of the murine norovirus genomea
| Band no. | Protein ID | Accession no. | Mass (kDa) | SC (%) | No. of unique peptides |
Identification probability (%) |
||
|---|---|---|---|---|---|---|---|---|
| 5′G | NR | 5′G | NR | |||||
| 1 | TLN1 (Talin-1) | P26039 | 270 | 8 | 15 | 0 | 100 | 0 |
| FASN | P19096 | 272 | 2 | 4 | 0 | 100 | 0 | |
| CNOT1 | Q6ZQ08 | 266 | 1 | 2 | 0 | 100 | 0 | |
| 2 | HDLBP (Vigilin) | Q8VDJ3 | 142 | 40 | 43 | 6 | 100 | 100 |
| DHX9 | O70133 | 149 | 10 | 12 | 28 | 100 | 100 | |
| VARS | Q9Z1Q9 | 140 | 3 | 4 | 16 | 100 | 100 | |
| DIAPH1 | O08808 | 139 | 2 | 2 | 2 | 99 | 100 | |
| 3 | hnRNPU | Q8VEK3 | 88 | 7 | 4 | 1 | 100 | 54 |
| hnRNPUL2 | Q00PI9 | 85 | 5 | 3 | 3 | 100 | 100 | |
| EIF3B | Q8JZQ9 | 109 | 3 | 2 | 0 | 100 | 0 | |
| USP5 | P56399 | 96 | 3 | 2 | 0 | 98 | 0 | |
| PDCD6IP | Q9WU78 | 97 | 3 | 2 | 0 | 100 | 0 | |
| 4 | HSP90AB1 | Q71LX8 | 83 | 34 | 21 | 3 | 100 | 100 |
| HSP90AA1 | P07901 | 85 | 24 | 6 | 0 | 100 | 0 | |
| KHSRP | Q3U0V1 | 77 | 10 | 6 | 24 | 100 | 100 | |
| NSUN2 | Q1HFZ0 | 85 | 9 | 4 | 3 | 100 | 100 | |
| Ncl (nucleolin) | P09405 | 77 | 6 | 3 | 4 | 100 | 100 | |
| PLA2G4A | P47713 | 85 | 4 | 2 | 1 | 100 | 50 | |
| 5 | LCP1 | Q61233 | 70 | 44 | 23 | 7 | 100 | 100 |
| DDX5 | A1L333 | 69 | 17 | 10 | 8 | 100 | 100 | |
| RPA1 | Q8VEE4 | 71 | 6 | 4 | 7 | 100 | 100 | |
| 6 | FUBP3 | A2AJ72 | 61 | 41 | 18 | 16 | 100 | 100 |
| SYNCRIP | Q7TMK9 | 70 | 24 | 12 | 1 | 100 | 71 | |
| MYEF2 | Q8C854 | 63 | 13 | 7 | 0 | 100 | 0 | |
| FUS | P56959 | 53 | 11 | 4 | 3 | 100 | 100 | |
| GNL3 | Q8CI11 | 61 | 10 | 5 | 0 | 100 | 0 | |
| PTPN6 | P29351 | 68 | 6 | 3 | 2 | 100 | 99 | |
| Ncl | P09405 | 77 | 3 | 2 | 0 | 99 | 0 | |
| 7 | hnRNPK | P61979 | 51 | 29 | 10 | 1 | 100 | 90 |
| USP14 | Q9JMA1 | 52 | 9 | 4 | 0 | 100 | 0 | |
| CCT2 | P80314 | 57 | 8 | 3 | 0 | 100 | 0 | |
| KPNA3 | O35344 | 58 | 4 | 2 | 0 | 100 | 0 | |
| PRPF19 | Q99KP6 | 55 | 4 | 2 | 0 | 100 | 0 | |
| PHGDH | Q61753 | 57 | 4 | 2 | 0 | 100 | 0 | |
| 8 | La | P32067 | 48 | 44 | 17 | 5 | 100 | 100 |
| Eef1a1 | P10126 | 50 | 30 | 10 | 12 | 100 | 100 | |
| YBX1 | P62960 | 36 | 20 | 4 | 0 | 100 | 0 | |
| ENO1 | P17182 | 47 | 18 | 5 | 1 | 100 | 80 | |
| PHAX | Q9JJT9 | 43 | 18 | 5 | 0 | 100 | 0 | |
| DAZAP1 | Q9JII5 | 43 | 16 | 4 | 3 | 100 | 100 | |
| hnRNPF | Q9Z2X1 | 46 | 12 | 3 | 10 | 100 | 100 | |
| DDX39B | Q9Z1N5 | 49 | 9 | 4 | 2 | 100 | 100 | |
| NCF1 | Q09014 | 45 | 7 | 2 | 1 | 100 | 80 | |
| PDIA6 | Q922R8 | 48 | 6 | 2 | 1 | 100 | 80 | |
| TUBB | B1B178 | 50 | 5 | 2 | 8 | 99 | 100 | |
| EEF1G | Q9D8N0 | 50 | 5 | 2 | 7 | 100 | 100 | |
| 9 | PCBP1 | P60335 | 37 | 56 | 13 | 11 | 100 | 100 |
| Unrip | Q9Z1Z2 | 38 | 55 | 14 | 0 | 100 | 0 | |
| PCBP2 | Q61990 | 38 | 34 | 5 | 3 | 100 | 100 | |
| hnRNPA2B1 | O88569 | 37 | 28 | 7 | 1 | 100 | 84 | |
| hnRNPAB | Q99020 | 37 | 27 | 6 | 5 | 100 | 100 | |
| hnRNPA3 | Q8BG05 | 40 | 25 | 10 | 6 | 100 | 100 | |
| CAPG | Q3UJ44 | 39 | 16 | 4 | 7 | 100 | 100 | |
| hnRNPD | Q60668 | 38 | 15 | 3 | 1 | 100 | 88 | |
| RTN4 | Q99P72 | 38 | 14 | 2 | 2 | 100 | 100 | |
| ANXA1 | P10107 | 39 | 12 | 3 | 3 | 100 | 100 | |
| GNAI2 | P08752 | 41 | 6 | 2 | 5 | 99 | 100 | |
Protein ID lists the UniProtKB accession number for each of the proteins identified in Fig. 2A. SC refers to the percent sequence coverage for the protein purified and identified using the 5′-extremity of MNV genomic RNA (5′G). NR refers to the data acquired when we used a nonrelated control RNA, as described in the text. The identification probability was calculated using the ProteinProphet algorithm. Samples were arranged per gel slice excised from the gel presented in Fig. 2A and then ranked based on the number of unique peptides identified.
Table 2.
Host factors from RAW264.7 cell S-100 extracts that interacted with the 3′ extremity of the murine norovirus genomea
| Band no. | Protein ID | Accession no. | Mass (kDa) | SC (%) | No. of unique peptides |
Identification probability (%) |
||
|---|---|---|---|---|---|---|---|---|
| 3′Ex | NR | 3′Ex | NR | |||||
| 10 | PABPC1 | P29341 | 71 | 31 | 16 | 5 | 100 | 100 |
| DDX3X | Q62167 | 73 | 13 | 7 | 14 | 100 | 100 | |
| FUBP1 | Q91WJ8 | 69 | 7 | 4 | 12 | 100 | 100 | |
| DHX58 | Q99J87 | 77 | 3 | 2 | 0 | 100 | 0 | |
| 11 | PTBP1 | Q5RJV5 | 59 | 33 | 13 | 3 | 100 | 100 |
| PKM2 | P52480 | 58 | 13 | 5 | 5 | 100 | 100 | |
| FARSA | Q8C0C7 | 58 | 5 | 2 | 2 | 100 | 100 | |
| 12 | PTBP1 | Q5RJV5 | 59 | 32 | 13 | 5 | 100 | 100 |
| hnRNPK | P61979 | 49 | 6 | 2 | 1 | 100 | 90 | |
| 13 | hnRNPF | Q9Z2X1 | 46 | 30 | 9 | 6 | 100 | 100 |
| DAZAP1 | Q9JII5 | 43 | 22 | 5 | 4 | 100 | 100 | |
| La | P32067 | 48 | 18 | 4 | 6 | 100 | 100 | |
| Eef1a1 | P10126 | 50 | 9 | 3 | 0 | 100 | 0 | |
| ENO1 | P17182 | 47 | 8 | 3 | 4 | 100 | 100 | |
| VAT1 | Q62465 | 43 | 6 | 2 | 1 | 100 | 82 | |
| 14 | GPD1L | Q3ULJ0 | 38 | 28 | 10 | 1 | 100 | 91 |
| hnRNPA1 | P49312 | 34 | 23 | 5 | 2 | 100 | 100 | |
| hnRNPA2B1 | O88569 | 37 | 22 | 6 | 1 | 100 | 69 | |
| GAPDH | P16858 | 36 | 15 | 2 | 1 | 99 | 91 | |
| hnRNPAB | Q99020 | 31 | 6 | 2 | 0 | 100 | 0 | |
| HuR | P70372 | 36 | 4 | 2 | 3 | 98 | 100 | |
Protein ID lists the UniProtKB accession number for each of the proteins identified in Fig. 2A. SC refers to the percent sequence coverage for the protein purified and identified using the 3′ extremity of the MNV genomic RNA (3′Ex). NR refers to the data acquired when we used a nonrelated control RNA, as described in the text. The identification probability was calculated using the ProteinProphet algorithm. Samples were arranged per gel slice excised from the gel presented in Fig. 2A and then ranked based on the number of unique peptides identified.
Table 3.
Host factors from a ribosomal salt wash of rabbit reticulocyte lysates that interacted with the 5′-extremity of the murine norovirus genomea
| Band no. | Protein ID | Accession no. | Mass (kDa) | SC (%) | No. of unique peptides |
Identification probability (%) |
||
|---|---|---|---|---|---|---|---|---|
| 5′G | NR | 5′G | NR | |||||
| 1 | HNRNPK | B2M1R6 | 49 | 23 | 7 | 0 | 100 | 0 |
| YBX1 | B5ABI3 | 36 | 14 | 2 | 2 | 100 | 99 | |
| CCT2 | P80314 | 57 | 7 | 2 | 0 | 75 | 0 | |
| TCP-1-ζ | O77622 | 58 | 6 | 3 | 0 | 100 | 0 | |
| CCT4 | A8K3C3 | 58 | 5 | 2 | 0 | 100 | 0 | |
| 2 | CSNK2A1 | Q61177 | 45 | 37 | 13 | 1 | 100 | 89 |
| 3 | PCBP1 | P60335 | 37 | 37 | 9 | 5 | 100 | 100 |
| CSNK2A1 | Q61177 | 45 | 34 | 9 | 0 | 100 | 0 | |
| PCBP2 | B2M1R7 | 38 | 21 | 2 | 0 | 100 | 0 | |
| YBX1 | B5ABI3 | 36 | 11 | 2 | 4 | 100 | 100 | |
| 4 | CSNK2B | Q5SRQ6 | 25 | 42 | 8 | 0 | 100 | 0 |
Protein ID lists the UniProtKB accession number for each of the proteins identified in Fig. 2B. SC refers to the percent sequence coverage for the protein purified and identified using the 5′ extremity of the MNV genomic RNA (5′G). NR refers to the data acquired when we used a nonrelated RNA, as described in the text. The identification probability was calculated using the ProteinProphet algorithm. Samples were arranged per gel slice excised from the gel presented in Fig. 2B and then ranked based on the number of unique peptides identified.
Table 4.
Host factors from a ribosomal salt wash of rabbit reticulocyte lysates that interacted with the 3′ extremity of the murine norovirus genomea
| Band no. | Protein ID | Accession no. | Mass (kDa) | SC (%) | No. of unique peptides |
Identification probability (%) |
||
|---|---|---|---|---|---|---|---|---|
| 3′Ex | NR | 3′Ex | N/R | |||||
| 5 | PABPC1 | P29341 | 71 | 42 | 19 | 8 | 100 | 100 |
| PABPC4 | Q6IQ30 | 72 | 21 | 4 | 0 | 100 | 0 | |
| HSPA8 | P63017 | 71 | 10 | 5 | 2 | 100 | 100 | |
| 15-LOX | P12530 | 75 | 9 | 4 | 6 | 100 | 100 | |
| FUBP1 | Q0P6B2 | 66 | 3 | 2 | 10 | 99 | 100 | |
| 6 | CCT2 | P80314 | 57 | 15 | 5 | 0 | 100 | 0 |
| PTBP1 | Q3U5I2 | 59 | 14 | 4 | 1 | 100 | 86 | |
| TCP-1-ζ | O77622 | 58 | 3 | 2 | 0 | 100 | 0 | |
| PABPC1 | P29341 | 71 | 16 | 9 | 4 | 100 | 100 | |
Protein ID lists the UniProtKB accession number for each of the proteins identified in Fig. 2B. SC refers to the percent sequence coverage for the protein purified and identified using the 3′ extremity of the MNV genomic RNA (3′Ex). NR refers to the data acquired when we used a nonrelated RNA, as described in the text. The identification probability was calculated using the ProteinProphet algorithm. Samples were arranged per gel slice excised from the gel presented in Fig. 2B and then ranked based on the number of unique peptides identified.
Although these experiments were not designed to be quantitative, it was clear that in some instances the protein (or proteins) that interacted with the viral sequence was entirely absent from the irrelevant RNA control, e.g., UNR-interacting protein (Unrip) (Table 1). However, in the majority of cases, host factors appeared to be simply enriched when purified on viral RNA compared to the nonrelated RNA, e.g., La protein (Table 1). This likely simply reflects the rather promiscuous nature of the nucleic acid-binding activities of many of the identified factors and the fact that the in vitro assay was performed under relatively mild conditions. As expected, we identified a number of host proteins previously reported to interact with either the MNV or human norovirus genome. Our previous studies confirmed that the cellular RNA chaperone PTB interacts with the variable polypyrimidine tract within the terminal noncoding stem-loop at the 3′ end of the MNV genome (3). This protein was readily detected in the samples purified using the 3′ extremity of the MNV genome. In addition to proteins previously reported to interact with the human norovirus RNA genome, e.g., La (SSB) and PCBP isoforms 1 and 2, we also identified a number of well-characterized host factors previously implicated in host cell mRNA translation or splicing control, including YBX1, DDX proteins, hnRNP proteins, PABP isoforms, and nucleolin. Ingenuity Pathway analysis, based on published data curated in the Ingenuity Knowledge Base, was used to identify possible interactions and associations between the proteins identified in the combined data set (Fig. 3). This approach allowed the identification of a number of highly connected proteins that may function as hub proteins, with many interacting partners, as well as a number of potential protein-protein interactions that may contribute to interactions between the 5′ and 3′ extremities of the norovirus genome.
Fig 3.

Network analysis of the host factors found to be associated with the extremities of the murine norovirus genome. The interaction network shows the proteins identified as being associated with the MNV genome by riboproteomics, as described in the text. The proteins shown were identified with a probability greater than 90% and with at least 2 unique peptides showing at least a 90% peptide identification probability. To simplify these interdependencies, host factors that interacted with themselves only or did not interact with other factors in the network are not shown. Proteins shaded in orange were identified using only the the 5′ extremity of MNV genomic RNA and those shaded in blue were identified using the 3′ extremity, while proteins shaded in gray were identified using both RNA targets. Proteins in brackets represent those further characterized during this study. Note that the network was generated using the data presented in Tables 1 and 2 as well as additional experimental data sets. Proteins not identified in the data set illustrated in Fig. 2 and Tables 1 and 2 are highlighted with an asterisk. The interaction network was generated using the Ingenuity Pathway Analysis program (Ingenuity Systems). Full lines represent direct interactions, while dashed lines indicate indirect interactions. An arrow pointing from one protein to another indicates that the first protein acts on or activates the second protein (at which the arrow is pointing).
Host factors interact with the norovirus genome during virus replication in cell culture.
To confirm that the proteins identified in the in vitro riboproteomics assay described above interact either directly or indirectly with the MNV genome during authentic virus replication, we use coimmunoprecipitation followed by RT-PCR. Although the riboproteomics approach identified host factors associated with in vitro-transcribed RNA, it is to be noted that during replication, the 5′ end of the MNV genome is covalently attached to the viral protein (VPg) protein, which might also affect binding of some of the identified proteins. The MNV-permissive murine macrophage cell line RAW264.7 was infected with MNV, and antibodies to a selection of host factors were used for immunoprecipitation. Host factors used for the validation were selected based on the availability of antibodies capable of immunoprecipitating the target antigen and whether or not there were previous data that indicated a role for these factors in the life cycle of other viruses. Talin-1 was also included, as to date there has been no reported role for this protein in the life cycle of any positive-strand RNA viruses, indicating that it may therefore represent a novel interaction. Based on these criteria, we performed coimmunoprecipitation RT-PCR using antisera to PTB, La, PCBP, DDX3, HuR, YBX1, HSP90α/β, and Talin-1. The presence of viral RNA in the immunoprecipitated complex was examined by RT-PCR amplification (Fig. 4). In each case, control immunopreciptations were also performed with antisera to the cellular protein GAPDH, rabbit IgG, or actin (data not shown). We were able to detect the presence of the viral RNA associated with all the target proteins tested, although to various degrees, but no signal was obtained using the control antisera (Fig. 4). The variations in the amount of viral RNA copurified with the host factor may simply reflect the variabilities in the efficiencies of the antisera to immunoprecipitate the target protein or possible variabilities in the dynamics of the interaction of the host proteins with viral RNA. Also, in some cases, we cannot rule out the possibility that the antisera obscure or hinder the relevant binding site required to pull down the viral RNA.
Fig 4.
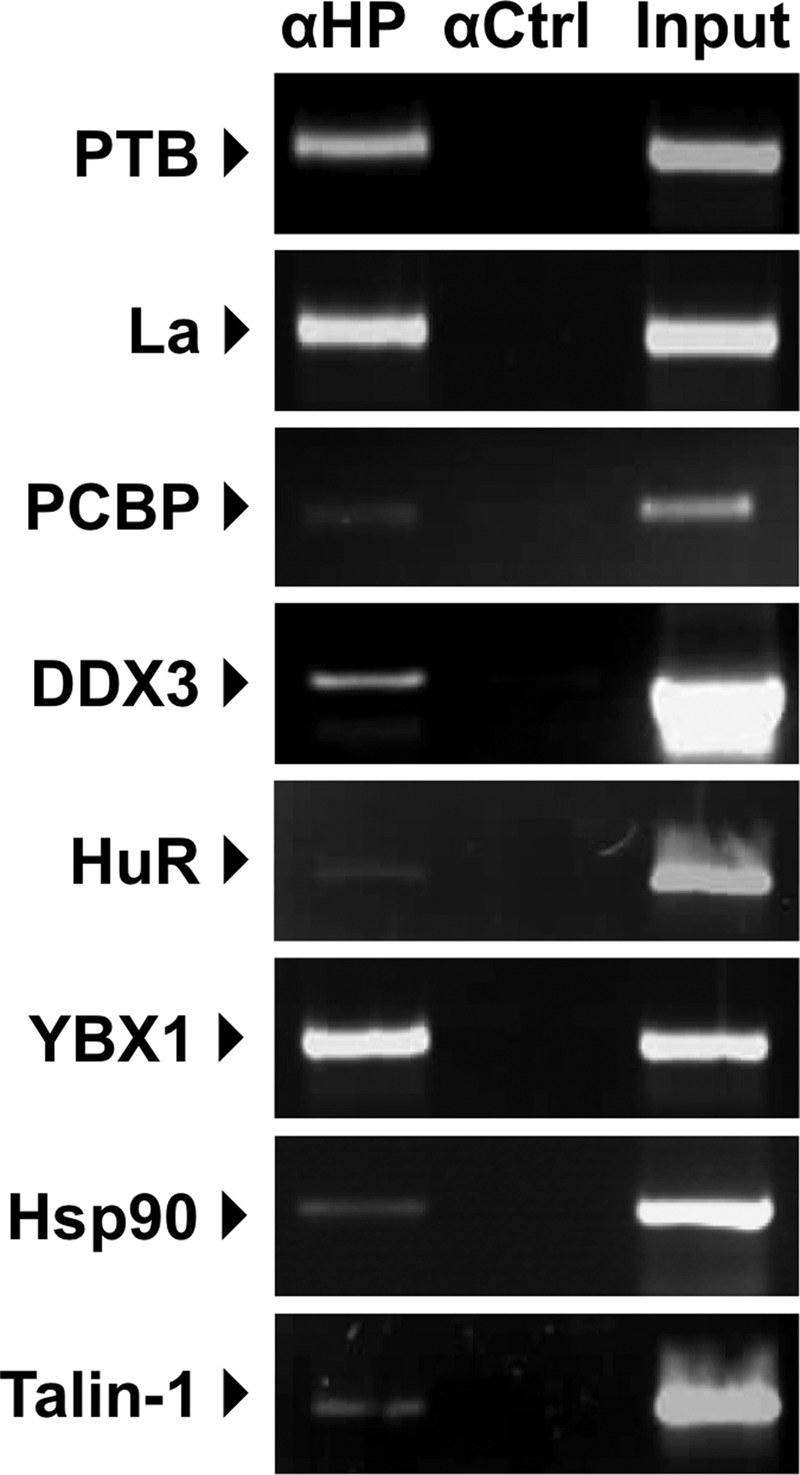
Host factors interact with the murine norovirus genome during virus replication in cell culture. RAW264.7 cells were infected with MNV-1 at a multiplicity of infection of 1 TCID50 per cell for 15 h, followed by lysis and immunoprecipitation using antisera to either host factors identified using the riboproteomics approach described in the text (anti-HP [αHP]), control rabbit IgG (αCtrl, used for La, PCBP, YBX1, and HSP90), or antisera to GAPDH (αCtrl, used for PTB, DDX3, HuR, and Talin-1). Following immunoprecipitation, any associated RNA was purified, and RT-PCR amplification was performed to identify the presence of viral RNA, as described in the text. An aliquot of the starting material (input) was used to confirm efficient infection and replication.
Cellular nucleic acid-binding proteins are associated with the viral replication complexes.
Further analysis of the localization of the identified host factors and the effect of virus infection on the distribution of the identified host factors was performed by using confocal microscopy. The murine microglial cell line BV-2 (5), previously shown to be highly permissive for MNV-1 infection (10), was chosen for microscopy, as its physical properties, e.g., better adherence and a larger cytoplasm, makes this cell line more suitable for imaging under the conditions used in this study. BV-2 cells were infected with MNV-1 and subsequently stained for viral antigen or viral dsRNA and costained for a variety of cellular factors identified as being associated with the viral RNA during replication, namely, PTB, DDX3, and La (Fig. 5). As previously reported, the localization of the majority of the viral antigen or dsRNA was found to have a perinuclear localization (30). Costaining of cells for VPg and PTB resulted in a large degree of overlap of the signal (Fig. 5A). The distribution of La was largely unaffected by MNV-1 replication; however, some degree of signal overlap was observed, with punctate foci of La protein found within the VPg-positive perinuclear region (Fig. 5B). Costaining of MNV-1-infected cells for dsRNA and DDX3 showed that DDX3 staining appeared to be at least partially associated with sites of dsRNA localization (Fig. 5C). Whereas in uninfected cells DDX3 distribution appeared largely diffuse through the cytoplasm of the cell, the DDX3 in infected cells formed aggregates and puncta, some of which localized within the region also staining positive for dsRNA. A possible increase in the levels of PTB and DDX3 was observed during the analysis of the infected cells by confocal microscopy; however, this was not evident by Western blotting (data not shown). This apparent increased staining was the likely result of increased epitope accessibility that resulted in increased primary antibody binding. Taken together with the previous RNA coimmunoprecipitation data (Fig. 4), we conclude that at least fractions of the cellular PTB, DDX3, and La are associated with viral replication during authentic MNV-1 replication in cell culture.
Fig 5.

Confocal analysis of the localization of host factors during murine norovirus replication in cell culture. The murine microglial cell line BV-2 was infected with MNV-1 at a multiplicity of infection of 5 TCID50 per cell, and the infection was allowed to proceed for 12 h. After infection, cells were fixed, permeabilized, and stained with antisera to either the viral protein VPg, viral double-stranded RNA (dsRNA), or various host factors. DAPI staining was used to stain the nuclear DNA. The “Col. points” column highlights the positions of overlapping signals between the viral and cellular antigens generated using the ImageJ colocalization plugin. Bar, 10 μm.
RNA interference-mediated reduction in the level of PTB, DDX3, or La inhibits the murine norovirus life cycle.
The role of three of the host factors identified as being associated with viral RNA in MNV-1-infected cells was further characterized using RNA interference to reduce the intracellular RNA levels, and the effect on the viral life cycle examined. Attempts to achieve robust and reproducible reductions in the levels of host factors in the MNV-1-permissive cell line RAW264.7 using transfection of either siRNAs or shRNA-expressing plasmids were largely unsuccessful (data not shown). In order to achieve reproducible knockdown of various host factors, it was necessary to use Neon-mediated electroporation of the murine microglial cell line BV-2 with siRNAs. Optimized transfection conditions were determined by using a green fluorescent protein expression plasmid; similar conditions were then used for siRNAs against PTB, La, and DDX3 to achieve knockdown of either protein to various levels (Fig. 6), and a time course of MNV-1 infection was determined. Figure 6 represents the effects of knockdown of La protein (Fig. 6A), PTB (Fig. 6B), and DDX3 (Fig. 6C) on levels of viral protein (NS7), RNA, and the viral titer at 18, 24, and 30 h postinfection. Knockdown of the La protein in BV-2 cells was inefficient, typically resulting in around a 50% reduction in the protein levels compared to cells transfected with a nonspecific siRNA. This reduction in the intracellular levels of La resulted in a similar reduction in the levels of the viral polymerase NS7, to ∼37 to 38% of the levels of those observed in control cells at 24 and 30 h postinfection, respectively. This also resulted in a substantial effect on viral titers and viral RNA synthesis levels, which were significantly reduced at all times (Fig. 6A). Reduction of PTB to around 21% of the normal intracellular levels resulted in a reduction in viral protein synthesis along with a >80% reduction in viral yield and viral RNA levels from 24 h postinfection onwards (Fig. 6B). siRNA-mediated reduction in intracellular DDX3 levels resulted in an 80 to 90% reduction in the levels of viral NS7, viral yield, and viral RNA levels (Fig. 6C). These data confirm a functional role for some of the proteins identified using the riboproteomics approach; however, it is worth noting that this siRNA-based approach does not discriminate between direct and indirect effects on the reduction in the target proteins on virus replication.
Fig 6.

siRNA-mediated reductions in intracellular levels of PTB, La, or DDX3 inhibit murine norovirus. The murine microglial cell line BV-2 was transfected with either nonspecific siRNA (Nsp) or siRNA duplexes targeting La (A), PTB (B), or DDX3 (C). At 12 h posttransfection, the cells were infected with MNV-1 at a multiplicity of infection of 0.5 TCID50 per cell. Total infectious virus production and the levels of viral RNA were then determined at various time points postinfection, as indicated by the TCID50 and RT-quantitative PCR results, respectively. The data were plotted versus the control cells, which were transfected with a nonspecific siRNA control (NSP). In each graph, the percentage value refers to the relative amount of viral RNA or infectious units, expressed as a percentage of the control at that time point. Each experiment was performed using three independent biological repeats and in at least three independent experiments. The data presented are from a single representative data set along with the standard deviations. Western blot analysis was performed using a chemiluminescence-based detection system, and protein expression was quantified using densitometry. In each case, samples were normalized to the levels of GAPDH and the viral NS7 levels expressed relative to the levels observed in cells transfected with nonspecific siRNA control (NSP). Statistical analyses were performed by two-way analysis of variance with Bonferroni's posttest. *, P < 0.05; ***, P < 0.001.
DISCUSSION
The genomes of positive-strand RNA viruses such as noroviruses not only function as mRNA for the production of viral proteins but also they contain additional information in the form of conserved cis-acting RNA structures required for various aspects of the viral life cycle (40). These RNA structures play many important roles, including regulating viral translation, replication, and encapsidation, as well as viral pathogenesis. While in the majority of instances the cis-acting RNA structures lay within the 5′ and 3′ noncoding regions of viral genomes, such regions are invariably small in members of the Caliciviridae, e.g., 5 and 78 nt, respectively, for the murine norovirus 5′ and 3′ noncoding regions. We previously used computational analysis to demonstrate that the genomes of members of the Caliciviridae family contain several regions of synonymous codon suppression near the 5′ extremity of the viral genomic and subgenomic RNAs but also at the 3′ extremity of the subgenomic RNA in the VP2 coding region (65). This feature is evolutionarily conserved across all members of the Caliciviridae; however, the structures that are adopted and the degree to which the RNA structures lay within the coding region differ. For example, the subgenomic RNA for murine norovirus encodes three proteins, namely, the structural proteins VP1 and VP2 and also a recently identified protein involved in viral virulence, VF1 (45). The presence of an additional overlapping open reading frame (ORF) results in a substantially increased amount of synonymous codon suppression in the MNV subgenomic RNA compared to other caliciviruses. Feline calicivirus also appears to have an additional region of conserved synonymous codon suppression within ORF1, in the region coding for p30 (NS4), the function of which has yet to be determined (65).
While to date there has been limited information regarding the role of host nucleic acid-binding proteins in the norovirus life cycle, previous studies demonstrated that a number of host factors interact with the Norwalk virus genome. Specifically, La, PTB, and PABP interact with the 3′ untranslated region (UTR) of the Norwalk virus genome (27), and La, PTB, PCBP-2, and hnRNPL interact with the 5′ end (25). While a functional role for these interactions is yet to be determined, due to the lack of an amenable functional assay, studies have indicated that cellular proteins may contribute to genome circularization via interactions with the 5′ and 3′ extremities of the viral genome (60). This type of genome circularization is a common emerging theme and has been reported for a number of positive-strand RNA viruses, including poliovirus (28) and dengue virus (1, 18). In this study, cell lysates from MNV-permissible cells, as well as rabbit reticulocyte lysates, known to support MNV viral RNA translation, were used to enable the identification of approximately 150 different host proteins that interacted with the 5′ and 3′ extremities of the viral positive-sense RNA genome. The interactions of some of these proteins were confirmed during infection using coimmunoprecipitation RT-PCR. Although we cannot rule out that some of these proteins might interact with the viral genome indirectly by virtue of protein-protein interactions, the association with viral RNA during infection is supportive of a role in the viral life cycle. Based on curated data available in the Ingenuity Knowledge Base, we were able to map a network of interactions between the various proteins identified by our riboproteomics approach (Fig. 3). Within this network, there were numerous example of factors that were purified using both the 5′ and 3′ extremities of the viral genome, e.g., PTB and a number of hnRNP proteins. In addition, there were clear examples of possible 5′-3′-bridging interactions that would occur via protein-protein interactions, e.g., hnRNPD and HuR. Whether or not these types of bridging interactions function in the MNV life cycle will require additional functional studies; however, this work provides new insights into the identity of possible host factors involved in these interactions. It is also worth noting that while this study was focused on the RNA structure present within the viral positive-sense genomic RNA, RNA structures are predicted to be present in the antisense RNA (65). It is therefore likely that structures present on both strands of the viral RNA interact with viral and host proteins, contributing to the viral life cycle, and therefore further studies are warranted.
While we identified a number of host factors already known to interact with the prototype human norovirus, namely, the Norwalk virus genome or the genomes of other RNA viruses, we also identified a number of proteins that have not yet been reported to play a functional role in the life cycle of positive-strand RNA viruses. For example, Talin-1, identified with 15 unique peptides as binding to the 5′ extremity of the MNV genome (Table 1), links focal adhesion formation with the cytoskeleton via interactions with multiple focal adhesion molecules, including vinculin and actin (reviewed in reference 50). To date, a direct nucleic acid-binding activity of Talin-1 has not been reported; however, this protein has been found associated with a number of nucleic acid-binding proteins also identified in this study, namely, YBX1, FUS, hnRNPK, hnRNP E1 (PCBP1), and PABP (13). Recent studies indicated that MNV replication requires components of the cytoskeleton network for correct localization of the viral replication complex (29). Further studies to dissect a possible role for Talin-1 in the norovirus life cycle are under way.
We identified high-density lipoprotein-binding protein (HDLBP), also known as vigilin, with high sequence coverage and that bound to the viral 5′ end (Table 1). HDLBP is a widely expressed cholesterol- and RNA-binding protein (reviewed in reference 17). It contains 15 copies of the highly conserved hnRNP K homology (KH) domain, and it is known that HDLBP is essential for the viability of both dividing and nondividing mammalian cells (22). HDLBP can be found associated with 80S ribosomes in the cytoplasm of cells (71), and it plays a role in the posttranscriptional regulation of c-fms proto-oncogene mRNA by binding to the 3′-UTR (76). HDLBP has been reported to interact with binding sites containing (A)nCU and UC(A)n (32), and although these sequences are absent from the 5′ extremity of the MNV genomic RNA, UCA was present at positions 170 and 194 (Fig. 1). Significantly, studies on human norovirus replication in cell culture have highlighted that cholesterol levels negatively regulate virus replication and that the use of cholesterol-sequestering statins stimulates norovirus replication in cell culture (6). In addition, epidemiological data suggest that the use of statins to reduce cholesterol levels in the elderly is a possible risk factor in the development of symptomatic norovirus infection (59). Expression of HDLBP is increased in response to cholesterol in murine macrophage cells (46); however, whether or not cholesterol levels and HDLBP play a role in MNV replication remains to be determined and requires further additional studies.
We previously demonstrated that PTB interacts with the 5′ end of feline calicivirus (FCV) genomic and subgenomic RNAs (34) and that during virus replication in cell culture PTB relocalizes to the cytoplasm, where it associates with the viral replication complex (35). Our published data would indicate that at least for FCV, PTB may play a role in the regulation of viral translation, as the addition of PTB to in vitro translation reactions inhibits FCV VPg-mediated translation. This led us to propose a model whereby during the later stages in the viral life cycle, the nucleus-cytoplasmic shuttling of PTB is specifically reduced and PTB remains largely cytoplasmic, where it contributes to the switch from viral translation to replication (35). In our current study, however, we found that PTB is largely cytoplasmic in murine microglial BV-2 cells (Fig. 5A) and macrophage cells (RAW264.7 cells) (data not shown); therefore, significant redistribution from the nucleus was not readily observed. We did however observe a significant relocalization of PTB to the perinuclear region, previously reported to be the site of active RNA synthesis (30). The precise role of PTB in the norovirus life cycle is yet to be determined and requires further studies; however, we have reported that PTB binding to a variable pyrimidine-rich [p(Y)] tract in the 3′-terminal stem-loop may contribute to viral virulence in the host (3). Importantly, while we demonstrated that a virus lacking the p(Y) tract, generated by reverse genetics, replicated efficiently in cell culture, it displayed a fitness cost, as it was readily outcompeted when coinoculated with wild-type virus in cell culture. While this virus (referred to as SL3 GNRA in reference 3) lacked the p(Y) tract, PTB binding to the 3′ extremity was not entirely abolished. It is also possible that any defect in replication that occurs due to the inability of the 3′-UTR to bind PTB may be at least partially compensated for in cell culture, as the expression levels of host cell nucleic acid-binding proteins may differ significantly from those observed in vivo. Significantly, siRNA-mediated knockdown of PTB resulted in a significant reduction of viral protein, viral titer, and viral RNA levels (Fig. 6A). Based on our previous findings with FCV, it is possible PTB also contributes to the regulation of translation and replication; therefore, any reduction in intracellular levels would have a combined effect of deregulating both steps, causing significant reductions in viral replication.
La, also known as SSB, is a widely expressed host cell RNA-binding protein originally identified as an autoantigen in Sjogren's syndrome and systemic lupus erythematosus (75). One of the primary functions of La in the host cell is the maturation of RNA polymerase III transcripts, including tRNAs, via an association with a (U)n-OH motif (4). La is also now widely recognized as an IRES trans-acting factor, as it is known to bind to and regulate the activity of the IRESs from a variety of positive-strand RNA viruses, including poliovirus (47) and coxsackievirus B3 (56). La interacts with the 5′ and 3′ ends of the Japanese encephalitis virus genome (69, 70) and contributes to the translation of a subset of cellular mRNAs known as TOP mRNAs (41). We identified La protein in purifications prepared using either the 5′ or 3′ extremities of the MNV genomic RNA (Table 1); these proteins have also been reported for Norwalk virus (25, 27), further confirming the utility of MNV as a model for studying aspects of human norovirus biology. La immunoprecipitation demonstrated an association, either direct or indirect, with the viral replication complex (Fig. 4),, and although confocal microscopy showed that some La was present within regions of cells containing viral antigen, a large sequestration of La to the sites of virus replication was not observed (Fig. 5B). It is worth highlighting that while a strong colocalization of La with the sites of viral replication was not detected, issues surrounding epitope availability or stability during the fixation process may have affected our ability to image La efficiently. Taken together with the other experimental approaches, however, the data suggest that at least some of the La present within the cell is associated with the viral replication complex and, importantly, plays a role in MNV replication. Surprisingly, while a modest reduction in La levels resulted in the reduction of viral antigen production to around 46% of the wild-type levels, more substantial effects on viral RNA (∼77% reduction) and viral titer (88% reduction) were observed (Fig. 6B). Based on our findings, it is clear that La plays a significant, but yet not fully defined, role in the norovirus life cycle.
DDX3, also referred to as DBX and DDX3X, is a multifunctional host cell RNA helicase that has been implicated in the life cycle of a number of viruses (reviewed in reference 61). DDX3 has not only been implicated in contributing the innate immune response (48, 51, 62, 79) but may also function in host cell translation initiation via an interaction with eukaryotic initiation factor 3 (eIF3) (38). Recent studies suggested that DDX3 contributes to HIV translation (39) and stress granule formation (64). eIF3 has been implicated to have a role in the unique mechanism of norovirus VPg-mediated translation initiation (11, 12). Our recent studies also suggest that DDX3 is found associated with VPg-containing translation initiation factor complexes (L. Chung and I. Goodfellow, unpublished data). Therefore, it is tempting to speculate that DDX3 also contributes to viral VPg-dependent translation initiation, although this will require additional studies and the development of a cell-based assay that allows norovirus translation to be studied in the absence of virus replication. siRNA targeting of DDX3 was particularly effective at reducing viral protein levels, viral titer, and viral RNA, with ∼10-fold reductions observed in all cases (Fig. 6C). DDX3 has recently been identified with a number of small-molecule inhibitors as a potential anti-HIV therapeutic (20, 43, 44, 55). Therefore, the identification of DDX3 as an intracellular factor required for norovirus replication provides an additional target for small-molecule intervention against norovirus infection.
In summary, we have provided in this report a network of host proteins that interact with the 5′ and 3′ ends of the MNV genome. After confirming the interaction of some of these host proteins during infection, we have provided evidence that these proteins play a role in the viral replication/life cycle. To our knowledge, this is the first report of the use of RNA interference to inhibit a host factor involved in the norovirus life cycle. Studies such as this one will undoubtedly facilitate a further understanding of the finer details of norovirus translation and replication. Given the reported disease burden, the case for the development of antiviral therapeutics (14, 58) and possible vaccines (2) is strong. Studies such as that described here provide additional intracellular targets for therapeutic interventions for these important pathogens.
Supplementary Material
ACKNOWLEDGMENTS
We thank Arvind Patel (Medical Research Council, University of Glasgow Centre for Virus Research, Glasgow) and Jennifer Pocock (University College London, London) for reagents, as well as Sylvie Laboissiere and the staff at the Génome Québec Innovation Centre for their help with mass spectrometry.
This work was supported by a Wellcome Trust Senior Fellowship, the Imperial College NIHR Biomedical Research Centre, and SENACYT-Panama. I.G. is a Wellcome Senior Fellow.
Footnotes
Published ahead of print 29 August 2012
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1. Alvarez DE, Lodeiro MF, Ludueña SJ, Pietrasanta LI, Gamarnik AV. 2005. Long-range RNA-RNA interactions circularize the dengue virus genome. J. Virol. 79:6631–6643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Atmar RL, et al. 2011. Norovirus vaccine against experimental human Norwalk virus illness. N. Engl. J. Med. 365:2178–2187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bailey D, et al. 2010. Functional analysis of RNA structures present at the 3′ extremity of the murine norovirus genome: the variable polypyrimidine tract plays a role in viral virulence. J. Virol. 84:2859–2870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bayfield MA, Yang R, Maraia RJ. 2010. Conserved and divergent features of the structure and function of La and La-related proteins (LARPs). Biochim. Biophys. Acta 1799:365–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Blasi E, Barluzzi R, Bocchini V, Mazzolla R, Bistoni F. 1990. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J. Neuroimmunol. 27:229–237 [DOI] [PubMed] [Google Scholar]
- 6. Chang K-O. 2009. Role of cholesterol pathways in norovirus replication. J. Virol. 83:8587–8595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chang K-O, George DW. 2007. Interferons and ribavirin effectively inhibit Norwalk virus replication in replicon-bearing cells. J. Virol. 81:12111–12118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chaudhry Y, et al. 2006. Caliciviruses differ in their functional requirements for eIF4F components. J. Biol. Chem. 281:25315–25325 [DOI] [PubMed] [Google Scholar]
- 9. Chaudhry Y, Skinner MA, Goodfellow IG. 2007. Recovery of genetically defined murine norovirus in tissue culture by using a fowlpox virus expressing T7 RNA polymerase. J. Gen. Virol. 88:2091–2100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cox C, Cao S, Lu Y. 2009. Enhanced detection and study of murine norovirus-1 using a more efficient microglial cell line. Virol. J. 6:196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Daughenbaugh KF, Fraser CS, Hershey JWB, Hardy ME. 2003. The genome-linked protein VPg of the Norwalk virus binds eIF3, suggesting its role in translation initiation complex recruitment. EMBO J. 22:2852–2859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Daughenbaugh KF, Wobus CE, Hardy ME. 2006. VPg of murine norovirus binds translation initiation factors in infected cells. Virol. J. 3:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. de Hoog CL, Foster LJ, Mann M. 2004. RNA and RNA binding proteins participate in early stages of cell spreading through spreading initiation centers. Cell 117:649–662 [DOI] [PubMed] [Google Scholar]
- 14. Dou D, et al. 2011. Design and synthesis of inhibitors of noroviruses by scaffold hopping. Bioorg. Med. Chem. 19:5749–5755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Duizer E, et al. 2004. Laboratory efforts to cultivate noroviruses. J. Gen. Virol. 85:79–87 [DOI] [PubMed] [Google Scholar]
- 16. Ettayebi K, Hardy ME. 2003. Norwalk virus nonstructural protein p48 forms a complex with the SNARE regulator VAP-A and prevents cell surface expression of vesicular stomatitis virus G protein. J. Virol. 77:11790–11797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fidge NH. 1999. High density lipoprotein receptors, binding proteins, and ligands. J. Lipid Res. 40:187–201 [PubMed] [Google Scholar]
- 18. Filomatori CV, et al. 2006. A 5′ RNA element promotes dengue virus RNA synthesis on a circular genome. Genes Dev. 20:2238–2249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fontanes V, Raychaudhuri S, Dasgupta A. 2009. A cell-permeable peptide inhibits hepatitis C virus replication by sequestering IRES transacting factors. Virology 394:82–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Garbelli A, et al. 2011. Targeting the human DEAD-box polypeptide 3 (DDX3) RNA helicase as a novel strategy to inhibit viral replication. Curr. Med. Chem. 18:3015–3027 [DOI] [PubMed] [Google Scholar]
- 21. Glass RI, Parashar UD, Estes MK. 2009. Norovirus gastroenteritis. N. Engl. J. Med. 361:1776–1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Goolsby KM, Shapiro DJ. 2003. RNAi-mediated depletion of the 15 KH domain protein, vigilin, induces death of dividing and non-dividing human cells but does not initially inhibit protein synthesis. Nucleic Acids Res. 31:5644–5653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guix S, et al. 2007. Norwalk virus RNA is infectious in mammalian cells. J. Virol. 81:12238–12248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gutiérrez AL, Denova-Ocampo M, Racaniello VR, del Angel RM. 1997. Attenuating mutations in the poliovirus 5′ untranslated region alter its interaction with polypyrimidine tract-binding protein. J. Virol. 71:3826–3833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gutiérrez-Escolano AL, Brito ZU, del Angel RM, Jiang X. 2000. Interaction of cellular proteins with the 5′ end of Norwalk virus genomic RNA. J. Virol. 74:8558–8562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gutiérrez-Escolano AL, Medina F, Racaniello VR, del Angel RM. 1997. Differences in the UV-crosslinking patterns of the poliovirus 5′-untranslated region with cell proteins from poliovirus-susceptible and -resistant tissues. Virology 227:505–508 [DOI] [PubMed] [Google Scholar]
- 27. Gutiérrez-Escolano AL, Vázquez-Ochoa M, Escobar-Herrera J, Hernández-Acosta J. 2003. La, PTB, and PAB proteins bind to the 3′ untranslated region of Norwalk virus genomic RNA. Biochem. Biophys. Res. Commun. 311:759–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Herold J, Andino R. 2001. Poliovirus RNA replication requires genome circularization through a protein-protein bridge. Mol. Cell 7:581–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hyde JL, Gillespie LK, Mackenzie JM. 2012. Mouse norovirus-1 utilizes the cytoskeleton network to establish localization of the replication complex proximal to the microtubule organizing center. J. Virol. 86:4110–4122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hyde JL, et al. 2009. Mouse norovirus replication is associated with virus-induced vesicle clusters originating from membranes derived from the secretory pathway. J. Virol. 83:9709–9719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kaminski A, Hunt SL, Patton JG, Jackson RJ. 1995. Direct evidence that polypyrimidine tract binding protein (PTB) is essential for internal initiation of translation of encephalomyocarditis virus RNA. RNA 1:924–938 [PMC free article] [PubMed] [Google Scholar]
- 32. Kanamori H, Dodson RE, Shapiro DJ. 1998. In vitro genetic analysis of the RNA binding site of vigilin, a multi-KH-domain protein. Mol. Cell. Biol. 18:3991–4003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kapikian AZ. 2000. The discovery of the 27-nm Norwalk virus: an historic perspective. J. Infect. Dis. 181(Suppl. 2):S295–S302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Karakasiliotis I, Chaudhry Y, Roberts LO, Goodfellow IG. 2006. Feline calicivirus replication: requirement for polypyrimidine tract-binding protein is temperature-dependent. J. Gen. Virol. 87:3339–3347 [DOI] [PubMed] [Google Scholar]
- 35. Karakasiliotis I, et al. 2010. Polypyrimidine tract binding protein functions as a negative regulator of feline calicivirus translation. PLoS One 5:e9562 doi:10.1371/journal.pone.0009562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Karst SM, Wobus CE, Lay M, Davidson J, Virgin HW. 2003. STAT1-dependent innate immunity to a Norwalk-like virus. Science 299:1575–1578 [DOI] [PubMed] [Google Scholar]
- 37. Lay MK, et al. 2010. Norwalk virus does not replicate in human macrophages or dendritic cells derived from the peripheral blood of susceptible humans. Virology 406:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lee C-S, et al. 2008. Human DDX3 functions in translation and interacts with the translation initiation factor eIF3. Nucleic Acids Res. 36:4708–4718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu J, Henao-Mejia J, Liu H, Zhao Y, He JJ. 2011. Translational regulation of HIV-1 replication by HIV-1 Rev cellular cofactors Sam68, eIF5A, hRIP, and DDX3. J. Neuroimmune Pharmacol. 6:308–321 [DOI] [PubMed] [Google Scholar]
- 40. Liu Y, Wimmer E, Paul AV. 2009. cis-acting RNA elements in human and animal plus-strand RNA viruses. Biochim. Biophys. Acta 1789:495–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Loreni F, Pierandrei-Amaldi P, Amaldi F. 2000. La protein has a positive effect on the translation of TOP mRNAs in vivo. Nucleic Acids Res. 28:2927–2934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lowman HBH, Draper DED. 1986. On the recognition of helical RNA by cobra venom V1 nuclease. J. Biol. Chem. 261:5396–5403 [PubMed] [Google Scholar]
- 43. Maga G, et al. 2008. Pharmacophore modeling and molecular docking led to the discovery of inhibitors of human immunodeficiency virus-1 replication targeting the human cellular aspartic acid-glutamic acid-alanine-aspartic acid box polypeptide 3. J. Med. Chem. 51:6635–6638 [DOI] [PubMed] [Google Scholar]
- 44. Maga G, et al. 2011. Toward the discovery of novel anti-HIV drugs. Second-generation inhibitors of the cellular ATPase DDX3 with improved anti-HIV activity: synthesis, structure-activity relationship analysis, cytotoxicity studies, and target validation. ChemMedChem 6:1371–1389 [DOI] [PubMed] [Google Scholar]
- 45. McFadden N, et al. 2011. Norovirus regulation of the innate immune response and apoptosis occurs via the product of the alternative open reading frame 4. PLoS Pathog. 7:e1002413 doi:10.1371/journal.ppat.1002413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. McKnight GL, et al. 1992. Cloning and expression of a cellular high density lipoprotein-binding protein that is up-regulated by cholesterol loading of cells. J. Biol. Chem. 267:12131–12141 [PubMed] [Google Scholar]
- 47. Meerovitch K, et al. 1993. La autoantigen enhances and corrects aberrant translation of poliovirus RNA in reticulocyte lysate. J. Virol. 67:3798–3807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mulhern O, Bowie AG. 2010. Unexpected roles for DEAD-box protein 3 in viral RNA sensing pathways. Eur. J. Immunol. 40:933–935 [DOI] [PubMed] [Google Scholar]
- 49. Nagy PD, Pogany J. 2012. The dependence of viral RNA replication on co-opted host factors. Nat. Rev. Microbiol. 10:137–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nayal A, Webb DJ, Horwitz AF. 2004. Talin: an emerging focal point of adhesion dynamics. Curr. Opin. Cell Biol. 16:94–98 [DOI] [PubMed] [Google Scholar]
- 51. Oshiumi H, Sakai K, Matsumoto M, Seya T. 2010. DEAD/H BOX 3 (DDX3) helicase binds the RIG-I adaptor IPS-1 to up-regulate IFN-beta-inducing potential. Eur. J. Immunol. 40:940–948 [DOI] [PubMed] [Google Scholar]
- 52. Pacheco A, López de Quinto S, Ramajo J, Fernández N, Martínez-Salas E. 2009. A novel role for Gemin5 in mRNA translation. Nucleic Acids Res. 37:582–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pacheco A, Reigadas S, Martínez-Salas E. 2008. Riboproteomic analysis of polypeptides interacting with the internal ribosome-entry site element of foot-and-mouth disease viral RNA. Proteomics 8:4782–4790 [DOI] [PubMed] [Google Scholar]
- 54. Perry JW, Wobus CE. 2010. Endocytosis of murine norovirus 1 into murine macrophages is dependent on dynamin II and cholesterol. J. Virol. 84:6163–6176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Radi M, et al. 2012. Discovery of the first small molecule inhibitor of human DDX3 specifically designed to target the RNA binding site: towards the next generation of HIV-1 inhibitors. Bioorg. Med. Chem. Lett. 22:2094–2098 [DOI] [PubMed] [Google Scholar]
- 56. Ray P. 2002. La autoantigen is required for the internal ribosome entry site-mediated translation of coxsackievirus B3 RNA. Nucleic Acids Res. 30:4500–4508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rodríguez Pulido M, Sobrino F, Borrego B, Sáiz M. 2009. Attenuated foot-and-mouth disease virus RNA carrying a deletion in the 3′ noncoding region can elicit immunity in swine. J. Virol. 83:3475–3485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rohayem J, et al. 2010. Antiviral strategies to control calicivirus infections. Antiviral Res. 87:162–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rondy M, et al. 2011. Norovirus disease associated with excess mortality and use of statins: a retrospective cohort study of an outbreak following a pilgrimage to Lourdes. Epidemiol. Infect. 139:453–463 [DOI] [PubMed] [Google Scholar]
- 60. Sandoval-Jaime C, Gutiérrez-Escolano AL. 2009. Cellular proteins mediate 5′-3′ end contacts of Norwalk virus genomic RNA. Virology 387:322–330 [DOI] [PubMed] [Google Scholar]
- 61. Schröder M. 2011. Viruses and the human DEAD-box helicase DDX3: inhibition or exploitation? Biochem. Soc. Trans. 39:679–683 [DOI] [PubMed] [Google Scholar]
- 62. Schröder M, Baran M, Bowie AG. 2008. Viral targeting of DEAD box protein 3 reveals its role in TBK1/IKKε-mediated IRF activation. EMBO J. 27:2147–2157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sharp TM, Guix S, Katayama K, Crawford SE, Estes MK. 2010. Inhibition of cellular protein secretion by Norwalk virus nonstructural protein p22 requires a mimic of an endoplasmic reticulum export signal. PLoS One 5:e13130 doi:10.1371/journal.pone.0013130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Shih J-W, et al. 2012. Critical roles of RNA helicase DDX3 and its interactions with eIF4E/PABP1 in stress granule assembly and stress response. Biochem. J. 441:119–129 [DOI] [PubMed] [Google Scholar]
- 65. Simmonds P, et al. 2008. Bioinformatic and functional analysis of RNA secondary structure elements among different genera of human and animal caliciviruses. Nucleic Acids Res. 36:2530–2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sosnovtsev SV, et al. 2006. Cleavage map and proteolytic processing of the murine norovirus nonstructural polyprotein in infected cells. J. Virol. 80:7816–7831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Syljuåsen RGR, Sørensen CSC, Bartek JJ, 6 2004. Inhibition of Chk1 by CEP-3891 accelerates mitotic nuclear fragmentation in response to ionizing radiation. Cancer Res. 64:9035–9040 [DOI] [PubMed] [Google Scholar]
- 68. Vashist S, Bailey D, Putics A, Goodfellow I. 2009. Model systems for the study of human norovirus biology. Future Virol. 4:353–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Vashist S, Anantpadma M, Sharma H, Vrati S. 2009. La protein binds the predicted loop structures in the 3′ non-coding region of Japanese encephalitis virus genome: role in virus replication. J. Gen. Virol. 90:1343–1352 [DOI] [PubMed] [Google Scholar]
- 70. Vashist S, Bhullar D, Vrati S. 2011. La protein can simultaneously bind to both 3′- and 5′-noncoding regions of Japanese encephalitis virus genome. DNA Cell Biol. 30:339–346 [DOI] [PubMed] [Google Scholar]
- 71. Vollbrandt T, Willkomm D, Stossberg H, Kruse C. 2004. Vigilin is co-localized with 80S ribosomes and binds to the ribosomal complex through its C-terminal domain. Int. J. Biochem. Cell Biol. 36:1306–1318 [DOI] [PubMed] [Google Scholar]
- 72. Ward VK, et al. 2007. Recovery of infectious murine norovirus using Pol II-driven expression of full-length cDNA. Proc. Natl. Acad. Sci. U. S. A. 104:11050–11055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wasiak S, et al. 2002. Enthoprotin: a novel clathrin-associated protein identified through subcellular proteomics. J. Cell Biol. 158:855–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wobus CE, et al. 2004. Replication of Norovirus in cell culture reveals a tropism for dendritic cells and macrophages. PLoS Biol. 2:e432 doi:10.1371/journal.pbio.0020432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wolin SL, Cedervall T. 2002. The La protein. Annu. Rev. Biochem. 71:375–403 [DOI] [PubMed] [Google Scholar]
- 76. Woo H-H, et al. 2011. Posttranscriptional suppression of proto-oncogene c-fms expression by vigilin in breast cancer. Mol. Cell. Biol. 31:215–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Xi JN, Graham DY, Wang KN, Estes MK. 1990. Norwalk virus genome cloning and characterization. Science 250:1580–1583 [DOI] [PubMed] [Google Scholar]
- 78. Yu L, Robert Putnak J, Pletnev AG, Markoff L. 2008. Attenuated West Nile viruses bearing 3′SL and envelope gene substitution mutations. Vaccine 26:5981–5988 [DOI] [PubMed] [Google Scholar]
- 79. Yu S, et al. 2010. Hepatitis B virus polymerase inhibits RIG-I- and Toll-like receptor 3-mediated beta interferon induction in human hepatocytes through interference with interferon regulatory factor 3 activation and dampening of the interaction between TBK1/IKKε and DDX3. J. Gen. Virol. 91:2080–2090 [DOI] [PubMed] [Google Scholar]
- 80. Yunus MA, Chung LMW, Chaudhry Y, Bailey D, Goodfellow I. 2010. Development of an optimized RNA-based murine norovirus reverse genetics system. J. Virol. Methods 169:112–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.