

Abstract
Antineutrophil cytoplasmic autoantibodies (ANCA) associated vasculitis (AAV) are a group of systemic vasculitis characterized by inflammation and necrosis of blood vessel walls. Genetic, epigenetic and environmental factors contribute to the etiology and pathogenesis of AAV. Based on currently available clinical and experimental evidence, it is reasonable to conceptualize that in predisposed patients, different triggers can lead to the production of autoantibodies (ANCA) that in the context of an inflammatory environment can cause tissue inflammation and vascular injury. Several different pathways and mechanisms in the pathogenesis of AAV are described in this contemporary review.
Keywords: ANCA, ANCA-associated vasculitis, Granulomatosis with polyangiitis, Microscopic polyangiitis, Eosinophilic granulomatosis with polyangiitis, Pathogenesis, Etiology
Introduction
Three systemic autoimmune small vessel vasculitis syndromes are associated with antineutrophil cytoplasmic autoantibodies (ANCA) and are collectively known as ANCA-associated vasculitides (AAV). (1). AAV comprises granulomatosis with polyangiitis (GPA, formerly known as Wegener’s granulomatosis), microscopic polyangiitis (MPA) and eosinophilic granulomatosis with polyangiitis (EGPA, formerly known as Churg-Strauss syndrome)(1). These syndromes have distinguishing clinical manifestations and histological findings. GPA and EGPA are characterized by granulomatous inflammation that usually affects the respiratory tract, which is – by definition – absent in MPA. The granulomatous inflammation of GPA is predominantly neutrophilic, whereas that of EGPA is eosinophil. In contrast, the clinical manifestations caused by necrotizing small vessel vasculitis and capillaritis, such as diffuse alveolar hemorrhage, mononeuritis multiplex or glomerulonephritis, are shared between all three syndromes.
Two different types of ANCA are associated with these syndromes(2). ANCA directed against the neutrophil serine protease, proteinase 3 (PR3) cause a cytoplasmic (C-ANCA) staining pattern on ethanol fixed neutrophils by indirect immunofluorescence microscopy. PR3-ANCA are the predominant ANCA type in GPA, less common in MPA, and the rare exception in EGPA(2). In contrast, ANCA directed against myeloperoxidase (MPO) generating a perinuclear (P-ANCA) fluorescence pattern are found in the majority of patients with MPA, but only in about 5–10% of patients with GPA(2). In EGPA, ANCA are usually of the MPO-ANCA type and can be detected in 30–70% of patients (3–5).
There is likely a contribution of genetic, epigenetic and environmental factors in the etiology and pathogenesis of AAV. In this review, we aim to summarize and discuss the current views and insights on the pathogenesis of AAV with particular focus on GPA and MPA.
Epidemiology of AAV
AAV have a reported incidence of 10–20 cases per million per year(6). The distribution of the specific syndromes varies geographically and with ethnicity(7). In different populations the annual incidence of GPA ranges between 4.9–10.6 cases/million and the reported point prevalence varies between 24–157 cases/million(6, 8–10). GPA is more common in Northern than in Mediterranean Europe, where MPA is more common(7). GPA is also exceedingly rare in African-Americans and in Japan, where it is almost exclusively encountered on the Northern island of Hokkaido (11, 12). A similar latitude-dependent incidence gradient has also been documented in New Zealand(13). Furthermore, in New Zealand as well as in France the incidence of AAV is higher in the populations of European ethnicity than among Non-Europeans, Asians or Pacific Islanders (13, 14).
Etiology and Pathogenesis
The etiology of the AAV syndromes remains unclear. Like for many other polygenic systemic autoimmune diseases, AAV are in all likelihood the result of complex interactions between genetic factors predisposing for the loss of self-tolerance and autoimmunity and triggering environmental exposures. These factors induce and maintain inappropriate lymphocyte activation and autoantibody production which lead to tissue injury and predispose to disease relapses. Table 1 summarizes different mechanisms and pathways that may contribute to the development of AAV.
Table 1.
Different potential factors in the pathogenesis of ANCA-associated vasculitis
| Mechanism | Evidence |
|---|---|
| Genetic predisposition | Genetic association studies of different SNPs of the following genes: HLA, SERPINA1, CTLA 4, PRTN3 and PTPN22 |
| Environmental trigger | Silica as inflammasome complex activator, drugs (propylthiouracil, hydralazine and penicillamine) which could have polyclonal B lymphocyte stimulatory properties that may induce the production of ANCA |
| Infectious trigger | Staphylococcus aureus eliciting molecular mimicry and T and B cell activation via superantigens. |
| B-cells | B-cells are precursors of ANCA forming plasma cells. Increased proportion and total number of B-cells have been identified in AAV patients. Elevated B-lymphocyte stimulator factors including BAFF have also been identified in AAV patients. |
| T-cells | T-cells are found within granulomas as well as in other lesions present in AAV. Elevated levels of markers of T-cell activity such as soluble interleukin-2 (IL-2) receptor, neopterin, and soluble CD30 have been shown to be associated with disease activity. Increased T-cell number and activity(CD4+CD25+ and CD4+CD25+CD134+/GITR+) have also been found. Functional defect in Tregs (CD4+CD25high FoxP3+CD127low) have been described in patients in remission. GPA patients in remission allow an increased amount of Th17 cells(CD4+IL-17+) reactive to PR3. |
| Autoantibodies (ANCA) | Pathogenic role of ANCA has been supported by several in vitro and also in vivo animal models. Activation of neutrophils and monocytes by ANCA antibodies causes generation of reactive oxygen species, release of proteases and cytokine production by which inflammation and tissue damage is created. ANCA antibodies also induce apoptosis in endothelial cells. |
| Complement pathway | Increased neutrophil activity via the alternative complement pathway→C5a primes neutrophils and enhances ANCA induced neutrophil activation. Complement activation and its resulting products promote inflammation and enhance tissue damage. |
SNP: single nucleotide polymorphism; HLA: human leukocyte antigen;CTLA4: Cytotoxic T-lymphocyte antigen 4; ANCA: anti-neutrophil cytoplasmic antibody; AAV: ANCA-associated vasculitis; BAFF: B cell activating factor; GPA: granulomatosis with polyangiitis; PR3: proteinase 3
Genetic Predisposition
There are studies of rare cases in which single gene defects can cause AAV-like phenotypes(15–17); however, the vast majority of genetic association studies in AAV have been in the context of single nucleotide polymorphisms (SNPs) with small effect size. A familial aggregation study found a relative risk of 1.56 for GPA among first degree relatives of patients with GPA, which is similar to the risk seen in rheumatoid arthritis(18). Familial clustering has also been reported with other AAV syndromes (19, 20). Several candidate genes have been investigated in different cohorts of patients with AAV, and SNPs have been found in several genes coding for proteins involved in the immune response(21).
Variants most strongly and most reproducibly associated with AAV are found in the human leukocyte antigen (HLA) and PTPN22 genes (21). HLA genes, located on the short arm of chromosome 6, have been associated with various autoimmune disorders such as rheumatoid arthritis, giant cell arteritis, Behçet’s disease, and antiglomerular basement membrane disease(22, 23). Multiple HLA genes have been confirmed to be prevalent in different AAV populations; for example, in a Caucasian population of German patients with GPA, a group of investigators reported that HLA-DRB1*04 was over-represented in these individuals(24), whereas another group located the risk alleles as HLA-DPB1*0401 in a cohort of 282 subjects with GPA(25). In the Asian population, HLA-DRB1*0901 has been found to be associated with MPA and MPO-ANCA-positive vasculitis patients in a Japanese population (26, 27).
Most candidate gene studies in AAV comprise only GPA cohorts. MPA and EGPA are usually underrepresented in many genetic association studies. Nevertheless, interesting genetic differences are emerging between the different syndromes. For instance, the HLA-DPB1*0401 variant represents a strong and reproducible genetic risk factor for GPA, but not for MPA or EGPA (28–30). Interestingly, African Americans with PR3-ANCA associated vasculitis had a 36 fold higher likelihood to have the HLA-DRB1*15 genotype than community-based controls, and the HLA-DRB*1501 allele, which is of Caucasian rather than African descent, conveyed a 73.3-fold higher risk for PR3-ANCA associated vasculitis (31)
In contrast, an increase in the PTPN22 620W allele, which has been associated with other autoantibody associated autoimmune diseases and has thus been implicated in the regulation of B lymphocyte activity, was found to be associated with both GPA and MPA(32–34).
Cytotoxic T-lymphocyte antigen 4 (CTLA4), expressed mostly on CD4 positive T lymphocytes, exerts an inhibitory function on T lymphocytes by binding to CD80 and CD86 on antigen presenting cells. CTLA4 competes with the co-stimulatory molecule CD28, which exerts a stimulatory effect on T lymphocytes, for binding to CD80 and CD86. If T lymphocytes are activated via the T cell receptor and CD28, CTLA4 expression is increased, probably as a regulatory mechanism. Via these mechanisms CTLA4 is thought to maintain peripheral self-tolerance(35). Elevated levels of CTLA4 have been found in GPA, undoubtedly a reflection of T lymphocyte activation(36). CTLA4 is coded for by the CTLA4 gene, and CTLA4 polymorphisms, which may negatively affect expression or function of CTLA4, have been associated with autoimmune diseases including GPA (33, 37, 38). Recently, a meta-analysis of 7 studies showed an increased susceptibility for AAV associated with CTLA-4 polymorphisms in European patients (39).
Another gene of interest is PRTN3, which codes for PR3, the most prominent target antigen for ANCA in GPA. Eight SNPs have been identified in the PRTN3 promotor region and exons, but their function for PR3 expression is unclear. An increased frequency of one promotor polymorphism (A546G) was found among patients with GPA(40). PR3 is not only stored in granules and released during neutrophil activation, but it is also expressed on the neutrophil membrane, where it may be engaged by PR3-ANCA. The membrane expression of PR3 varies between individuals, remains constant over time, and appears genetically determined(41, 42). One study found a link between membrane PR3 expression and HLA antigens(43). Individuals with high membrane PR3 expression are significantly more frequent among patients with GPA than in the normal population, and patients with GPA and high membrane PR3 expression are at higher risk for relapses than those with low membrane PR3 expression(44, 45).
The major natural inhibitor for PR3 is α1-antitrypsin, coded for by the SERPINA1 gene(AAT). Of the many polymorphisms of AAT, the Z- and S alleles are responsible for the majority of α1-antitrypsin deficiency cases. An increased frequency of heterozygosity for the Z- and S-alleles in association with GPA, but not MPA, have been reported by several groups in small studies and recently confirmed in larger studies(46, 47). The clinical significance of this association is not entirely clear, but heterozygosity for the Z-allele has been associated with a worse prognosis of the disease(48). This may be because Z-allele carriers, but not S-allele carriers, have increased levels of pro-inflammatory polymers circulating in their blood (47). Deposits of these polymers have been documented in kidney biopsy specimens from patients with active GPA, and these polymers can prime neutrophils and augment the activation of neutrophils by PR3-ANCA in vitro(47).
Unconfirmed or conflicting results have been reported for the association with either GPA or MPA and polymorphisms of other genes including those coding for the high affinity soluble interleukin 2 receptor (IL-2RA), interleukin 10 (IL-10), leukocyte immunoglobulin-like receptor A2 (LILRA2) and CD226 (CD226) as well as for the Fc receptors FcRIIa and FcRIIIb(21).
Another, yet unclear, factor to be considered in the interpretation of genetic SNP association studies is the issue of copy number variation(49). Copy number variations as a result of gene duplication, triplication or exon shuffling may occur at a higher frequency in the genome than SNPs and may be more important for evolution(49). The study of effects of copy number variations on the disease phenotype of complex autoimmune diseases including the AAV syndromes is just beginning. Genome-wide association studies (GWAS) have produced valuable insights into the genetic background of several complex immunologic and non-immunologic disorders. Recently, the first GWAS in AAV (GPA and MPA, no EGPA) was performed in the United Kingdom in a discovery cohort of 1233 AAV patients and 5884 controls; and was replicated in 1454 northern European case patients and 1666 controls(50••). This study confirms previous preliminary data that favor heterogenic genetic backgrounds for GPA and MPA, even though the study was relatively underpowered for MPA cases. The strongest associations found in this study were with the antigenic specificity of ANCA (PR3-ANCA versus MPO-ANCA) rather than with the clinical diagnosis (GPA versus MPA). This GWAS confirmed strong associations with the HLA-DP region, the serpin A1 gene (SERPINA1, which encodes α1-antitrypsin), and with the PRTN3 gene (encodes proteinase 3) in patients with PR3 antibodies, and HLA-DQ in patients with MPO antibodies. Other previously reported associations were not confirmed in this study.
In summary, the predisposing genetic factors in AAV are most likely heterogeneous and differ between patients. Genetic factors may result in alterations in HLA mediated antigen presentation, global T- and B-lymphocyte activation, defective immune regulation and abnormal target antigen structure and/or function. Whether any of these predisposing genetic factors by itself or in combination is sufficient to cause the disease remains to be determined.
Environmental Exposures and Drugs as Triggers
Environmental triggers for the onset of AAV in susceptible individuals remain unknown for most patients. A significant association between MPA with MPO-ANCA and exposure to silica has been reported(51). Silica is a potential activator of the inflammasome complex that generates, among other mediators, interleukin 1 (IL-1)(52).
The onset of AAV (predominantly MPA) has also been reported following exposure to a variety of therapeutic agents including propylthiouracil, hydralazine and penicillamine (53, 54). These agents have polyclonal B lymphocyte stimulatory properties, which may induce the production of ANCA. However, there are several features distinguishing drug-induced AAV from typical AAV. First, the ANCA response is often targeting several different antigens at the same time, whereas in typical AAV; ANCA target either PR3 or MPO, but not both, and not other additional antigens such as human neutrophil elastase or lactoferrin. Second, drug-induced AAV usually subsides after discontinuation of the offending agent. Thus, the cases of drug-induced AAV support the hypothesis of the pathogenic role of ANCA in the development of vasculitis, but they do not explain the loss of tolerance to ANCA target antigens characterizing GPA and MPA.
Infectious Triggers
Infectious pathogens such as mycobacterial and fungal organisms represent the most frequent cause of granulomatous inflammation of the upper respiratory tract. Therefore the presence of granulomatous inflammation in patients with GPA resulted in an initial extensive search for a causative microorganism (55–57). So far almost all attempts to culture, stain or treat for infectious pathogens failed. There are however certain indicators that imply a role of microorganisms in GPA. Patients with GPA commonly report symptoms of upper respiratory tract infections immediately preceding their initial presentation or a disease relapse (58–61).
Infections have also been implicated as triggers as well as persistent drivers of various autoimmune diseases including AAV. A variety of different and often interrelated mechanisms by which infections trigger and perpetuate the disease in predisposed patients have been proposed. Many infectious agents have been reported to induce an ANCA response. In most instances these ANCA are directed against target antigens other than MPO or PR3. Furthermore, these infection-associated ANCA usually disappear once the infection resolves (2, 62, 63). Thus, it seems that two conditions need to be met for infections to trigger AAV. First, tolerance to self-antigens needs to be broken, i.e. host conditions permissive for the development of autoimmunity need to allow for the ongoing production of antibodies directed against self antigens (ANCA). Second, the persistent specific autoantibodies (ANCA) need to have pathogenic potential for the development of tissue injury characteristic of AAV.
One concept by which infections can elicit an autoimmune response in susceptible hosts is molecular mimicry(64). Subsequent diversification of T and B lymphocyte responses (“epitope spreading”) may lead to reactivities with different epitopes on the same target molecule (intramolecular spreading) or even extend to other molecules (intermolecular spreading)(65, 66). The only example of direct epitope mimicry leading to ANCA in patients with pauci-immune glomerulonephritis has been reported by Kain and co-workers(67). They found ANCA targeting lysosomal membrane protein-2 (LAMP-2) in the majority of patients with pauci-immune focal necrotizing glomerulonephritis (67). These ANCA recognized an epitope on LAMP-2 and cross-reacted with the homologous bacterial adhesin FimH (67). Antibodies to LAMP-2 transferred to rats caused pauci-immune glomerulonephritis in the recipients (67). Rats immunized with FimH developed antibodies to FimH that cross-reacted with human LAMP-2 and also developed pauci-immune glomerulonephritis (67). The frequency of LAMP-2 specific ANCA has recently been confirmed in different patient cohorts from Europe(68), but not from the United States(69). It also appears that the LAMP-2 specific ANCA disappear quickly after initiation of immunosuppressive therapy, and methods for their detection are not as robust as those used for the detection of PR3-ANCA and MPO-ANCA(68). Consequently, the clinical relevance of this finding remains controversial.
An indirect mechanism of molecular mimicry leading to typical PR3-ANCA has been proposed by Pendergraft and colleagues(70). They demonstrated that in selected patients ANCA may represent anti-idiotype antibodies(71). Antibodies are formed against complementary peptides (antisense peptide sequence) to PR3 (cPR3)(70). The cPR3 peptides, which may represent mimics of microbial peptide sequences, are the target of the primary immune response. Indeed, several bacterial peptide sequences including Staphylococcus aureus (S. aureus) sequences were found to have homologies with cPR3(70). True PR3-ANCA are subsequently the result of a secondary immune response mounted against the idiotype of these anti-cPR3 antibodies(70). Pendergraft et al. were able to show that mice immunized with cPR3 developed antibodies against both cPR3 and PR3, and they found antibodies against cPR3 in 7 of 34 PR3-ANCA positive patients(70). However, the same group was unable to demonstrate a similar scenario for MPO-ANCA(72). Furthermore, Tadema et al. were unable to reproduce the increased frequency of anti-cPR3 antibodies in patients with GPA compared to healthy volunteers or MPO-ANCA positive patients(73). Interestingly, portions of the cPR3 sequence have homology with portions of the plasminogen sequence, and anti-plasminogen antibodies were detected in some patients with AAV, potentially contributing, among other potential mechanisms, to the well recognized increased risk for thromboembolic events(74, 75).
Whereas direct or indirect epitope mimicry may account for the development of an antibody response, these phenomena do not fully explain the sustained loss of self tolerance observed in the majority of these patients. A variety of mechanisms by which infections promote the autoimmune response as well as the propensity for chronic relapses in AAV have recently been studied. Nearly two thirds of patients presenting with GPA are nasal carriers of S. aureus(76). This is much higher than in the general population. S. aureus carriers are at higher risk for relapse of GPA than non-carriers, and treatment with trimethoprim-sulfamethoxazole resulted in a significant reduction of the relapse rate (76–78). Multiple mechanisms may contribute to the increased risk of relapse conveyed by S. aureus. S. aureus produces superantigens known to be powerful non-specific (antigen-independent) T- and B lymphocyte activators able to induce significant cell proliferation and cytokine release(79, 80). Patients colonized with superantigen-producing strains of S. aureus are at higher risk for disease flares than those colonized with superantigen-negative strains (81). Compared to healthy controls, patients with GPA were found to have an expansion of T lymphocytes expressing Vbeta segments specific for S. aureus superantigen(82). However, a direct association between the presence of S. aureus producing superantigens and the expansion of T lymphocytes reactive to these superantigens in individual patients could not be confirmed(82).
S. aureus derived superantigens and peptidoglycans as well as fungal beta-glucans can induce the expansion of IL-17 producing CD4 positive T cells, so called Th17 cells, in an IL-23 dependent manner(83–85). Th17 cells, which are now recognized as central players in the development of autoimmunity. They are highly potent inflammatory cells that initiate and maintain tissue inflammation by recruiting other inflammatory cells while generating a milieu that makes them resistant to control by T-regulatory (Treg) cells(86, 87). Persistent IL-23 production by antigen-presenting cells appears necessary to maintain Th17 cells at the site of inflammation(87). Both elevated IL-23 and IL-17 levels have been found in patients with active disease and remained elevated despite treatment during clinical remission(88•). An increased frequency of Th17 cells responding to staphylococcal enterotoxin B was found in GPA patients in remission compared to healthy controls, regardless of ANCA status, whereas an increased frequency of PR3-responsive Th17 cells was restricted to PR3-ANCA positive patients(89). Chronic infections may thus set the stage for chronic ongoing inflammation and the loss of self tolerance.
Necrotizing granulomatous inflammation consisting of monocytes, macrophages, neutrophils, T cells, B cells and plasma cells, predominantly located in the respiratory tract, sets GPA clinically apart from MPA(1). Structures resembling germinal centers have been documented within the granulomatous inflammatory tissue of GPA, and the tissue also stained positive for PR3(90). The immunoglobulin (VH) gene mutational patterns obtained from granulomatous nasal tissue of patients with GPA suggest that the selection and maturation of PR3-ANCA producing B lymphocytes may start within the granulomatous lesions(90).
Cytosine-phosphate-guanine (CPG) motifs are pathogen-associated molecular patterns (PAMPs) that are recognized by the pattern recognition receptor Toll-like receptor 9 (TLR9). Unmethylated CPG oligodeoxynucleotides (CpG-ODN) are potent immune-stimulants. B lymphocytes isolated from patients with AAV with active disease as well as during remission can be induced to produce ANCA when exposed to CpG-ODN and IL-2 (91, 92•). Significantly, more PR3-ANCA patients than MPO-ANCA patients produced ANCA in vitro in response to CpG exposure (92•). These observations provide another link between S. aureus infection, granulomatous inflammation of the respiratory tract and the higher relapse rate of patients with PR3-ANCA or GPA compared to patients with MPO-ANCA or MPA.
Neutrophils are key players of the innate immune defense against microorganisms. An additional antimicrobial defense mechanism of neutrophils has been described recently – a unique type of cell death distinct from apoptosis and necrosis that is associated with the formation and release of neutrophil extracellular traps (NETs)(93). NETs are extracellular structures containing chromatin and granule proteins (including ANCA target antigens) in which invading microbes are trapped and killed. The formation of NETs depends on reactive oxygen species generated by NADPH oxidase (94). ANCA, which are known to induce a respiratory burst in primed neutrophils can also induce the formation of NETs, and ANCA antigens bound to NETs are accessible to ANCA(95••). NETs containing PR3 and MPO are detectable in the kidneys of patients with AAV in the absence of infection. Interestingly, S. aureus can rapidly and strongly induce NETs formation even without causing neutrophil death (96••). Chromatin-immunoglobulin complexes are thought to lead to the loss of tolerance and autoantibody (ANCA) production in a TLR9-dependent manner (97). Taken together, these observations suggest that ANCA may induce a vicious cycle of self-perpetuated NETs formation and more ANCA production, particularly in the presence of S. aureus infection. This hypothesis is further supported by observations of increased TLR9 expression by monocytes of patients with GPA who were S. aureus nasal carriers (98•).
To summarize, current evidence suggests that infections and/or colonization with infectious organisms such as S. aureus are contributing to the pathogenesis of AAV, especially GPA. Pathogens may provide the initial antigen sources (molecular mimicry), pro-inflammatory signals (e.g. stimulation of pathogen-associated molecular pattern receptors by CpG, peptidoglycans and fungal beta-glucans) and triggers for innate (NET formation) and adaptive (superantigens) immune cells activation. In genetically susceptible individuals and/or in the context of other environmental exposures these factors may trigger a break in immune tolerance and the development of sustained or recurrent autoimmunity.
Role of B and T Lymphocytes
The role of B lymphocytes has gained increased interest after efficacy in AAV of the B-cell depleting agent, rituximab, was demonstrated (99). B lymphocytes have been found in affected tissues including kidneys and most significantly in granulomatous lesions of the respiratory tract, where they are located in close proximity to numerous PR3-positive cells and where the selection and maturation into PR3-ANCA producing B lymphocytes may occur(90). Antigen-specific B lymphocytes are the progenitor cells of short-lived plasma cells thought to be the source of autoantibodies including ANCA (100, 101). The proportion of circulating activated B lymphocytes is increased in patients with GPA compared to healthy controls, and it is higher in patients with active disease versus those in remission and in patients with generalized disease versus those with limited disease(102). Moreover, elevated serum levels of B lymphocyte stimulator (BLyS, also known as B cell activating factor, BAFF), a cytokine belonging to the TNF superfamily and known to promote B cell survival, differentiation and proliferation, were found to be increased in patients with active AAV(103,104•,105).
T cells are usually found within granulomas as well as other lesions present in AAV. Circulating T cells in AAV have shown different abnormalities with disturbed homeostasis that is skewed towards memory and pro-inflammatory T-cell types. ANCA are high-affinity class-switched antibodies(106). This implies that ANCA production is dependent on T lymphocyte help, that autoreactive T lymphocytes are present, and that there is insufficient counter-regulation by regulatory T cells (Tregs). Indeed, T lymphocyte abnormalities have long been suspected to be the main reason for the chronic relapsing nature of GPA(102). Markers of T cell activity such as soluble IL-2 receptor, neopterin and soluble CD30 have been shown to be associated with disease activity(107, 108). Patients with GPA in remission have an increased percentage of circulating CD4 positive effector memory T lymphocytes as well as a functional defect of circulating CD4 positive CD25 positive Tregs (109–111••). Moreover, patients with reduced numbers of Tregs required more prolonged treatment to achieve remission, and their relapse rates were higher (111••).
As previously discussed, another T cell subset that has drawn attention recently in autoimmunity is the IL-17-producing T cell subset (Th17)(112). The number of Th17 cells reactive to the autoantigen PR3 was found to be increased in patients with GPA in remission (88, 113). Furthermore, Ordonez et al. found that AAV patients exhibit an expanded CD45RC positive T helper cell population that is a source of IL-17 (114). IL-17 acts on endothelial cells, epithelial cells and antigen presenting cells causing release of chemokines that will activate neutrophils, such as IL-8 and CXCL-1(115). IL-17 can also induce IL1-beta and TNF-alpha production and release by macrophages, and these cytokines, in turn, prime neutrophils and monocytes resulting in the expression of ANCA-target antigens on their surface(116).
Pathogenic Role of ANCA
Clinical observations and a large body of experimental data support a pathogenic role of ANCA for the development of small vessel vasculitis. The small vessel injury of AAV appears to be caused by activated leukocytes. The pro-inflammatory pathogenic effects of ANCA are all contingent on their interactions with their target antigens expressed on the surface of primed neutrophils and monocytes. When primed with inflammatory cytokines such as tumor necrosis factor (TNF)-α or microbial products in vitro, leukocytes express proteinase 3 (PR3) and myeloperoxidase (MPO) on their surface (117, 118). PR3-ANCA and MPO-ANCA can activate primed neutrophils and monocytes by binding directly to their antigens expressed on the surface or by Fc-receptor engagement; these interactions initiate signal transduction cascades via multiple pathways that are similar in neutrophils and monocytes (119–123).
Several pro-inflammatory effects derived from the activation of neutrophils and monocytes by ANCA are responsible for the tissue injury seen in AAV. Fully activated neutrophils degranulate and release toxic proteases and enzymes including elastase, PR3, MPO and others(117). ANCA also induce a respiratory burst resulting in the release of oxygen radical species (117, 124). In addition, ANCA induce the expression of cell adhesion molecules on neutrophils and endothelial cells leading to an increased adhesion of neutrophils to endothelial cells (125–129). Moreover, the binding of ANCA to primed leukocytes induces the production and release of chemotactic cytokines including IL-1, MCP-1 and IL-8(130–133). These cytokines attract more neutrophils and monocytes to the site of inflammation. Thus, when the ANCA-induced cytokine release occurs at the endothelial interface, the normal chemotactic gradient that draws neutrophils out of the vasculature into the tissues is lost. This causes further accumulation of fully activated neutrophils in the vessel wall, where they cause more injury. Figure 1 summarizes several complex interactions between ANCA and inflammatory and endothelial cells thought to be instrumental during the formation of granulomatous inflammation and vasculitis.
Figure 1.
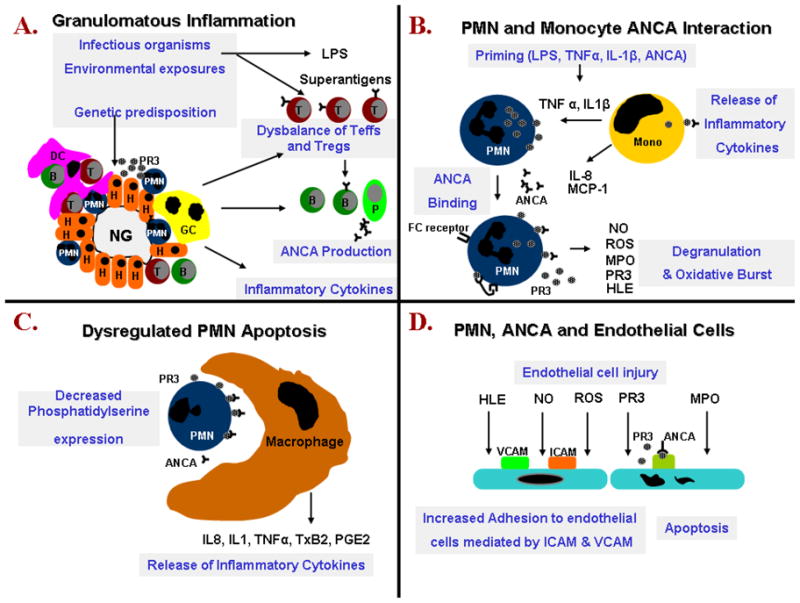
A. Necrotizing granulomatous inflammation develops in response to an unknown insult. This inflammatory reaction as well as the original insult result in the release of various cytokines, proteinase 3 (PR3) and potentially bacterial superantigens. Granulomatous inflammation in conjunction with the original insult, secreted cytokines and possibly bacterial superantigen promote the production of ANCA by PR3 specific B-lymphocytes and the selection of PR3 specific T-cells in predisposed individuals. There is a dysbalance between effector T cells (Teffs) and regulatory T cells (Tregs) with further release of proinflammatory cytokines promoting neutrophil priming.
B. ANCA have been shown in vitro to bind to PR3 expressed on the surface of neutrophils and monocytes. ANCA binding results in cellular activation, release of pro-inflammatory cytokines and neutrophil degranulation.
C. In vitro ANCA also opsonize apoptotic neutrophils by binding to PR3 expressed on their cell surface. In addition in the presence of ANCA apoptotic neutrophils exhibit decreased phosphatidylserine expression on their cell surface. Neutrophil opsonization and perturbed phosphatidylserine expression result in the inappropriate release of pro-inflammatory cytokines by macrophages clearing apoptotic neutrophils.
D. Endothelial damage is promoted by various mediators released by neutrophils. In addition the increased expression of cell adhesion molecules and presence of ANCA facilitates the binding of neutrophils to endothelial cells. PR3 is internalized by endothelial cells and promotes apoptosis.
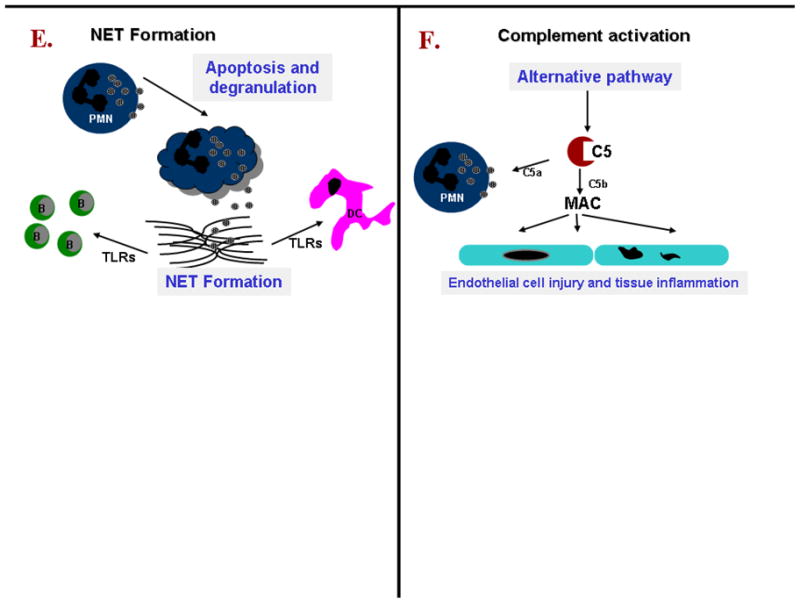
E. Neutrophil extracellular trap (NET) formation occurs in lesions as a consequence of neutrophil apoptosis and degranulation. NET-derived products activate dendritic cells and B-cells by sensing via Toll-like receptors. Interferon (IFN-a) production by dendritic might have an impact on local immune regulation and it has been shown to impair regulatory T cells function.
F. Endothelial damage and tissue inflammation caused by localized complement activation. Complement is activated predominantly by the alternative pathway resulting in cleavage of C5 into C5b which causes the assembly of MAC. C5a is able to prime neutrophils to enhance ANCA-induced neutrophil activation.
The ANCA target antigens released from activated or dying neutrophils can also directly bind to endothelial cells(134). This may result in apoptosis of endothelial cells and in localized immune complex formation with circulating ANCA (135). Low levels of localized immune-complex deposition, which has been documented in early vasculitic skin lesions as well as renal lesions, can in turn induce localized complement activation(136, 137).
In vitro studies have also suggested that ANCA modify the clearance of apoptotic cells. Opsonization of pre-apoptotic cells by ANCA is associated with an increased production of inflammatory cytokines by phagocytosing macrophages(138). Moreover, pre-apoptotic cells have a decreased cell surface expression of phosphatidylserine (the recognition signal for macrophages) in the presence of ANCA(139). Consequently, in the presence of ANCA the non-inflammatory clearance of apoptotic cells by macrophages may be perturbed in favor of inflammation and necrosis.
The pathogenic role of ANCA has also been supported by several in vivo animal models. These models are based on the transfer of antibodies generated against ANCA target antigens into healthy recipient animals. The transfer of anti-MPO IgG or splenocytes obtained from MPO-knockout mice, which were immunized with murine MPO, into Rag 2 knock-out mice (lacking mature T- and B-lymphocytes) as well as into wild-type mice resulted in pauci-immune crescentic necrotizing glomerulonephritis similar to that found in humans(140). A direct augmenting effect of MPO-ANCA on neutrophil - endothelial interactions causing microvascular injury was documented in a rat anti-MPO antibody transfer model (141). Murine anti-PR3 antibodies generated in a similar fashion only caused an increased inflammatory response at the site of tissue injury, but not a vasculitic phenotype, when transferred into wild-type mice(142).
The inflammatory lesions of AAV are referred to as “pauci-immune”, implying that only few immune complexes or complement factors can be identified by immunofluorescence microscopy. Furthermore, in contrast to classic immune-complex mediated disease, patients with active AAV have normal serum complement levels. Nevertheless, low grade localized immune-complex formation and complement activation may play a role, and there is growing evidence suggesting that the complement pathway is involved in the pathogenesis of AAV (137, 143–145). Moreover, complement activation might also contribute to the increased risk of venous thromboembolism observed in active AAV, as activated complement factors trigger the coagulation cascade(146, 147).
It has recently been recognized that activation of the alternative complement pathway by ANCA may represent an important amplification loop of inflammation that contributes to renal (and other tissue) injury in AAV. In the murine anti-MPO antibody transfer model the development of necrotizing glomerulonephritis is dependent on activation of the alternative complement pathway, and the development of lesions can be prevented and treated with an antibody that inhibited complement factor 5 (C5) activation(148, 149). Mice lacking the receptor for activated C5 on neutrophils also do not develop the renal lesions (150). In vitro studies showed that supernatants from ANCA-activated neutrophils can cause the production of C5a in normal serum, C5a receptor dependent priming of normal neutrophils, and the increased neutrophil membrane expression of PR3. Even though the renal lesions in humans are called “pauci-immune”, components of the alternative complement pathway can be detected in patients with AAV, but not normal controls or patients with minimal change disease(151).
Despite all this evidence, proof that ANCA alone can cause disease in humans has remained elusive. One case study in which an infant was born to a mother with active MPA is often quoted as such evidence (152). The infant developed a pulmonary-renal syndrome 48 hours after delivery and was found to have serum MPO-ANCA titers similar to the mother (152). The child was treated with glucocorticoids and plasma exchange and recovered. However, this observation is countered by another report of a case where MPO-ANCA were also transferred from the mother to the newborn, but the newborn remained perfectly healthy despite persistence of the transferred MPO-ANCA in the newborn for several weeks(153). The two contrasting case studies are consistent with observations made in large cohort studies. The development of severe vasculitic disease manifestations and severe flares usually do not occur in the absence of ANCA, but not all patients with persistent ANCA inevitably suffer such flares (76, 154–156).
Conclusions
Based on currently available clinical and experimental evidence it is reasonable to conceptualize that in predisposed patients, different triggers can lead to the production of autoantibodies (ANCA) that in the context of an inflammatory environment can cause tissue inflammation and vascular injury. However, many of the proposed mechanisms behind the pathophysiology of AAV may only apply to certain clinical subsets of patients. Moreover, despite the several different pathways and mechanisms described in this review, there is not a one-size-fits all cases, or even most of the cases. Despite the substantial advances in our understanding of the pathogenesis of AAV, many open questions still remain. As our knowledge regarding the pathogenesis of AAV evolves, new therapeutic targeted strategies are emerging for the continued quest to control disease activity with the minimum of adverse effects in individual patients.
Abbreviations
- NG
necrotizing granuloma
- H
histiocyte
- PMN
polymorphonuclear neutrophil
- T
T-lymphocyte
- B
B-lymphocyte
- DC
dendritic cell
- GC
giant cell
- P
plasma cell
- Mono
monocyte
- PR3
proteinase 3
- LPS
lipopolysaccaride
- ROS
reactive oxygen species
- ANCA
anti-neutrophil cytoplasmic antibodies
- HLE
human leukocyte elastase
- NO
nitric oxide
- MPO
myeloperoxidase
- IL-8
interleukin 8
- MCP-1
monocyte chemoattractant protein-1
- TNFα
tumor necrosis factor α
- IL-1β
interleukin 1β
- IL-1
interleukin 1
- ICAM
intercellular adhesion molecule
- VCAM
vascular cell adhesion molecule
- PGE2
prostaglandin E2
- TxB2
thromboxane B2
- MAC
Membrane attack complex
- TLRs
Toll-like receptors
Footnotes
Disclosure No potential conflicts of interest relevant to this article were reported.
References
Papers of particular interest, published since 2009, have been highlighted as:
• of importance
•• of major importance
- 1.Jennette JC, Falk RJ, Andrassy K, Bacon BA, Churg J, Gross WL, et al. Nomenclature of systemic vasculitides: The proposal of an international consensus conference. Arthritis Rheum. 1994;37:187–92. doi: 10.1002/art.1780370206. [DOI] [PubMed] [Google Scholar]
- 2.Hoffman GS, Specks U. Anti-neutrophil cytoplasmic antibodies. Arthritis Rheum. 1998;41:1521–37. doi: 10.1002/1529-0131(199809)41:9<1521::AID-ART2>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 3.Keogh KA, Specks U. Churg-Strauss syndrome. clinical presentation, antineutrophil cytoplasmic antibodies, and leukotriene receptor antagonists. Am J Med. 2003 Sep;115(4):284–90. doi: 10.1016/s0002-9343(03)00359-0. [DOI] [PubMed] [Google Scholar]
- 4.Sinico RA, Di Toma L, Maggiore U, Bottero P, Radice A, Tosoni C, et al. Prevalence and clinical significance of antineutrophil cytoplasmic antibodies in Churg-Strauss syndrome. Arthritis Rheum. 2005 Sep;52(9):2926–35. doi: 10.1002/art.21250. [DOI] [PubMed] [Google Scholar]
- 5.Sable-Fourtassou R, Cohen P, Mahr A, Pagnoux C, Mouthon L, Jayne D, et al. Antineutrophil cytoplasmic antibodies and the Churg-Strauss syndrome. Ann Intern Med. 2005 Nov 1;143(9):632–8. doi: 10.7326/0003-4819-143-9-200511010-00006. [DOI] [PubMed] [Google Scholar]
- 6.Watts RA, Lane SE, Bentham G, Scott DG. Epidemiology of systemic vasculitis: a ten-year study in the United Kingdom. Arthritis Rheum. 2000;43(2):414–9. doi: 10.1002/1529-0131(200002)43:2<414::AID-ANR23>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 7.Watts RA, Gonzalez-Gay MA, Lane SE, Garcia-Porrua C, Bentham G, Scott DG. Geoepidemiology of systemic vasculitis: comparison of the incidence in two regions of Europe. Ann Rheum Dis. 2001;60(2):170–2. doi: 10.1136/ard.60.2.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reinhold-Keller E, Herlyn K, Wagner-Bastmeyer R, Gutfleisch J, Peter HH, Raspe HH, et al. No difference in the incidences of vasculitides between north and south Germany: first results of the German vasculitis register. Rheumatology (Oxford) 2002;41(5):540–9. doi: 10.1093/rheumatology/41.5.540. [DOI] [PubMed] [Google Scholar]
- 9.Watts RA, Lane SE, Scott DG, Koldingsnes W, Nossent H, Gonzalez-Gay MA, et al. Epidemiology of vasculitis in Europe. Annals of the Rheumatic Diseases. 2001;60(12):1156–7. doi: 10.1136/ard.60.12.1156a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Watts RA, Scott DG. Epidemiology of the vasculitides. Semin Respir Crit Care Med. 2004 Oct;25(5):455–64. doi: 10.1055/s-2004-836139. [DOI] [PubMed] [Google Scholar]
- 11.Cotch MF, Hoffman GS, Yerg DE, Kaufman GI, Targonski P, Kaslow RA. The epidemiology of Wegener’s granulomatosis. Estimates of the five-year period prevalence, annual mortality, and geographic disease distribution from population-based data sources. Arthritis Rheum. 1996;39:87–92. doi: 10.1002/art.1780390112. [DOI] [PubMed] [Google Scholar]
- 12.Watts RA, Scott DG, Jayne DR, Ito-Ihara T, Muso E, Fujimoto S, et al. Renal vasculitis in Japan and the UK--are there differences in epidemiology and clinical phenotype? Nephrol Dial Transplant. 2008 Dec;23(12):3928–31. doi: 10.1093/ndt/gfn354. [DOI] [PubMed] [Google Scholar]
- 13.O’Donnell JL, Stevanovic VR, Frampton C, Stamp LK, Chapman PT. Wegener’s granulomatosis in New Zealand: evidence for a latitude-dependent incidence gradient. Intern Med J. 2007 Apr;37(4):242–6. doi: 10.1111/j.1445-5994.2006.01297.x. [DOI] [PubMed] [Google Scholar]
- 14.Mahr A, Guillevin L, Poissonnet M, Ayme S. Prevalences of polyarteritis nodosa, microscopic polyangiitis, Wegener’s granulomatosis, and Churg-Strauss syndrome in a French urban multiethnic population in 2000: a capture-recapture estimate. Arthritis Rheum. 2004 Feb 15;51(1):92–9. doi: 10.1002/art.20077. [DOI] [PubMed] [Google Scholar]
- 15.Moins-Teisserenc HT, Gadola SD, Cella M, Dunbar PR, Exley A, Blake N, et al. Association of a syndrome resembling Wegener’s granulomatosis with low surface expression of HLA class-I molecules. Lancet. 1999 Nov 6;354(9190):1598–603. doi: 10.1016/s0140-6736(99)04206-3. [Research Support, Non-U.S. Gov’t] [DOI] [PubMed] [Google Scholar]
- 16.Villa-Forte A, de la Salle H, Fricker D, Hentges F, Zimmer J. HLA class I deficiency syndrome mimicking Wegener’s granulomatosis. Arthritis and rheumatism. 2008 Aug;58(8):2579–82. doi: 10.1002/art.23675. [Case Reports Research Support, Non-U.S. Gov’t] [DOI] [PubMed] [Google Scholar]
- 17.Moins-Teisserenc HT, Gadola SD, Cella M, Dunbar PR, Exley A, Blake N, et al. Association of a syndrome resembling Wegener’s granulomatosis with low surface expression of HLA class-I molecules. Lancet. 1999;354(9190):1598–603. doi: 10.1016/s0140-6736(99)04206-3. [DOI] [PubMed] [Google Scholar]
- 18.Knight A, Sandin S, Askling J. Risks and relative risks of Wegener’s granulomatosis among close relatives of patients with the disease. Arthritis Rheum. 2008 Jan;58(1):302–7. doi: 10.1002/art.23157. [DOI] [PubMed] [Google Scholar]
- 19.Rottem M, Cotch MF, Fauci AS, Hoffman GS. Familial vasculitis: report of 2 families. J Rheumatol. 1994 Mar;21(3):561–3. [PubMed] [Google Scholar]
- 20.Manganelli P, Giacosa R, Fietta P, Zanetti A, Neri TM. Familial vasculitides: Churg-Strauss syndrome and Wegener’s granulomatosis in 2 first-degree relatives. J Rheumatol. 2003 Mar;30(3):618–21. [PubMed] [Google Scholar]
- 21.Willcocks LC, Lyons PA, Rees AJ, Smith KG. The contribution of genetic variation and infection to the pathogenesis of ANCA-associated systemic vasculitis. Arthritis Res Ther. 2010;12(1):202. doi: 10.1186/ar2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shiina T, Inoko H, Kulski JK. An update of the HLA genomic region, locus information and disease associations: 2004. Tissue antigens. 2004 Dec;64(6):631–49. doi: 10.1111/j.1399-0039.2004.00327.x. [Review] [DOI] [PubMed] [Google Scholar]
- 23.Yang R, Cui Z, Zhao J, Zhao MH. The role of HLA-DRB1 alleles on susceptibility of Chinese patients with anti-GBM disease. Clinical Immunology. 2009 Nov;133(2):245–50. doi: 10.1016/j.clim.2009.07.005. [Research Support, Non-U.S. Gov’t] [DOI] [PubMed] [Google Scholar]
- 24.Gencik M, Borgmann S, Zahn R, Albert E, Sitter T, Epplen JT, et al. Immunogenetic risk factors for anti-neutrophil cytoplasmic antibody (ANCA)-associated systemic vasculitis. Clin Exp Immunol. 1999;117(2):412–7. doi: 10.1046/j.1365-2249.1999.00969.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heckmann M, Holle JU, Arning L, Knaup S, Hellmich B, Nothnagel M, et al. The Wegener’s granulomatosis quantitative trait locus on chromosome 6p21.3 as characterised by tagSNP genotyping. Ann Rheum Dis. 2008 Jul;67(7):972–9. doi: 10.1136/ard.2007.077693. [DOI] [PubMed] [Google Scholar]
- 26.Tsuchiya N, Kobayashi S, Hashimoto H, Ozaki S, Tokunaga K. Association of HLA-DRB1*0901-DQB1*0303 haplotype with microscopic polyangiitis in Japanese. Genes and immunity. 2006 Jan;7(1):81–4. doi: 10.1038/sj.gene.6364262. [Comparative Study Research Support, Non-U.S. Gov’t] [DOI] [PubMed] [Google Scholar]
- 27.Tsuchiya N, Kobayashi S, Kawasaki A, Kyogoku C, Arimura Y, Yoshida M, et al. Genetic background of Japanese patients with antineutrophil cytoplasmic antibody-associated vasculitis: association of HLA-DRB1*0901 with microscopic polyangiitis. J Rheumatol. 2003 Jul;30(7):1534–40. [PubMed] [Google Scholar]
- 28.Vaglio A, Martorana D, Maggiore U, Grasselli C, Zanetti A, Pesci A, et al. HLA-DRB4 as a genetic risk factor for Churg-Strauss syndrome. Arthritis Rheum. 2007 Sep;56(9):3159–66. doi: 10.1002/art.22834. [DOI] [PubMed] [Google Scholar]
- 29.Wieczorek S, Hellmich B, Gross WL, Epplen JT. Associations of Churg-Strauss syndrome with the HLA-DRB1 locus, and relationship to the genetics of antineutrophil cytoplasmic antibody-associated vasculitides: comment on the article by Vaglio et al. Arthritis Rheum. 2008 Jan;58(1):329–30. doi: 10.1002/art.23209. [DOI] [PubMed] [Google Scholar]
- 30.Arning L, Holle JU, Harper L, Millar DS, Gross WL, Epplen JT, et al. Are there specific genetic risk factors for the different forms of ANCA-associated vasculitis? Ann Rheum Dis. 2011 Apr;70(4):707–8. doi: 10.1136/ard.2010.130971. [DOI] [PubMed] [Google Scholar]
- 31.Cao Y, Schmitz JL, Yang J, Hogan SL, Bunch D, Hu Y, et al. DRB1*15 allele is a risk factor for PR3-ANCA disease in African Americans. J Am Soc Nephrol. 2011 Jun;22(6):1161–7. doi: 10.1681/ASN.2010101058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jagiello P, Aries P, Arning L, Wagenleiter SE, Csernok E, Hellmich B, et al. The PTPN22 620W allele is a risk factor for Wegener’s granulomatosis. Arthritis Rheum. 2005 Nov 30;52(12):4039–43. doi: 10.1002/art.21487. [DOI] [PubMed] [Google Scholar]
- 33.Carr EJ, Niederer HA, Williams J, Harper L, Watts RA, Lyons PA, et al. Confirmation of the genetic association of CTLA4 and PTPN22 with ANCA-associated vasculitis. BMC Med Genet. 2009;10:121. doi: 10.1186/1471-2350-10-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gregersen PK, Lee HS, Batliwalla F, Begovich AB. PTPN22: setting thresholds for autoimmunity. Semin Immunol. 2006 Aug;18(4):214–23. doi: 10.1016/j.smim.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 35.Bluestone JA. Is CTLA-4 a master switch for peripheral T cell tolerance? J Immunol. 1997 Mar 1;158(5):1989–93. [PubMed] [Google Scholar]
- 36.Steiner K, Moosig F, Csernok E, Selleng K, Gross WL, Fleischer B, et al. Increased expression of CTLA-4 (CD152) by T and B lymphocytes in Wegener’s granulomatosis. Clin Exp Immunol. 2001;126(1):143–50. doi: 10.1046/j.1365-2249.2001.01575.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gough SC, Walker LS, Sansom DM. CTLA4 gene polymorphism and autoimmunity. Immunol Rev. 2005 Apr;204:102–15. doi: 10.1111/j.0105-2896.2005.00249.x. [DOI] [PubMed] [Google Scholar]
- 38.Wang XB, Zhao X, Giscombe R, Lefvert AK. A CTLA-4 gene polymorphism at position -318 in the promoter region affects the expression of protein. Genes Immun. 2002 Jun;3(4):233–4. doi: 10.1038/sj.gene.6363869. [DOI] [PubMed] [Google Scholar]
- 39.Lee YH, Choi SJ, Ji JD, Song GG. CTLA-4 and TNF-alpha promoter-308 A/G polymorphisms and ANCA-associated vasculitis susceptibility: a meta-analysis. Mol Biol Rep. 2012 Jan;39(1):319–26. doi: 10.1007/s11033-011-0741-2. [DOI] [PubMed] [Google Scholar]
- 40.Gencik M, Meller S, Borgmann S, Fricke H. Proteinase 3 gene polymorphisms and Wegener’s granulomatosis. Kidney Int. 2000;58(6):2473–7. doi: 10.1046/j.1523-1755.2000.00430.x. [DOI] [PubMed] [Google Scholar]
- 41.Halbwachs-Mecarelli L, Bessou G, Lesavre P, Lopez S, Witko-Sarsat V. Bimodal distribution of proteinase 3 (PR3) surface expression reflects a constitutive heterogeneity in the polymorphonuclear neutrophil pool. FEBS. 1995;374:29–33. doi: 10.1016/0014-5793(95)01073-n. [DOI] [PubMed] [Google Scholar]
- 42.Schreiber A, Busjahn A, Luft FC, Kettritz R. Membrane expression of proteinase 3 is genetically determined. J Am Soc Nephrol. 2003;14(1):68–75. doi: 10.1097/01.asn.0000040751.83734.d1. [DOI] [PubMed] [Google Scholar]
- 43.von Vietinghoff S, Busjahn A, Schonemann C, Massenkeil G, Otto B, Luft FC, et al. Major histocompatibility complex HLA region largely explains the genetic variance exercised on neutrophil membrane proteinase 3 expression. J Am Soc Nephrol. 2006 Nov;17(11):3185–91. doi: 10.1681/ASN.2006050522. [DOI] [PubMed] [Google Scholar]
- 44.Witko-Sarsat V, Lesavre P, Lopez S, Bessou G, Hieblot C, Prum B, et al. A large subset of neutrophils expressing membrane proteinase 3 is a risk factor for vasculitis and rheumatoid arthritis. J Am Soc Nephrol. 1999;10(6):1224–33. doi: 10.1681/ASN.V1061224. [DOI] [PubMed] [Google Scholar]
- 45.Rarok AA, Stegeman CA, Limburg PC, Kallenberg CG. Neutrophil membrane expression of proteinase 3 (PR3) is related to relapse in PR3-ANCA-associated vasculitis. J Am Soc Nephrol. 2002;13(9):2232–8. doi: 10.1097/01.asn.0000028642.26222.00. [DOI] [PubMed] [Google Scholar]
- 46.Mahr AD, Edberg JC, Stone JH, Hoffman GS, St Clair EW, Specks U, et al. Alpha 1-antitrypsin deficiency-related alleles Z and S and the risk for Wegener’s granulomatosis. Arthritis Rheum. 2010 Sep 8; doi: 10.1002/art.27742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morris H, Morgan MD, Wood AM, Smith SW, Ekeowa UI, Herrmann K, et al. ANCA-associated vasculitis is linked to carriage of the Z allele of alpha antitrypsin and its polymers. Ann Rheum Dis. 2011 Oct;70(10):1851–6. doi: 10.1136/ard.2011.153569. [DOI] [PubMed] [Google Scholar]
- 48.Segelmark M, Elzouki AN, Wieslander J, Eriksson S. The PiZ gene of alpha 1-antitrypsin as a determinant of outcome in PR3-ANCA-positive vasculitis. Kidney Int. 1995 Sep;48(3):844–50. doi: 10.1038/ki.1995.360. [DOI] [PubMed] [Google Scholar]
- 49.Stankiewicz P, Lupski JR. Structural variation in the human genome and its role in disease. Annu Rev Med. 2010;61:437–55. doi: 10.1146/annurev-med-100708-204735. [DOI] [PubMed] [Google Scholar]
- ••50.Lyons PA, Rayner TF, Trivedi S, Holle JU, Watts RA, Jayne DR, et al. Genetically Distinct Subsets within ANCA-Associated Vasculitis. N Engl J Med [Research Support, Non-US Gov’t] 2012 Jul 19;367(3):214–23. doi: 10.1056/NEJMoa1108735. First AAV GWAS that confirmed that the pathogenesis of AAV has a genetic component with genetic distinctions between GPA and MPA that are associated with ANCA specificity where the response against the autoantigen PR3 is a central feature of PR3 AAV patients. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hogan SL, Cooper GS, Savitz DA, Nylander-French LA, Parks CG, Chin H, et al. Association of silica exposure with anti-neutrophil cytoplasmic autoantibody small-vessel vasculitis: a population-based, case-control study. Clin J Am Soc Nephrol. 2007 Mar;2(2):290–9. doi: 10.2215/CJN.03501006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Franchi L, Eigenbrod T, Nunez G. Cutting edge: TNF-alpha mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. Journal of Immunology. 2009 Jul 15;183(2):792–6. doi: 10.4049/jimmunol.0900173. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Short AK, Lockwood CM. Antigen specificity in hydralazine associated ANCA positive systemic vasculitis. Q J Med. 1995;88:775–83. [PubMed] [Google Scholar]
- 54.Choi HK, Merkel PA, Walker AM, Niles JL. Drug-associated antineutrophil cytoplasmic antibody-positive vasculitis: prevalence among patients with high titers of antimyeloperoxidase antibodies. Arthritis Rheum. 2000;43(2):405–13. doi: 10.1002/1529-0131(200002)43:2<405::AID-ANR22>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 55.Carrington CB, Liebow A. Limited forms of angiitis and granulomatosis of Wegener’s type. American Journal of Medicine. 1966;41(4):497–527. doi: 10.1016/0002-9343(66)90214-2. [DOI] [PubMed] [Google Scholar]
- 56.Fahey J, EL, JC, GCG Wegener’s Granulomatosis. American Journal of Medicine. 1954;17:168–79. doi: 10.1016/0002-9343(54)90255-7. [DOI] [PubMed] [Google Scholar]
- 57.Walton E. Giant-cell granuloma of the respiratory tract (Wegener’s granulomatosis) British Medical Journal. 1958;2:497–527. doi: 10.1136/bmj.2.5091.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fauci AS, Haynes BF, Katz P, Wolff SM. Wegener’s granulomatosis: prospective clinical and therapeutic experience with 85 patients for 21 years. Annals of Internal Medicine. 1983;98(1):76–85. doi: 10.7326/0003-4819-98-1-76. [DOI] [PubMed] [Google Scholar]
- 59.Hoffman GS, Kerr GS, Leavitt RY, Hallahan CW, Lebovics RS, Travis WD, et al. Wegener granulomatosis: an analysis of 158 patients.[see comment] Annals of Internal Medicine. 1992;116(6):488–98. doi: 10.7326/0003-4819-116-6-488. [DOI] [PubMed] [Google Scholar]
- 60.Pinching AJ, Rees AJ, Pussell BA, Lockwood CM, Mitchison RS, Peters DK. Relapses in Wegener’s granulomatosis: the role of infection. British Medical Journal. 1980;281(6244):836–8. doi: 10.1136/bmj.281.6244.836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Raynauld JP, Bloch DA, Fries JF. Seasonal variation in the onset of Wegener’s granulomatosis, polyarteritis nodosa and giant cell arteritis. Journal of Rheumatology. 1993;20(9):1524–6. [PubMed] [Google Scholar]
- 62.Choi HK, Lamprecht P, Niles JL, Gross WL, Merkel PA. Subacute bacterial endocarditis with positive cytoplasmic antineutrophil cytoplasmic antibodies and anti-proteinase 3 antibodies. Arthritis Rheum. 2000;43(1):226–31. doi: 10.1002/1529-0131(200001)43:1<226::AID-ANR27>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 63.Capizzi SA, Specks U. Does infection play a role in the pathogenesis of pulmonary vasculitis? Semin Respir Infect. 2003 Mar;18(1):17–22. doi: 10.1053/srin.2003.50002. [DOI] [PubMed] [Google Scholar]
- 64.Albert LJ, Inman RD. Molecular mimicry and autoimmunity. N Engl J Med. 1999;341(27):2068–74. doi: 10.1056/NEJM199912303412707. [DOI] [PubMed] [Google Scholar]
- 65.Craft J, Fatenejad S. Self antigens and epitope spreading in systemic autoimmunity. Arthritis Rheum. 1997;40:1374–82. doi: 10.1002/art.1780400803. [DOI] [PubMed] [Google Scholar]
- 66.Vanderlugt CJ, Miller SD. Epitope spreading. Curr Opin Immunol. 1996;8:831–6. doi: 10.1016/S0952-7915(96)80012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kain R, Exner M, Brandes R, Ziebermayr R, Cunningham D, Alderson CA, et al. Molecular mimicry in pauci-immune focal necrotizing glomerulonephritis. Nat Med. 2008 Oct;14(10):1088–96. doi: 10.1038/nm.1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kain R, Tadema H, McKinney EF, Benharkou A, Brandes R, Peschel A, et al. High Prevalence of Autoantibodies to hLAMP-2 in Anti-Neutrophil Cytoplasmic Antibody-Associated Vasculitis. J Am Soc Nephrol. 2012 Mar;23(3):556–66. doi: 10.1681/ASN.2011090920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Roth AJ, Brown MC, Smith RN, Badhwar AK, Parente O, Chung HC, et al. Anti-LAMP-2 Antibodies Are Not Prevalent in Patients With Antineutrophil Cytoplasmic Autoantibody Glomerulonephritis. J Am Soc Nephrol. 2012 Mar;23(3):545–55. doi: 10.1681/ASN.2011030273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pendergraft WF, 3rd, Preston GA, Shah RR, Tropsha A, Carter CW, Jr, Jennette JC, et al. Autoimmunity is triggered by cPR-3(105-201), a protein complementary to human autoantigen proteinase-3. Nat Med. 2004 Jan;10(1):72–9. doi: 10.1038/nm968. [DOI] [PubMed] [Google Scholar]
- 71.Shoenfeld Y. Idiotypic induction of autoimmunity: a new aspect of the idiotypic network. FASEB J. 1994 Dec;8(15):1296–301. doi: 10.1096/fasebj.8.15.8001742. [DOI] [PubMed] [Google Scholar]
- 72.Astern JM. Myeloperoxidase in vascular disease and autoimmunity. Chapel Hill: University of North Carolina; 2007. [Google Scholar]
- 73.Tadema H, Kallenberg CG, Stegeman CA, Heeringa P. Reactivity against complementary proteinase-3 is not increased in patients with PR3-ANCA-associated vasculitis. PLoS One. 2011;6(3):e17972. doi: 10.1371/journal.pone.0017972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bautz DJ, Preston GA, Lionaki S, Hewins P, Wolberg AS, Yang JJ, et al. Antibodies with Dual Reactivity to Plasminogen and Complementary PR3 in PR3-ANCA Vasculitis. J Am Soc Nephrol. 2008 Aug 13; doi: 10.1681/ASN.2008030270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Merkel PA, Lo GH, Holbrook JT, Tibbs AK, Allen NB, Davis JC, Jr, et al. High incidence of venous thrombotic events among patients with Wegener granulomatosis: the Wegener’s Clinical Occurrence of Thrombosis (WeCLOT) Study. Ann Intern Med. 2005 Apr 19;142(8):620–6. doi: 10.7326/0003-4819-142-8-200505030-00011. [DOI] [PubMed] [Google Scholar]
- 76.Stegeman CA, Cohen Tervaert JW, Sluiter WJ, Manson WL, de Jong PE, Kallenberg CGM. Association of chronic nasal carriage of Staphylococcus aureus and higher relapse rates in Wegener granulomatosis. Ann Intern Med. 1994;120:12–7. doi: 10.7326/0003-4819-120-1-199401010-00003. [DOI] [PubMed] [Google Scholar]
- 77.Stegeman CA, Cohen Tervaert JW, de Jong PE, Kallenberg CG. Trimethoprim-sulfamethoxazole (co-trimoxazole) for the prevention of relapses of Wegener’s granulomatosis. N Engl J Med. 1996;335(1):16–20. doi: 10.1056/NEJM199607043350103. [DOI] [PubMed] [Google Scholar]
- 78.Zycinska K, Wardyn KA, Zielonka TM, Krupa R, Lukas W. Co-trimoxazole and prevention of relapses of PR3-ANCA positive vasculitis with pulmonary involvement. Eur J Med Res. 2009 Dec 7;14( Suppl 4):265–7. doi: 10.1186/2047-783X-14-S4-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Proft T, Fraser JD. Bacterial superantigens. Clin Exp Immunol. 2003 Sep;133(3):299–306. doi: 10.1046/j.1365-2249.2003.02203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zouali M. Exploitation of host signaling pathways by B cell superantigens--potential strategies for developing targeted therapies in systemic autoimmunity. Ann N Y Acad Sci. 2007 Jan;1095:342–54. doi: 10.1196/annals.1397.038. [DOI] [PubMed] [Google Scholar]
- 81.Popa ER, Stegeman CA, Abdulahad WH, van der Meer B, Arends J, Manson WM, et al. Staphylococcal toxic-shock-syndrome-toxin-1 as a risk factor for disease relapse in Wegener’s granulomatosis. Rheumatology (Oxford) 2007 Jun;46(6):1029–33. doi: 10.1093/rheumatology/kem022. [DOI] [PubMed] [Google Scholar]
- 82.Popa ER, Stegeman CA, Bos NA, Kallenberg CG, Tervaert JW. Staphylococcal superantigens and T cell expansions in Wegener’s granulomatosis. Clin Exp Immunol. 2003 Jun;132(3):496–504. doi: 10.1046/j.1365-2249.2003.02157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li H, Nooh MM, Kotb M, Re F. Commercial peptidoglycan preparations are contaminated with superantigen-like activity that stimulates IL-17 production. J Leukoc Biol. 2008 Feb;83(2):409–18. doi: 10.1189/jlb.0807588. [DOI] [PubMed] [Google Scholar]
- 84.Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, et al. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol. 2007 Jun;8(6):639–46. doi: 10.1038/ni1467. [DOI] [PubMed] [Google Scholar]
- 85.Gerosa F, Baldani-Guerra B, Lyakh LA, Batoni G, Esin S, Winkler-Pickett RT, et al. Differential regulation of interleukin 12 and interleukin 23 production in human dendritic cells. J Exp Med. 2008 Jun 9;205(6):1447–61. doi: 10.1084/jem.20071450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fouser LA, Wright JF, Dunussi-Joannopoulos K, Collins M. Th17 cytokines and their emerging roles in inflammation and autoimmunity. Immunol Rev. 2008 Dec;226:87–102. doi: 10.1111/j.1600-065X.2008.00712.x. [DOI] [PubMed] [Google Scholar]
- 87.Oukka M. Th17 cells in immunity and autoimmunity. Ann Rheum Dis. 2008 Dec;67(Suppl 3):iii26–9. doi: 10.1136/ard.2008.098004. [DOI] [PubMed] [Google Scholar]
- •88.Nogueira E, Hamour S, Sawant D, Henderson S, Mansfield N, Chavele KM, et al. Serum IL-17 and IL-23 levels and autoantigen-specific Th17 cells are elevated in patients with ANCA-associated vasculitis. Nephrol Dial Transplant. 2010 Jul;25(7):2209–17. doi: 10.1093/ndt/gfp783. T cells producing IL-17 (Th17) have been implicated in the pathogenesis of several autoimmune diseases and this study identified that serum IL-17 levels and autoantigen-specific Th17 cells were elevated in patients with active AAV as compared to helathy individuals. [DOI] [PubMed] [Google Scholar]
- 89.Abdulahad WH, Stegeman CA, Limburg PC, Kallenberg CG. Skewed distribution of Th17 lymphocytes in patients with Wegener’s granulomatosis in remission. Arthritis Rheum. 2008 Jul;58(7):2196–205. doi: 10.1002/art.23557. [DOI] [PubMed] [Google Scholar]
- 90.Voswinkel J, Mueller A, Kraemer JA, Lamprecht P, Herlyn K, Holl-Ulrich K, et al. B lymphocyte maturation in Wegener’s granulomatosis: a comparative analysis of VH genes from endonasal lesions. Ann Rheum Dis. 2006 Jul;65(7):859–64. doi: 10.1136/ard.2005.044909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hurtado PR, Jeffs L, Nitschke J, Patel M, Sarvestani G, Cassidy J, et al. CpG oligodeoxynucleotide stimulates production of anti-neutrophil cytoplasmic antibodies in ANCA associated vasculitis. BMC Immunol. 2008;9:34. doi: 10.1186/1471-2172-9-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •92.Tadema H, Abdulahad WH, Lepse N, Stegeman CA, Kallenberg CG, Heeringa P. Bacterial DNA motifs trigger ANCA production in ANCA-associated vasculitis in remission. Rheumatology (Oxford) 2011 Apr;50(4):689–96. doi: 10.1093/rheumatology/keq375. This in vitro study showed that bacterial DNA motifs(CpG) trigger the production of ANCA by B lymphocytes in patients with AAV in remission. [DOI] [PubMed] [Google Scholar]
- 93.Brinkmann V, Zychlinsky A. Beneficial suicide: why neutrophils die to make NETs. Nat Rev Microbiol. 2007 Aug;5(8):577–82. doi: 10.1038/nrmicro1710. [DOI] [PubMed] [Google Scholar]
- 94.Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007 Jan 15;176(2):231–41. doi: 10.1083/jcb.200606027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••95.Kessenbrock K, Krumbholz M, Schonermarck U, Back W, Gross WL, Werb Z, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med. 2009 Jun;15(6):623–5. doi: 10.1038/nm.1959. Neutrophil extracellular traps (NETs) are released by ANCA-stimulated neutrophils in the absence of microbial infection and contain proteinase-3 and myeloperoxidase. Deposition of NETs in inflammed tissue suggest that NET formation triggers vasculitis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••96.Pilsczek FH, Salina D, Poon KK, Fahey C, Yipp BG, Sibley CD, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol. 2010 Dec 15;185(12):7413–25. doi: 10.4049/jimmunol.1000675. Neutrophils responded uniquely to Staphylococcus aureus via a novel process of NET formation that did not require neutrophil lysis. S. aureus strogly induce NETs and S. aureus infections are linked to relapses of AAV. [DOI] [PubMed] [Google Scholar]
- 97.Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 2002 Apr 11;416(6881):603–7. doi: 10.1038/416603a. [DOI] [PubMed] [Google Scholar]
- •98.Tadema H, Abdulahad WH, Stegeman CA, Kallenberg CG, Heeringa P. Increased expression of Toll-like receptors by monocytes and natural killer cells in ANCA-associated vasculitis. PLoS One. 2011;6(9):e24315. doi: 10.1371/journal.pone.0024315. Toll-like receptors (TLRs) sense pathogen associated patterns and bacterial infections are known to be associated with AAV. In patients with AAV, monocytes and NK cells were shown to have an increased TLR expression probably resulting from increased activation, which could play a role in disease reactivation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Stone JH, Merkel PA, Spiera R, Seo P, Langford CA, Hoffman GS, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010 Jul 15;363(3):221–32. doi: 10.1056/NEJMoa0909905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Odendahl M, Mei H, Hoyer BF, Jacobi AM, Hansen A, Muehlinghaus G, et al. Generation of migratory antigen-specific plasma blasts and mobilization of resident plasma cells in a secondary immune response. Blood. 2005 Feb 15;105(4):1614–21. doi: 10.1182/blood-2004-07-2507. [DOI] [PubMed] [Google Scholar]
- 101.Huang H, Benoist C, Mathis D. Rituximab specifically depletes short-lived autoreactive plasma cells in a mouse model of inflammatory arthritis. Proc Natl Acad Sci U S A. 2010 Mar 9;107(10):4658–63. doi: 10.1073/pnas.1001074107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Popa ER, Stegeman CA, Bos NA, Kallenberg CG, Tervaert JW. Differential B- and T-cell activation in Wegener’s granulomatosis. J Allergy Clin Immunol. 1999;103(5 Pt 1):885–94. doi: 10.1016/s0091-6749(99)70434-3. [DOI] [PubMed] [Google Scholar]
- 103.Krumbholz M, Specks U, Wick M, Kalled SL, Jenne D, Meinl E. BAFF is elevated in serum of patients with Wegener’s granulomatosis. J Autoimmun. 2005 Dec;25(4):298–302. doi: 10.1016/j.jaut.2005.08.004. [DOI] [PubMed] [Google Scholar]
- •104.Schneeweis C, Rafalowicz M, Feist E, Buttgereit F, Rudolph PE, Burmester GR, et al. Increased levels of BLyS and sVCAM-1 in anti-neutrophil cytoplasmatic antibody (ANCA)-associated vasculitides (AAV) Clinical and experimental rheumatology. 2010 Jan-Feb;28(1 Suppl 57):62–6. Levels of endothelial cell activation (sVCAM-1) were elevated in patients with AAV as compared to healthy controls. [PubMed] [Google Scholar]
- 105.Bader L, Koldingsnes W, Nossent J. B-lymphocyte activating factor levels are increased in patients with Wegener’s granulomatosis and inversely correlated with ANCA titer. Clinical rheumatology. 2010 Sep;29(9):1031–5. doi: 10.1007/s10067-010-1526-z. [DOI] [PubMed] [Google Scholar]
- 106.Mellbye OJ, Mollnes TE, Steen LS. IgG subclass distribution and complement activation ability of autoantibodies to neutrophil cytoplasmic antigens (ANCA) Clin Immunol Immunopathol. 1994 Jan;70(1):32–9. doi: 10.1006/clin.1994.1007. [DOI] [PubMed] [Google Scholar]
- 107.Cohen Tervaert JW, Mulder L, Stegeman C, Elema J, Huitema M, The H, et al. Occurrence of autoantibodies to human leucocyte elastase in Wegener’s granulomatosis and other inflammatory disorders. Ann Rheum Dis. 1993;52:115–20. doi: 10.1136/ard.52.2.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Caux C, Dezutter-Dambuyant C, Schmitt D, Bandhereau J. GM-CSF and TNF- cooperate in the generation of dendritic Langerhans cells. Nature. 1992;360(6401):258–61. doi: 10.1038/360258a0. [DOI] [PubMed] [Google Scholar]
- 109.Abdulahad WH, van der Geld YM, Stegeman CA, Kallenberg CG. Persistent expansion of CD4+ effector memory T cells in Wegener’s granulomatosis. Kidney Int. 2006 Sep;70(5):938–47. doi: 10.1038/sj.ki.5001670. [DOI] [PubMed] [Google Scholar]
- 110.Abdulahad WH, Stegeman CA, van der Geld YM, Doornbos-van der Meer B, Limburg PC, Kallenberg CG. Functional defect of circulating regulatory CD4+ T cells in patients with Wegener’s granulomatosis in remission. Arthritis Rheum. 2007 Jun;56(6):2080–91. doi: 10.1002/art.22692. [DOI] [PubMed] [Google Scholar]
- ••111.Morgan MD, Day CJ, Piper KP, Khan N, Harper L, Moss PA, et al. Patients with Wegener’s granulomatosis demonstrate a relative deficiency and functional impairment of T-regulatory cells. Immunology. 2010 May;130(1):64–73. doi: 10.1111/j.1365-2567.2009.03213.x. Immune balance is important in controlling autoimmune diseases and in this study the percentage of regulatory T cells (Foxp3 positive cells) were found to be decreased in patients with GPA as compared to healthy controls. In addition, the percentage of regulatory T cells was found to be inversely related to the rate of disease relapse. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bettelli E, Oukka M, Kuchroo VK. T(H)-17 cells in the circle of immunity and autoimmunity. Nature immunology. 2007 Apr;8(4):345–50. doi: 10.1038/ni0407-345. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Review] [DOI] [PubMed] [Google Scholar]
- 113.Abdulahad WH, Stegeman CA, Kallenberg CG. Review article: The role of CD4(+) T cells in ANCA-associated systemic vasculitis. Nephrology (Carlton) 2009 Feb;14(1):26–32. doi: 10.1111/j.1440-1797.2008.01069.x. [DOI] [PubMed] [Google Scholar]
- 114.Ordonez L, Bernard I, L’Faqihi-Olive FE, Tervaert JW, Damoiseaux J, Saoudi A. CD45RC isoform expression identifies functionally distinct T cell subsets differentially distributed between healthy individuals and AAV patients. PloS one. 2009;4(4):e5287. doi: 10.1371/journal.pone.0005287. [Research Support, Non-U.S. Gov’t] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Altmann F, Staudacher E, Wilson IB, Marz L. Insect cells as hosts for the expression of recombinant glycoproteins. Glycoconj J. 1999 Feb;16(2):109–23. doi: 10.1023/a:1026488408951. [DOI] [PubMed] [Google Scholar]
- 116.Jovanovic DV, Di Battista JA, Martel-Pelletier J, Jolicoeur FC, He Y, Zhang M, et al. IL-17 stimulates the production and expression of proinflammatory cytokines, IL-beta and TNF-alpha, by human macrophages. J Immunol. 1998 Apr 1;160(7):3513–21. [PubMed] [Google Scholar]
- 117.Falk RJ, Terrell RS, Charles LA, Jennette JC. Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc Natl Acad Sci USA. 1990;87:4115–9. doi: 10.1073/pnas.87.11.4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Csernok E, Ernst M, Schmitt W, Bainton DF, Gross WL. Activated neutrophils express proteinase 3 on their plasma membrane in vitro and in vivo. Clin Exp Immunol. 1994;95(2):244–50. doi: 10.1111/j.1365-2249.1994.tb06518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Franssen CF, Huitema MG, Muller Kobold AC, Oost-Kort WW, Limburg PC, Tiebosch A, et al. In vitro neutrophil activation by antibodies to proteinase 3 and myeloperoxidase from patients with crescentic glomerulonephritis. J Am Soc Nephrol. 1999;10(7):1506–15. doi: 10.1681/ASN.V1071506. [DOI] [PubMed] [Google Scholar]
- 120.Kettritz R, Jennette JC, Falk RJ. Crosslinking of ANCA-antigens stimulates superoxide release by human neutrophils. J Am Soc Nephrol. 1997;8:386–94. doi: 10.1681/ASN.V83386. [DOI] [PubMed] [Google Scholar]
- 121.Weidner S, Neupert W, Goppelt-Struebe M, Rupprecht HD. Antineutrophil cytoplasmic antibodies induce human monocytes to produce oxygen radicals in vitro. Arthritis Rheum. 2001;44(7):1698–706. doi: 10.1002/1529-0131(200107)44:7<1698::AID-ART294>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 122.Hewins P, Williams JM, Wakelam MJ, Savage CO. Activation of Syk in neutrophils by antineutrophil cytoplasm antibodies occurs via Fcgamma receptors and CD18. J Am Soc Nephrol. 2004 Mar;15(3):796–808. doi: 10.1097/01.asn.0000113241.98702.77. [DOI] [PubMed] [Google Scholar]
- 123.van der Veen BS, Chen M, Muller R, van Timmeren MM, Petersen AH, Lee PA, et al. Effects of p38 mitogen-activated protein kinase inhibition on anti-neutrophil cytoplasmic autoantibody pathogenicity in vitro and in vivo. Ann Rheum Dis. 2010 Nov 9; doi: 10.1136/ard.2010.129106. [DOI] [PubMed] [Google Scholar]
- 124.Radford DJ, Lord JM, Savage CO. The activation of the neutrophil respiratory burst by anti-neutrophil cytoplasm autoantibody (ANCA) from patients with systemic vasculitis requires tyrosine kinases and protein kinase C activation. Clin Exp Immunol. 1999;118(1):171–9. doi: 10.1046/j.1365-2249.1999.01043.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mayet WJ, Schwarting A, Orth T, Duchmann R, Meyer zum Buschenfelde KH. Antibodies to proteinase 3 mediate expression of vascular cell adhesion molecule-1 (VCAM-1) Clin Exp Immunol. 1996;103(2):259–67. doi: 10.1046/j.1365-2249.1996.d01-626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.De Bandt M, Meyer O, Hakim J, Pasquier C. Antibodies to proteinase-3 mediate expression of intercellular adhesion molecule-1 (ICAM-1, CD 54) Br J Rheumatol. 1997;36(8):839–46. doi: 10.1093/rheumatology/36.8.839. [DOI] [PubMed] [Google Scholar]
- 127.Muller Kobold AC, van Wijk RT, Franssen CF, Molema G, Kallenberg CG, Tervaert JW. In vitro up-regulation of E-selectin and induction of interleukin-6 in endothelial cells by autoantibodies in Wegener’s granulomatosis and microscopic polyangiitis. Clin Exp Rheumatol. 1999;17(4):433–40. [PubMed] [Google Scholar]
- 128.Radford DJ, Savage CO, Nash GB. Treatment of rolling neutrophils with antineutrophil cytoplasmic antibodies causes conversion to firm integrin-mediated adhesion. Arthritis Rheum. 2000;43(6):1337–45. doi: 10.1002/1529-0131(200006)43:6<1337::AID-ANR16>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 129.Taekema-Roelvink ME, Kooten C, Kooij SV, Heemskerk E, Daha MR. Proteinase 3 enhances endothelial monocyte chemoattractant protein-1 production and induces increased adhesion of neutrophils to endothelial cells by upregulating intercellular cell adhesion molecule-1. J Am Soc Nephrol. 2001;12(5):932–40. doi: 10.1681/ASN.V125932. [DOI] [PubMed] [Google Scholar]
- 130.Casselman BL, Kilgore KS, Miller BF, Warren JS. Antibodies to neutrophil cytoplasmic antigens induce monocyte chemoattractant protein-1 secretion from human monocytes. J Lab Clin Med. 1995;126(5):495–502. [PubMed] [Google Scholar]
- 131.Berger SP, Seelen MAJ, Hiemstra PS, Gerritsma JSJ, Heemskerk E, van der Woude FJ, et al. Proteinase 3, the major autoantigen of Wegener’s granulomatosis, enhances IL-8 production by endothelial cells in vitro. J Am Soc Nephrol. 1996;7:694–701. doi: 10.1681/ASN.V75694. [DOI] [PubMed] [Google Scholar]
- 132.Brooks CJ, King WJ, Radford DJ, Adu D, McGrath M, Savage COS. IL-1b production by human polymorphonuclear leucocytes stimulated by anti-neutrophil cytoplasmic autoantibodies: relevance to systemic vasculitis. Clin Exp Immunol. 1996;106:273–9. doi: 10.1046/j.1365-2249.1996.d01-835.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ralston DR, Marsh CB, Lowe MP, Wewers MD. Antineutrophil cytoplasmic antibodies induce monocyte IL-8 release. Role of surface proteinase-3, alpha1-antitrypsin, and Fcgamma receptors. J Clin Invest. 1997;100(6):1416–24. doi: 10.1172/JCI119662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhou Z, Dionne A, Richard C, Menard HA. On the origin of surface proteinase 3 of nonmyeloid cells: evidence favoring an exogenous source. Clin Immunol. 2000 Nov;97(2):171–81. doi: 10.1006/clim.2000.4922. [DOI] [PubMed] [Google Scholar]
- 135.Yang JJ, Preston GA, Pendergraft WF, Segelmark M, Heeringa P, Hogan SL, et al. Internalization of proteinase 3 is concomitant with endothelial cell apoptosis and internalization of myeloperoxidase with generation of intracellular oxidants. Am J Pathol. 2001;158(2):581–92. doi: 10.1016/S0002-9440(10)64000-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Brons RH, de Jong MC, de Boer NK, Stegeman CA, Kallenberg CG, Cohen Tervaert JW. Detection of immune deposits in skin lesions of patients with Wegener’s granulomatosis. Ann Rheum Dis. 2001;60(12):1097–102. doi: 10.1136/ard.60.12.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Haas M, Eustace JA. Immune complex deposits in ANCA-associated crescentic glomerulonephritis: a study of 126 cases. Kidney Int. 2004 Jun;65(6):2145–52. doi: 10.1111/j.1523-1755.2004.00632.x. [DOI] [PubMed] [Google Scholar]
- 138.Harper L, Cockwell P, Adu D, Savage CO. Neutrophil priming and apoptosis in anti-neutrophil cytoplasmic autoantibody-associated vasculitis. Kidney Int. 2001;59(5):1729–38. doi: 10.1046/j.1523-1755.2001.0590051729.x. [DOI] [PubMed] [Google Scholar]
- 139.Harper L, Ren Y, Savill J, Adu D, Savage CO. Antineutrophil cytoplasmic antibodies induce reactive oxygen-dependent dysregulation of primed neutrophil apoptosis and clearance by macrophages. Am J Pathol. 2000;157(1):211–20. doi: 10.1016/S0002-9440(10)64532-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Xiao H, Heeringa P, Hu P, Liu Z, Zhao M, Aratani Y, et al. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest. 2002;110(7):955–63. doi: 10.1172/JCI15918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Little MA, Smyth CL, Yadav R, Ambrose L, Cook HT, Nourshargh S, et al. Antineutrophil cytoplasm antibodies directed against myeloperoxidase augment leukocyte-microvascular interactions in vivo. Blood. 2005 Sep 15;106(6):2050–8. doi: 10.1182/blood-2005-03-0921. [DOI] [PubMed] [Google Scholar]
- 142.Pfister H, Ollert M, Frohlich LF, Quintanilla-Martinez L, Colby TV, Specks U, et al. Antineutrophil cytoplasmic autoantibodies against the murine homolog of proteinase 3 (Wegener autoantigen) are pathogenic in vivo. Blood. 2004 Sep 1;104(5):1411–8. doi: 10.1182/blood-2004-01-0267. [DOI] [PubMed] [Google Scholar]
- 143.European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of multifocal motor neuropathy. Report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society. J Peripher Nerv Syst. 2006 Mar;11(1):1–8. doi: 10.1111/j.1085-9489.2006.00058.x. [DOI] [PubMed] [Google Scholar]
- 144.Haas M, Jafri J, Bartosh SM, Karp SL, Adler SG, Meehan SM. ANCA-associated crescentic glomerulonephritis with mesangial IgA deposits. Am J Kidney Dis. 2000;36(4):709–18. doi: 10.1053/ajkd.2000.17615. [DOI] [PubMed] [Google Scholar]
- 145.Mentzel HJ, Neumann T, Fitzek C, Sauner D, Reichenbach JR, Kaiser WA. MR Imaging in Wegener granulomatosis of the spinal cord. AJNR Am J Neuroradiol. 2003 Jan;24(1):18–21. [PMC free article] [PubMed] [Google Scholar]
- 146.Stassen PM, Derks RP, Kallenberg CG, Stegeman CA. Venous thromboembolism in ANCA-associated vasculitis--incidence and risk factors. Rheumatology (Oxford) 2008 Apr;47(4):530–4. doi: 10.1093/rheumatology/ken035. [DOI] [PubMed] [Google Scholar]
- 147.Markiewski MM, Nilsson B, Ekdahl KN, Mollnes TE, Lambris JD. Complement and coagulation: strangers or partners in crime? Trends in immunology. 2007 Apr;28(4):184–92. doi: 10.1016/j.it.2007.02.006. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Review] [DOI] [PubMed] [Google Scholar]
- 148.Xiao H, Schreiber A, Heeringa P, Falk RJ, Jennette JC. Alternative complement pathway in the pathogenesis of disease mediated by anti-neutrophil cytoplasmic autoantibodies. Am J Pathol. 2007 Jan;170(1):52–64. doi: 10.2353/ajpath.2007.060573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Huugen D, van Esch A, Xiao H, Peutz-Kootstra CJ, Buurman WA, Tervaert JW, et al. Inhibition of complement factor C5 protects against anti-myeloperoxidase antibody-mediated glomerulonephritis in mice. Kidney Int. 2007 Apr;71(7):646–54. doi: 10.1038/sj.ki.5002103. [DOI] [PubMed] [Google Scholar]
- 150.Schreiber A, Xiao H, Jennette JC, Schneider W, Luft FC, Kettritz R. C5a receptor mediates neutrophil activation and ANCA-induced glomerulonephritis. J Am Soc Nephrol. 2009 Feb;20(2):289–98. doi: 10.1681/ASN.2008050497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Xing GQ, Chen M, Liu G, Heeringa P, Zhang JJ, Zheng X, et al. Complement activation is involved in renal damage in human antineutrophil cytoplasmic autoantibody associated pauci-immune vasculitis. J Clin Immunol. 2009 May;29(3):282–91. doi: 10.1007/s10875-008-9268-2. [DOI] [PubMed] [Google Scholar]
- 152.Schlieben DJ, Korbet SM, Kimura RE, Schwartz MM, Lewis EJ. Pulmonary-renal syndrome in a newborn with placental transmission of ANCAs. Am J Kidney Dis. 2005 Apr;45(4):758–61. doi: 10.1053/j.ajkd.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 153.Silva F, Specks U, Sethi S, Irazabal MV, Fervenza FC. Successful Pregnancy and Delivery of a Healthy Newborn Despite Transplacental Transfer of Antimyeloperoxidase Antibodies From a Mother With Microscopic Polyangiitis. Am J Kidney Dis. 2009 Apr 22; doi: 10.1053/j.ajkd.2009.02.016. [DOI] [PubMed] [Google Scholar]
- 154.Sinico RA, Di Toma L, Maggiore U, Tosoni C, Bottero P, Sabadini E, et al. Renal involvement in Churg-Strauss syndrome. Am J Kidney Dis. 2006 May;47(5):770–9. doi: 10.1053/j.ajkd.2006.01.026. [DOI] [PubMed] [Google Scholar]
- 155.Finkielman JD, Lee AS, Hummel AM, Viss MA, Jacob GL, Homburger HA, et al. ANCA are detectable in nearly all patients with active severe Wegener’s granulomatosis. Am J Med. 2007 Jul;120(7):643, e9–14. doi: 10.1016/j.amjmed.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 156.Finkielman JD, Merkel PA, Schroeder D, Hoffman GS, Spiera R, St Clair EW, et al. Antiproteinase 3 Antineutrophil Cytoplasmic Antibodies and Disease Activity in Wegener Granulomatosis. Ann Intern Med. 2007 Nov 6;147(9):611–9. doi: 10.7326/0003-4819-147-9-200711060-00005. [DOI] [PubMed] [Google Scholar]