

Abstract
Recent pilot studies found natural chlorophyll (Chl) to inhibit carcinogen uptake and tumorigenesis in rodent and fish models, and to alter uptake and biodistribution of trace 14C-aflatoxin B1 in human volunteers. The present study extends these promising findings, using a dose-dose matrix design to examine Chl-mediated effects on dibenzo(def,p)chrysene (DBC)-induced DNA adduct formation, tumor incidence, tumor multiplicity, and changes in gene regulation in the trout. The dose-dose matrix design employed an initial 12,360 rainbow trout, which were treated with 0–4000 ppm dietary Chl along with 0 – 225 ppm DBC for up to 4 weeks. Dietary DBC was found to induce dose-responsive changes in gene expression that were abolished by Chl co-treatment, whereas Chl alone had no effect on the same genes. Chl co-treatment provided a dose-responsive reduction in total DBC-DNA adducts without altering relative adduct intensities along the chromatographic profile. In animals receiving DBC alone, liver tumor incidence (as logit) and tumor multiplicity were linear in DBC dose (as log) up to their maximum-effect dose, and declined thereafter. Chl co-treatment substantially inhibited incidence and multiplicity at DBC doses up to their maximum-effect dose. These results show that Chl concentrations encountered in Chl-rich green vegetables can provide substantial cancer chemoprotection, and suggest that they do so by reducing carcinogen bioavailability. However, at DBC doses above the optima, Chl co-treatments failed to inhibit tumor incidence and significantly enhanced multiplicity. This finding questions the human relevance of chemoprevention studies carried out at high carcinogen doses that are not proven to lie within a linear, or at least monotonic, endpoint dose-response range.
Keywords: chlorophyll; chlorophyllin; dibenzo(def, p)chrysene; tumor incidence; bioavailability; liver cancer; stomach cancer; chemoprevention
1 Introduction
The polycyclic aromatic hydrocarbon dibenzo(def,p)chrysene (DBC), formerly known as dibenzo(a,l)pyrene or DBP, can be readily detected on particulate matter from wood charcoal, coal and fuel-oil combustion (Deraat et al., 1987; Mumford et al., 1987; Mumford et al., 1995) in soil and sediment samples (Kozin et al., 1995), in vehicle exhaust condensate (Seidel et al., 2004) and in tobacco smoke condensate (Snook et al., 1977; Hoffmann and Hoffmann, 1998). DBC displays strong in vitro mutagenicity in bacterial and animal cell assays (Busby et al., 1995; Durant et al., 1999), forms multiple DNA adducts in vitro and in vivo (Mahadevan et al., 2005), and may act in vivo through promotional as well as tumor initiation mechanisms (Baird et al., 2005). Comparative studies in rodent models have demonstrated DBC to be the most carcinogenic polycyclic aromatic hydrocarbon tested to date, significantly exceeding the potencies of the well-known carcinogens benzo[a]pyrene and 7,12-dimethylbenz[a]anthracene (Cavalieri et al., 1989; Cavalieri et al., 1991; Higginbotham et al., 1993; LaVoie et al., 1993). Dermal application of DBC to mice produced tumors of the skin and lung, and malignant lymphoma involving multiple organs (Higginbotham et al., 1993). Intra-peritoneal administration of DBC to mice produced lung tumors (Prahalad et al., 1997), and intra-mammary injection in rats yielded mammary tumors (Cavalieri et al., 1991). In the rainbow trout, dietary exposure to DBC resulted in tumors of the stomach, liver and swim bladder (Reddy et al., 1999; Pratt et al., 2007; Simonich et al., 2008).
The exceptionally high carcinogenic potency of DBC (Bailey et al., 2009), along with unavoidable and potentially significant human exposure in urban areas, makes measures to mitigate its adverse effects particularly attractive. Experiments in animal models have identified many natural and synthetic chemopreventive agents that can dramatically inhibit carcinogen-induced damage and the resulting incidence of cancer (reviewed in (Barrett, 2002; Guyton and Kensler, 2002; Hirose et al., 2002; Kensler, 2004; Aggarwal and Shishodia, 2006; Kelloff et al., 2006; Wright et al., 2006)). One such agent, chlorophyllin (CHL), a sodium-copper derivative of chlorophyll (Chl), appears promising as a non-toxic agent that can protect against several classes of dietary carcinogen, including DBC, in animal models. In vivo mechanistic studies in mammals and fish indicate that dietary CHL acts to block tumor initiation (Reddy et al., 1999; Pratt et al., 2007; Simonich et al., 2008), and in vitro studies suggest this may occur in part by the formation of tight complexes with such carcinogens as aflatoxins, DBC, and some of the heterocyclic amines (Breinholt et al., 1995; Dashwood et al., 1996; Hayashi et al., 1999), which share partially planar, aromatic structural elements. Whether through complex formation or some other undefined transport mechanism, CHL co-treatment effectively inhibits absorption of these carcinogens, thereby reducing bioavailability to the target tissue, DNA-adduction, and ultimate tumor response (Dashwood et al., 1998). Translational experiments have demonstrated that CHL can significantly reduce biomarkers of effective carcinogen exposure in humans in China with chronic, unavoidably high aflatoxin B1 (AFB1) exposure and a high incidence of liver cancer (Qian et al., 1994; Jacobson et al., 1997; Egner et al., 2001; Egner et al., 2003). A recent micro-dosing study indicated that CHL co-exposure accomplishes this by significantly altering AFB1 pharmacokinetic parameters and reducing its bioavailability in humans (Jubert et al., 2009), as it does in animal models. Further support for the utility of CHL in human intervention is provided by a recent study by Shaughnessy (Shaughnessy et al.), who found that CHL tablets in combination with cruciferous vegetables and yogurt reduced colorectal DNA damage and systemic genotoxicity in humans consuming meat cooked at high temperature.
While these results suggest substantial potential for CHL supplements in the reduction of human cancer risk, relatively little information is available regarding the chemoprotective potential of natural chlorophylls as they may occur in the human diet. We recently reported that, in the rainbow trout carcinogenesis model, dietary co-exposure for one month to 1000–6000 ppm of highly purified Chl dramatically reduced the incidence of liver and stomach tumors initiated by DBC (Simonich et al., 2008). We also reported a similar degree of protection against early and late pre-cancerous biomarkers in the rat when 2000 ppm of purified Chl was co-administered with 250 ppb AFB1 (Simonich et al., 2007). Finally, our recent microdosing study showed that co-treatment with purified Chl was as effective as CHL at altering AFB1 pharmacokinetic parameters and reducing its uptake in human volunteers (Jubert et al., 2009). Although these results suggest that natural chlorophylls may have protective efficacy comparable to the derivative CHL, stability and cost issues currently limit use of highly purified chlorophylls as a long-term human intervention strategy. One purpose of the present study was to compare the chemopreventive efficacy of a more readily obtainable Chl-enriched spinach extract with those of purified Chl, and of commercial CHL. An additional and important aim of the study was to determine if our previous observation of carcinogen dose-dependency in cancer chemoprevention (Pratt et al., 2007) could be confirmed. The pertinent question is whether chemopreventive effects, or non-effects, observed in a given animal model at high carcinogen dose may extrapolate to lower carcinogen exposures and tumor responses pertinent to humans. To address this we employed a 12,000-animal dose-dose matrix design, which provides a quantitative examination of chemopreventive efficacy as a function of carcinogen as well as Chl dose. The endpoints and biomarkers assessed were initial DBC- DNA adduction, changes in gene expression profiles and final tumor responses for liver and stomach.
2 Materials and Methods
2.1.1 Chemicals
Dibenzo[def,p]chrysene (99.9% purity as determined by HPLC) was obtained from the NCI Chemical Carcinogen Reference Standard Repository at Midwest Research Institute (Kansas City, MO). DBC was used without further purification and concentration was confirmed by absorbance in ethanol at 316 nm (ε316 = 4.75 × 104 M−1). Proteinase K was from AMRESCO (Solon, OH). RNAse A and T1 cocktail was purchased from Ambion (Austin, TX). Alpha-amylase was from MP Biomedicals (Aurora, OH). CHL was purchased from Sigma (St. Louis, MO). The chlorin content was based on the manufacturer's assay of 4.5% copper and assertion that all copper was present as copper-chlorins. The dose of Sigma CHL was corrected to the actual chlorin content of 51.3%. Chlorophyll was prepared as described below.
2.1.2 Preparation of Chlorophyll
Chl and Chl-enriched spinach extracts were prepared, and Chl was quantified spectrophotometrically, as described (Jubert and Bailey, 2007). Briefly, organic spinach was purchased from a local supplier, washed with cold water, and freeze-dried after stem removal. The dried leaves were washed twice with petroleum ether (b.p. 30–60°C) and then extracted twice using methanol/petroleum ether (3:1, v/v). The combined extracts were transferred to a separatory funnel and washed with saturated sodium chloride. The aqueous layer was extracted again with petroleum ether and the extracts combined and washed with saturated sodium chloride. The final extract was filtered and evaporated in vacuo (T<30°C). This crude Chl extract (90% pure by HPLC) contained other pigments such as carotenoids as well as oils, fats, and waxes derived from the spinach. This was the main product used in the dose-dose matrix design. Purified Chl was obtained from the enriched extract by counter-current chromatography (CCC) using an Ito multilayer-coil separator (P.C., Potomac, MD), as described (Simonich et al., 2007). Analyses of CCC fractions were performed by HPLC and by 1H-NMR. Absence of any residual solvents from the extraction/purification was verified by spectral analysis. Purity was estimated to be > 95% compared to Chl-a standards (Sigma Chemical Co.) which were shown to be 90–92% pure based on spectroscopic measurements. The yield from 30 grams of freeze-dried spinach was 300 mg of Chl-a and 100 mg of Chl-b (Jubert and Bailey, 2007) which were recombined for use in the dietary intervention experiment.
2.1.3 Animals
Shasta strain rainbow trout were reared in the Sinnhuber Aquatic Research Laboratory of Oregon State University as published elsewhere (Sinnhuber et al., 1978) under animal treatment protocols approved by the Oregon State University Institutional Animal Care and Use Committee. Trout were maintained on a semi-synthetic Oregon Test Diet (OTD) formula (Lee et al., 1991). Fry (2.5 g) were fed control diet until experimental diet initiation. In total 12,360 trout were selected and distributed, at 120 fish per tank, to 103 tanks in random order. The fish were acclimatized to their tanks one week prior to commencement of test agent feeding.
2.2.1 Study Design
Table I shows the overview of the dose-dose matrix design of the study. The chemopreventive test agents (Chl extract at 500, 1000, 2000 and 4000 ppm Chl; CCC-purified Chl or CHL at 2000 ppm) were fed to the trout 3 days prior to commencing carcinogen treatment exposure. The co-exposure of chemopreventive test agent plus carcinogen was carried out for an additional 4 weeks followed by 3 days post-exposure feeding of the chemopreventive test agent alone. At each dose of Chl extract (hereafter referred to simply as Chl), multiple doses of DBC were tested. For example, treatment groups 13–16 were fed 1000 ppm of Chl for 3 days, then co-fed 1000 ppm of Chl with increasing doses of DBC (28, 56, 112 or 224 ppm) for 4 weeks followed by 3 days of 1000 ppm of Chl alone. Higher doses of Chl required higher dose ranges of DBC to compensate for the anticipated substantial inhibition of tumor response (Pratt et al., 2007). Groups 1–2 were negative controls with expected outcomes of spontaneous tumors <1%. Groups 3–7 were positive controls for tumor response from the DBC alone. Groups 8–24 were the main dose-dose matrix component of the study. Groups 25 and 26 were positive controls for tumor inhibition from CHL (Pratt et al., 2007) and CCC-purified Chl (Simonich et al., 2008).
Table I.
Chlorophyll DBC Dose-Dose Matrix Experimental Design
| Group | tanks | 3 days pre-fed | 4 weeks co-fed | 3 days post-fed |
|---|---|---|---|---|
| 1 | 2 | OTD | OTD | OTD |
| 2 | 2 | Chl 4000 ppm | Chl 4000 ppm | Chl 4000 ppm |
| 3 | 3 | OTD | DBC 14 ppm | OTD |
| 4 | 6 | OTD | DBC 28 ppm | OTD |
| 5 | 3 | OTD | DBC 56 ppm | OTD |
| 6 | 6 | OTD | DBC 112 ppm | OTD |
| 7 | 3 | OTD | DBC 224 ppm | OTD |
| 8 | 6 | Chi 500 ppm | Chi 500 ppm & DBC 14 ppm | Chi 500 ppm |
| 9 | 6 | Chi 500 ppm | Chi 500 ppm & DBC 28 ppm | Chi 500 ppm |
| 10 | 3 | Chi 500 ppm | Chi 500 ppm & DBC 56 ppm | rChl 500 ppm |
| 11 | 3 | Chi 500 ppm | Chi 500 ppm & DBC 112 ppm | Chi 500 ppm |
| 12 | 3 | Chi 500 ppm | Chi 500 ppm & DBC 224 ppm | Chi 500 ppm |
| 13 | 6 | Chl 1000 ppm | Chi 1000 ppm & DBC 28 ppm | Chl 1000 ppm |
| 14 | 3 | Chl 1000 ppm | Chi 1000 ppm & DBC 56 ppm | Chl 1000 ppm |
| 15 | 3 | Chl 1000 ppm | Chi 1000 ppm & DBC 112 ppm | Chl 1000 ppm |
| 16 | 3 | Chl 1000 ppm | Chi 1000 ppm & DBC 224 ppm | Chl 1000 ppm |
| 17 | 6 | Chl 2000 ppm | Chl 2000 ppm & DBC 28 ppm | Chl 2000 ppm |
| 18 | 3 | Chl 2000 ppm | Chl 2000 ppm & DBC 56 ppm | Chl 2000 ppm |
| 19 | 3 | Chl 2000 ppm | Chl 2000 ppm & DBC 112 ppm | Chl 2000 ppm |
| 20 | 3 | Chl 2000 ppm | Chl 2000 ppm & DBC 224 ppm | Chl 2000 ppm |
| 21 | 6 | Chl 4000 ppm | Chl 4000 ppm & DBC 56 ppm | Chl 4000 ppm |
| 22 | 3 | Chl 4000 ppm | Chl 4000 ppm & DBC 112 ppm | Chl 4000 ppm |
| 23 | 3 | Chl 4000 ppm | Chl 4000 ppm & DBC 224 ppm | Chl 4000 ppm |
| 24 | 3 | Chl 4000 ppm | Chl 4000 ppm & DBC 448 ppm | Chl 4000 ppm |
| 25 | 6 | CHL 2000 ppm | CHL 2000 ppm & DBC 112 ppm | CHL 2000 ppm |
| 26 | 3 | CCC Chl 2000 ppm | CCC Chl 2000 ppm & DBC 112 ppm | CCC Chl 2000 ppm |
After 2 and 4 weeks of DBC exposure, 5 fish were removed from each tank and sacrificed by buffered-MS-222 overdose to obtain tissues for DBC-DNA adduct analysis as possible biomarkers. Livers from an additional 5 fish were taken at week 4 for gene expression analysis. The remaining trout in each tank were fed OTD for the next nine months until the fish were overdosed and liver and stomach tumors analyzed by histopathology (see Tumor Histology).
2.2.2 Diet Preparation
Lipophilic test agents (DBC, Chl) were added to the oil component of the OTD diet formulation, whereas the hydrophilic CHL was added to the aqueous portion. Final dietary concentrations of DBC, Chl, and CHL (expressed in ppm) are mg of agent/kg of dry weight portion of the diet. Quantifications for DBC, Chl, and CHL were performed as described (Pratt et al., 2007; Simonich et al., 2007; Simonich et al., 2008). During treatment and prior to sampling, diet was fed at a rate of 2% body weight per day. Dose ranges for DBC and Chl concentrations were chosen based on previous studies (Reddy et al., 1999; Pratt et al., 2007; Simonich et al., 2008). (Note: DBC is a potent carcinogen; it was handled, stored, and disposed in compliance with NIH and Oregon State University guidelines for extreme hazard class of carcinogens). DBC, Chl and CHL are light-sensitive compounds; therefore, all diets were prepared under subdued lighting or under lighting with a 400 nm cut-off. Diets were prepared every two weeks and stored in the dark at −20°C until one day prior to feeding when they were thawed at 4°. DBC in OTD trout diet was previously determined to be stable when stored at −20°C for up to 3 years, in the presence and absence of CHL (Loveland et al., 2001).
2.2.3 Isolation of hepatic DNA and determination of DBC-DNA adduction
Fourteen days after commencement of carcinogen feeding, 5 fish were removed from each tank and euthanized. Livers were removed and pooled by tank to provide a minimum of N=3 tank samples for each Chl-DBC treatment. The samples were quick-frozen in liquid nitrogen and stored at −80°C. DNA was purified from selected livers as described (Reddy et al., 1999). The DBC adducts in the liver DNA were analyzed by 33P-postlabeling followed by HPLC as described elsewhere (Ralston et al., 1995; Pratt et al., 2007). An exhaustive determination of DBC-DNA adducts among both target organs and all treatment groups was carried out in our previous DBC-CHL dose-dose matrix study (Pratt et al., 2007), and was not repeated here.
2.2.4 RNA isolation and quality determination
Trout were sub-sampled at week 4 from the various treatment groups, and total hepatic RNA was extracted in TRIzol reagent according to manufacturer's instructions. Equal amounts of RNA from each individual liver sample were pooled by tank (five fish per tank) resulting in three biological replicates for each treatment. A reference RNA pool was made by combining equal amounts of RNA from control (OTD) liver samples. Following cleanup with the RNeasy Mini kit (Qiagen, Valencia, CA), RNA quantity and quality were estimated using the Bioanalyzer 2100 (Agilent, Palo Alto, CA).
2.2.5 Rainbow trout oligonucleotide microarray
Details on the development, manufacture and quality control assessment of the rainbow trout oligonucleotide microarray (OSUrbt array) have been provided previously (Tilton et al., 2005) and will be summarized here. The OSUrbt ver. 5 array contains 1661 elements representing approximately 1450 genes important for carcinogenesis, environmental toxicology, comparative immunology, stress physiology and endocrinology. Array printing and quality control analysis were conducted at the Center for Genome Research and Biocomputing at Oregon State University. Each element was printed in duplicate onto Corning UltraGap slides (Acton, MA). Additionally, sixteen replicate sets (one for each array block) of 10 SpotReport Alien Oligos (Stratagene, La Jolla, CA) were also printed on each array. Buffer-only spots were included as negative controls. Altogether, each array consists of 4096 spots. Array print quality was assessed by scanning for red reflectance using a ScanArray 4000 scanner (PerkinElmer, Boston, MA), and one array per printing batch was stained with Syto 61 (Molecular Probes, Eugene, OR) and scanned at 633 nm. Arrays were stored for ≤ 6 months prior to hybridization. Array hybridization, scanning, gene annotation and expression change confirmation by quantitative PCR were performed as previously described in (Benninghoff and Williams, 2008). Mean treatment-related fold changes represent background corrected, Lowess normalized signal ratios. Only genes that were regulated at least two-fold consistently in all features from biological replicates, and had a p value <0.05 by Welches t-test, are reported.
2.2.6 Determination of Tumor Response
Two tanks of trout treated with 112 ppm DBC were pre-sacrificed to evaluate the extent of tumor formation and to optimize the timing of the final tumor sampling. From this it was estimated that adequate tumor development would occur by 10 months after commencing DBC treatment. Based on previous experience, the final termination and necropsy of the remaining 100 tanks of approximately 100 animals each was estimated to require 6–7 weeks for completion. To avoid tumor-development time as a confounding variable, a sampling schedule was devised so that the mean sampling day for all treatment groups would be day 24 (± 2) after commencement of sampling at the end of month 10. This strategy proved effective, as there was no evidence that treatment effects differed among tank lots terminated before and after 24 days (P>0.5 for treatment by period interactions). Sampling was conducted in three blocks of time with each block containing a third of the tanks for each treatment. Within the blocks, the order was randomized with the restriction that overall mean sampling day was similar for all treatment groups (as described above. The daily ration of food during sampling was reduced to maintenance rations (1% body weight) in order to minimize the effect of sampling time on final tumor incidence (Pratt et al., 2007).
2.2.7 Tumor Histology
Tissues were examined under a dissecting scope for gross tumors (≥0.5 mm diameter), fixed in Bouin's solution and processed by routine histological procedures. Numerous studies over the past 40 years have shown that 100% of stomach and approximately 95% of liver tumors are surface-oriented outgrowths that are easily detected at gross necropsy. From each organ having one or more suspect tumors at necropsy, one slide was prepared for histology. Liver and stomach neoplasms were classified according to criteria established by Hendricks et al. (Hendricks et al., 1984; Hendricks et al., 1995). Percentages for each different histological type of liver neoplasms were calculated from the total number of each type divided by the total number of all hepatic neoplasms in the group. Tumor incidence is expressed as the percentage of fish with one or more confirmed tumors at each dose. Apparent tumor multiplicity was calculated by dividing the total number of tumors observed grossly by the total number of tumor-bearing fish. This endpoint is termed apparent tumor multiplicity because not every lesion in organs exhibiting multiple lesions at gross necropsy was examined.
2.2.8 Statistical methods
For all analyses the unit of analysis was the tank within a treatment group. Logistic regression for grouped binomial data with quasilikelihood to account for any overdispersion between replicate tanks (dscale option in the Genmod procedure of SAS 9.2) was used to model binary responses (mortality, tumor phenotype and tumor incidence for both liver and stomach) as functions of carcinogen and test agent doses. When trends in sampling date explained a significant part of the residual variation in the responses (mortality and liver tumor incidence), all statistical inferences (hypothesis tests, standard errors, etc.) were made after adjusting for sampling date (quasilikelihood approximate F based). Because the experimental design was set up with near-orthogonal blocking on sampling order, the addition of the date covariate reduced the magnitude of the overdispersion, but had negligible effect on the point estimates of model parameters (not shown). Therefore, for simplicity of presentation, the observed pooled tumor incidences and fitted curves for liver tumor incidence (Figure 1) were without adjustment for date of sampling. Linear models were used to model body weight and liver somatic index as functions of DBC and CHL doses with sampling date included as a covariate. Apparent multiplicity was compared between treatment groups with nonparametric rank tests (Kruskal-Wallis and Wilcoxon tests) when problems were found with residuals (e.g., heavy tails) for all parametric analyses (including conditionally distributions such as negative binomial and other overdispersed Poisson-type models).
Figure 1.
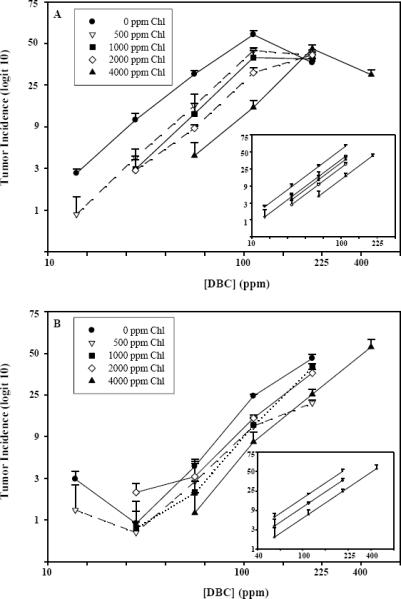
DBC-initiated liver and stomach tumor response as a function of Chl dose. Rainbow trout were treated by dietary exposure for 4 weeks. Diets contained 14, 28, 56, 112, 224 or 448 ppm DBC and 0, 500, 1000, 2000 or 4000 ppm Chl. Trout were examined for tumor formation 9 months after DBC exposure ceased. Panel A shows the un-modeled liver tumor data as observed means (± SE), and the inset shows the modeled liver tumor dose-response curves without the highest DBC dose for each Chl curve. Panel B shows the un-modeled stomach tumor data as observed means (± SE), and the inset shows modeled stomach tumor dose-response curves for 0, 2000 and 4000 ppm Chl without data from the two lowest DBC doses. SE calculated for each data point prior to transformation to logit-scale. Lower error bars have been omitted due to distortion at lower end of logit scale and crowding of data points at higher incidence levels.
2.2.9 Animal growth, mortality, and sampling date effects
Table II gives the carcinogen dose, modulator dose, number of replicate tanks, and distribution by treatment of the 9,470 trout sampled for tumors and reports mean body weight, liver somatic index, and tumor incidence for each treatment. There was no difference in mortality rates between DBC or Chl doses and negative controls (p>0.2). Liver somatic indices showed a small decrease over time (p<0.01) but were not affected by DBC or Chl doses (p>0.1) except for the 2000 ppm dose of Chl which showed a small, unexplained decrease (p = 0.04). As noted above, a long necropsy and sampling time is unavoidable in a tumor study of this magnitude, which makes it essential that the sampling schedule be designed to account for potentially increasing odds of tumor with time. The sampling schedule avoided confounding of treatment effects and day of sampling by using near-orthogonal randomized blocks based on sampling order, so that the average sample date for the replicate tanks was similar for every treatment. For liver tumors (but not stomach tumors) there was evidence of linearly increasing log-odds of tumor with day of sampling (p<0.0001 quasilikelihood F-test with treatments in the logistic regression model). The previously noted increase in body weight with sampling day does not by itself explain the time effect because after adjusting for linear in body weight effect there was still evidence of a linear in sampling day effect (p=0.0048) on the log-odds of liver tumor. The block design was effective because, as noted above, inclusion of linear in sampling day as a covariate in the logistic improved the precision (reduced overdispersion), but had very little effect on parameter estimates (due to near-orthogonal blocks).
Table II.
Tumor Incidence, liver somatic index, and tumor type among animals fed varying doses of DBC and Chlorophyll
| [DBC] (ppm) | [Chla] (ppm) | N | Tumor Incidencea |
Mean Body Weight (g)a | Liver Somatic Indexa,b | # Livers with tumors Examined | % of Liver Tumors Examinedc |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Liver | Stomach | HCC | HCA | MC | MA | CCC | CCA | ||||||
| 0 | 0 | 186 | _ | _ | 122.5 (8.1) | 0.73 (0.01) | _ | _ | _ | _ | _ | _ | _ |
| 0 | 4000 | 194 | _ | 1.6 (0.6) | 117.1 (8.7) | 0.70 (0.02) | _ | _ | _ | _ | _ | _ | _ |
| 14 | 0 | 297 | 2.7 (0.3) | 3.0 (0.6) | 116.5 (3.3) | 0.71 (0.01) | 8 | 62.5 | 25.0 | 0 | 12.5 | 0 | 0 |
| 28 | 0 | 575 | 10.8 (1.8) | 0.9 (0.3) | 117.9 (2.9) | 0.74 (0.02) | 62 | 69.4 | 16.1 | 11.3 | 1.6 | 0 | 1.6 |
| 56 | 0 | 284 | 29.9 (1.8) | 4.2 (1.1) | 118.9 (2.6) | 0.72 (0.04) | 85 | 45.9 | 37.6 | 10.6 | 3.5 | 0 | 2.4 |
| 112 | 0 | 574 | 56.2 (2.7) | 23.5 (1.0) | 115.0 (3.4) | 0.72 (0.01) | 322 | 57.5 | 23.6 | 17.7 | 0.9 | 0.3 | 0 |
| 224 | 0 | 289 | 37.1 (2.6) | 46.5 (3.1) | 114.4 (2.3) | 0.73 (0.02) | 107 | 58.9 | 34.6 | 4.7 | 1.9 | 0 | 0 |
| 14 | 500 | 568 | 0.9 (0.4) | 1.3 (0.8) | 118.8 (3.6) | 0.72 (0.01) | 5 | 20.0 | 80.0 | 0 | 0 | 0 | 0 |
| 28 | 500 | 560 | 4.1 (0.9) | 0.7 (0.2) | 115.0 (1.7) | 0.72 (0.02) | 23 | 87.0 | 8.7 | 4.3 | 0 | 0 | 0 |
| 56 | 500 | 282 | 15.2 (4.0) | 2.8 (1.3) | 117.2 (1.9) | 0.71 (0.01) | 43 | 65.1 | 18.6 | 16.3 | 0 | 0 | 0 |
| 112 | 500 | 286 | 45.2 (1.9) | 11.8 (2.2) | 115.9 (3.4) | 0.71 (0.01) | 129 | 42.6 | 39.5 | 16.3 | 0.8 | 0 | 0.8 |
| 224 | 500 | 284 | 42.1 (2.2) | 20.2 (1.6) | 114.3 (5.1) | 0.74 (0.02) | 119 | 57.1 | 32.8 | 9.2 | 0 | 0.8 | 0 |
| 28 | 1000 | 566 | 3.0 (1.0) | 0.8 (0.5) | 117.9 (2.7) | 0.72 (0.02) | 17 | 41.2 | 41.2 | 11.8 | 5.9 | 0 | 0 |
| 56 | 1000 | 296 | 12.8 (3.2) | 2.1 (0.6) | 115.1 (5.8) | 0.72 (0.03) | 37 | 54.1 | 29.7 | 13.5 | 0 | 2.7 | 0 |
| 112 | 1000 | 250* | 39.1 (4.5) | 12.2 (2.2) | 116.8 (5.5) | 0.67 (0.05) | 101 | 45.5 | 38.6 | 12.9 | 1.0 | 2.0 | 0 |
| 224 | 1000 | 274 | 39.1 (3.1) | 40.8 (2.1) | 119.0 (3.0) | 0.71 (0.04) | 108 | 44.4 | 34.3 | 18.5 | 2.8 | 0 | 0 |
| 28 | 2000 | 584 | 2.9 (0.8) | 2.1 (0.5) | 116.9 (1.9) | 0.71 (0.01) | 17 | 11.8 | 58.8 | 29.4 | 0 | 0 | 0 |
| 56 | 2000 | 217* | 9.0 (0.7) | 3.2 (1.2) | 118.1 (1.8) | 0.64 (0.02) | 19 | 47.4 | 36.8 | 10.5 | 0 | 0 | 5.3 |
| 112 | 2000 | 279 | 30.8 (3.1) | 14.3 (1.0) | 120.2 (1.2) | 0.69 (0.01) | 86 | 57.0 | 33.7 | 8.1 | 1.2 | 0 | 0 |
| 224 | 2000 | 292 | 42.1 (3.4) | 36.7 (2.3) | 117.5 (3.9) | 0.69 (0.01) | 122* | 56.6 | 28.5 | 13.1 | 0.8 | 0.8 | 0 |
| 56 | 4000 | 580 | 4.2 (1.4) | 1.2 (0.6) | 114.8 (1.8) | 0.72 (0.01) | 25 | 32.0 | 40.0 | 12.0 | 16.0 | 0 | 0 |
| 112 | 4000 | 295 | 14.5 (2.3) | 8.1 (2.0) | 114.7 (2.6) | 0.70 (0.03) | 44 | 40.9 | 38.6 | 15.9 | 2.3 | 0 | 2.3 |
| 224 | 4000 | 287 | 46.3 (3.5) | 24.4 (2.4) | 115.8 (2.6) | 0.70 (0.02) | 133 | 51.9 | 30.8 | 15.8 | 1.5 | 0 | 0 |
| 448 | 4000 | 300 | 29.6 (3.0) | 54.3 (4.2) | 111.3 (1.1) | 0.71 (0.01) | 89 | 50.6 | 38.2 | 11.2 | 0 | 0 | 0 |
| 112 | CCC | 303 | 14.2 (2.5) | 6.6 (1.2) | 113.6 (3.2) | 0.69 (0.01) | 43 | 53.5 | 18.6 | 18.6 | 9.3 | 0 | 0 |
| 112 | CHL | 568 | 12.3 (2.3) | 3.4 (0.7) | 113.8 (2.6) | 0.70 (0.01) | 69 | 55.1 | 33.3 | 10.1 | 0 | 0 | 1.4 |
Mean (SE);
Somatic index is defined as mean liver weight (g)/ mean body weight (g).
HCC, hepatocellular carcinoma; HCA, hepatocellular adenoma; MC, mixed hepatocellular/ cholangiocellular carcinoma; MA, mixed hepatocellular/cholangiocellular carcinoma; CCC, cholangiocellular carcinoma; CCA, cholangiocellular adenoma.
'_' indicates that no tumors were observed.
Loss of trout due to error in husbandry.
122 of the 123 livers with tumors were classified as to tumor type.
3 Results
3.1 DBC-initiated tumor formation
Dietary administration of DBC to juvenile rainbow trout for 4 weeks resulted in dose-dependent formation of liver and stomach tumors when sampled 9 months after cessation of dietary DBC treatment (Table II). Tumor responses in liver and stomach for this study agree closely with those in our 40,800-animal mega dose-response ED01 study with DBC (Reddy et al., 1999; Bailey et al., 2009)(Reddy et al., 1999) and demonstrate distinct differences in DBC dose-response in the two major target organs. As seen in Figure 1A, incidence in liver increased in direct proportion to DBC dose on the logit scale, in the 14–112 ppm range, whereas response at the next higher dose of 224 ppm was lower than that at 112 ppm (p<0.0001, quasilikelihood f-test with scale parameter estimated from entire data set). Thus the dose-response to DBC in this model and target organ is non-monotonic and exhibits a maximum-effect dose . This is in distinct contrast to AFB1, which produces a response with incidence in direct proportion to dose over a broad exposure range in the trout model (Dashwood et al., 1998). In stomach, responses to DBC were not significantly above background at the two lowest doses, as anticipated from our previous study (Bailey et al., 2009). This precludes examination of chemoprevention effects in this organ at those two doses. We do not consider this apparent lag in detectable response as evidence for a threshold in this organ, rather it is an example of having insufficient animal numbers to distinguish carcinogen-driven from background response at these ultra-low doses (Bailey et al., 2009). We also note that DBC toxicity to juvenile trout (Bailey et al., 2009) precludes testing at doses much above 224 ppm, unless an effective chemoprotective agent such as Chl is also given (e.g. as in treatment group 24, Table 1). Stomach tumor incidence did appear to increase in direct proportion to dose on the logit scale at doses in the 56–224 ppm range, providing a useful range for examining chemopreventive effects of chlorophylls in this organ.
3.2 Inhibition of tumor response by dietary chlorophylls
The potential for dietary co-treatment with Chl extract to inhibit tumor response to DBC in liver is shown in Figure 1A and Table II. The extent of inhibition appeared to increase with Chl dose, and was observable at all DBC doses up to and including the maximum-effect dose at 112 ppm DBC. At 224 ppm DBC, however, which lies above the maximum-effect dose, there was no evidence for tumor inhibition at any Chl dose (Figure 1A). In stomach (Figure 1B), there was clear evidence for inhibition of tumor incidence at each of the four Chl doses tested, for the above-background DBC doses of 56, 112, and 224 ppm .Visual inspection of the liver and stomach data plotted on the logit incidence versus log [DBC] scale (Figures 1A and 1B respectively) give a general impression of the DBC dose-tumor response curves shifting further right with increasing doses of Chl. That is, each Chl dose shifted the carcinogen-only dose response curve in both organs such that higher DBC doses would be necessary to achieve any particular tumor response. As noted, stomach tumor incidence at the lowest DBC doses, 14 & 28 ppm, did not differ significantly from background (p = 0.06 and p = 0.3, respectively). At higher DBC doses, Chl shifted the DBC-only dose-response curve to the right. We note, however that the maximal degree of shift at 4000 ppm Chl was only about half that observed in liver. As a consequence the extents of shift among the intermediate Chl doses of 500–2000 ppm Chl in stomach are less readily distinguished than in liver. Overall, however, visual inspection of the DBC dose-response curves at 0, intermediate, and 4000 ppm Chl supports a progressive shift to the right (inhibition of tumor response) with increasing Chl co-treatment (Figure 1B inset). Unlike liver, the DBC-only dose-response curve in stomach does not pass through an optimum within the DBC dose range tested here, consequently there is no evidence for absence of Chl protection at highest DBC dose.
There is also no evidence for an effect of DBC or Chl dose on tumor phenotype. The liver tumors initiated by DBC were primarily hepatocellular carcinomas (HCC) and adenomas (HCA), with relatively few mixed hepatocellular/cholangiocellular carcinomas (MC) or adenomas (MA) (Table II) consistent with previous findings (Reddy et al., 1999; Pratt et al., 2007). There was no DBC dose-dependent change in relative percentages of HCC, HCA, or MC, the three most abundant trout liver phenotypes (P≥ 0.1, all three phenotypes). The addition of Chl at any dose did not alter the phenotypic response (P≥ 0.1 for HCC, HCA and MC) compared to the response from DBC alone. All stomach tumors were of a single phenotype, papillary adenoma, irrespective of DBC or Chl dose.
3.3 Modeling of tumor responses
The study was designed in part to determine if the magnitude of Chl protection remained constant over the DBC dose range. This information is vital if we are to consider extrapolating the degree of Chl protection at high-carcinogen doses and incidences, such as would be typical in rodent chemoprevention study designs, down to lower doses and incidences more representative of human cancer risks. Utilization of the dose-dose matrix design allows the magnitude of cancer chemoprevention protection for any of several anti-carcinogen doses to be assessed by modeling the effect of each dose on the shape and position of the entire dose-response curve (Pratt et al., 2007). As we have previously shown, the extent of protection at any dose of chemopreventive agent will be independent of carcinogen dose under those conditions, if any, where the logit [incidence] versus log [carcinogen] dose-response curves with and without agent have the same shape but are offset horizontally (Dashwood et al., 1989). These data sets can also be used to generate quantitative estimates of Chl potency to inhibit tumor formation at any or all the DBC doses. For this purpose, the dose of carcinogen required to produce ρ% tumor incidence is defined as the TDρ value. For instance, at ρ= 25%, the response curve for each dose of Chl will provide a TD25 estimate which can be used to calculate the magnitude of tumor inhibition for that dose of Chl using the following: % inhibition = 100 (1-(TD25Chlo/TDChlx)) where Chlo refers to the treatment with no Chl and Chlx refers to the treatment at any dose X (Dashwood et al., 1989). When the logit response-log dose curves are linear and parallel, the percent inhibition calculated from any tumor incidence within the linear range will be equivalent, that is, degree of inhibition is independent of carcinogen dose. This would be the circumstance where there is greater justification for extrapolation of inhibition data from high carcinogen doses and tumor responses down to exposures and cancer rates more relevant to humans.
The liver tumor response to various DBC doses was sufficient (Table II) to provide a full set of five un-modeled dose-response curves (Figure 1A). Using the data up to and including the maximum-effect dose, the curves were successfully modeled by logistic regression as a series of linear and parallel dose-response curves (overall lack-of-fit (11df) p>0.45), with each increase in Chl providing successive offsets toward higher TD values (Figure 1A, inset). This finding supports the conclusion that the magnitude of liver tumor inhibition increased with Chl dose and was independent of DBC dose at each of the four Chl doses studied, and over all DBC doses up to the optimum. By contrast, inclusion of the data points beyond the optimum (above 112 ppm DBC for [Chl] of 2000 ppm and lower; and above 224 ppm DBC at 4000 ppm) results in rejection of the parallel offset model (Figure 1A). This condition precludes extrapolation of the data at highest DBC dose to predict anything about possible Chl chemopreventive effects at lower carcinogen exposures and cancer incidences of potential human relevance.
Since DBC is a multi-organ carcinogen in the trout, we were able to compare Chl chemopreventive efficacy in stomach as well as liver. There are, however, some limitations due to reduced DBC and Chl sensitivity in stomach (Bailey et al., 2009). As seen in Table II, the stomach tumor response to various DBC doses also gave five dose response curves. If the entire stomach tumor data set among all DBC doses is examined, it does not fit the regression model with linear and parallel lines (p<0.013). However, the DBC dose range was already known to be indistinguishable from background in the second organ for the two lowest DBC doses (Bailey et al., 2009), so that these DBC data points would not be of use in modeling of parallel-offset responses. An additional problem occurs because the potency for Chl inhibition is less in stomach than in liver. This means that the design, which was optimized for liver, has less sensitivity for demonstrating incremental inhibition with increasing Chl dose in this organ. In particular, there is extensive overlap in the observed tumor incidences among the 500–2000 ppm Chl curves (Figure 1B). This occurs as a natural consequence of the diminished inhibition by Chl in stomach compared with liver, and the statistical limitations of detecting subtle modulator differences despite using over 9000 animals to assess tumor response. These limitations were accommodated by modeling a simplified data set matrix of above-background DBC doses (56, 112, 224 ppm) arrayed against 0, 2000, and 4000 ppm Chl. For this subset of data, the dose-response curves successfully fit a regression model with linear and parallel lines reasonably well, lack of fit, p=0.66 (Figure 1B, inset).
After the data were successfully fit to parallel-offset models, % inhibition could be determined as indicated above. For liver, this calculation yields dose-response potencies for Chl % inhibition (± S.E.) of 29 (± 7), 36 (± 7), 46 (±8), and 63 (±7) %, at Chl doses of 500, 1000, 2000 and 4000 ppm, respectively (see Figure 2A). For stomach, the Chl % inhibition is 24 (± 9) % for 2000 ppm Chl and 45 (± 7)% for 4000 ppm Chl (Figure 2B). Therefore DBC shows less potency for tumor initiation in stomach than in liver, and dietary Chl is less effective as a blocking agent against DBC in this organ. Taken together, these results support the idea that Chl concentrations commonly encountered in Chl-rich green vegetables such as spinach may provide substantial cancer chemoprotection against such multi-organ genotoxins as DBC. They also show clearly that a dose-dose matrix design optimized for one organ (liver) may not give optimal results for another organ.
Figure 2.
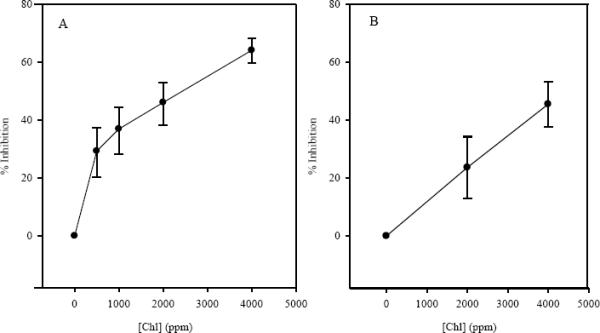
Percent inhibition of final tumor incidence in liver (panel A) and stomach (panel B) at varying concentrations of dietary Chl. Percent inhibition was calculated for each Chl dose based on its alteration in the DBC TD25 value (see text). Liver tumor inhibition values were determined as described in the results section. Diets contained 14, 28, 56, 112, 224 or 448 ppm DBC and 500, 1000, 2000, or 4000 ppm Chl. Error bars represent 95% confidence intervals.
3.4 Chl effects on apparent liver tumor multiplicity
Due to the magnitude of the study, we were unable to conduct exhaustive pathology on every lesion within livers bearing multiple grossly observable lesions. However, it was feasible to examine the gross lesion data for possible effects of DBC and Chl dose on “apparent tumor multiplicity”. Similar to the tumor incidence data, the apparent liver tumor multiplicity exhibited a maximal response which occurred near 56 ppm DBC for this endpoint (Figure 3). Increasing co-treatment doses of Chl shifted the dose response curve further to the right toward higher DBC doses. However, the dose-response for tumor multiplicity is not a simple linear or monotonic function of DBC dose, and as a consequence the effects of Chl intervention are profoundly dependent on DBC dose. At the three lowest DBC doses tested (14, 28, 56 ppm), Chl concentrations of 500–4000 ppm reduced the observed apparent tumor multiplicity compared to DBC alone, significantly so at 56 ppm DBC (p<0.02). The effects of Chl additions at DBC doses above its optimum at 56 ppm, however, produced distinctly different results. For example, addition of 500, 1000, or 2000 ppm Chl failed to reduce apparent multiplicity, but instead significantly enhanced it compared with 112 ppm DBC alone (p == 0.059, 0.014, 0.008, resp.; pairwise exact Wilcoxin test). This finding recapitulates and reinforces our previous observations with CHL co-treatment, which demonstrated the same pattern as Chl of reducing apparent liver tumor multiplicity at low DBC doses, but enhancing it at the higher DBC doses (Pratt et al., 2007).
Figure 3.
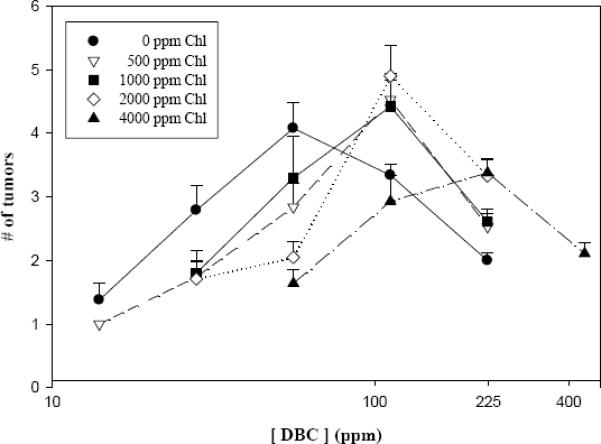
Chl effects on apparent tumor multiplicity in liver. Trout were examined for tumor formation 9 months after DBC exposure ceased. Apparent tumor multiplicity is used to denote the inclusion of some gross as well as histologically confirmed lesions among individuals with more than one lesion. Error bars represent standard deviation.
3.5 Relative chemopreventive efficacies of CHL and Chl preparations
All chlorophyll species when tested at 2000 ppm significantly reduced liver tumor incidence (Figure 4). The well-established positive control for chemoprevention, 2000 ppm CHL, showed a substantial and significant reduction from 57% incidence for DBC alone to 12 %(p<0.0001). Chl purified by CCC gave a similar response, 57% to 14% (p<0.05). The Chl extract used in the dose-dose matrix part of the study reduced liver tumor incidence from 57% to 31% (p<0.005). The Chl extract, the CCC purified Chl, and CHL all showed significant reductions in DBC-initiated stomach tumor incidence as well (p=0.002; p<0.0001, p<0.0001, respectively). Based on these results the Chl extract containing 2000 ppm Chl appears to be less effective than 2000 ppm purified Chl (p<0.0001) or 2000 ppm CHL (p<0.0001) in reducing tumor incidence. The basis for the reduced Chl efficacy in the extract is presently not known, but may reflect some degree of Chl sequestration by other components in the extract. In any case, additional protection is readily achieved simply by increasing the dosage of the Chl extract in the diet (Figures 1, 3; Table II).
Figure 4.
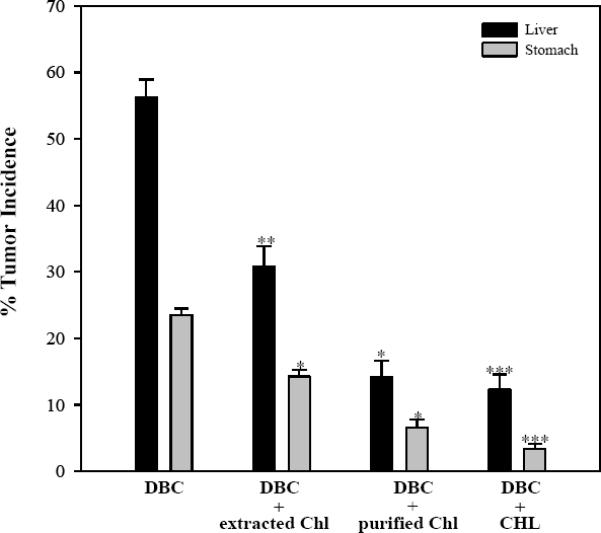
Reduction of DBC-induced liver tumors by Chl preparations. Diets contained 112 ppm DBC and 2000 ppm Chl or CHL. Trout were examined for tumor formation 9 months after DBC exposure ceased. Tumor incidence was reported as observed mean (± SE).
3.6 DBC-DNA adduct inhibition by Chl
The profiles of DBC-DNA adducts recovered from trout liver were virtually identical to those reported in our earlier studies using the same 33P-postlabeling protocol (Harttig and Bailey, 1998; Pratt et al., 2007). A profile of liver DBC-DNA adducts (Figure 5A) have polar adduct peaks eluting at 10–20 min and less polar peaks eluting at 35–65 min. Addition of 2000 ppm Chl did not change the DBC adduct profile but did change the magnitude of the response signifying a reduction in total adduct amounts (Figure 5B). Using HPLC, the total concentration of the major DBC-DNA adducts at 112 ppm DBC was determined to be 26 ± 2 pmoles adducts/mg DNA; the addition of 2000 ppm Chl at this same [DBC] reduced DBC-DNA adduct levels to 12 ± 1 pmoles adducts/mg DNA. This is more than a 50% reduction in DBC-DNA adducts by the intervention of natural Chl at 2000 ppm, and comparable to the approximate halving of final tumor incidence by this treatment.
Figure 5.
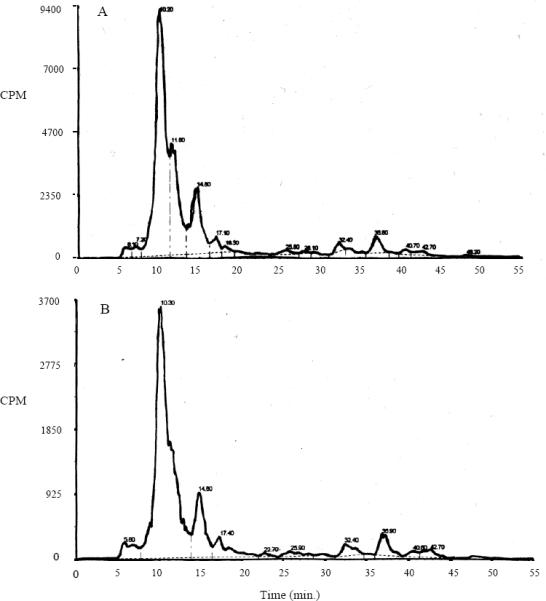
Chromatograms of in vivo DBC-DNA adducts from rainbow trout after 33P post-labeling and HPLC analysis. Liver adduct profiles were generated by 2-week exposure to either 112 ppm DBC alone (A) or 112 ppm DBC and 2000 ppm Chl (B). Note the same profile with and without Chl. The reduction is shown by the difference in the y-axis scales.
3.7 Gene regulation by chlorophylls
Previous studies (Fahey et al., 2005) have shown that chlorophylls can modulate gene expression patterns in cultured cells, in a direction favoring carcinogen detoxication pathways. Since this could constitute an important chemoprotection mechanism, we carried out a preliminary examination of the effects of dietary chlorophylls, and DBC, on expression of hepatic genes in the whole animal. Exposure to 224 ppm dietary DBC alone resulted in two-fold or greater up-regulation of 25 annotated genes in 11 functional categories in trout liver, relative to no-treatment controls (Table III). Fewer genes were up-regulated (13) in the liver of animals treated at 80 ppm DBC (data not shown), indicating that DBC-induced alterations in expression were dose-responsive. Co-administration of 2000 ppm Chl essentially abolished the DBC-induced up-regulated gene expression. Of equal importance, Chl alone had essentially no inductive effect on transcription, as only gene one among the 4000 elements in this Oncorhynchus library showed evidence for up-regulation. The overall effects of DBC on down-regulation were less extensive, but followed the same trend. As shown in Table IV, treatment with 224 ppm DBC produced a two-fold or greater down-regulation of four annotated trout genes in four functional categories. Again, these effects were abolished by co-treatment with 2000 ppm Chl, whereas Chl alone had no suppressive effect on transcription. We interpret these results to indicate that the mechanism of Chl chemoprevention is consistent with Chl-mediated reduction in carcinogen bioavailability and target organ access, and does not reflect global alterations in hepatic gene expression by Chl.
Table III.
Genes upregulated by dietary exposure to DBC compared to chemo-intervention
| Mean fold change in gene expressiona a | ||||
|---|---|---|---|---|
| Gene | Annotation (DFCI accession / GenBank accession) | DBC b | DBC+Chl c | Chl d |
| Extracellular, environmental response | ||||
| ARNT | TC142567 / U73841.1 | 1.76 | - | - |
| Growth factor | ||||
| EGFR | FM865854 | 1.43 | - | - |
| Immune response | ||||
| IFITM3 | TC146120 / AJ320157 | 1.29 | - | - |
| PSME1 | BT074326 | 1.00 | - | - |
| PSME2 | BT074299 | 1.00 | - | - |
| cf H | TC162759 / AJ627206 | 1.12 | - | - |
| CD63 | TC137037 / AY593998 | 1.00 | - | - |
| MHC I | TC132378 / AB012064 | 1.24 | - | - |
| HAMP | TC164847 / AF281354 | 1.02 | ||
| Cell proliferation/apoptosis | ||||
| P53 | TC132531 / NM_001124692 | 2.28 | - | - |
| PCNA | NM_131404 | 1.43 | - | - |
| MAPK8 | CX028516 | 1.34 | - | - |
| Metabolism | ||||
| CYP1A1 | TC158463 / U62796 | 2.79 | - | - |
| SULT1A2 | TC135750 / BX867991 | 1.02 | - | - |
| GAPD | TC137453 / NM_001124246 | 1.00 | - | - |
| DLAT | TC132429 / J04051 M29099 | 1.16 | - | - |
| SOD1 | NP543739 / AF469663 | 1.39 | - | - |
| Transport proteins | ||||
| FXYD | TC165305 / BT074290 | 1.81 | - | - |
| Elongation factors | ||||
| EF-1 alpha | TC166688 / AF498320 | 1.31 | - | - |
| Extracellular matrix | ||||
| C-type lectin | NM_001160495 | 1.00 | ||
| MMP9 | TC132759 / AJ320533 | 1.12 | - | - |
| Bone and cartilage maintenance | ||||
| Cathepsin K | TC8255 / U61499 | 1.45 | - | - |
| Function in trout unknown | ||||
| Cathepsin S | TC8256 / AY622858 | 1.17 | - | - |
| General cell signaling | ||||
| RhoG | TC169097 / BE859102 | 1.03 | - | - |
Mean fold changes in gene expression were log2 transformed. Only probes that differed by an average of at least twofold between treatment and control groups and were significant (P<0.05) are reported.
224 ppm DBC was administered via the diet for 20 days and liver samples for RNA were collected at the end of the 20 day exposure period.
224 ppm DBC + 2000 ppm Chl were co-administered via the diet for 20 days and liver samples for RNA were collected at the end of the 20 day exposure period.
2000 ppm Chl were co-administered via the diet for 20 days and liver samples for RNA were collected at the end of the 20 day exposure period.
Table IV.
Genes downregulated by dietary exposure to DBC compared to chemo-intervention
| Mean fold change in gene expressiona | ||||
|---|---|---|---|---|
| Gene | Annotation (DFCI accession / GenBank accession) | DBC b | DBC+Chl c | Chl d |
| Growth factor | ||||
| FGF6 | NP544420 / Y16850 | −1.00 | - | - |
| Immune response | ||||
| IGF1 | NP544251 / M95183 | −1.25 | - | - |
| LECT2 | TC154068 / AF363272 | −1.15 | - | |
| Metabolism | ||||
| Vtg1 | TC132491 / X92804 | −1.20 | - | - |
| Extracellular matrix | ||||
| MMP | TC147381 / AB043536 | −1.26 | - | - |
Mean fold changes in gene expression were log2 transformed. Only probes that differed by an average of at least twofold between treatment and control groups and were significant (P<0.05) are reported.
224 ppm DBC was administered via the diet for 20 days and liver samples for RNA were collected at the end of the 20 day exposure period.
224 ppm DBC + 2000ppm Chl were co-administered via the diet for 20 days and liver samples for RNA were collected at the end of the 20 day exposure period.
2000 ppm Chl were co-administered via the diet for 20 days and liver samples for RNA were collected at the end of the 20 day exposure period.
4 Discussion
4.1 Cancer chemoprevention by dietary chlorophylls
The present chemoprevention study employed a 12,000-animal dose-dose matrix design to evaluate the dose-responsive effects of dietary Chl co-treatment over a range of DBC exposures (Table II). Co-exposure with the Chl-enriched spinach extract was shown to provide dose-dependent reduction in final tumor incidence in stomach at all DBC doses, and in liver at all but the highest DBC dose (Figure 1). Chl chemopreventive potency over the lower DBC exposures was estimated through a successful modeling of the data sets as a series of parallel dose-response curves displaced toward higher TD25 with increasing [Chl] (Figure 1, inset panels). By this analysis, 500–4000 ppm dietary Chl reduced liver tumor incidence to DBC from 29–64% and stomach tumor response from 24–45% (Figure 2). These results are similar to our previous dose-dose matrix study with the derivative CHL, which inhibited liver tumor incidence by 62–82% and stomach tumor incidence from 30–68% using a greater, 1500–6000 ppm CHL dose range (Pratt et al., 2007). Although direct comparisons are difficult, these two studies suggest that synthetic CHL and partially purified natural Chl have at least comparable molar chemopreventive potencies in this model. A more direct comparison of chemopreventive efficacies among the various chlorophyll preparations is provided in Figure 4. By this result, CHL and CCC-purified Chl were comparable at reducing tumor response in stomach and liver, whereas the Chl extract used for the bulk of the dose-dose matrix study exhibited a reduced potency, requiring additional dietary concentration to achieve comparable chemoprevention.
4.2 Mechanisms of chemoprevention by chlorophylls
Previous pharmacokinetic experiments in rats, trout, and human volunteers have provided ample evidence that oral co-treatments with CHL or purified Chl interfere with oral absorption of such carcinogens as DBC, AFB1, and certain heterocyclic amines, thus reducing their systemic bioavailability and potential for genetic damage. As seen in this and previous studies, reduced carcinogen bioavailability is accompanied by substantial reductions in target organ carcinogen-DNA damage, and tumor initiation. Alternative or additional blocking mechanisms could include alterations in the expression and/or the catalytic activities of xenobiotic metabolizing enzymes such that detoxification reactions are favored. Indeed, various chlorophylls have been shown in cell culture to alter expression of genes important to carcinogen metabolism. However, attempts to demonstrate CHL- or Chl-mediated alterations in the expression of such genes in the whole animal, in pertinent target organs, under chemopreventive exposure conditions, have so far proven negative (Simonich et al., 2007). The present small scale microarray experiment lends additional support to the hypothesis that Chl chemoprevention occurs simply, by a sequestration of carcinogen within the GI tract for rapid elimination from the body. The net effect from a transcriptional standpoint is that the already low bioavailability of DBC is further reduced by chlorophyll co-treatments, whereas chlorophylls alone failed to alter expression in any of the trout genes and gene families available for analysis. The attractiveness of a mechanistically simple, carcinogen-sequestration mechanism for human intervention is obvious. Additional, post-initiation effects for Chl might be equally important for human protection. We have recently determined that a diet containing 10% spinach substantially suppressed tumor development in multiple target organs (small intestine, colon, skin, spleen, liver, lung) in the rat, when this diet was fed for 34 weeks following carcinogen treatment (Dashwood et al., unpublished results). Mechanisms associated with this tumor suppression clearly have nothing to do with Chl-carcinogen interactions, but data are presently lacking to indicate that suppression can be ascribed to Chl itself.
4.3 Limitations for chemoprevention at high carcinogen dose
Earlier pilot studies demonstrated the ability of purified natural Chl to protect against DBC-initiated liver and stomach tumorigenesis in the trout (Simonich et al., 2008) and AFB1 carcinogenicity in the rat (Simonich et al., 2007). However, our initial Chl studies, like nearly all other chemoprevention studies reported, used a single high concentration of carcinogen aimed at providing a high tumor incidence in the positive controls. This is done routinely in order to maximize statistical power to detect significant reduction of endpoints (biomarkers, pre-cancerous lesions and tumors) with an affordable number of animals. The central assumption of such experiments, however, is that intervention effects at high carcinogen dose and high endpoint responses will apply equally at lower carcinogen doses and incidences more relevant to human exposures and risk levels. This assumption has now been examined twice, using 10,000 and 12,000--animal dose-dose matrix designs with the low-cost rainbow trout model. These studies used graded dietary doses of DBC arrayed against graded dietary doses of either CHL (Pratt et al., 2007) or natural Chl-enriched spinach extract (present study). As expected, the results demonstrated that Chl, as well as CHL, efficacy in both target organs were dependent on inhibitor dose. However, contrary to the usual assumption, the outcomes in the major target organ were strikingly dependent on carcinogen dose. At lower DBC doses, graded CHL or Chl co-treatments provided strong protection against initial liver DBC-DNA adduct formation, tumor incidence, and tumor multiplicity. At high DBC doses, however, co-treatments with either agent failed to protect against tumor incidence, and in fact significantly increased tumor multiplicity. We stress that CHL failed to protect against tumor response at high DBC dose, despite protection against initial DBC-DNA adduct biomarkers over the entire DBC dose range (Pratt et al., 2007). This phenomenon occurred because the incidence and multiplicity dose-responses for DBC in this target organ are not linear on any modeling scale, nor even monotonic, but reach an optimum at moderate dose and decline at higher dose. Since the net effect of Chl treatment was to reduce effective DBC exposures, and thus shift the dose-response curves to higher and higher DBC treatment levels, Chl co-treatments that were inhibitory at lower DBC treatments became ineffective or enhancing at high DBC exposure. Had this particular experiment been carried out at the traditional, single high DBC dose, the conclusion would have been that CHL is either ineffective (incidence) or co-carcinogenic (multiplicity) and therefore not useful for reduction of human cancer risk at environmentally relevant carcinogen exposures. This would be an unfortunate conclusion, and a missed opportunity, since recent human studies show clear promise for protection by Chl as well as CHL (Dashwood et al., 1989; Hayashi et al., 1999; Jubert et al., 2009), at environmental aflatoxin exposures.
4.4 Conclusions
The results of the present study demonstrate that increasing dietary doses of Chl-enriched spinach extract provide increasing and potent protection against initial DBC-initiated tumor response in two target organs, that protection by the extract was moderately reduced compared with equivalent doses of CHL or purified Chl, and that protection occurred in the absence of demonstrable changes in gene expression patterns. We also determined that the protective efficacy of dietary Chl co-exposure was strongly dependent on the concentration of DBC in the diet, with good protection in both target organs at low carcinogen dose and tumor response but apparent enhancement in liver tumor response at high carcinogen doses and tumor responses not encountered in human populations. These findings emphasize the necessity in the design of cancer chemoprevention studies to select carcinogen doses within a known linear or monotonic dose-response range. In the absence of this information, results derived at high carcinogen doses and high tumor responses may be irrelevant for human intervention.
Chlorophyll inhibited PAH induced liver and stomach tumorigenesis in trout.
Chlorophyll abolished dibenzo(def, p)chrysene induced changes in gene expression.
Chlorophyll provides cancer chemoprotection by reducing carcinogen bioavailability.
Above DBC optima, chl failed to inhibit tumor incidence and enhanced multiplicity.
For human relevance, a high carcinogen dose may mislead in chemoprevention studies.
5 Acknowledgements
We would like to thank Eric Johnson, Greg Gonnerman, and Shelia Cleveland of the Sinnhuber Aquatic Research Laboratory for their excellence in fish rearing, necropsy and histology. The laboratory of William Baird provided additional guidance in 33P-postlabeling of DNA adducts. We also wish to thank Gina Miller for extracting chlorophyll from spinach for the study. This project was performed, in part using compound provided by the National Cancer Institute's Chemical Carcinogen Reference Standards Repository operated under contract by Midwest Research Institute, NO. NO2-CB-66600. This work was supported by NIH grants CA90890, ES00210, and ES03850.
Abbreviations
- Chl
chlorophyll
- CHL
chlorophyllin
- DBC
dibenzo(def, p)chrysene
- AFB1
aflatoxin B1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
6 References
- Aggarwal BB, Shishodia S. Molecular targets of dietary agents for prevention and therapy of cancer. Biochem Pharmacol. 2006;71:1397–1421. doi: 10.1016/j.bcp.2006.02.009. [DOI] [PubMed] [Google Scholar]
- Bailey GS, Reddy AP, Pereira CB, Harttig U, Baird W, Spitsbergen JM, Hendricks JD, Orner GA, Williams DE, Swenberg JA. Nonlinear cancer response at ultralow dose: a 40800-animal ED(001) tumor and biomarker study. Chem Res Toxicol. 2009;22:1264–1276. doi: 10.1021/tx9000754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird WM, Hooven LA, Mahadevan B. Carcinogenic polycyclic aromatic hydrocarbon-DNA adducts and mechanism of action. Environ Mol Mutagen. 2005;45:106–114. doi: 10.1002/em.20095. [DOI] [PubMed] [Google Scholar]
- Barrett JR. Cancer. Plants provide prevention. Environ Health Perspect. 2002;110:A180. doi: 10.1289/ehp.110-a180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benninghoff AD, Williams DE. Identification of a transcriptional fingerprint of estrogen exposure in rainbow trout liver. Toxicol Sci. 2008;101:65–80. doi: 10.1093/toxsci/kfm238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breinholt V, Schimerlik M, Dashwood R, Bailey G. Mechanisms of chlorophyllin anticarcinogenesis against aflatoxin B1: complex formation with the carcinogen. Chem Res Toxicol. 1995;8:506–514. doi: 10.1021/tx00046a004. [DOI] [PubMed] [Google Scholar]
- Busby WF, Jr., Smith H, Crespi CL, Penman BW. Mutagenicity of benzo[a]pyrene and dibenzopyrenes in the Salmonella typhimurium TM677 and the MCL-5 human cell forward mutation assays. Mutat Res. 1995;342:9–16. doi: 10.1016/0165-1218(95)90085-3. [DOI] [PubMed] [Google Scholar]
- Cavalieri EL, Higginbotham S, RamaKrishna NV, Devanesan PD, Todorovic R, Rogan EG, Salmasi S. Comparative dose-response tumorigenicity studies of dibenzo[alpha,l]pyrene versus 7,12-dimethylbenz[alpha]anthracene, benzo[alpha,l]pyrene and two dibenzo[alpha,l]pyrene dihydrodiols in mouse skin and rat mammary gland. Carcinogenesis. 1991;12:1939–1944. doi: 10.1093/carcin/12.10.1939. [DOI] [PubMed] [Google Scholar]
- Cavalieri EL, Rogan EG, Higginbotham S, Cremonesi P, Salmasi S. Tumor-initiating activity in mouse skin and carcinogenicity in rat mammary gland of dibenzo[a]pyrenes: the very potent environmental carcinogen dibenzo[a, l]pyrene. J Cancer Res Clin Oncol. 1989;115:67–72. doi: 10.1007/BF00391602. [DOI] [PubMed] [Google Scholar]
- Dashwood R, Negishi T, Hayatsu H, Breinholt V, Hendricks J, Bailey G. Chemopreventive properties of chlorophylls towards aflatoxin B1: a review of the antimutagenicity and anticarcinogenicity data in rainbow trout. Mutat Res. 1998;399:245–253. doi: 10.1016/s0027-5107(97)00259-5. [DOI] [PubMed] [Google Scholar]
- Dashwood R, Yamane S, Larsen R. Study of the forces of stabilizing complexes between chlorophylls and heterocyclic amine mutagens. Environ Mol Mutagen. 1996;27:211–218. doi: 10.1002/(SICI)1098-2280(1996)27:3<211::AID-EM6>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Dashwood RH, Arbogast DN, Fong AT, Pereira C, Hendricks JD, Bailey GS. Quantitative inter-relationships between aflatoxin B1 carcinogen dose, indole-3-carbinol anti-carcinogen dose, target organ DNA adduction and final tumor response. Carcinogenesis. 1989;10:175–181. doi: 10.1093/carcin/10.1.175. [DOI] [PubMed] [Google Scholar]
- Deraat WK, Kooijman SALM, Gielen JWJ. Concentrations of Polycyclic-Hydrocarbons in Airborne Particles in the Netherlands and Their Correlation with Mutagenicity. Science of the Total Environment. 1987;66:95–114. doi: 10.1016/0048-9697(87)90080-5. [DOI] [PubMed] [Google Scholar]
- Durant JL, Lafleur AL, Busby WF, Jr., Donhoffner LL, Penman BW, Crespi CL. Mutagenicity of C24H14 PAH in human cells expressing CYP1A1. Mutat Res. 1999;446:1–14. doi: 10.1016/s1383-5718(99)00135-7. [DOI] [PubMed] [Google Scholar]
- Egner PA, Munoz A, Kensler TW. Chemoprevention with chlorophyllin in individuals exposed to dietary aflatoxin. Mutat Res. 2003;523–524:209–216. doi: 10.1016/s0027-5107(02)00337-8. [DOI] [PubMed] [Google Scholar]
- Egner PA, Wang JB, Zhu YR, Zhang BC, Wu Y, Zhang QN, Qian GS, Kuang SY, Gange SJ, Jacobson LP, Helzlsouer KJ, Bailey GS, Groopman JD, Kensler TW. Chlorophyllin intervention reduces aflatoxin-DNA adducts in individuals at high risk for liver cancer. Proc Natl Acad Sci U S A. 2001;98:14601–14606. doi: 10.1073/pnas.251536898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahey JW, Stephenson KK, Dinkova-Kostova AT, Egner PA, Kensler TW, Talalay P. Chlorophyll, chlorophyllin and related tetrapyrroles are significant inducers of mammalian phase 2 cytoprotective genes. Carcinogenesis. 2005;26:1247–1255. doi: 10.1093/carcin/bgi068. [DOI] [PubMed] [Google Scholar]
- Guyton KZ, Kensler TW. Prevention of liver cancer. Curr Oncol Rep. 2002;4:464–470. doi: 10.1007/s11912-002-0057-4. [DOI] [PubMed] [Google Scholar]
- Harttig U, Bailey GS. Chemoprotection by natural chlorophylls in vivo: inhibition of dibenzo[a,l]pyrene-DNA adducts in rainbow trout liver. Carcinogenesis. 1998;19:1323–1326. doi: 10.1093/carcin/19.7.1323. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Schimerlik M, Bailey G. Mechanisms of chlorophyllin anticarcinogenesis: dose-responsive inhibition of aflatoxin uptake and biodistribution following oral co-administration in rainbow trout. Toxicol Appl Pharmacol. 1999;158:132–140. doi: 10.1006/taap.1999.8695. [DOI] [PubMed] [Google Scholar]
- Hendricks JD, Meyers TR, Shelton DW. Histological progression of hepatic neoplasia in rainbow trout (Salmo gairdneri) Natl Cancer Inst Monogr. 1984;65:321–336. [PubMed] [Google Scholar]
- Hendricks JD, Shelton DW, Loveland PM, Pereira CB, Bailey GS. Carcinogenicity of dietary dimethylnitrosomorpholine, N-methyl-N'-nitro-N-nitrosoguanidine, and dibromoethane in rainbow trout. Toxicol Pathol. 1995;23:447–457. doi: 10.1177/019262339502300402. [DOI] [PubMed] [Google Scholar]
- Higginbotham S, RamaKrishna NV, Johansson SL, Rogan EG, Cavalieri EL. Tumor-initiating activity and carcinogenicity of dibenzo[a,l]pyrene versus 7,12- dimethylbenz[a]anthracene and benzo[a]pyrene at low doses in mouse skin. Carcinogenesis. 1993;14:875–878. doi: 10.1093/carcin/14.5.875. [DOI] [PubMed] [Google Scholar]
- Hirose M, Nishikawa A, Shibutani M, Imai T, Shirai T. Chemoprevention of heterocyclic amine-induced mammary carcinogenesis in rats. Environ Mol Mutagen. 2002;39:271–278. doi: 10.1002/em.10066. [DOI] [PubMed] [Google Scholar]
- Hoffmann D, Hoffmann I. Tobacco smoke components. Beitr. Tabakforsch. 1998;18:49–52. [Google Scholar]
- Jacobson LP, Zhang BC, Zhu YR, Wang JB, Wu Y, Zhang QN, Yu LY, Qian GS, Kuang SY, Li YF, Fang X, Zarba A, Chen B, Enger C, Davidson NE, Gorman MB, Gordon GB, Prochaska HJ, Egner PA, Groopman JD, Munoz A, Helzlsouer KJ, Kensler TW. Oltipraz chemoprevention trial in Qidong, People's Republic of China: study design and clinical outcomes. Cancer Epidemiol Biomarkers Prev. 1997;6:257–265. [PubMed] [Google Scholar]
- Jubert C, Bailey G. Isolation of chlorophylls a and b from spinach by counter-current chromatography. J Chromatogr A. 2007;1140:95–100. doi: 10.1016/j.chroma.2006.11.063. [DOI] [PubMed] [Google Scholar]
- Jubert C, Mata J, Bench G, Dashwood R, Pereira C, Tracewell W, Turteltaub K, Williams D, Bailey G. Effects of chlorophyll and chlorophyllin on low-dose aflatoxin B(1) pharmacokinetics in human volunteers. Cancer Prev Res (Phila Pa) 2009;2:1015–1022. doi: 10.1158/1940-6207.CAPR-09-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelloff GJ, Lippman SM, Dannenberg AJ, Sigman CC, Pearce HL, Reid BJ, Szabo E, Jordan VC, Spitz MR, Mills GB, Papadimitrakopoulou VA, Lotan R, Aggarwal BB, Bresalier RS, Kim J, Arun B, Lu KH, Thomas ME, Rhodes HE, Brewer MA, Follen M, Shin DM, Parnes HL, Siegfried JM, Evans AA, Blot WJ, Chow WH, Blount PL, Maley CC, Wang KK, Lam S, Lee JJ, Dubinett SM, Engstrom PF, Meyskens FL, Jr., O'Shaughnessy J, Hawk ET, Levin B, Nelson WG, Hong WK. Progress in chemoprevention drug development: the promise of molecular biomarkers for prevention of intraepithelial neoplasia and cancer--a plan to move forward. Clin Cancer Res. 2006;12:3661–3697. doi: 10.1158/1078-0432.CCR-06-1104. [DOI] [PubMed] [Google Scholar]
- Kensler TW, T. P. Inducers of enzymes that protect against carcinogens and oxidants. In: Kelloff GJ, Hawk ET, Sigman CC, editors. Cancer Chemoprevention, Vol 1: Promising Cancer Chemoprevention Agents. Humana Press Inc.; Totowa, NJ: 2004. pp. 3–20. [Google Scholar]
- Kozin IS, Gooijer C, Velthorst NH. Direct Determination of Dibenzo[a,L]Pyrene in Crude Extracts of Environmental-Samples by Laser-Excited Shpolskii Spectroscopy. Analytical Chemistry. 1995;67:1623–1626. [Google Scholar]
- LaVoie EJ, He ZM, Meegalla RL, Weyand EH. Exceptional tumor-initiating activity of 4-fluorobenzo[j]-fluoranthene on mouse skin: comparison with benzo[j]-fluoranthene, 10-fluoro-benzo[j]fluoranthene, benzo[a]pyrene, dibenzo[a,l]pyrene and 7,12-dimethylbenz[a]anthracene. Cancer Lett. 1993;70:7–14. doi: 10.1016/0304-3835(93)90068-k. [DOI] [PubMed] [Google Scholar]
- Lee BC, Hendricks JD, Bailey GS. Toxicity of mycotoxins in the feed of fish. Mycotoxins and Animal Feedingstuff: Natural Occurrence, Toxicity and Control. 1991:607–626. [Google Scholar]
- Loveland PM, Reddy AP, Pereira CB, Field JA, Bailey GS. Application of matrix solid-phase dispersion in the determination of dibenzo[a,l]pyrene content of experimental animal diets used in a large-scale tumor study. Journal of Chromatography A. 2001;932:33–41. doi: 10.1016/s0021-9673(01)01207-9. [DOI] [PubMed] [Google Scholar]
- Mahadevan B, Luch A, Bravo CF, Atkin J, Steppan LB, Pereira C, Kerkvliet NI, Baird WM. Dibenzo[a,l]pyrene induced DNA adduct formation in lung tissue in vivo. Cancer Lett. 2005;227:25–32. doi: 10.1016/j.canlet.2004.11.056. [DOI] [PubMed] [Google Scholar]
- Mumford JL, Harris DB, Williams K, Chuang JC, Cooke M. Indoor Air Sampling and Mutagenicity Studies of Emissions from Unvented Coal Combustion. Environmental Science & Technology. 1987;21:308–311. doi: 10.1021/es00157a014. [DOI] [PubMed] [Google Scholar]
- Mumford JL, Li X, Hu F, Lu XB, Chuang JC. Human exposure and dosimetry of polycyclic aromatic hydrocarbons in urine from Xuan Wei, China with high lung cancer mortality associated with exposure to unvented coal smoke. Carcinogenesis. 1995;16:3031–3036. doi: 10.1093/carcin/16.12.3031. [DOI] [PubMed] [Google Scholar]
- Prahalad AK, Ross JA, Nelson GB, Roop BC, King LC, Nesnow S, Mass MJ. Dibenzo[a,l]pyrene-induced DNA adduction, tumorigenicity, and Ki-ras oncogene mutations in strain A/J mouse lung. Carcinogenesis. 1997;18:1955–1963. doi: 10.1093/carcin/18.10.1955. [DOI] [PubMed] [Google Scholar]
- Pratt MM, Reddy AP, Hendricks JD, Pereira C, Kensler TW, Bailey GS. The importance of carcinogen dose in chemoprevention studies: quantitative interrelationships between, dibenzo[a,l]pyrene dose, chlorophyllin dose, target organ DNA adduct biomarkers and final tumor outcome. Carcinogenesis. 2007;28:611–624. doi: 10.1093/carcin/bgl174. [DOI] [PubMed] [Google Scholar]
- Qian GS, Ross RK, Yu MC, Yuan JM, Gao YT, Henderson BE, Wogan GN, Groopman JD. A follow-up study of urinary markers of aflatoxin exposure and liver cancer risk in Shanghai, People's Republic of China. Cancer Epidemiol Biomarkers Prev. 1994;3:3–10. [PubMed] [Google Scholar]
- Ralston SL, Seidel A, Luch A, Platt KL, Baird WM. Stereoselective activation of dibenzo[a,l]pyrene to (−)-anti (11R,12S,13S,14R)- and (+)-syn(11S,12R,13S,14R)-11,12-diol-13,14-epoxides which bind extensively to deoxyadenosine residues of DNA in the human mammary carcinoma cell line MCF-7. Carcinogenesis. 1995;16:2899–2907. doi: 10.1093/carcin/16.12.2899. [DOI] [PubMed] [Google Scholar]
- Reddy AP, Harttig U, Barth MC, Baird WM, Schimerlik M, Hendricks JD, Bailey GS. Inhibition of dibenzo[a,l]pyrene-induced multi-organ carcinogenesis by dietary chlorophyllin in rainbow trout. Carcinogenesis. 1999;20:1919–1926. doi: 10.1093/carcin/20.10.1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel A, Frank H, Behnke A, Schneider D, Jacob J. Determination of dibenzo[a]pyrene and other fjord-region PAH isomers with MW 302 in environmental samples. Polycyclic Aromatic Compounds. 2004;24:759–771. [Google Scholar]
- Shaughnessy DT, Gangarosa LM, Schliebe B, Umbach DM, Xu Z, Macintosh B, Knize MG, Matthews PP, Swank AE, Sandler RS, Demarini DM, Taylor JA. Inhibition of fried meat-induced colorectal DNA damage and altered systemic genotoxicity in humans by crucifera, chlorophyllin, and yogurt. PLoS One. 6:e18707. doi: 10.1371/journal.pone.0018707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonich MT, Egner PA, Roebuck BD, Orner GA, Jubert C, Pereira C, Groopman JD, Kensler TW, Dashwood RH, Williams DE, Bailey GS. Natural chlorophyll inhibits aflatoxin B1-induced multi-organ carcinogenesis in the rat. Carcinogenesis. 2007;28:1294–1302. doi: 10.1093/carcin/bgm027. [DOI] [PubMed] [Google Scholar]
- Simonich MT, McQuistan T, Jubert C, Pereira C, Hendricks JD, Schimerlik M, Zhu B, Dashwood RH, Williams DE, Bailey GS. Low-dose dietary chlorophyll inhibits multi-organ carcinogenesis in the rainbow trout. Food Chem Toxicol. 2008;46:1014–1024. doi: 10.1016/j.fct.2007.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnhuber RO, Hendricks JD, Wales JH, Putnam GB. Neoplasms in rainbow trout, a sensitive animal model for environmental carcinogenesis. Ann N Y Acad Sci. 1978;298:389–408. doi: 10.1111/j.1749-6632.1977.tb19280.x. [DOI] [PubMed] [Google Scholar]
- Snook ME, Severson RF, Arrendale RF, Higman HC, Chortyk OT. Identification of High Molecular-Weight Polynuclear Aromatic-Hydrocarbons in a Biologically-Active Fraction of Cigarette-Smoke Condensate. Beitrage Zur Tabakforschung International. 1977;9:79–101. [Google Scholar]
- Tilton SC, Gerwick LG, Hendricks JD, Rosato CS, Corley-Smith G, Givan SA, Bailey GS, Bayne CJ, Williams DE. Use of a rainbow trout oligonucleotide microarray to determine transcriptional patterns in aflatoxin B1-induced hepatocellular carcinoma compared to adjacent liver. Toxicol Sci. 2005;88:319–330. doi: 10.1093/toxsci/kfi309. [DOI] [PubMed] [Google Scholar]
- Wright TI, Spencer JM, Flowers FP. Chemoprevention of nonmelanoma skin cancer. J Am Acad Dermatol. 2006;54:933–946. doi: 10.1016/j.jaad.2005.08.062. [DOI] [PubMed] [Google Scholar]