

Abstract
HIV-1 resistance testing was performed in 47 antiretroviral (ARV)-treated subjects with low-level viremia (LLV) of <1,000 copies/ml. The median viral load was 267 copies/ml. In those with ≥2 LLV episodes, 44% accumulated additional resistance mutations. Fewer active ARVs and longer elapsed time were associated with an increased risk of resistance accumulation after controlling for adherence and viral load. Virologic failure followed 16% of LLV time points. Strategies for early intervention after LLV episodes should be further studied.
TEXT
Current guidelines recommend optimal suppression of HIV plasma viral load (pVL) to below the level of detection of conventional assays. The significance of low-level viremia (LLV) remains controversial, as some studies have associated episodes of LLV with an increased risk of virologic failure (1, 4), while other studies have not (8). It is hypothesized that an increased risk of virologic failure may be mediated by accumulating drug resistance mutations during periods of incomplete viral suppression, but this remains speculative, as most commercial genotyping assays are optimized for pVL of ≥1,000 copies/ml. Several cross-sectional studies have found that genotypic testing of LLV samples can detect drug resistance mutations, but these studies could not evaluate when the mutations arose (6, 9). In the modern era of highly active antiretroviral therapy (HAART), the accumulation of drug resistance during LLV has been demonstrated specifically in subjects initiating a first-line HAART regimen (10), in those participating in a raltegravir treatment study (3), and within a French clinical cohort (2). A recent study using genotypic assays to optimize treatment regimens resulted in excellent rates of virologic suppression (7). In the present study, we evaluated the emergence of HIV drug resistance during periods of LLV and subsequent clinical outcomes in a cohort of chronically infected and mostly treatment-experienced HIV-infected adults.
(This study was presented in part as an oral presentation at the 2012 International Workshop on HIV & Hepatitis Virus Drug Resistance and Curative Strategies, 5 to 9 June 2012.)
Participants were part of the University of California, San Francisco, SCOPE cohort, a prospective study of HIV-infected adults. Participants with LLV (detectable pVL of <1,000 copies/ml) while on HAART were selected. In participants with multiple LLV time points, genotyping of the first and last time points was attempted, and other time points were studied primarily if new resistance mutations were detected at the last time point. If an LLV sample was unavailable, we attempted genotypic resistance testing of the sample closest to that time point. Medication adherence in the previous 30 days was recorded based upon patient self-report. Stored plasma samples were ultracentrifuged at 28,000 × g for 1 h to pellet virus prior to RNA extraction (QIAamp viral RNA minikit). The coding regions of HIV-1 PR, RT, IN, and gp41 were amplified by nested gene-specific primers. Population sequencing was performed on purified amplicons with an ABI 3730 automated DNA sequencer and processed using Sequencher (Genecodes). Phylogenetic analysis using PhyML was used to confirm sequence identity and exclude PCR contamination. When available, historical genotyping results during episodes of LLV were also included. A new mutation was defined as any mutation (major or minor) that decreased the activity of the participant's antiretroviral (ARV) regimen. A fully active ARV was defined as having a Stanford resistance mutation score of <10 (11). In the analysis of baseline patient characteristics, the earliest LLV time point was used. The association between HIV-1 pVL and the number of active ARVs was evaluated using the Kruskal-Wallis test, and comparison of samples with and without new resistance mutations was performed with the Wilcoxon rank sum test. Repeated-measures multivariable logistic regression was used to evaluate predictors of resistance mutation accumulation. The variables included in the multivariable model were selected a priori based upon their suspected association with risk of resistance mutation emergence. Each LLV sample was grouped into three outcomes: (i) virologic suppression below the detection of the assay used, (ii) virologic failure with a pVL of ≥1,000 copies/ml without preceding virologic suppression, or (iii) lost to follow-up or change in ARV regimen before either virologic suppression or failure. The exact χ2 test was used to compare the virologic outcome with numbers of fully active ARVs. This study was approved by the institutional review board of the University of California, San Francisco.
Drug resistance genotyping was successful for samples collected at 82 time points between 2001 and 2010 in 47 participants, including 18 (38%) participants with LLV time points of ≥2. Demographic and treatment characteristics of the patients and samples are shown in Tables 1 and 2. The majority of participants (89%) were treatment experienced at baseline, and the median pVL of the LLV episodes was 267 copies/ml. The most commonly utilized protease inhibitors included ritonavir-boosted lopinavir (45%), nelfinavir (19%), ritonavir-boosted darunavir (14%), and ritonavir-boosted atazanavir (12%). The median number of fully active ARVs at the LLV episode was 2 (interquartile range [IQR] of 1 to 3). Median 30-day adherence prior to the LLV episode was 98% (n = 81; IQR, 93 to 100%), and 72% of participants reported ≥95% adherence in the prior 30 days.
Table 1.
Demographic and treatment characteristics of SCOPE participants with low-level viremiaa
| Variable | Valueb |
|---|---|
| Age, median (IQR) | 49 (42–54) |
| % male | 87 |
| Race (%) | |
| White | 64 |
| Black | 15 |
| Hispanic | 13 |
| Other | 8 |
| Median CD4 count (IQR) | 367 (202–494) |
| Median no. of yrs since HIV diagnosis (IQR) | 13 (8–19) |
| % of participants that were treatment-experienced at study entry | 89 |
| ARV regimen (% of participants) | |
| PI | 66 |
| NNRTI | 11 |
| Both PI and NNRTI | 23 |
| No. of LLV time points with genotyping (% of participants) | |
| One | 62 |
| Two | 15 |
| ≥Three | 23 |
IQR, interquartile range; ARV, antiretroviral medication; PI, protease inhibitor; NNRTI, nonnucleoside reverse transcriptase inhibitor; LLV, low-level viremia. For those with ≥2 LLV episodes, characteristics at the first time point are included in this table.
n = 47 participants.
Table 2.
Characteristics of the low-level viremia time points evaluated
| Variable | Valueb |
|---|---|
| No. of HIV-1 RNA copies/ml (IQRa) | 267 (174–414) |
| Median no. of fully active ARVs (IQR) | 2 (1–3) |
| % of new mutations vs. historical genotyping (n = 39) | 46 |
| Median % of 4-day adherence (IQR) (n = 81) | 100 (100–100) |
| Median % of 30-day adherence (IQR) (n = 81) | 98.3 (93.3–100) |
IQR, interquartile range.
n = 82 samples.
There was no significant association between pVL and the number of fully active ARVs (Fig. 1) (Kruskal-Wallis P = 0.69). Compared to a prior genotype, 46% (18/39) of LLV samples had a new resistance mutation to the participant's current ARV regimen. Of those on a PI-based regimen, 57% (24/42) had detectable NRTI resistance, and 45% (19/42) had PI resistance at the first LLV sample. For those on an NNRTI-based regimen, 20% (3/15) had detectable NRTI resistance and 33% (5/15) had NNRTI resistance. In those with ≥2 LLV time points, 44% (8/18) were found to have accumulated additional resistance mutations to the ARV regimen over a median of 11.4 months (IQR, 4.4 to 20.0), and in 3 of those participants, the new resistance mutation resulted in a decrease in the number of fully active ARVs (Table 3). In a multivariable repeated-measures logistic regression analysis, fewer fully active ARVs at the prior time point (β = −1.3; 95% confidence interval [CI] of −2.2 to −0.5; P = 0.003) and longer elapsed time (months, β = 0.08; 95% CI of 0.01 to 0.14; P = 0.02) were associated with an increased risk of resistance accumulation after controlling for 30-day medication adherence and pVL. Virologic suppression occurred after 76% (62/82) of the LLV episodes, while virologic failure occurred after 16% (13/82) of LLV time points. Lost to follow-up or change in ART regimen occurred in 9% (7/82) of LLV episodes before either virologic suppression or failure.
Fig 1.
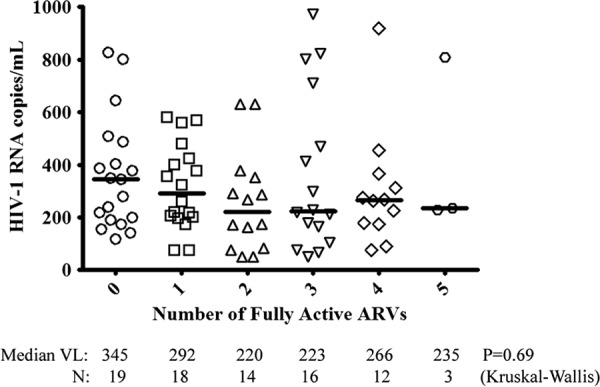
No significant association was detected between plasma HIV viral load and the number of fully active ARVs present during episodes of detectable low-level viremia. A fully active ARV was defined as having a Stanford HIV DB resistance mutation score of <10.
Table 3.
Participants of the SCOPE cohort with ≥2 LLV episodes and emerging drug resistance mutationsa
| PID | Resistance class | Resistance mutations | ARV regimen | No. of active ARVsb | Interval (no. of days) |
|---|---|---|---|---|---|
| 2070 | NRTI, PI | M184V, L10I | D4T, 3TC, NFV | 2 | |
| PI | A71V, L90 M | D4T, 3TC, NFV | 1 | 526 | |
| NRTI | D67N, K70R, K219Q | D4T, 3TC, NFV | 0 | 1,195 | |
| 3022 | None | None | ABC, 3TC, NVP, LPV/r | 4 | |
| NNRTI | Y188C | ABC, 3TC, NVP, LPV/r | 3 | 21 | |
| 3033 | NRTI, PI | D67N, M184V, D30N, N88D, A71T | D4T, 3TC, NFV | 0 | |
| NRTI, PI | K70R, L10R | D4T, 3TC, NFV | 0 | 826 | |
| 3041 | NRTI, PI | L74V, M184V, D30N, N88D, L10F | ABC, 3TC, NFV | 0 | |
| NRTI | Y115F | ABC, 3TC, NFV | 0 | 511 | |
| 3072 | NNRTI, PI | Y181C, L10F, V32I, M46I, I47V, V82A | AZT, NVP, LPV/r | 1 | |
| NNRTI | K103N | AZT, NVP, LPV/r | 1 | 115 | |
| PI | I54 M | AZT, NVP, LPV/r | 1 | 746 | |
| 3141 | NRTI | M41L, D67N, K70R, M184V, T215Y, K219E | ABC, D4T, LPV/r | 1 | |
| PI | M46I, I54V | ABC, D4T, LPV/r | 0 | 343 | |
| PI | L10F | ABC, D4T, LPV/r | 0 | 485 | |
| 6036 | NRTI, PI | M41L, L74V, M184V, T215F, M46I, I54V, G73S, V82A, L90 M | TDF, ABC, 3TC, LPV/r | 0 | |
| NRTI | Y115F | TDF, ABC, 3TC, LPV/r | 0 | 1,430 | |
| 6054 | NRTI, NNRTI | D67N, K70R, K219E, G190A | ABC, D4T, NVP | 0 | |
| NRTI | M41L | ABC, D4T, NVP | 0 | 231 |
PID, patient identification number; ARV, antiretroviral medication; LLV, low-level viremia. For each participant, the first row represents resistance at the first LLV time point. The second row shows emerging resistance mutations between LLV time points 1 and 2, and the third row represents emerging resistance between LLV time points 2 and 3. Resistance mutations listed at time points 2 and 3 represent additive mutations in addition to those detected at time point 1.
“Active ARVs” refers to the number of fully active ARVs (Stanford resistance score of <10). New resistance mutations may arise against ARVs that are already not fully active due to the presence of preexisting resistance.
In this study of mainly treatment-experienced individuals, new drug resistance mutations were frequently discovered at the time of initial LLV, and accumulating resistance was commonly detected over time. These results provide an explanation for the observation that recurrent LLV episodes increase the risk of virologic failure (5) and are consistent with several previous studies that also demonstrate the presence of viral evolution during recurrent episodes of LLV (2, 3, 10, 12, 13). Limitations of this study include the limited availability of multiple LLV time points, the use of older ARVs in a subset of participants, and various ARV regimens in this real-world cohort of HIV-infected patients. There also remains the possibility that some of the accumulating resistance mutations seen in this study represent the emergence of archived mutations as opposed to de novo resistance mutations generated during viral evolution.
Interestingly, the number of fully active ARVs during the prior LLV episode and time since that episode were both predictive of emerging resistance. These results support early resistance genotyping or regimen switches to prevent the accumulation of resistance mutations and virologic failure. However, most LLV episodes were followed by subsequent virologic suppression, suggesting either a stochastic process or subsequent improvement in medication adherence in a majority of instances. Strategies for early intervention during LLV episodes (e.g., adherence counseling, resistance genotyping at low viral loads, regimen switch) should receive further study.
ACKNOWLEDGMENTS
We thank the participants of the SCOPE cohort. The authors are grateful to Christina Kitchen for her assistance in the sequence analysis and to Heather Ribaudo for statistical advice.
D.R.K. has served as a consultant to and/or has received research grant support from Abbott, Avexa, Boehringer-Ingelheim, Bristol-Myers Squibb, Gilead, GlaxoSmithKline, Merck, Oncolys, Pfizer, Roche, Tobira, Vertex, ViroStatistics, and ViiV Healthcare. H.H. has received research grant support from Roche Molecular Diagnostics. T.D.D. is an employee of Roche Molecular Diagnostics. Plasma viral load testing was performed by Roche Molecular Diagnostics at no cost to the study. No potential conflicts of interest relevant to this article were reported.
J.Z.L. is the recipient of a Clinical Investigator Training Program Fellowship: Harvard/MIT Health Sciences and Technology—Beth Israel Deaconess Medical Center, in collaboration with Pfizer Inc. and Merck & Co. The UCSF-based SCOPE cohort was supported by the NIAID (RO1 AI087145, R01 HL095130, P01 AI071713, K24AI069994), the UCSF Center for AIDS Research (CFAR) (PO AI27763), the UCSF Clinical and Translational Science Institute (UL1 RR024131), and CFAR Network of Integrated Clinical Systems (CNICS; grant 1 R24 AI067039-1).
Footnotes
Published ahead of print 13 August 2012
REFERENCES
- 1. Cohen C. 2009. Low-level viremia in HIV-1 infection: consequences and implications for switching to a new regimen. HIV Clin. Trials 10:116–124 [DOI] [PubMed] [Google Scholar]
- 2. Delaugerre C, et al. 2012. Impact of low-level-viremia on HIV-1 drug-resistance evolution among antiretroviral treated-patients. PLoS One 7:e36673 doi:10.1371/journal.pone.0036673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gallien S, et al. 2011. Emerging integrase inhibitor resistance mutations in raltegravir-treated HIV-1-infected patients with low-level viremia. AIDS 25:665–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Geretti AM, et al. 2008. Determinants of virological failure after successful viral load suppression in first-line highly active antiretroviral therapy. Antivir. Ther. 13:927–936 [PubMed] [Google Scholar]
- 5. Greub G, et al. 2002. Intermittent and sustained low-level HIV viral rebound in patients receiving potent antiretroviral therapy. AIDS 16:1967–1969 [DOI] [PubMed] [Google Scholar]
- 6. Mackie NE, Phillips AN, Kaye S, Booth C, Geretti AM. 2010. Antiretroviral drug resistance in HIV-1-infected patients with low-level viremia. J. Infect. Dis. 201:1303–1307 [DOI] [PubMed] [Google Scholar]
- 7. McConnell MJ, et al. 2011. Improved viral suppression after treatment optimization in HIV-infected patients with persistent low-level viremia. J. Acquir. Immune Defic. Syndr. 58:446–449 [DOI] [PubMed] [Google Scholar]
- 8. Nettles RE, et al. 2005. Intermittent HIV-1 viremia (Blips) and drug resistance in patients receiving HAART. JAMA 293:817–829 [DOI] [PubMed] [Google Scholar]
- 9. Nettles RE, et al. 2004. Genotypic resistance in HIV-1-infected patients with persistently detectable low-level viremia while receiving highly active antiretroviral therapy. Clin. Infect. Dis. 39:1030–1037 [DOI] [PubMed] [Google Scholar]
- 10. Taiwo B, et al. 2011. Antiretroviral drug resistance in HIV-1-infected patients experiencing persistent low-level viremia during first-line therapy. J. Infect. Dis. 204:515–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tang MW, Liu TF, Shafer RW. 2012. The HIVdb system for HIV-1 genotypic resistance interpretation. Intervirology 55:98–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tobin NH, et al. 2005. Evidence that low-level viremias during effective highly active antiretroviral therapy result from two processes: expression of archival virus and replication of virus. J. Virol. 79:9625–9634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. von Wyl V, et al. 2012. Incidence of HIV-1 drug resistance among antiretroviral treatment-naive individuals starting modern therapy combinations. Clin. Infect. Dis. 54:131–140 [DOI] [PubMed] [Google Scholar]