

Abstract
Nitroheterocyclic prodrugs are used to treat infections caused by Trypanosoma cruzi and Trypanosoma brucei. A key component in selectivity involves a specific activation step mediated by a protein homologous with type I nitroreductases, enzymes found predominantly in prokaryotes. Using data from determinations based on flavin cofactor, oxygen-insensitive activity, substrate range, and inhibition profiles, we demonstrate that NTRs from T. cruzi and T. brucei display many characteristics of their bacterial counterparts. Intriguingly, both enzymes preferentially use NADH and quinones as the electron donor and acceptor, respectively, suggesting that they may function as NADH:ubiquinone oxidoreductases in the parasite mitochondrion. We exploited this preference to determine the trypanocidal activity of a library of aziridinyl benzoquinones against bloodstream-form T. brucei. Biochemical screens using recombinant NTR demonstrated that several quinones were effective substrates for the parasite enzyme, having Kcat/Km values 2 orders of magnitude greater than those of nifurtimox and benznidazole. In tests against T. brucei, antiparasitic activity mirrored the biochemical data, with the most potent compounds generally being preferred enzyme substrates. Trypanocidal activity was shown to be NTR dependent, as parasites with elevated levels of this enzyme were hypersensitive to the aziridinyl agent. By unraveling the biochemical characteristics exhibited by the trypanosomal NTRs, we have shown that quinone-based compounds represent a class of trypanocidal compound.
INTRODUCTION
Over 10 million people are infected with Trypanosoma cruzi and Trypanosoma brucei, the causative agents of Chagas' disease and human African trypanosomiasis, respectively (8, 52). Together, these pathogens are responsible for around 40,000 deaths per year and represent a major public health problem in the regions of the world least able to deal with the associated economic burden (54). As a consequence of concerted surveillance, insect vector control, and improved housing programs, there has been a significant decline in the prevalence of infections by both species. However, as a result of blood transfusion, organ transplantation, illicit drug usage, and population migration, these trypanosomiases are emerging as a problem in areas of nonendemicity, including in Europe and the United States (5, 22, 23, 60). Currently, drugs are the only viable option to treat these diseases. For T. cruzi, treatment is based on nifurtimox and benznidazole, whereas suramin, pentamidine, melarsoprol, eflornithine, and nifurtimox-eflornithine combinational therapy are used against HAT (64). In addition to the problems represented by toxicity, resistance, and cost, therapy is further complicated by the fact that the effectiveness of some drugs is dependent on the subspecies and the disease stage. Therefore, there is an urgent requirement for alternative chemotherapies.
Quinone-based compounds encompass a range of molecules, characterized by two carbonyl groups linked to a carbocyclic backbone. They are widely distributed in nature and are able to participate in a number of crucial biological oxidoreductase cascades. Additionally, both natural and synthetic quinones have been extensively used in medicine as antimicrobial and anticancer agents, functioning either as inhibitors of essential redox pathways or as prodrugs. In the latter case, the compound must undergo activation by quinone reductases before mediating their cytotoxic effects. Quinone reductases can broadly be divided into two groups based on the number of reducing equivalents transferred to their target substrate. Some enzymes mediate the 1e− reduction of the quinone carbonyl oxygens to form an unstable semiquinone radical. Under hypoxic conditions, the radical then undergoes further reduction to produce the hydroquinone derivative. However, in the presence of O2, formation of the hydroquinone product is inhibited, with the semiquinone radical undergoing futile cycling to produce superoxide and regeneration of the parent compound (42, 47). In contrast, other quinone reductases, typified by NAD(P)H:quinone oxidoreductase, catalyze the 2e− reduction of the quinone to form the hydroquinone directly (15, 33, 57). Interestingly, expression of these enzymes in normal tissue is low but is elevated in cancer cells, a difference that has been exploited in the development of anticancer therapies (34). During the 2e− reduction of the quinone to its hydroquinone form, a redistribution of electrons occurs within the compound's backbone that can lead to exposure of a prodrug's cytotoxic component. For azirinidyl anticancer agents, such as mitomycin C, EO9, and AZQ, the cytotoxic agent remains attached to the hydroquinone core, whereas in other compounds, for example, phosphoramidate mustards and indolequinone-camptothecins, this “electronic switch” promotes cleavage of specific bonds in the structure's backbone, resulting in compound fragmentation and drug release (11).
Trypanosomal screening programs using natural and synthetic quinones have identified a number of lead compounds (31, 46). However, the mechanism(s) by which such agents mediate their antiparasitic activity is unclear. Based on in vitro enzyme studies and the belief that protozoan parasites have a limited enzymatic capacity to metabolize reactive oxygen species, it had been postulated that some quinones may function by inhibiting key essential enzymes such as trypanothione reductase or lipoamide dehydrogenase, while others are thought to induce oxidative stress within the parasite (18, 19, 21, 29, 37, 51). However, no demonstration of a functional link between the antiparasitic activity of any quinone-based agent and a proposed protein target within the pathogen has been reported and it is now well established that trypanosomatids possess a series of novel enzymatic oxidative defense pathways.
In many regards, the situation concerning quinone reduction mirrors the confusion that surrounded investigations of the trypanocidal mechanism of action of nifurtimox and benznidazole. It is now clear that trypanosomes activate such nitro-based drugs using a mitochondrially targeted, oxygen-insensitive type I NTR, leading to the formation of cytotoxic reduction products (26, 27, 65). This class of oxidoreductase is largely restricted to bacteria and absent from most eukaryotes, with a subset of protozoan parasites being major exceptions. The differences between the pathogens and human host in NTR distribution are believed to form the basis for the drug selectivity of nitroheterocyclic prodrugs. Here, we show that TbNTR and TcNTR display many of the characteristics displayed by their bacterial counterparts and reveal that both parasite enzymes have a preference for quinone-based substrates. Exploiting this property, we evaluated the trypanocidal activities of an aziridinyl 1,4-benzoquinone series, identifying several compounds that display high activity against bloodstream-form T. brucei.
MATERIALS AND METHODS
Abbreviations.
AZQ, 2,5-diaziridinyl-3,6-bis(carboethoxy-amino)-1,4-benzoquinone: BCA, bicinchoninic acid; BSF, bloodstream form; DCPIP, dichlorophenolindolephenol; DZQ, 2,5-diaziridinyl-1,4-benzoquinone; FAD, flavin adenine dinucleotide; FMN, flavin mononuclotide; GPDH, glycerol-3-phosphate dehydrogenase; HAT, human African trypanosomiasis; IC50, drug concentration that inhibits cell growth by 50%; IPTG, isopropyl-β-d-thiogalactopyranoside; MeDZQ, 2,5 dimethyl-3,6 diaziridinyl-1,4-benzoquinone; Ni-NTA, nickel-nitrilotriacetic acid; NTR, nitroreductase; NQO1, NAD(P)H:quinone oxidoreductase; RH1, 2,5-diaziridinyl-3-(hydroxymethyl)-6-methyl-1,4-benzoquinone; TbNTR, type I NTR from T. brucei; TcNTR, type I NTR from T. cruzi; TZQ, triaziquone [2,3,5-tris(aziridin-1-yl)cyclohexa-2,5-diene-1,4-dione].
Compounds.
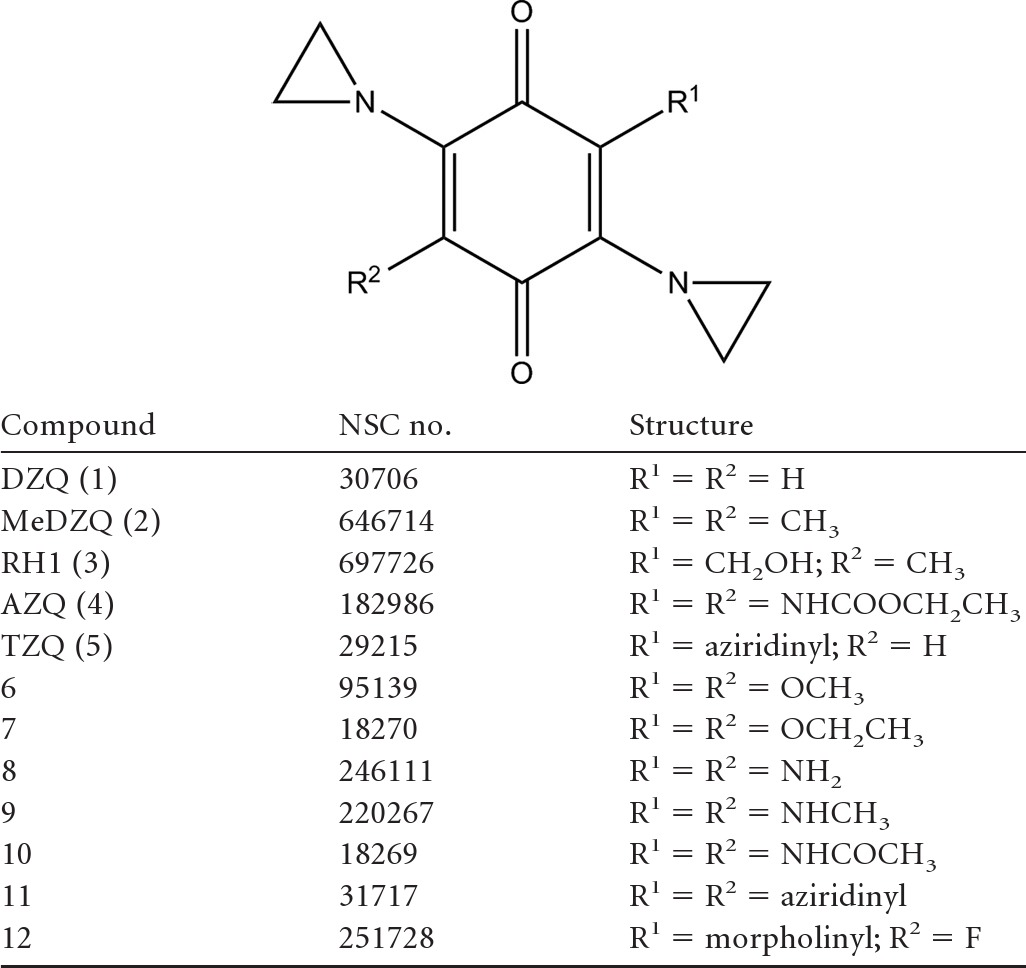
The compounds used in this study were obtained from the following sources: nitrofurazone, nitrofurantoin, CB1954, ubiquinone 5, duroquinone, and DCPIP were purchased from Sigma-Aldrich. Metronidazole was a gift from Nubia Boechat (Far Manguinhos, Rio de Janeiro, Brazil), megazol from Mike Barrett (University of Glasgow), LH7 and LH37 from Longqin Hu (Rutgers University), and amino-824 from Clifton Barry III (National Institute of Allergy and Infectious Diseases) and Ujjini Manjunatha (Novartis Institute for Tropical Diseases, Singapore). Benznidazole and nifurtimox were provided by Simon Croft (London School of Hygiene and Tropical Medicine). The aziridinyl benzoquinones (Table 1) were supplied by the Department of Therapeutics, NCI, except for RH1, which was donated by Frank Guziec, Jr. (Southwestern University, Georgetown, TX).
Table 1.
Structure of aziridinyl 1,4-benzoquinones used in this study
Parasite culturing.
T. brucei brucei (Lister 427; clone 221a and a derivative [2T1] engineered to constitutively express the tetracycline repressor protein) BSF parasites were grown in modified Iscove's medium at 37°C under a 5% CO2 atmosphere (2, 30). The 2T1 parasites transformed to overexpress TbNTR were grown in the presence of 1 μg ml−1 phleomycin and 2.5 μg ml−1 hygromycin (65).
Growth inhibition assays.
T. brucei brucei BSF parasites were seeded at 1 × 103 cells ml−1 in 200 μl of growth medium containing different concentrations of aziridinyl benzoquinone. After incubation at 37°C for 3 days, 20 μl alamarBlue (Invitrogen, Paisley, United Kingdom) was added to each well and the plates were incubated for a further 8 to 16 h. Cell densities were determined by monitoring the fluorescence of each culture using a Gemini Fluorescent Plate Reader [Molecular Devices (UK) Ltd., Wokingham, United Kingdom] at excitation λ = 530 nm, emission λ = 585 nm, and a filter cutoff at 550 nm, and the IC50 was established.
Expression constructs.
A DNA fragment encoding the catalytic domain of TcNTR (amino acids 78 to 312) was amplified from T. cruzi Sylvio X10/6 genomic DNA using the primers ggatccTGGATGCCATGAAACGTGTA and aagcttTCAAAACTTTCCCCACCGAAC (lowercase type corresponds to restriction sites incorporated into the primers to facilitate cloning). The product was digested using BamHI and HindIII and cloned into the corresponding sites of pTrcHisC. Construction of the expression vector containing the equivalent region from TbNTR is described elsewhere (28).
Oligonucleotide site-directed mutagenesis was carried out using a Stratagene QuikChange mutagenesis kit (Agilent Technologies, Stockport, United Kingdom) with pTrcHis-TcNTR as the template for DNA. Amplifications were performed in accordance with the manufacturer's instructions using primers (Eurofins) to generate each of the desired mutations. The forward primer sequences were R90A (GTGTAGTACACGAGGCACGCTCCTGCAAGCG), K94A (GAGCGTCGCTCCTGCGCGCGATTTGACCCAAC), Q124A (GCTCCTACCGCCCTGAACTTAGCGCCATGGGTGG), and R302A (CCGTACGCAGCCCAGCATTTCCCACGAAGC). The relevant substitution sites, incorporating the required base change, are underlined.
Protein purification.
His-tagged TcNTR (wild type and mutants) and TbNTR were expressed and purified as described previously (28). Briefly, overnight cultures of Escherichia coli BL21(+) containing the expression plasmid were diluted 1:50 in NZCYM (Sigma-Aldrich) medium containing 100 μg ml−1 ampicillin and grown for 2 to 4 h at 37°C. The culture was transferred to 16°C for 30 min, and then protein expression was induced by the addition of 100 μM IPTG. Cultures were incubated at 16°C for a further 20 h. Cells were formed into pellets and then lysed in a reaction mixture consisting of 50 mM NaH2PO4 (pH 7.8) and 500 mM NaCl (buffer A) containing 1% (vol/vol) Triton X-100, 1 mg ml−1 lysozyme, and a cocktail of protease inhibitors (Roche Diagnostics, Burgess Hill, United Kingdom). The clarified lysate was passed through a Ni-NTA column (Qiagen, Crawley, United Kingdom) and recombinant protein eluted using buffer A supplemented with 500 mM imidazole and 0.5% (vol/vol) Triton X-100. The protein concentration was determined using a BCA assay system (Thermo Fisher Scientific, Cramlington, United Kingdom) and the purity level confirmed by SDS-PAGE.
Flavin characterization.
The flavin cofactor bound to trypanosomal NTRs was established by determining the fluorescence spectrum in acidic and neutral buffers (16). Purified protein was desalted and boiled for 5 min. In a total volume of 100 μl, clarified supernatants (90 μl) containing 0.5 mg NTR were mixed with 10 μl 50 mM NaH2PO4 (pH 7.6) or 1 M HCl (final pH = 2.2). The fluorescence profile for each treatment was then determined using a Gemini Fluorescent Plate Reader [Molecular Devices (UK) Ltd., Wokingham, United Kingdom] with excitation λ = 450 nm and emission λ = 480 to 600 nm. The resultant patterns were compared with profiles obtained using FMN and FAD standards.
Enzyme activity.
NTR activity was measured using several electron acceptors as the substrate. A standard reaction mixture (1 ml) containing 50 mM Tris-Cl (pH 7.5), 100 μM NADH, and 100 μM electron acceptor was incubated at room temperature for 5 min. The background reaction rate was determined and the assay initiated by addition of the trypanosomal NTR (20 μg). For reaction mixtures containing nitroimidazoles, nitrobenzyl phosphoramides, or most quinones, activity was measured by following the change in absorbance at 340 nm corresponding to NADH oxidation (ε = 6,200 M−1 cm−1), whereas for assays involving nitrofurans, the direct reduction of the substrate itself was monitored at 435 nm for nifurtimox (ε = 18,000 M−1 cm−1) or at 400 nm for nitrofurazone and nitrofurantoin (ε = 12,000 and 15,000 M−1 cm−1, respectively). DCPIP reduction was measured by following the decrease in absorbance at 600 nm (ε = 21,000), and CB1954 activity was determined by the increase in absorbance at 420 nm, corresponding to production of the hydroxylamine (ε = 1,200 M−1 cm−1). Kinetic analysis was performed using GraphPad Prism 5 software.
RESULTS
Trypanosomal type I nitroreductases are FMN binding proteins.
Based on cofactor and oxygen sensitivity, NTRs can be divided into two groups: type I and type II (45). To evaluate which class the trypanosomal enzymes fall into, recombinant protein containing the catalytic domains from T. brucei or T. cruzi NTR was expressed in E. coli and purified after one round of affinity chromatography (Fig. 1A). Elutions containing recombinant proteins were yellow, and analysis of their absorbance spectra identified peaks at 370 and 461 nm (Fig. 1B), both characteristic of flavin binding type I NTRs (62, 67, 68). To determine the nature of the NTR/cofactor interaction and identify which flavin was present, recombinant protein (TbNTR and TcNTR) was denatured and the fluorescence profile of supernatants analyzed. Flavin was released from both enzymes, indicating a noncovalent association with the protein backbone (9, 62, 68), and the fluorescence profiles under conditions of neutral and acidic pHs identified FMN as a cofactor (Fig. 1C); at neutral pH and excitation at 450 nm, flavin derived from TbNTR and TcNTR had a peak emission at 535 nm, representing a signal quenched under acidic conditions, typical of FMN and distinct from FAD (Fig. 1C) (16).
Fig 1.

Trypanosomal NTRs contain FMN as a cofactor. (A) Coomassie-stained SDS-PAGE gel (10%) containing purified, recombinant TbNTR (lane 2). Lane 1, size standards. (B) Absorption spectrum (300 to 550 nm) of purified TbNTR (2 mg) in 50 mM NaH2PO. (C) Fluorescence spectra of FMN and FAD (both 25 μM) and of supernatant from boiled and purified recombinant TbNTR (0.45 μg) at pH 7.6 (solid line) and pH 2.2 (dashed line) with excitation λ at 450 nm and emission λ between 480 and 600 nm. All the fluorescence analyses were carried out in triplicate; the profiles are derived from the mean values. When TcNTR was used in place of TbNTR, similar absorption and fluorescence profiles were observed.
The overall level of conservation among type I NTRs is low, although certain key residues are highly conserved. Comparison of the trypanosomal enzymes with their bacterial counterparts identified several such amino acids, including R90, K94, and R302, which may play a role in FMN binding, and Q124, which could influence interactions with the substrate (note that the numbering is in accordance with the TcNTR sequence) (Fig. 2A) (44, 49). To evaluate the importance of these four residues in TcNTR activity, they were converted to alanine and His-tagged mutant proteins purified by affinity chromatography on a Ni2+-NTa column. After determination of the size and purity of each protein by SDS-PAGE, activity was monitored using nifurtimox as a substrate (Fig. 2B). In one mutant (R90A), a complete loss of NTR activity was observed, and this correlated with a total loss of FMN binding (Fig. 2C). The other three mutants all showed a partial (60% to 75%) inhibition of reductase activity but no significant change in their FMN binding capacity (Fig. 2B and C). Presumably, the K94, Q124, and R302 residues play some role in substrate (either NADH or nitroaromatic) binding.
Fig 2.

Site-directed mutagenesis of TcNTR. (A) Schematic of TcNTR from T. cruzi Silvio X10/6 showing the amino-terminal extension (residues 1 to 79), FMN binding motifs (residues 87 to 97 and 297 to 305), and substrate binding regions (residues 119 to 129) (44, 48, 65). Residues selected for mutagenesis are in bold, with arrows indicating the new amino acid substitution. Numbers refer to positions of amino acids in the TcNTR sequence (GenBank accession no. EFZ30370). (B) NTR activity was determined by following the reduction of nifurtimox (0 to 100 μM) at a fixed concentration of NADH (100 μM) in the presence of purified wild-type (WT) or mutant TcNTR protein (4 μg). All assays were carried out in triplicate. The apparent Vmax for each protein was calculated, and the results are presented as a percentage of the wild-type activity ± standard deviation. (C) The flavin recovered from the boiled, clarified supernatants of purified wild-type or mutant TcNTR (0.45 μg) was identified as described for Fig. 1. The fluorescence of each supernatant was measured at pH 7.6 using excitation λ = 450 nm and emission λ = 535 nm. For each protein, the fluorescence value from three independent readings was taken, and the data are presented as a percentage of the wild-type value ± standard deviation.
Substrate preference of the trypanosomal NTRs.
To investigate their substrate specificity, trypanosomal NTR activity was monitored by following NADH oxidation at 340 nm or, when a compound's absorbance spectra precluded this, by following reduction of the substrate itself (Fig. 3A). This demonstrated that both TbNTR and TcNTR functioned as typical bacterial type I NTRs, able to catalyze the aerobic reduction of a wide range of nitroaromatics and quinones (Table 2). For some 5-nitroimidazole compounds, such as the metronidazole-like drugs and a PA-824-based antituberculosis agent, no or low activity was detected using either of the parasite enzymes whereas other structures, such as those of the anti-Chagas' disease drugs nifurtimox and benznidazole, were metabolized at equivalent rates (Table 2). The biggest difference between the two trypanosomal enzymes related to their ability to reduce nitrobenzyl-based substrates. The aziridinyl dinitrobenzamidine CB1954 and the two nitrobenzyl phosphoramide mustards LH7 and LH37 were more readily metabolized by TbNTR than by TcNTR. In the case of the mustards, this property has been shown to extend to trypanocidal activity: LH37 displays considerable growth-inhibitory activity against BSF T. brucei, yielding an IC50 of 7 nM, whereas in studies of its activity against intracellular T. cruzi, an IC50 140-fold higher (0.99 μM) has been reported (28, 32).
Fig 3.

Investigating the kinetic properties of trypanosomal type I nitroreductase toward quinones. (A) Proposed scheme for the reduction of quinone- and nitroaromatic-based substrates by NTRs using NADH as the electron donor; “red” represents the reduced and “oxid” the oxidized form of the compound. The interactions of the trypanosomal NTR with NADH (reaction I) and the substrate (reaction II) are indicated. (B) NTR activity was assayed by following the reduction of DCPIP at 600 nm. In reaction I, assays were carried out using different concentrations of NADH (0 to 100 μM) in the presence of DCPIP (0.5 [●], 1.0 [■], and 2 [▼] μM) and TcNTR (20 μg). In reaction II, NTR activity was assayed by following the reduction of various concentrations of DCPIP (0 to 2 μM) in the presence of NADH (20 [♦], 40 [▼], and 60 [▲] μM) and TcNTR (20 μg). When TbNTR was used in place of TcNTR, similar enzyme kinetic profiles were observed.
Table 2.
Substrate specificity of trypansomal NTRsa
| Substrate | TbNTR |
TcNTR |
||||
|---|---|---|---|---|---|---|
| Apparent Vmax | Apparent Km | Kcat/Km | Apparent Vmax | Apparent Km | Kcat/Km | |
| Nitroimidazoles | ||||||
| Benznidazole | 419.0 ± 35.0 | 60.0 ± 13.0 | 3.0 × 103 | 564.0 ± 19 | 43.0 ± 4.0 | 6.1 × 103 |
| Megazol | 476.0 ± 21.0 | 5.0 ± 1.0 | 4.0 × 104 | 1,358.0 ± 78.0 | 22.0 ± 3.0 | 2.7 × 104 |
| Metronidazole | 93.0 ± 9.0 | 55.0 ± 13.0 | 7.3 × 102 | 0 | ||
| 6-Amino PA824 | 0 | 0 | ||||
| Nitrofurans | ||||||
| Nifurtimox | 349.0 ± 22.0 | 41.0 ± 7.0 | 3.6 × 103 | 457.0 ± 9.0 | 22.0 ± 2.0 | 9.2 × 103 |
| Nitrofurazone | 1,167.0 ± 51.0 | 10.0 ± 1.0 | 5.2 × 104 | 439.0 ± 20.0 | 13.0 ± 2.0 | 3.3 × 103 |
| Nitrofurantoin | 772.0 ± 37.0 | 1.3 ± 0.4 | 2.5 × 105 | 1,059.0 ± 10.0 | 11.0 ± 1.0 | 4.2 × 104 |
| Nitrobenzyls | ||||||
| CB1954 | 318.0 ± 103.0 | 41.0 ± 25.0 | 3.5 × 103 | 56.0 ± 4.0 | 151.0 ± 2.0 | 1.5 × 102 |
| LH7 | 245.0 ± 35.0 | 80.3 ± 25.0 | 1.3 × 103 | 25.0 ± 2.0 | 20.0 ± 8.0 | 5.3 × 102 |
| LH37 | 1,238.0 ± 48.0 | 2.8 ± 0.4 | 1.8 × 105 | 151.0 ± 13.0 | 5.0 ± 3.0 | 1.3 × 104 |
| Quinones | ||||||
| Ubiquinone 5 | 1,227.0 ± 1.0 | 0.3 ± 0.0 | 1.6 × 106 | 963.0 ± 52.0 | 4.0 ± 0.5 | 1.1 × 105 |
| Duroquinone | 852.0 ± 56.0 | 2.0 ± 0.5 | 1.5 × 105 | 384.0 ± 27.0 | 10.0 ± 2.0 | 1.6 × 104 |
| DCPIP | 999.0 ± 183.0 | 3.0 ± 1.5 | 1.4 × 105 | 1,172.0 ± 40.0 | 0.4 ± 0.1 | 1.2 × 106 |
The apparent Vmax and Km (± standard deviation) TbNTR and TcNTR (20 μg) values for various nitroaromatic and quinone-based substrates (0 to 200 μM) were determined in the presence of NADH (100 μM). For all nitroimidazoles, LH7, LH37, ubiquinone 5, and duroquinone, activity was followed by monitoring the rate of NADH oxidation and the kinetic values were calculated using ε = 6,220 M−1cm−1. For nitrofurans, assays were carried out by following the direct reduction of the nitroheterocycle. Kinetic values were calculated using ε = 18,000, 12,000, or 15,000 M−1cm−1 for nifurtimox, nitrofurazone, or nitrofurantoin, respectively, and the amount of reductant consumed per reaction was determined, assuming that 4 molecules of NADH are oxidized per molecule of nitrofuran reduced (26). In reactions using DCPIP as the substrate, direct reduction of the indophenol was followed and activity determined using ε = 21,000 M−1cm−1, assuming that 2 molecules of NADH are metabolized during reduction of 1 molecule of DCPIP (3). For CB1954, activity was monitored by detecting production of the hydroxylamine. Kinetic values were determined using ε = 1,200 M−1cm−1, and the amount of NADH oxidized per reaction was determined, assuming that 4 molecules of reductant are turned over per molecule of CB1954 reduced (7, 50). The apparent Vmax values are expressed as nanomoles of NADH oxidized per minute per milligram and apparent Km values in micromoles. The specificity constant (Kcat/Km), expressed in per molar per second, was determined and assumed one catalytic site per 30-kDa monomer. Metronidazole was not a substrate for TcNTR (n/a = no activity).
When the specific activity generated by a given substrate is compared with its affinity toward the enzyme (Kcat/Km), both trypanosomal NTRs appear to have a preference for quinones over nitroaromatics: TbNTR exhibited an apparent Kcat/Km for ubiquinone 5 (coenzyme Q1) of 1.6 × 106, whereas for nifurtimox and benznidazole, values of 9.2 × 103 and 6.1 × 103 M−1s−1 were calculated, respectively. Such preferential reduction of quinoid compounds is common among bacterial type I NTRs (9, 40, 67, 68). Interestingly, TbNTR could use NADPH and NADH as electron donors, although the apparent Km values indicate a preference for NADH: using nifurtimox (100 μM) as the substrate, TbNTR had an apparent Km value of 71 μM for NADH compared to 198 μM for NADPH. In contrast, TcNTR could use only NADH as a reductant, with a Km of 86 μM (65).
To further investigate the type of kinetics that the parasite NTRs display toward NADH, assays were carried out using various concentrations of reductant against a fixed concentration of substrate (Fig. 3B shows data relating to TcNTR reduction of DCPIP; similar results were obtained using TbNTR). For most substrates, double-reciprocal plots were linear at all concentrations of electron acceptor, with the slopes remaining parallel, a pattern characteristic of a ping-pong mechanism. Likewise, reciprocal assays using a fixed concentration of NADH and various amounts of substrate generated a similar pattern of parallel slopes (Fig. 3B). This mechanism of kinetics, typical for oxidoreductase cascades, has previously been observed when using benznidazole as an electron acceptor under normoxic conditions but not when employing nifurtimox as the substrate (26, 27). When reduced by the trypanosomal NTRs, this 5-nitrofuran forms a cytotoxic open chain nitrile while undergoing limited futile cycling. No oxygen consumption was detected during the reduction of the quinone derivatives studied here (data not shown), suggesting that such reactions, if they do occur, are negligible when using these compounds as the substrate.
Like NAD(P)H:quinone oxidoreductases, bacterial type I NTRs are highly susceptible to inhibition by dicoumarol (36, 40, 56). When using NADH and nifurtimox (both at 100 μM) as the electron donor/acceptor, dicoumarol inhibited TbNTR activity with a Ki of 14 nM (Fig. 4A), indicating that this parasite enzyme is particularly sensitive to the 4-hydroxycoumarin derivative. TcNTR was less sensitive, with a Ki of 258 nM. Double-reciprocal plots indicated that inhibition was competitive for NADH (Fig. 4B).
Fig 4.

Inhibition of trypanosomal NTR quinone reductase activity by dicoumarol. (A) DCPIP (5 μM) reduction by TbNTR (20 μg) in the presence of NADH (100 μM) was readily inhibited by dicoumarol (0 to 200 nM). Inhibitory activity is expressed as a percentage compared to uninhibited control results. (B) NTR activity was assayed by following DCPIP (5 μM) reduction in the presence of NADH (0 to 100 μM) and 0 (●), 5 (■), 10 (▲), and 20 (▼) nM dicoumarol. All reactions were initiated by addition of TbNTR (20 μg), and activity (v) is expressed in micromoles of DCPIP reduced per minute per milligram.
Taken together, the FMN binding, aerobic activity, substrate range, kinetics, and inhibition profiles of the two trypanosomal NTRs confirm that both function as classic type I NTRs.
Aziridinyl benzoquinones as TbNTR substrates and trypanocidal agents.
Aziridinyl benzoquinones have long been of interest in the treatment of cancer. These compounds are activatable by the enzyme NQO1, which is rarely expressed in most normal human tissues but is highly upregulated in many tumor cells. RH1 (compound 3), AZQ (compound 4), and TZQ (compound 5) have been shown to be effective anticancer agents in preclinical studies, with RH1 recently completing phase I trials (4, 14, 61). All appear to induce cell damage primarily by DNA alkylation and cross-linking following reductive activation (13, 66). To determine whether the trypanosomal NTR could activate aziridinyl benzoquinones, a series of compounds (Table 1) were biochemically screened to determine whether they could drive NADH oxidation (fixed at 100 μM) in the presence of TbNTR (20 μg) (Table 3). Under these conditions, seven compounds (compounds 1 to 6 and 12) were metabolized by TbNTR at respectable rates, with some displaying high affinities for the enzyme, resulting in Kcat/Km values ranging from 1.6 × 104 (compound 4) to 6.4 × 105 (compound 3). For the remaining compounds, two (compounds 9 and 10) generated relatively low activities, resulting in Kcat/Km values equivalent to those determined using benznidazole and nifurtimox, while three (compounds 7, 8, and 11) were not metabolized.
Table 3.
Activity of TbNTR toward aziridinyl benzoquinonesa
| Compound | Apparent Vmax | Apparent Km | Kcat/Km |
|---|---|---|---|
| DZQ (1) | 3,340.0 ± 267.0 | 2.9 ± 0.3 | 5.0 × 105 |
| MeDZQ (2) | 2,334.0 ± 221.0 | 6.9 ± 1.5 | 1.5 × 105 |
| RH1 (3) | 3,080.0 ± 110.0 | 2.1 ± 0.2 | 6.4 × 105 |
| AZQ (4) | 750.0 ± 30.0 | 20.6 ± 2.3 | 1.6 × 104 |
| TZQ (5) | 1,799.0 ± 180.0 | 6.5 ± 2.1 | 1.2 × 105 |
| 6 | 1,080.0 ± 30.0 | 9.9 ± 0.9 | 4.7 × 104 |
| 7 | n/a | ||
| 8 | n/a | ||
| 9 | 120.0 ± 20.0 | 30.2 ± 11.5 | 1.7 × 103 |
| 10 | 430.0 ± 50.0 | 66.7 ± 16.3 | 2.8 × 103 |
| 11 | n/a | ||
| 12 | 2,377.0 ± 231.0 | 5.2 ± 1.3 | 2.0 × 105 |
The apparent Vmax and Km (± standard deviation) TbNTR (20 μg) values for various aziridinyl benzoquinone substrates (0 to 100 μM) were determined in the presence of NADH (100 μM). TbNTR activity was followed by monitoring NADH oxidation, and the kinetic values were calculated using ε = 6,220 M−1 cm−1. The apparent Vmax values are expressed as nanomoles of NADH oxidized per minute per milligram, and apparent Km values are expressed in micromoles. The specificity constant (Kcat/Km), expressed in per molar per second, was determined by assuming one catalytic site per 30-kDa monomer. Compounds 7, 8, and 11 were not metabolized by TbNTR (n/a = no activity).
All 12 compounds were then screened to determine whether they displayed growth-inhibitory activity toward BSF T. brucei (Table 4). For all except one (compound 8), IC50s < 10 μM were calculated. Those compounds that were poor TbNTR substrates (compounds 7 to 11) generally displayed the highest IC50s, with only the tetra-aziridinyl benzoquinone (compound 11) showing significant antiparasitic activity (IC50 of 87 ± 6 nM). All the other compounds found to be effective TbNTR substrates (compounds 1 to 6 and 12) were also shown to have trypanocidal activities, with IC50s ranging from 21 nM (RH1 [compound 3]) to 1.5 μM (AZQ [compound 4]). To evaluate whether TbNTR plays a role in activating the six most potent aziridinyl benzoquinones (DZQ [compound 1], MeDZQ [compound 2], RH1 [compound 3], TZQ [compound 5], compound 6, and compound 11) within the parasite, we made use of a cell line manipulated to express elevated levels of the enzyme (65). This revealed that, with one exception, cells induced to overexpress NTR were 2- to 6-fold more sensitive to the prodrug (DZQ [compound 1], MeDZQ [compound 2], RH1 [compound 3], TZQ [compound 5], and compound 6) than noninduced controls (Table 4, Fig. 5). In only one case (compound 11) did elevation of the NTR level fail to alter susceptibility, presumably because that compound is not a substrate for this enzyme (Tables 3 and 4).
Table 4.
Trypanocidal activity of aziridinyl benzoquinonesa
| Compound |
T. brucei IC50 (nM) |
Ratio (− tet/+ tet) | ||
|---|---|---|---|---|
| Wild type | Transformed (− tet) | Transformed (+ tet) | ||
| DZQ (1) | 272 ± 1 | 318 ± 38 | 90 ± 3 | 3.5 |
| MeDZQ (2) | 698 ± 57 | 740 ± 18 | 115 ± 3 | 6.4 |
| RH1 (3) | 21 ± 1 | 12 ± 2 | 3 ± 1 | 4.0 |
| AZQ (4) | 1,590 ± 110 | |||
| TZQ (5) | 179 ± 1 | 184 ± 10 | 60 ± 9 | 3.1 |
| 6 | 283 ± 41 | 310 ± 40 | 140 ± 20 | 2.2 |
| 7 | 7,733 ± 1030 | |||
| 8 | >10,000 | |||
| 9 | 2,450 ± 328 | |||
| 10 | 6,293 ± 15 | |||
| 11 | 87 ± 6 | 121 ± 21 | 148 ± 9 | 0.8 |
| 12 | 1,253 ± 77 | |||
Data represent the growth-inhibitory effect (as judged by their IC50s) of aziridinyl benzoquinones on wild-type T. brucei (Lister 427; clone 221a). For the six most potent compounds, IC50s (± standard deviations [SD]) against parasites transformed to express elevated levels of TbNTR under tetracycline (1 μg ml−1) control were also determined. The data in columns 2 to 4 represent mean IC50s (± SD) determined in growth assays performed in triplicate.
Fig 5.

Susceptibility of bloodstream-form T. brucei to aziridinyl benzoquinones. IC50 values of the most potent aziridinyl benzoquinones on T. brucei cells induced to express elevated levels of TbNTR (black bars) compared to noninduced control values (white bars) are shown. Data represent means ± standard deviations (SD) of the results of four experiments, and the differences in susceptibility for DZQ (compound 1), MeDZQ (compound 2), RH1 (compound 3), TZQ (compound 5), and compound 6 were statistically significant (P < 0.01), as assessed by Student's t test.
DISCUSSION
Type I NTRs are a group of enzymes initially believed to be restricted to bacteria. They typically contain FMN as a cofactor and metabolize a wide range of nitroaromatic and quinone substrates in a reaction that is not affected by oxygen (45, 62, 67, 68). It is now clear that several eukaryotic protozoan parasites also express type I NTRs, with these orthologs playing a key role in the activation of the clinically used nitroaromatic prodrugs nifurtimox, benznidazole, and metronidazole (38, 41, 43, 65). Here, we report that the trypanosomal enzymes noncovalently bind FMN, utilize NADH as a reductant, and catalyze the reduction of a wide range of substrates via a ping-pong mechanism, an activity readily inhibited by dicoumarol. These properties are characteristic of their bacterial counterparts.
A distinct feature displayed by all bacterial type I NTRs is their ability to bind FMN as a cofactor, a characteristic shared by TbNTR and TcNTR (Fig. 1) (9, 62, 68). Overall sequence conservation between type I NTRs from different species (pro- and eukaryotic) is low, although some key residues are shared, with many being implicated in cofactor binding (44, 49, 65). For the E. coli nfsB sequence, nine residues located across five sites are involved in cofactor interactions (44). Two of these sites, including a characteristic R[S/T/A]X[R/H] amino-terminal cluster, mediate FMN binding via the ribityl chain. Two further regions mediate interactions via the isoalloxazine tricyclic ring, and the fifth attaches to FMN′s phosphate grouping. Of these, five amino acids (R90, S92, K94, E260, and R302 [numbering based on TcNTR]) found at three locations are also present in the two trypanosomal enzymes. Site-directed mutagenesis of TcNTR revealed that only one of the mutations, R90A, prevented cofactor interaction and generated an inactive enzyme (Fig. 2). This is the only invariable residue in an amino-terminal cluster present in all nfsB-like type I NTRs from different origins, suggesting the universal importance of this arginine in FMN binding and NTR activity. In bacterial nfsB-like enzymes, reduced NTR activity in vivo contributes to drug resistance, with residues equivalent to K94, Q124, and R302 implicated in this process (53, 63). Alterations at these sites generated mutant proteins that bound FMN at levels equivalent to those seen with the wild type but exhibited significantly reduced enzymatic activities (Fig. 2). As K94 and R302 represent two of the residues conserved between nfsB and TcNTR, as noted above, we initially predicted that conversion of the lysine or arginine to alanine at either site would affect cofactor binding. This clearly is not the case, and our results suggest that if K94 or R302 or both are involved in TcNTR/FMN binding, then their contribution to this interaction is secondary to that of R90, with a single mutation at either site insufficient to block attachment.
The spectrum of compounds that TcNTR and TbNTR can metabolize is typical of that reported for other type I NTRs, although there are slight differences in substrate specificity (Table 2) (9, 40, 62, 67, 68). For example, 5-nitroimidazoles such as metronidazole are not readily metabolized by either of the trypanosome enzymes whereas they are efficiently reduced by bacterial and other protozoal type I NTRs (24, 39, 43). When the substrate ranges of the two trypanosomal reductases are contrasted, the two enzymes yielded equivalent specificity constant values for 2-nitroimidazoles, 5-nitrofurans, and DCPIP. However, they can be distinguished based on their ability to reduce nitrobenzyls and benzoquinones, with TbNTR generating specificity constant values ∼10-fold higher than TcNTR toward the azirinidyl dinitrobenzamidine CB1954, the nitrobenzyl phosphoramide mustards LH7 and LH37, and the 1,4-benzoquinones ubiquinone 5 and duroquinone. Such disparities are not unexpected, as relatively small sequence variations can have a profound impact on substrate preference: alteration of two amino acids in the E. coli nfsB sequence is sufficient to enhance its ability to activate CB1954 by 100-fold (35). Despite these differences, based on specificity constant values, quinone-based compounds generally represent the preferred substrate for both TcNTR and TbNTR (Table 2).
The biological role of the trypanosomal NTRs is unknown. However, targeted gene deletion studies have clearly shown that they are essential to mammalian infective trypanosomes, as BSF TbNTR null mutants cannot be generated (65). Likewise, T. cruzi insect-form TcNTR null mutants are unable to differentiate into infectious metacyclic trypomastigotes and cannot infect mammalian cells (65). Based on their mitochondrial location and preference for quinone-based substrates, coupled with their similarity to FMN-dependent NADH dehydrogenases, enzymes that mediate reduction of ubiquinone to ubiquinol, the trypanosomal type I NTRs may function as ubiquinone reductases. T. brucei insect forms are reported to express several distinct ubiquinone reductases, but only two activities have been identified in BSF parasites (17–20, 25, 55). One is attributed to GPDH and the second to an uncharacterized enzyme. The difference between the parasite forms is associated with the lack of cytochrome-dependent respiratory chains in the mammalian infective stage (6, 58, 59). However, T. brucei BSFs do possess a non-cytochrome-dependent terminal oxidase, the trypanosomal alternative oxidase (TAO), which, in concert with GPDH, functions to transfer reducing equivalents from cytosol/glycosome-generated NADH via ubiquinone to O2, to produce H2O (6, 10, 12). This cascade plays a key role in energy metabolism by maintaining the cytosolic/glycosomal NADH/NAD+ balance. Potentially, TbNTR may fulfill a similar role in the parasite's single mitochondrion, helping to maintain the NADH/NAD+ balance in this organelle. As such, it may represent the “enigmatic” BSF NADH:ubiquinone oxidoreductase activity whose presence has been vigorously debated (55). Interestingly, deep sequence analysis of T. brucei populations selected for nifurtimox resistance using a whole-genome loss-of-function screen provided further evidence to link TbNTR activity with ubiquinone availability, giving additional credence to the hypothesis that the biological function of TbNTR is as a ubiquinone reductase (1). Whether this pathway functions in T. cruzi is unclear. The mitochondrial energetics of this parasite is less well characterized than that of T. brucei, particularly regarding the intracellular stage. T. cruzi does not express a direct homologue of TAO but does possess a gene that has the potential to encode a mitochondrially targeted alternative oxidase-like protein (GenBank accession number EAN97989). However, this enzyme lacks certain key features that typify this class of oxidase, and its activity has yet to be confirmed.
The marked preference of the trypanosomal NTRs for quinone substrates suggests that this class of compound might represent an alternative to nitroheterocycles for the treatment of Chagas' disease and/or HAT. Quinones are widely used in medicine as anti-infection and anticancer agents. Trypanocidal screens using natural and synthetic quinones have shown that compounds of this class have potential as antiparasitic agents, although how these mediate their activity remains unclear. Here, we evaluated whether a series of aziridinyl benzoquinone derivatives, anticancer prodrugs designed to undergo activation by the human NQO1, displayed growth inhibitory properties against BSF T. brucei and then determined if TbNTR played a role in this activity. All compounds tested contained a 1,4-benzoquinone backbone and two aziridinyl groups at the 2 and 5 positions (Table 1). For most structures, the 3 and 6 positions contained equivalent substitutions (e.g., H for DZQ [compound 1], CH3 for MeDZQ [compound 2], etc.), although in three cases divergent groupings were present (e.g., CH2OH and CH3 in RH1 [compound 3], aziridinyl and H in TZQ [compound 5], or morpholinyl and F in compound 12). Several compounds were identified as the substrates for the parasite enzyme, generating specificity constants 100-fold higher than those of nifurtimox and benznidazole (Tables 2 and 3), with RH1 (compound 3) yielding the highest specificity constant. Although no structure activity relationship could be determined from this limited biochemical screen, compounds containing amine-linked substitutions at positions 3 and 6 on the benzyl backbone were generally the poorest substrates for the parasite enzyme. In tests performed with T. brucei, the biochemical data translated into trypanocidal activity (Table 4) such that the best TbNTR substrates were the most potent against the parasite: the most effective aziridinyl quinone was RH1 (compound 3), which yielded an IC50 of 21 nM. The exception to this pattern was the tetra-aziridinyl benzoquinone (compound 11), which, although inactive in the biochemical assays, displayed significant potency (IC50 of 87 nM) against infection-form parasites. To conclusively demonstrate the link between reductase and trypanocidal activities, susceptibility to the most effective compounds was investigated using parasites engineered to overexpress TbNTR (Table 4; Fig. 5). For five of the six compounds tested, trypanosomes with elevated levels of the TbNTR were more susceptible to the aziridinyl benzoquinone than controls, indicating that this enzyme can activate those agents in vivo. The only quinone that failed to elicit a difference was the tetra-aziridinyl benzoquinone (compound 11), which was not metabolized by TbNTR in vitro.
We have now shown that the trypanosome enzymes which activate the clinically used prodrugs nifurtimox and benznidazole display many of the characteristics typical of bacterial type I NTRs: they are FMN-containing proteins that reduce a wide range of substrates via an oxygen-insensitive activity that can be readily inhibited by dicoumarol. Based on substrate preference, the trypanosomal type I NTRs can be regarded as quinone (possibly ubiquinone) reductases. We have exploited this characteristic to identify several aziridinyl benzoquinones that display significant antitrypanosomal properties. Understanding how these parasite enzymes mediate their biological function may therefore contribute to the development of novel prodrugs with improved selectivity and pharmacokinetic properties compared to those currently available.
ACKNOWLEDGMENTS
We thank John Kelly (London School of Hygiene and Tropical Medicine) for valuable discussions and comments on the manuscript.
E.L.M. is a recipient of a BBSRC Doctorial Training Studentship.
Footnotes
Published ahead of print 4 September 2012
REFERENCES
- 1. Alsford S, et al. 2012. High-throughput decoding of antitrypanosomal drug efficacy and resistance. Nature 482:232–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alsford S, Kawahara T, Glover L, Horn D. 2005. Tagging a T. brucei rRNA locus improves stable transfection efficiency and circumvents inducible expression position effects. Mol. Biochem. Parasitol. 144:142–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Armstrong JM. 1964. The molar extinction coefficient of 2,6-dichlorophenol indophenol. Biochim. Biophy. Acta 86:194–197 [DOI] [PubMed] [Google Scholar]
- 4. Begleiter A. 2000. Clinical applications of quinone-containing alkylating agents. Front. Biosci. 5:E153–E171 [DOI] [PubMed] [Google Scholar]
- 5. Bern C, Montgomery SP. 2009. An estimate of the burden of Chagas disease in the United States. Clin. Infect. Dis. 49:e52–e54 [DOI] [PubMed] [Google Scholar]
- 6. Bienen EJ, Maturi RK, Pollakis G, Clarkson AB., Jr 1993. Non-cytochrome mediated mitochondrial ATP production in bloodstream form Trypanosoma brucei brucei. Eur. J. Biochem. 216:75–80 [DOI] [PubMed] [Google Scholar]
- 7. Bot C, et al. 2010. Trypanocidal activity of aziridinyl nitrobenzamide prodrugs. Antimicrob. Agents Chemother. 54:4246–4252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brun R, Blum J, Chappuis F, Burri C. 2010. Human African trypanosomiasis. Lancet 375:148–159 [DOI] [PubMed] [Google Scholar]
- 9. Bryant C, DeLuca M. 1991. Purification and characterization of an oxygen-insensitive NAD(P)H nitroreductase from Enterobacter cloacae. J. Biol. Chem. 266:4119–4125 [PubMed] [Google Scholar]
- 10. Chaudhuri M, Ott RD, Hill GC. 2006. Trypanosome alternative oxidase: from molecule to function. Trends Parasitol. 22:484–491 [DOI] [PubMed] [Google Scholar]
- 11. Chen Y, Hu L. 2009. Design of anticancer prodrugs for reductive activation. Med. Res. Rev. 29:29–64 [DOI] [PubMed] [Google Scholar]
- 12. Clarkson AB, Jr, Bienen EJ, Pollakis G, Grady RW. 1989. Respiration of bloodstream forms of the parasite Trypanosoma brucei brucei is dependent on a plant-like alternative oxidase. J. Biol. Chem. 264:17770–17776 [PubMed] [Google Scholar]
- 13. Danson S, Ranson M, Denneny O, Cummings J, Ward TH. 2007. Validation of the comet-X assay as a pharmacodynamic assay for measuring DNA cross-linking produced by the novel anticancer agent RH1 during a phase I clinical trial. Cancer Chemother. Pharmacol. 60:851–861 [DOI] [PubMed] [Google Scholar]
- 14. Danson SJ, et al. 2011. Phase I pharmacokinetic and pharmacodynamic study of the bioreductive drug RH1. Ann. Oncol. 22:1653–1660 [DOI] [PubMed] [Google Scholar]
- 15. Ernster L, Danielson L, Ljunggren M. 1962. DT diaphorase. I. Purification from the soluble fraction of rat-liver cytoplasm, and properties. Biochim. Biophys. Acta 58:171–188 [DOI] [PubMed] [Google Scholar]
- 16. Faeder EJ, Siegel LM. 1973. A rapid micromethod for determination of FMN and FAD in mixtures. Anal. Biochem. 53:332–336 [DOI] [PubMed] [Google Scholar]
- 17. Fang J, Beattie DS. 2003. Identification of a gene encoding a 54 kDa alternative NADH dehydrogenase in Trypanosoma brucei. Mol. Biochem. Parasitol. 127:73–77 [DOI] [PubMed] [Google Scholar]
- 18. Fang J, Beattie DS. 2002. Novel FMN-containing rotenone-insensitive NADH dehydrogenase from Trypanosoma brucei mitochondria: isolation and characterization. Biochemistry 41:3065–3072 [DOI] [PubMed] [Google Scholar]
- 19. Fang J, Beattie DS. 2002. Rotenone-insensitive NADH dehydrogenase is a potential source of superoxide in procyclic Trypanosoma brucei mitochondria. Mol. Biochem. Parasitol. 123:135–142 [DOI] [PubMed] [Google Scholar]
- 20. Fang J, Wang Y, Beattie DS. 2001. Isolation and characterization of complex I, rotenone-sensitive NADH: ubiquinone oxidoreductase, from the procyclic forms of Trypanosoma brucei. Eur. J. Biochem. 268:3075–3082 [DOI] [PubMed] [Google Scholar]
- 21. Garavaglia PA, et al. 2010. Identification, cloning and characterization of an aldo-keto reductase from Trypanosoma cruzi with quinone oxido-reductase activity. Mol. Biochem. Parasitol. 173:132–141 [DOI] [PubMed] [Google Scholar]
- 22. Gascon J, Bern C, Pinazo MJ. 2009. Chagas disease in Spain, the United States and other non-endemic countries. Acta Trop. 115:22–27 [DOI] [PubMed] [Google Scholar]
- 23. Gautret P, et al. for EuroTravNet 2009. Imported human African trypanosomiasis in Europe, 2005–2009. Euro. Surveill. 14:pii=19327. http://www.eurosurveillance.org/ViewArticle.aspx? ArticleId=19327 [PubMed] [Google Scholar]
- 24. Goodwin A, et al. 1998. Metronidazole resistance in Helicobacter pylori is due to null mutations in a gene (rdxA) that encodes an oxygen-insensitive NADPH nitroreductase. Mol. Microbiol. 28:383–393 [DOI] [PubMed] [Google Scholar]
- 25. Guerra DG, Decottignies A, Bakker BM, Michels PA. 2006. The mitochondrial FAD-dependent glycerol-3-phosphate dehydrogenase of Trypanosomatidae and the glycosomal redox balance of insect stages of Trypanosoma brucei and Leishmania spp. Mol. Biochem. Parasitol. 149:155–169 [DOI] [PubMed] [Google Scholar]
- 26. Hall BS, Bot C, Wilkinson SR. 2011. Nifurtimox activation by trypanosomal type I nitroreductases generates cytotoxic nitrile metabolites. J. Biol. Chem. 286:13088–13095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hall BS, Wilkinson SR. 2012. Activation of benznidazole by trypanosomal type I nitroreductases results in glyoxal formation. Antimicrob. Agents Chemother. 56:115–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hall BS, Wu X, Hu L, Wilkinson SR. 2010. Exploiting the drug-activating properties of a novel trypanosomal nitroreductase. Antimicrob. Agents Chemother. 54:1193–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Henderson GB, et al. 1988. “Subversive” substrates for the enzyme trypanothione disulfide reductase: alternative approach to chemotherapy of Chagas disease. Proc. Natl. Acad. Sci. U. S. A. 85:5374–5378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hirumi H, Hirumi K. 1989. Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J. Parasitol. 75:985–989 [PubMed] [Google Scholar]
- 31. Hoet S, Opperdoes F, Brun R, Quetin-Leclercq J. 2004. Natural products active against African trypanosomes: a step towards new drugs. Nat. Prod. Rep. 21:353–364 [DOI] [PubMed] [Google Scholar]
- 32. Hu LQ, et al. 2011. Synthesis and structure-activity relationships of nitrobenzyl phosphoramide mustards as nitroreductase-activated prodrugs. Bioorg. Med. Chem. Lett. 21:3986–3991 [DOI] [PubMed] [Google Scholar]
- 33. Iyanagi T, Yamazaki I. 1970. One-electron-transfer reactions in biochemical systems. V. Difference in the mechanism of quinone reduction by the NADH dehydrogenase and the NAD(P)H dehydrogenase (DT-diaphorase). Biochim. Biophys. Acta 216:282–294 [DOI] [PubMed] [Google Scholar]
- 34. Jaiswal AK. 2000. Regulation of genes encoding NAD(P)H:quinone oxidoreductases. Free Radic. Biol. Med. 29:254–262 [DOI] [PubMed] [Google Scholar]
- 35. Jarrom D, et al. 2009. Steady-state and stopped-flow kinetic studies of three Escherichia coli NfsB mutants with enhanced activity for the prodrug CB1954. Biochemistry 48:7665–7672 [DOI] [PubMed] [Google Scholar]
- 36. Koder RL, Miller AF. 1998. Steady-state kinetic mechanism, stereospecificity, substrate and inhibitor specificity of Enterobacter cloacae nitroreductase. Biochim. Biophys. Acta 1387:395–405 [DOI] [PubMed] [Google Scholar]
- 37. Kubata BK, et al. 2002. A key role for old yellow enzyme in the metabolism of drugs by Trypanosoma cruzi. J. Exp. Med. 196:1241–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Müller J, Wastling J, Sanderson S, Muller N, Hemphill A. 2007. A novel Giardia lamblia nitroreductase, GlNR1, interacts with nitazoxanide and other thiazolides. Antimicrob. Agents Chemother. 51:1979–1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nillius D, Muller J, Muller N. 2011. Nitroreductase (GlNR1) increases susceptibility of Giardia lamblia and Escherichia coli to nitro drugs. J. Antimicrob. Chemother. 66:1029–1035 [DOI] [PubMed] [Google Scholar]
- 40. Nivinskas H, et al. 2002. Two-electron reduction of quinones by Enterobacter cloacae NAD(P)H:nitroreductase: quantitative structure-activity relationships. Arch. Biochem. Biophys. 403:249–258 [DOI] [PubMed] [Google Scholar]
- 41. Nixon JE, et al. 2002. Evidence for lateral transfer of genes encoding ferredoxins, nitroreductases, NADH oxidase, and alcohol dehydrogenase 3 from anaerobic prokaryotes to Giardia lamblia and Entamoeba histolytica. Eukaryot. Cell 1:181–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. O'Brien PJ. 1991. Molecular mechanisms of quinone cytotoxicity. Chem.-Biol. Interact. 80:1–41 [DOI] [PubMed] [Google Scholar]
- 43. Pal D, et al. 2009. Giardia, Entamoeba, and Trichomonas enzymes activate metronidazole (nitroreductases) and inactivate metronidazole (nitroimidazole reductases). Antimicrob. Agents Chemother. 53:458–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Parkinson GN, Skelly JV, Neidle S. 2000. Crystal structure of FMN-dependent nitroreductase from Escherichia coli B: a prodrug-activating enzyme. J. Med. Chem. 43:3624–3631 [DOI] [PubMed] [Google Scholar]
- 45. Peterson FJ, Mason RP, Hovsepian J, Holtzman JL. 1979. Oxygen-sensitive and -insensitive nitroreduction by Escherichia coli and rat hepatic microsomes. J. Biol. Chem. 254:4009–4014 [PubMed] [Google Scholar]
- 46. Pinto AV, de Castro SL. 2009. The trypanocidal activity of naphthoquinones: a review. Molecules 14:4570–4590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Powis G. 1989. Free radical formation by antitumor quinones. Free Rad. Biol. Med. 6:63–101 [DOI] [PubMed] [Google Scholar]
- 48. Purkayastha A, McCue LA, McDonough KA. 2002. Identification of a Mycobacterium tuberculosis putative classical nitroreductase gene whose expression is coregulated with that of the acr aene within macrophages, in standing versus shaking cultures, and under low oxygen conditions. Infect. Immun. 70:1518–1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Race PR, et al. 2005. Structural and mechanistic studies of Escherichia coli nitroreductase with the antibiotic nitrofurazone. Reversed binding orientations in different redox states of the enzyme. J. Biol. Chem. 280:13256–13264 [DOI] [PubMed] [Google Scholar]
- 50. Race PR, et al. 2007. Kinetic and structural characterisation of Escherichia coli nitroreductase mutants showing improved efficacy for the prodrug substrate CB1954. J. Mol. Biol. 368:481–492 [DOI] [PubMed] [Google Scholar]
- 51. Ramos EI, et al. 2009. 2,3-Diphenyl-1,4-naphthoquinone: a potential chemotherapeutic agent against Trypanosoma cruzi. J. Parasitol. 95:461–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rassi A, Marin-Neto JA. 2010. Chagas disease. Lancet 375:1388–1402 [DOI] [PubMed] [Google Scholar]
- 53. Stein DC, Carrizosa E, Dunham S. 2009. Use of nfsB, encoding nitroreductase, as a reporter gene to determine the mutational spectrum of spontaneous mutations in Neisseria gonorrhoeae. BMC Microbiol. 9:239 doi:10.1186/1471-2180-9-239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stuart K, et al. 2008. Kinetoplastids: related protozoan pathogens, different diseases. J. Clin. Invest. 118:1301–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Surve S, Heestand M, Panicucci B, Schnaufer A, Parsons M. 2012. Enigmatic presence of mitochondrial complex I in Trypanosoma brucei bloodstream forms. Eukaryot. Cell 11:183–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tatsumi K, et al. 1981. Studies on oxygen-insensitive nitrofuran reductase in Escherichia coli B/r. J. Biochem. 89:855–859 [DOI] [PubMed] [Google Scholar]
- 57. Tedeschi G, Chen S, Massey V. 1995. Active site studies of DT-diaphorase employing artificial flavins. J. Biol. Chem. 270:2512–2516 [DOI] [PubMed] [Google Scholar]
- 58. Torri AF, Bertrand KI, Hajduk SL. 1993. Protein stability regulates the expression of cytochrome c during the developmental cycle of Trypanosoma brucei. Mol. Biochem. Parasitol. 57:305–315 [DOI] [PubMed] [Google Scholar]
- 59. Torri AF, Hajduk SL. 1988. Posttranscriptional regulation of cytochrome c expression during the developmental cycle of Trypanosoma brucei. Mol. Cell. Biol. 8:4625–4633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Urech K, Neumayr A, Blum J. 2011. Sleeping sickness in travelers—do they really sleep? PLoS Negl. Trop. Dis. 5:e1358 doi:10.1371/journal.pntd.0001358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ward TH, et al. 2005. Preclinical evaluation of the pharmacodynamic properties of 2,5-diaziridinyl-3-hydroxymethyl-6-methyl-1,4-benzoquinone. Clin. Cancer Res. 11:2695–2701 [DOI] [PubMed] [Google Scholar]
- 62. Watanabe M, Nishino T, Takio K, Sofuni T, Nohmi T. 1998. Purification and characterization of wild-type and mutant “classical” nitroreductases of Salmonella typhimurium. L33R mutation greatly diminishes binding of FMN to the nitroreductase of S. typhimurium. J. Biol. Chem. 273:23922–23928 [DOI] [PubMed] [Google Scholar]
- 63. Whiteway J, et al. 1998. Oxygen-insensitive nitroreductases: analysis of the roles of nfsA and nfsB in development of resistance to 5-nitrofuran derivatives in Escherichia coli. J. Bacteriol. 180:5529–5539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wilkinson SR, Kelly JM. 2009. Trypanocidal drugs: mechanisms, resistance and new targets. Expert Rev. Mol. Med. 11:e31 http://dx.doi.org/10.1017/S1462399409001252 [DOI] [PubMed] [Google Scholar]
- 65. Wilkinson SR, Taylor MC, Horn D, Kelly JM, Cheeseman I. 2008. A mechanism for cross-resistance to nifurtimox and benznidazole in trypanosomes. Proc. Natl. Acad. Sci. U. S. A. 105:5022–5027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yan C, Kepa JK, Siegel D, Stratford IJ, Ross D. 2008. Dissecting the role of multiple reductases in bioactivation and cytotoxicity of the antitumor agent 2,5-diaziridinyl-3-(hydroxymethyl)-6-methyl-1,4-benzoquinone (RH1). Mol. Pharmacol. 74:1657–1665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zenno S, et al. 1996. Biochemical characterization of NfsA, the Escherichia coli major nitroreductase exhibiting a high amino acid sequence homology to Frp, a Vibrio harveyi flavin oxidoreductase. J. Bacteriol. 178:4508–4514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zenno S, Koike H, Tanokura M, Saigo K. 1996. Gene cloning, purification, and characterization of NfsB, a minor oxygen-insensitive nitroreductase from Escherichia coli, similar in biochemical properties to FRase I, the major flavin reductase in Vibrio fischeri. J. Biochem. 120:736–744 [DOI] [PubMed] [Google Scholar]