

Abstract
The mechanism of action of AFN-1252, a selective inhibitor of Staphylococcus aureus enoyl-acyl carrier protein reductase (FabI), which is involved in fatty acid biosynthesis, was confirmed by using biochemistry, macromolecular synthesis, genetics, and cocrystallization of an AFN-1252–FabI complex. AFN-1252 demonstrated a low propensity for spontaneous resistance development and a time-dependent reduction of the viability of both methicillin-susceptible and methicillin-resistant S. aureus, achieving a ≥2-log10 reduction in S. aureus counts over 24 h, and was extremely potent against clinical isolates of S. aureus (MIC90, 0.015 μg/ml) and coagulase-negative staphylococci (MIC90, 0.12 μg/ml), regardless of their drug resistance, hospital- or community-associated origin, or other clinical subgroup. AFN-1252 was orally available in mouse pharmacokinetic studies, and a single oral dose of 1 mg/kg AFN-1252 was efficacious in a mouse model of septicemia, providing 100% protection from an otherwise lethal peritoneal infection of S. aureus Smith. A median effective dose of 0.15 mg/kg indicated that AFN-1252 was 12 to 24 times more potent than linezolid in the model. These studies, demonstrating a selective mode of action, potent in vitro activity, and in vivo efficacy, support the continued investigation of AFN-1252 as a targeted therapeutic for staphylococcal infections.
INTRODUCTION
Methicillin-resistant Staphylococcus aureus (MRSA) is a major cause of patient morbidity and mortality and associated health care costs (1, 31). The emergence of new pathogenic strains has led to the recognition of community-associated MRSA (CA-MRSA), as well as hospital-associated MRSA (HA-MRSA) (11, 49). The often multidrug resistant CA-MRSA strains, e.g., USA300, now cause more pneumonia and invasive infections than nosocomial HA-MRSA strains do (17, 18, 23, 31, 35). The ongoing evolution of drug-resistant pathogenic staphylococci and the resulting clinical burden require the development of a new mechanism of action and, ideally, oral antibacterials to provide effective treatment.
Over the last decade, genomics-based novel antibacterial target discovery programs identified processes that are essential for the viability of pathogens (21, 37). Disruption of the bacterial fatty acid (FASII) biosynthetic pathway (Fig. 1) emerged as an early focus and has been extensively reviewed (e.g., see references 10, 27, 32, 33, and 38). The fatty acid biosynthetic cycle results in elongation of the fatty acid by two carbons, an acetate unit, during each cycle. FabI performs the last step in the pathway, and its inhibition prevents elongation of the acyl chain, disrupting both saturated and unsaturated fatty acid biosynthesis and preventing bacterial growth. In bacteria, the fatty acid biosynthetic enzymes are discrete proteins encoded by separate genes. In mammals, the activities are fused into a single protein (2). This organizational and structural difference between bacteria and mammals provided an a priori reason why inhibitors of bacterial fatty acid biosynthesis should be selective.
Fig 1.

Bacterial fatty acid biosynthesis.
The identification of enoyl-acyl carrier protein (ACP) reductase (FabI) as the target of diazaborines in Escherichia coli, of isoniazid and ethionamide in Mycobacterium tuberculosis, and of triclosan in S. aureus provided evidence that FabI is essential in vivo in at least some bacterial species and demonstrated that FabI is amenable to inhibition by small, drug-like compounds (3, 5, 6, 8, 24, 27, 28, 33, 36, 42, 52, 54).
Comparative bacterial genomics revealed additional and unexpected complexity in bacterial strategies used to synthesize fatty acids. In some bacterial species, FabI was absent and was replaced, e.g., by FabK in Streptococcus pneumoniae or by FabV in Vibrio cholerae (34). Sometimes FabI and FabK were both present, as in Enterococcus faecalis and Pseudomonas aeruginosa (25), or FabI and FabL were both present, as in Bacillus subtilis (26). A consequence of this genetic complexity and enzymatic heterogeneity and redundancy was that a specific FabI inhibitor would have a relatively narrow spectrum of activity.
FabI is the sole form of enoyl-ACP reductase present in S. aureus, S. epidermidis, and other staphylococci. No alternative enzyme or rescue pathway, e.g., exogenous fatty acids, for FabI in staphylococci has been identified, which suggests that FabI is essential to cell viability in Staphylococcus spp. and raises the possibility that FabI inhibition could be considered a targeted antistaphylococcal therapy.
An iterative structure-guided optimization of a collection of FabI inhibitors (38, 41) was initiated (45, 48), evaluating 500 additional compounds and delivering a 4- to 8-fold increase in antistaphylococcal potency before nominating AFN-1252 as a lead development candidate (Fig. 2) (29, 30, 40). AFN-1252 is composed of a 3-methylbenzofuran and an oxotetrahydronaphthyridine linked by an N-methylpropenamide. It demonstrates exceptional potency and specificity against staphylococcal isolates, with typical S. aureus MIC ranges of 0.002 to 0.12 μg/ml and MIC90s of ≤0.015 μg/ml (29, 30). It is not active against a wide range of other Gram-positive or Gram-negative bacteria. FabI was genetically confirmed as the primary in vivo target of AFN-1252 through the isolation and characterization of spontaneous AFN-1252-resistant S. aureus strains containing FabI mutations at a frequency of 1 × 10−10 to 2 × 10−9 (40).
Fig 2.
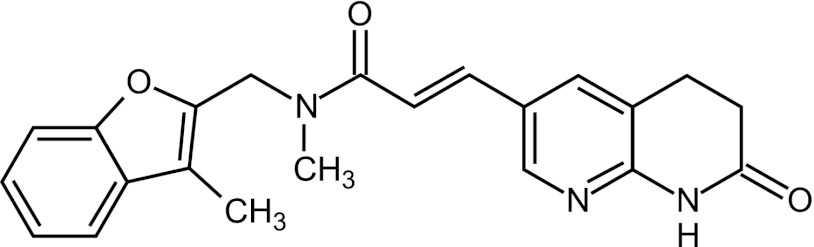
Structure of AFN-1252.
The present study focused on the characterization of the mechanism of action, target interaction, and spontaneous resistance frequency of AFN-1252; its activity against panels of well-characterized staphylococcal isolates, including CA-MRSA, HA-MRSA, and coagulase-negative strains; and the demonstration of the in vivo efficacy of AFN-1252 in a murine model of staphylococcal sepsis.
MATERIALS AND METHODS
Antibacterial agents.
AFN-1252, (2E)-N-methyl-N-[(3-methyl-1-benzofuran-2-yl)methyl]-3-(7-oxo-5,6,7,8-tetrahydro-1,8-naphthyridin-3-yl)prop-2-enamide (C22H21N3O3; relative molecular weight, 375.42), was prepared as a free base (AFN-12520000) by Affinium Pharmaceuticals. In some experiments, the tosylate anhydrate form (AFN-12520300) or a tosylate monohydrate salt (AFN-12520301) was used. In all cases, AFN-1252 concentrations are reported as AFN-1252 free-base equivalents. Vancomycin, rifampin, levofloxacin, tetracycline, daptomycin, and triclosan were purchased from Sigma-Aldrich (St. Louis, MO), and linezolid was obtained from Pfizer, Inc. (Kalamazoo, MI).
Enoyl-ACP (FabI) expression and purification. (i) Expression.
S. aureus fabI was cloned from genomic DNA of S. aureus ATCC 35556 into the multiple cloning site of expression vector pPW2, a derivative of pET15b (Novagen, Milwaukee, WI), to create a His-tagged FabI protein to enable rapid purification (26). The cloned fabI gene sequence was verified by PCR sequencing. Following expression of the cloned protein from E. coli cells, the expression of the correct protein size was verified by mass spectrometry of the excised gel band to determine its molecular weight.
(ii) Purification.
Cells containing His-tagged S. aureus FabI were thawed on ice, lysed in the presence of 0.5% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS), 0.2 mM phenylmethylsulfonyl fluoride, 0.5 mM benzamidine, and 250 U of Benzonase (Novagen, WI) in binding buffer (50 mM HEPES [pH 7.5], 500 mM NaCl, 5 mM imidazole, 5% glycerol) by sonication (Branson; VWR Scientific, Radnor, PA) on ice, and centrifuged at 24,000 rpm (Beckman J-25I, GA-25.50 rotor), and the clarified cell lysates were applied to a 20-ml DE52 column (Whatman, Maidstone, Kent, United Kingdom). The flowthrough from the ion-exchange column was applied to 5 ml of metal chelate chromatography resin (Superflow; Qiagen) charged with Ni2+. The column was washed with 4 column volumes of binding buffer and 20 column volumes of wash buffer (binding buffer containing 30 mM imidazole) and eluted with 4 to 6 column volumes of elution buffer (binding buffer containing 250 mM imidazole). The elution fractions containing FabI protein were pooled and dialyzed overnight against 10 mM HEPES–500 mM NaCl and concentrated to greater than 20 mg/ml.
Enoyl-ACP reductase (FabI) assay.
NADPH consumption was monitored by UV absorbance at 340 nm (41). Briefly, the assay mixture contained 100 mM Na–N-(2-acetamido)-2-iminodiacetic acid (pH 6.5), 50 μM NADPH, 25 μM crotonyl-ACP, 4% glycerol, 3 nM FabI, 10% dimethyl sulfoxide, 0.1% Pluronic F-68, and 30 nM recombinant S. aureus FabI at 30°C. Crotonyl-ACP and FabI concentrations were varied according to the experimental design.
Human FAS1 cloning, expression, and purification.
The construction of a His-tagged human FAS1 baculovirus expression vector, expression of the His-tagged FAS1 protein in Sf9 insect cells, and its purification by nickel affinity column chromatography were achieved by using generic protocols (2).
Human FAS1 enzyme assay.
NADPH consumption was monitored by UV absorbance at 340 nm. The assay mixture contained 50 mM potassium phosphate (pH 6.5), 4 mM dithiothreitol (DTT), 4% glycerol, 20 μM acetyl coenzyme A (acetyl-CoA), 100 μM NADPH, 75 μM malonyl-CoA, 25 mM ammonium acetate, 0.5% pluronic F-68, and 20 nM recombinant human FAS1 at 30°C. Cerulenin was used as a positive control for inhibition of human FAS1 and demonstrated a 50% inhibitory concentration (IC50) of 21 μM. Compounds were evaluated at 10 concentrations ranging from 2.6 to 100 μM. Wells containing insoluble compound were eliminated from the data analysis.
Macromolecular synthesis inhibition.
Overnight S. aureus ATCC 29213 cells were washed, resuspended in M9 minimal medium, and adjusted to 1.5 × 108 CFU/ml (optical density at 600 nm [OD600], ∼0.25). Test cultures were exposed to antibacterials, typically at 2 times the MIC, for 10 min prior to 20 min of incubation with 3H-radiolabeled precursors (3H-labeled N-acetylglucosamine, 3H-labeled thymidine, 3H-labeled uridine, and 3H-labeled isoleucine [Amersham]) or 3H-labeled acetic acid (Perkin-Elmer). The cultures were precipitated in ice-cold 5% trichloroacetic acid at 4°C for 1 to 2 h, and the macromolecular radioactivity in the precipitate was collected by vacuum filtration and counted by scintillation. Normalized results (averages of two independent experiments) were calculated by dividing the radioactivity values of the treated samples by the control (no antibacterial) values and are represented as a percentage of the control, where 100% equals no inhibition. Known reference antibacterials were used as controls for inhibition of the different pathways. The radiolabeled precursors (target pathway and control antibacterials) used were N-acetylglucosamine (cell wall, vancomycin), thymidine (DNA, levofloxacin), uridine (RNA, rifampin), isoleucine (protein, tetracycline), and acetate (lipid, triclosan).
Cocrystallization and structure solution.
Crystals suitable for X-ray experimentation were obtained by sitting-drop vapor diffusion against a 100-μl reservoir solution containing 32% (wt/vol) polyethylene glycol 400, 0.1 M Na HEPES (pH 7.4), and 0.2 M calcium acetate in a 96-well sitting-plate format. A 1.5-μl volume of a 15-mg/ml concentration of FabI protein mixed with 1.5 mM 3′NADPH, 0.5 mM DTT, and 1.5 mM AFN-1252 and a 1.5-μl reservoir solution volume were set in each drop. All data were collected on crystals frozen in 1 part Paratone and 1 part mineral oil at 100 K in a cold gas stream. Data were collected at the Advanced Photon Source at Argonne National Labs, X26C beamline. Data were indexed, integrated, and scaled by using HKL2000 (39). The S. aureus FabI complex structure was solved by molecular replacement by using the coordinates of E. coli FabI (Protein Data Bank entry 1MFP) (51) as the search model for molecular replacement with the CCP4 program suite (14). Alternate cycles of restrained refinement and manual rebuilding were performed with the program Coot (19). Five percent of the reflections were randomly excluded from the refinement and used to monitor the free residual-factor (Rfree) value. A summary of the data reduction and structure refinement statistics is provided in Table S1 in the supplemental material. Coordinates of the X-ray structure have been deposited in the Protein Data Bank (accession code PDB 4FS3).
Bacterial strains.
S. aureus ATCC 29213 (methicillin-susceptible S. aureus [MSSA]), S. aureus ATCC 35556 (MSSA), and S. aureus ATCC 43300 (MRSA) were obtained from the American Type Culture Collection; S. epidermidis 1024939 (methicillin-susceptible S. epidermidis [MSSE], clinical isolate) was from Affinium Pharmaceuticals Inc. (Toronto, Ontario, Canada); and S. aureus 2293 (CA-MRSA), 1137 (HA-MRSA), 2012 (vancomycin-intermediate MRSA), and 1725 (linezolid-resistant MRSA) were from Micromyx LLC (Kalamazoo, MI).
The following panels of isolates were used for evaluation of AFN-1252 activity against diverse staphylococci. Panel 1 included 100 S. aureus isolates, comprising 62 CA-MRSA, 17 HA-MRSA, and 21 MSSA strains, that were selected from the University of California, San Francisco, strain collection according to their sequence type (ST), the presence or absence of mecA and Panton-Valentine leucocidin (PVL) genes, a staphylococcal chromosomal cassette element (SCCmec), pulsed-field gel electrophoresis (PFGE) profiles, and antibacterial phenotypes (Table 1). Panel 2 included 199 clinical S. aureus isolates from the Canadian Bacterial Surveillance Network, comprising 104 MSSA and 86 MRSA strains as determined by mecA gene PCR, of which 22 were CA-MRSA (presence of type IV SCCmec) and 64 were HA-MRSA strains (presence of type II or III SCCmec). Panel 3 included 100 coagulase-negative staphylococcal clinical isolates from the Mount Sinai Hospital (Toronto, Ontario, Canada) that were characterized according to their methicillin resistance by measurement of their oxacillin MIC (methicillin-resistant Staphylococcus, MRS; methicillin-sensitive Staphylococcus, MSS) and identified to the species level by sequencing of their 16S rRNA, comprising S. lugdunensis (52 MSS strains), S. epidermidis (n = 27; 16 MRS strains, 11 MSS strains), S. schleiferi subsp. coagulans (5 MSS strains), S. schleiferi subsp. schleiferi (5 MSS strains), S. hominis (n = 4; 2 MRS strains, 2 MSS strains), S. pasteuri subsp. warneri (n = 4; 1 MRS strain, 3 MSS strains), S. haemolyticus (2 MRS strains), and S. capitis (1 MSS strain).
Table 1.
Genotypic characterization of the 100 unique S. aureus strains in this study
| Isolate group (no. of isolates) and CDC PFGE profile | MLST(s)a | No. of strains | No. of PFGE subtypes | SCCmec type | No. PVL+ |
|---|---|---|---|---|---|
| CA-MRSA (62) | |||||
| USA300 | ST8 | 26 | 10 | IV | 26 |
| USA400 | ST1 | 4 | 2 | IV | 4 |
| USA500 | ST8 | 6 | 4 | IV | 0 |
| USA700 | ST72 | 5 | 2 | IV | 0 |
| USA1100 | ST30 | 11 | 5 | IV | 11 |
| USA1000 | ST59 | 3 | 1 | IV | 0 |
| USA1000 | ST12, ST729 | 4 | 2 | 0 | |
| USA1000 | ST87, ST97, ST231 | 3 | 0 | ||
| HA-MRSA (17) | |||||
| USA100 | ST5 | 12 | 11 | II | 0 |
| USA200 | ST3 | 4 | 3 | II | 0 |
| USA600 | ST45 | 1 | 1 | II | 0 |
| MSSA (21) | ST5 | 2 | |||
| ST8 | 9 | ||||
| ST12 | 1 | ||||
| ST45 | 3 | ||||
| ST36 | 1 | ||||
| ST88 | 1 | ||||
| ST730 | 1 | ||||
| ST188 | 1 | ||||
| ST474 | 1 | ||||
| ST731 | 1 |
MLST, multilocus ST.
Antibacterial susceptibility testing.
MICs were determined in accordance with the appropriate Clinical and Laboratory Standards Institute (CLSI) methodology (13) by the broth microdilution method, by using cation-adjusted Mueller-Hinton broth (MH). CLSI and European Committee on Antimicrobial Susceptibility Testing breakpoint criteria for AFN-1252 have not been established to date.
Spontaneous resistance frequency.
Selection of spontaneous resistant mutants from seven parent strains was performed at 4, 8, 16, and 128 times the MICs of AFN-1252 and the comparator linezolid. Plates were incubated at 35°C in ambient air for 48 h (S. aureus ATCC 29213 and ATCC 43300 and S. epidermidis 1024939) or for 72 h (S. aureus 1137, 1725, 2012, and 2293). Resistant colonies were counted and restreaked on compound-containing plates to confirm uniformity and the resistance phenotype. The spontaneous mutation frequency at a particular drug concentration was determined by counting the confirmed resistant-colonies growing on drug-containing plates and dividing by the total inoculum size across all of the plates at that concentration.
Serial-passage studies.
Selection of S. aureus ATCC 43300 mutants with reduced susceptibility to AFN-1252, vancomycin, and linezolid was performed by serial passage in 96-well plates without purification of single colonies between passages. At each passage step, the inoculum was taken from the tube containing the highest concentration of the agent supporting visible growth (typically 1 dilution below the MIC). This process was repeated for 24 consecutive days.
Time-kill studies.
An inoculum was prepared from 5 to 10 single colonies of S. aureus ATCC 29213 (MSSA) or ATCC 43300 (MRSA) in 5 ml of cation-adjusted MH at 35°C with agitation for 2 h. This culture was then diluted in MH to a concentration of 106 CFU/ml in the test wells. Time-kill studies were performed with 2 ml of MH in 24-well plates with constant shaking at 35°C for 24 h. Antibacterial solutions (0.1 ml) at 10 times the desired final drug concentrations were added to 0.9 ml of the 106-CFU/ml bacterial inocula in MH. Each well received growth medium alone (negative control); growth medium and the inoculum (growth control); or growth medium, an antibacterial compound at a multiple of the MIC, and the inoculum (test). At 0, 4, 8, and 24 h of incubation, a 10-μl sample of each culture was serially diluted 10-fold in 0.9% NaCl. Twenty-microliter samples were plated in triplicate tryptic soy agar plates. These plates were then incubated at 35°C overnight, and the colonies were counted to determine the bacterial titer of each sample. The lower limit of detection was 50 CFU/ml.
fabI gene DNA sequencing.
The fabI genes from wild-type and AFN-1252-resistant spontaneous mutants were amplified by PCR, and the DNA was sequenced by using oligonucleotide primers. The primers used to sequence S. aureus fabI were designed with reference to the published sequence of S. aureus N315 (GenBank accession no. 14349174), and those used for S. epidermidis fabI were designed with reference to the sequence of S. epidermidis 12228 (GenBank accession no. 27315075). Three pairs of primers (forward and reverse) were designed for each species, each consisting of approximately 20 bp and spaced about 450 bp from each other (see Table S2 in the supplemental material). DNA sequencing was performed by ACTG DNA Technologies Corporation (Toronto, Ontario, Canada) by using all six primers for each amplification product. Sequence alignments, consensus sequence construction, and translation to amino acid sequences were all performed by using BioEdit V 7.04. Aligned primer sequences resulted in at least 3-fold coverage over the entire fabI gene.
Measuring the level of FabI expression.
The level of FabI protein expression in S. aureus strains was quantified by Western blotting by using polyclonal rabbit antibodies against recombinant S. aureus FabI and S. aureus FtsZ (Cedarlane Labs Ltd., Burlington, Ontario, Canada). All of the samples tested were normalized for cell mass (OD) and protein content, and FabI expression levels were normalized against the constitutively expressed cell division maintenance protein FtsZ.
Six to 10 colonies of each strain were inoculated into 3 ml of cation-adjusted MH in sterile 15-ml Falcon tubes and grown at 37°C with shaking at 250 rpm for 3 to 4 h. All of the cultures were then normalized to an OD600 of 1.
One milliliter of each normalized culture was collected by centrifugation in a sterile Eppendorf tube, and the pellet was resuspended in 25 μl of Triton X mix (50 mM KCl, 0.1% Triton X-100, 10 mM Tris-HCl [pH 9]) and 25 μl of lysostaphin mix (10 mM Tris-HCl [pH 9], 1 mM EDTA, 0.35 M sucrose, 0.2 mg/ml lysostaphin). Thirty units of Benzonase was added to the samples and incubated at 37°C for 10 min, and SDS was added to a final concentration of 2%. The samples were heated at 95°C for 10 min and then centrifuged at 14,000 rpm for 10 min in a benchtop centrifuge. The supernatants were then transferred to fresh Eppendorf tubes.
The protein concentration in the lysates was determined by using a Bio-Rad DC kit in accordance with the manufacturer's instructions. The lysates were adjusted to the desired concentration by using 2% SDS. NuPAGE sample buffer was added, and the samples were heated at 95°C for 10 min prior to being loaded onto NuPAGE gels.
Western immunoblotting was performed with the TE 70 Series SemiPhor Semi-Dry Transfer Unit in accordance with the manufacturer's instructions. The nitrocellulose membrane was blocked with 5% (wt/vol) skim milk powder in phosphate-buffered saline–1% Tween 80 (PBS-T) for 1 h on a rocking platform. The primary antibody was then added to the blocking buffer, and the membrane was incubated overnight at 4°C with rocking. The following day, the membrane was washed three times with PBS-T and incubated with the appropriate horseradish peroxidase-conjugated secondary antibody (diluted in PBS-T) for 1 h at room temperature with rocking. The membrane was washed three more times with PBS-T before it was developed with the ECL Western blotting detection system in accordance with the manufacturer's instructions. The images were captured and densitometry was performed with the Labworks software on the EpiChemi II Darkroom.
The densitometric analysis was calibrated such that a sample loaded typically gave a FabI or FtsZ signal within the linear response range of the respective recombinant purified protein.
Pharmacokinetics.
The pharmacokinetics of AFN-1252 formulated in 1% Poloxamer 407 (Sigma-Aldrich, St. Louis, MO) and administered to mice by oral gavage were determined at three different dose levels (0.3, 0.6, and 1 mg/kg). Blood was collected by cardiac puncture at 0.25, 0.5, 1, 2, 4, 6, 8, 10, and 24 h into Na-heparin tubes (three mice per time point, collected individually), stored on ice, and centrifuged to collect plasma. Plasma samples were kept frozen at −80°C. Plasma samples were analyzed for the drug and the internal standard via liquid chromatography-tandem mass spectrometry.
Acute lethal infection model.
The in vivo efficacy of AFN-1252 was demonstrated in a systemic mouse infection model performed in an American Association for Laboratory Animal Science- and USDA-accredited facility and under an IACUC-approved protocol (12, 22). Female 5- to 6-week-old CD-1 mice (18 to 22 g) were housed in groups of five with free access to food and water during the study. Mice were challenged by intraperitoneal injection (0.5 ml) of a predetermined S. aureus Smith bacterial inoculum suspended in hog gastric mucin. The 50% lethal dose (LD50) of the S. aureus strain was 5.07 × 103 CFU. The animals in the infected groups received an inoculum equivalent to 35 LD50s, which killed the nontreated control animals within 24 to 48 h. Antibacterial doses of AFN-1252 were prepared in aqueous 1% Poloxamer 407 formulations at doses of 3, 1, 0.3, 0.1, and 0.03 mg/kg (five doses). A linezolid stock solution was diluted in 5% dextrose in water and used as the positive-control compound in all tests. Compounds were administered as a single oral dose at 0.5 h postinfection. The protective effects of the antibacterial agents were measured by comparing the survival of the infected untreated animals with that of the treated animals. For this determination, animals were observed for 5 days after treatment. A census of survivors was taken twice daily, and at that time, dead and moribund animals were removed. Tests were performed three times for each dose on different days. The results of these tests were used to determine the median effective dose (ED50) by using a computerized program for Probit analysis (46, 47).
RESULTS
Biochemical characterization of the inhibition of S. aureus FabI by AFN-1252.
Confirmation that AFN-1252 is a potent and selective inhibitor of bacterial enoyl-ACP reductase was achieved through detailed biochemical characterization of its effects on the S. aureus FabI-catalyzed reduction of crotonyl-ACP (40). First the IC50 of AFN-1252 for S. aureus FabI was determined to be 14 nM (Not shown). Then the kinetics of FabI inhibition was followed by measuring the rate of NADPH oxidation to determine the Km of S. aureus FabI for crotonyl-ACP. The concentration of crotonyl-ACP was varied from 0 to 25 μM in the presence of 30 nM FabI. The Km of S. aureus FabI for crotonyl-ACP was 3.1 ± 1.9 μM. The Ki of AFN-1252 was determined by varying the concentrations of both AFN-1252 and crotonyl-ACP (15). Plots of [crotonyl-ACP]/V versus [AFN-1252], where V is the rate (M/s), confirmed that AFN-1252 was a potent inhibitor of enoyl-ACP reductase with an estimated Ki of 12.8 ± 0.5 nM (Fig. 3). In contrast, no inhibition of human FAS1 by AFN-1252 concentrations of up to 67 μM was observed (not shown). The FabI/FAS1 selectivity ratio of the bacterial enzyme is >4,800.
Fig 3.

Determination of the Ki of AFN-1252 for S. aureus FabI. Crotonyl-ACP (substrate) concentrations [S]/reaction velocity (V) were plotted against AFN-1252 (inhibitor) concentrations at three different substrate concentrations, i.e., 6.1 μM (▲), 25 μM (■), and 50 μM (⧫).
Macromolecular synthesis inhibition by AFN-1252 in S. aureus.
Confirmation that the fatty acid biosynthetic pathway was the primary in vivo target of AFN-1252 was achieved by demonstrating that the incorporation of radiolabeled acetate into fatty acids was particularly sensitive to exposure to AFN-1252. A summary of the effects of AFN-1252 and reference antibacterials on the incorporation of radiolabeled precursors of macromolecule synthesis is shown in Table 2. All reference control antibacterials selectively inhibited the macromolecule synthesis pathway, consistent with their known mechanism of action. Following treatment with AFN-1252, incorporation of acetate into lipids was 24% of the control whereas the levels of incorporation of N-acetylglucosamine (cell wall), thymidine (DNA), uridine (RNA), and isoleucine (protein) were 90, 101, 106, and 61% of the control values, respectively, showing that AFN-1252 selectively inhibits fatty acid biosynthesis, consistent with its mechanism of action at the enzyme level. Selective inhibition of acetate incorporation was also observed with triclosan, another inhibitor of FabI.
Table 2.
Effects of AFN-1252 and reference antibacterials on incorporation of radiolabeled precursors into S. aureus 29213 cells
| Precursor | Synthesis pathway | Normalized % precursor incorporationa |
|||||
|---|---|---|---|---|---|---|---|
| Vancomycin | Levofloxacin | Rifampin | Tetracycline | Triclosan | AFN-1252 | ||
| N-Acetylglucosamine | Cell wall | 19 | 95 | 73 | 82 | 85 | 90 |
| Thymidine | DNA | 101 | 35 | 113 | 75 | 89 | 101 |
| Uridine | RNA | 58 | 93 | 22 | 103 | 99 | 106 |
| Isoleucine | Protein | 62 | 81 | 61 | 22 | 59 | 61 |
| Acetate | Lipid | 42 | 86 | 97 | 55 | 36 | 24 |
A value of 100% represents the untreated control. Bold values are reference or specific antibacterials for each synthesis pathway.
Cocrystal structure of the AFN-1252–FabI complex.
The 1.8 Å structure for residues 1 to 256 of the S. aureus FabI–3′NADPH–AFN-1252 ternary complex was solved by molecular replacement (Protein Data Bank accession code PDB 4FS3). The use of the unnatural substrate 3′NADPH allowed the ternary complex to crystallize easily. The overall structure was similar to the S. aureus FabI apo-crystal (Protein Data Bank accession codes 3GNS and 3GNT) and the S. aureus FabI–NADP–triclosan ternary complex (Protein Data Bank accession code 3GR6 (43). In addition to the FabI protein, both 3′NADPH and AFN-1252 were visible in the active site. As described by Priyadarshi et al. (43), FabI forms a single domain composed of a seven-stranded parallel β-sheet flanked on each side by three α-helices with a further helix lying at the C terminus of the β-sheet. The cofactor is bound in an extended conformation at the COOH-terminal end of the β-sheet, with the nicotinamide ring lying deep in a pocket on the enzyme surface. A loop of the protein, residues 194 to 204, covers the AFN-1252 molecule in the binding pocket. It is known that this flexible loop can adopt different conformations in the formation of a ternary complex (43, 50). FabI has a cavity available for inhibitor or substrate binding above the NADPH (Fig. 4a). One side of the cavity is open and exposed to solvent (above the adenine ribose of the NADPH), and the other side contains a small opening (above the nicotinamide ring of NADPH). Overall, the pocket is hydrophobic in nature, with many aromatic residues clustered in the binding pocket (Fig. 4b).
Fig 4.
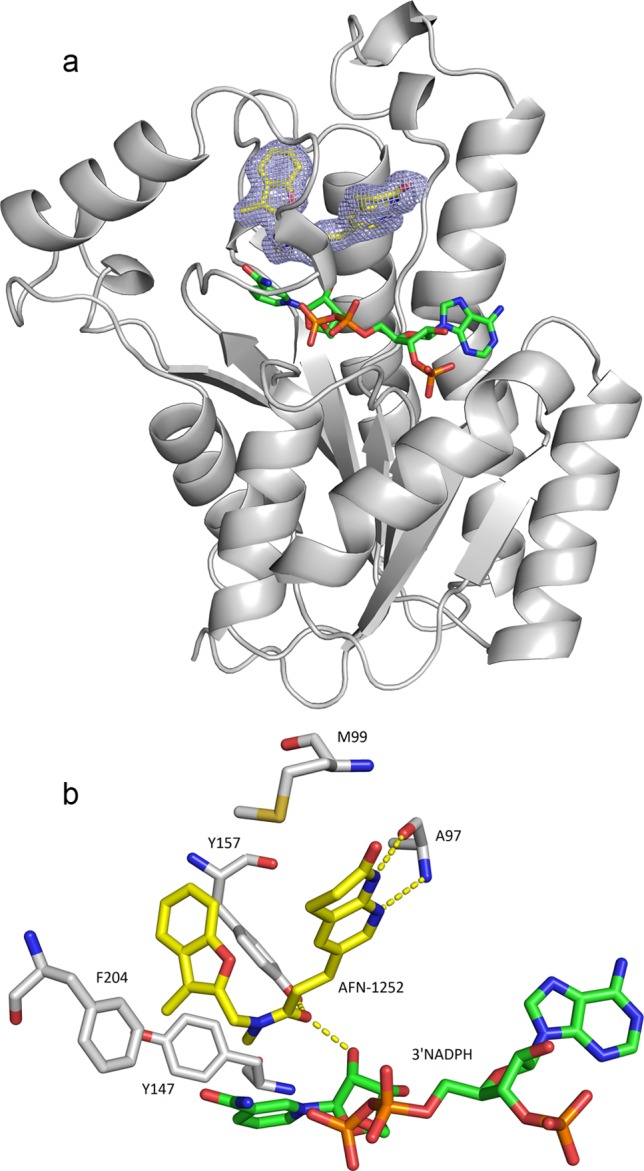
Cocrystal structure of AFN-1252–S. aureus FabI complex and schematic of key interactions. (a) Ribbon representation of the FabI structure with AFN-1252 and 3′NADPH in the binding pocket. Sigma A-weighted 2Fo − Fc electron density is shown for AFN-1252 in the region of the catalytic site at 1.8 Å resolution. The 3-methylbenzofuran side of AFN-1252 nestles in the hydrophobic pocket of the catalytic site, while the oxotetrahydronaphthyridine part of the molecule is more solvent exposed. AFN-1252 and 3′NADPH are shown as sticks, with carbon in yellow (AFN-1252) or green (3′NADPH), nitrogen in blue, oxygen in red, and sulfur in green. (b) Residues of FabI interacting with AFN-1252 and 3′NADPH. The hydroxyl of the ribose ring of 3′NADPH, A97, and Y157 form hydrogen bonds with the inhibitor, while M99, Y147, and F204 form a hydrophobic pocket. These images were generated with PyMOL (16).
The binding specificity of AFN-1252 is evident in the interaction between the inhibitor and FabI (Fig. 4b). The carbonyl of the linking cis-amide of the inhibitor is positioned to accept hydrogen bond interactions from the 2′-hydroxyl of NADPH and the hydroxyl of Y157. The cis-amide also appears to participate in a π-stacking interaction with the nicotinamide portion of NADPH. The oxotetrahydronaphthyridine moiety of the molecule makes two hydrogen bonds to the protein. The peptide backbone of A97 is involved in hydrogen bond interactions that bind both the pyridyl nitrogen and the N-acyl hydrogen of the oxotetrahydronaphthyridine. The 3-methylbenzofuran moiety of AFN-1252 is nestled in a hydrophobic pocket delineated by the lipophilic side chains of residues F204, Y157, and Y147, creating strong hydrophobic interactions. Ordered solvent molecules are found on either side of the inhibitor, with solvent molecules abutting the 3-methylbenzofuran forming a hydrogen-bonding network with the protein.
Determination of the AFN-1252 spontaneous resistance frequency and characterization of AFN-1252-resistant FabI mutants.
AFN-1252 demonstrated a low and dose-dependent propensity for spontaneous resistance development in all seven strains of staphylococci tested (Table 3). At 4 times the MIC (n = 7 strains), frequencies ranged from <6.6 × 10−10 to 2.1 × 10−9; at 8 times the MIC (n = 4 strains), the average frequency was 4.8 × 10−10; and at higher multiples of the MIC, it was near or below the limit of detection; e.g., at 16 times the MIC (n = 4 strains) or 128 times the MIC (n = 3 strains), the frequency was ≤5.7 × 10−10, which is comparable to that observed with linezolid. The fabI genes from 12 spontaneous AFN-1252-resistant S. aureus strains derived from S. aureus MSSA ATCC 29213 (n = 2 strains), S. aureus MRSA ATCC 43300 (n = 8 strains), and MSSE 1024939 (n = 2 strains) have been sequenced, and single amino acid substitutions have been identified. Seven resistant mutants, representing all three parent strains, possessed M99T, a methionine-to-threonine change at amino acid 99. Mutation M99T maps to a flexible loop adjacent to the active site (Fig. 4b) and increases the AFN-1252 MIC 2-fold but has no effect on the activity of triclosan, which also binds to FabI and overlaps the 3-methylbenzofuran side of AFN-1252. The second mutation, Y147H, a tyrosine-to-histidine change at position 147 found in resistant mutants of both S. aureus parents, maps to the active site of FabI (Fig. 4b) and had the strongest effect on potency. AFN-1252 MICs were elevated 8- to 16-fold, and triclosan MICs were elevated 64- to 128-fold in FabI Y147H mutant strains. No FabI amino acid substitutions were found in the fabI gene of S. aureus ATCC 43300 strain 4-B3. As modest increases in the level of FabI protein expression have been correlated with a reduction in sensitivity to FabI inhibitors, e.g., triclosan (20), FabI protein expression levels of several S. aureus strains were determined by Western blotting with FabI antibodies (Fig. 5). The expression level of FtsZ, a cell division maintenance protein, was also determined to enable normalization of expression levels between strains. AFN-1252-insensitive S. aureus strain 4-B3 expressed 2-fold-increased levels of FabI expression compared to that of the parent strain and may be responsible for the 2-fold increase in the MICs of AFN-1252 and triclosan. There was no increase in FabI expression among the other seven isolates tested.
Table 3.
Frequencies of spontaneous staphylococcal strain resistance to AFN-1252 and linezolid
| Strain | MIC (μg/ml) |
MIC multiple | Resistance frequency |
||
|---|---|---|---|---|---|
| AFN-1252 | Linezolid | AFN-1252 | Linezolid | ||
| S. aureus 29213 MSSA | 0.008 | 2 | 4 | 6.6E-10 | <5.8E-10 |
| 128 | <3.3E-10 | <5.8E-10 | |||
| S. aureus 43300 MRSA | 0.008 | 2 | 4 | 2.4E-09 | <4.3E-10 |
| 128 | <3.0E-10 | <4.3E-10 | |||
| S. aureus 2293 CA-MRSA | 0.008 | 4 | 4 | 8.6E-10 | <4.4E-11 |
| 8 | 4.6E-10 | <4.4E-11 | |||
| 16 | <6.6E-10 | <4.4E-11 | |||
| S. aureus 1137 HA-MRSA | 0.008 | 4 | 4 | <6.6E-10 | <6.6E-10 |
| 8 | 6.6E-10 | <6.6E-10 | |||
| 16 | <6.6E-10 | <6.6E-10 | |||
| S. aureus 2012 VISA MRSA | 0.008 | 1 | 4 | 1.4 × E-9 | <1.1E-10 |
| 8 | 4.0E-10 | <1.1E-10 | |||
| 16 | 3.3E-10 | <1.1E-10 | |||
| S. aureus 1725 LinR MRSA | 0.008 | >32 | 4 | 9.0E-09 | Not done |
| 8 | 4.0E-10 | Not done | |||
| 16 | 6.6E-10 | Not done | |||
| S. epidermidis 1024939 MSSE | 0.008 | 2 | 4 | 2.1E-09 | <2.9E-10 |
| 128 | <1.1E-09 | <2.9E-10 | |||
Fig 5.

Western blot assays of S. aureus ATCC 43300 and its AFN-1252-insensitive derivatives. Top panel shows detection of FabI and bottom panel shows FtsZ.
Serial-passage studies.
The consequence of prolonged exposure to subinhibitory concentrations of AFN-1252 for the MIC and development of resistant mutants of S. aureus ATCC 43300 (MRSA) was assessed and compared with those of vancomycin and ciprofloxacin in serial-passage studies for 24 days (Fig. 6). A 4-fold increase in the AFN-1252 MIC was observed after 10 days of exposure, but the MIC did not increase further and was similar to that of vancomycin. In contrast, the MIC of ciprofloxacin increased 16- to 64-fold during the experiment.
Fig 6.

Increase in the relative MIC for S. aureus MRSA ATCC 43300 serially passaged in the presence of subinhibitory concentrations of AFN-1252, vancomycin, or ciprofloxacin. On day 1, the AFN-1252, vancomycin, and ciprofloxacin MICs were 0.004, 1, and 0.25 μg/ml, respectively.
Mode of action.
Time-kill studies were used to explore the bactericidal activity of AFN-1252 and to compare it with those of vancomycin and linezolid by using S. aureus ATCC 29213 (MSSA) and S. aureus ATCC 43300 (MRSA) (Fig. 7). AFN-1252 caused a time-dependent reduction in the viability of both strains. Similar magnitudes and rates of killing were obtained at 4 and 128 times the MIC, with 2.0- and 2.9-log10 reductions in the numbers of CFU/ml observed at 24 h for the MSSA and MRSA strains, respectively. The rate of reduction of viability by AFN-1252 was similar to that observed with linezolid. Vancomycin demonstrated faster killing and a greater degree of killing.
Fig 7.

Time-kill kinetics of AFN-1252, linezolid, and vancomycin at 4 times the MIC with S. aureus ATCC 29213 (MSSA) and ATCC 43300 (MRSA). Each symbol represents the mean of two to four independent experiments.
In vitro activity against diverse and well-characterized clinical isolates.
Panels of isolates were selected to examine the extent of AFN-1252 potency against genetically well-defined representatives of major staphylococcal subgroups and North American locations.
The first panel included 100 genetically well-characterized S. aureus isolates (Table 1) comprising CA-MRSA, HA-MRSA, and MSSA subgroups and representing 100 unique genotypic/phenotypic profiles (see Table 5). The 79 MRSA strains belonged to 12 STs (SCCmec type IV, n = 62; SCCmec II, n = 17; PVL+, n = 35) and 47 unique PFGE profiles, including USA100 to USA1100. The 21 MSSA strains represented nine STs and 20 unique PFGE profiles. Seventy-nine percent of the strains originated from nonsterile sites. The strains, and particularly the HA-MRSA subgroup, displayed significant resistance, e.g., fluoroquinolones, 88%; clarithromycin, 94%; and clindamycin, 88% (data not shown). AFN-1252 exhibited potent activity against all S. aureus strains with an MIC90 of 0.015 μg/ml and a MIC range of 0.008 to 0.060 μg/ml for USA300, an MIC90 of 0.03 μg/ml and an MIC range of 0.004 to 0.06 μg/ml for other CA-MRSA, an MIC90 of 0.015 μg/ml and an MIC range of 0.008 to 0.125 μg/ml for HA-MRSA and an MIC90 of 0.015 μg/ml and an MIC range of 0.004 to 0.015 μg/ml for MSSA.
Table 5.
Pharmacokinetics of AFN-1252 (free base) in mice following oral administration
| AFN-1252 dose (mg/kg) | Cmax (ng/ml) | Tmaxa (h) | AUC0-t (ng-h/ml) | AUC0-∞b (ng-h/ml) | Elimination half-life (h) |
|---|---|---|---|---|---|
| 0.3 | 154 | 0.5 | 367 | 545 | 4.6 |
| 0.6 | 196 | 0.5 | 806 | 1,402 | 7.2 |
| 1 | 304 | 1 | 1,030 | 1,683 | 6.8 |
Tmax, time to maximum concentration of drug in serum.
AUC0-∞, AUC from 0 h to infinity.
The second panel comprised a diverse set of 190 Canadian S. aureus clinical isolates with significant levels of drug resistance; e.g., within the MSSA subpanel, 30% were clindamycin resistant and 31% were erythromycin resistant and in the MRSA subpanel, 86% were ciprofloxacin resistant, 77% were clindamycin resistant, and 91% were erythromycin resistant (data not shown). AFN-1252 was highly potent against this panel, with an MSSA MIC90 of 0.008 μg/ml, an MSSA MIC range of 0.004 to 0.06 μg/ml, an MRSA MIC90 of 0.016 μg/ml, and an MRSA MIC range of 0.004 to 0.12 μg/ml (see Table 5).
The third panel comprised 100 Canadian coagulase-negative staphylococcal clinical isolates with significant levels of drug resistance; e.g., of the MRS S. epidermidis strains, 94% were clindamycin resistant, 31% were mupirocin resistant, 38% were fusidic acid resistant, and 94% were erythromycin resistant, and of the MRS “other” strains, 60% were clindamycin resistant, 40% were ciprofloxacin resistant, and 100% were erythromycin resistant (data not shown). AFN-1252 was potent against these diverse strains, with an MIC90 and an MIC range, respectively, of 0.016 μg/ml and 0.002 to >0.25 μg/ml for S. lugdunensis, 0.12 μg/ml and ≤0.001 to >0.25 μg/ml for S. epidermidis, and 0.25 μg/ml and 0.002 to >0.25 μg/ml for the other coagulase-negative staphylococci (Table 4).
Table 4.
MICs of AFN-1252 and comparators for panels of diverse staphylococci
| Panel, isolate group (no. of isolates), and agent | MIC (μg/ml) |
||
|---|---|---|---|
| 50%d | 90%e | Range | |
| 1, genotyped U.S. S. aureus isolatesa | |||
| USA300 (26) | |||
| AFN-1252 | 0.015 | 0.015 | 0.008–0.06 |
| Vancomycin | 0.5 | 0.5 | 0.5–0.5 |
| Daptomycin | 0.25 | 0.25 | 0.12–0.25 |
| Linezolid | 2 | 2 | 1–2 |
| Other CA-MRSAa isolates (36) | |||
| AFN-1252 | 0.008 | 0.03 | 0.004–0.06 |
| Vancomycin | 0.5 | 1 | 0.5–1 |
| Daptomycin | 0.25 | 0.25 | 0.12–0.25 |
| Linezolid | 1 | 2 | 1–2 |
| HA-MRSA isolates (17) | |||
| AFN-1252 | 0.008 | 0.015 | 0.008–0.125 |
| Vancomycin | 1 | 1 | 0.5–1 |
| Daptomycin | 0.12 | 0.25 | 0.12–0.5 |
| Linezolid | 2 | 2 | 1–2 |
| MSSAb isolates (21) | |||
| AFN-1252 | 0.008 | 0.015 | 0.004–0.015 |
| Vancomycin | 0.5 | 1 | 0.5–1 |
| Daptomycin | 0.12 | 0.25 | 0.06–0.5 |
| Linezolid | 1 | 2 | 0.5–2 |
| 2, Canadian S. aureus isolatesb | |||
| MSSA (104) | |||
| AFN-1252 | 0.008 | 0.008 | 0.004–0.06 |
| Vancomycin | — | — | ≤2 |
| Daptomycin | 0.25 | 0.5 | ≤0.06–0.5 |
| Linezolid | 2 | 2 | ≤1–4 |
| MRSA isolates (86) | |||
| AFN-1252 | 0.008 | 0.016 | 0.004–0.12 |
| Vancomycin | — | — | ≤2 |
| Daptomycin | 0.25 | 0.5 | 0.25–0.5 |
| Linezolid | 2 | 2 | ≤1–4 |
| 3, 100 Canadian coagulase-negative Staphylococcus isolatesc | |||
| S. lugdunensis (52) | |||
| AFN-1252 | 0.008 | 0.016 | 0.002–>0.25 |
| Vancomycin | ≤2 | ≤2 | ≤2 |
| Daptomycin | 0.25 | 0.25 | ≤0.06–1 |
| Linezolid | ≤1 | ≤1 | ≤1 |
| S. epidermidis isolates (27) | |||
| AFN-1252 | 0.016 | 0.12 | ≤0.001–>0.25 |
| Vancomycin | ≤2 | ≤2 | ≤2 |
| Daptomycin | 0.5 | 1.0 | 0.25–1 |
| Linezolid | ≤1 | ≤1 | ≤1–2 |
| Other coagulase-negative Staphylococcus isolates (21) | |||
| AFN-1252 | 0.016 | 0.25 | 0.002–>0.25 |
| Vancomycin | ≤2 | ≤2 | ≤2 |
| Daptomycin | 0.12 | 1.0 | 0.12–1 |
| Linezolid | ≤1 | ≤1 | ≤1–2 |
Pharmacology.
AFN-1252 plasma exposure is clearly demonstrated in the mouse following oral administration of doses of 0.3, 0.6, and 1.0 mg/kg (Fig. 8), achieving maximum serum drug concentrations (Cmaxs) of 154, 196, and 304 ng/ml, based upon total drug levels, respectively. Derived pharmacokinetic parameters, indicate corresponding areas under the concentration-time curve from 0 h to the last measurable concentration (AUC0-ts) of 367, 806, and 1,030 ng-h/ml and long elimination half-lives of 5 to 7 h in this mouse model (Table 5). There was a linear relationship between the dose (mg/kg) and the observed Cmax and AUC0-t (ng-h/ml), with correlation coefficients (R2) of 0.9725 and 0.9320, respectively.
Fig 8.

Pharmacokinetics of orally administered AFN-1252 in the mouse. Shown are plasma AFN-1252 concentrations after the administration of oral doses of 0.3, 0.6, and 1.0 mg/kg in mice. Each symbol represents the mean level of AFN-1252 determined by liquid chromatography-tandem mass spectrometry in three mice.
Murine acute lethal septicemia.
The in vivo efficacy of a single oral AFN-1252 administration was evaluated and compared to that of linezolid in a systemic mouse model of S. aureus Smith infection (Fig. 9). Infection was initiated by the intraperitoneal injection of an otherwise lethal dose of S. aureus Smith, and AFN-1252 (0.03 to 3.0 mg/kg) or linezolid (1.0 to 16.0 mg/kg) was administered 0.5 h later by oral gavage. AFN-1252 demonstrated significant in vivo activity, with 100% survival obtained with a single oral dose of 1 mg/kg. The calculated ED50s (95% confidence limits) of AFN-1252 and linezolid were 0.15 (0.11 to 0.21) and 3.6 (2.8 to 4.7) mg/kg, respectively, indicating that AFN-1252 was approximately 12 to 24 times more potent than linezolid, on a weight-for-weight basis, in this model of S. aureus infection.
Fig 9.

Efficacy of AFN-1252 in a mouse sepsis model. The in vivo efficacy of AFN-1252 was demonstrated in an acute lethal mouse S. aureus Smith infection model. Thirty minutes after intraperitoneal inoculation with a lethal dose of S. aureus Smith, an oral dose of AFN-1252 or linezolid was administered and percent survival was recorded over 5 days.
DISCUSSION
The increasing prevalence of multidrug-resistant staphylococcal infections in the community, their progression to invasive disease, and the associated impact on hospital health care continue to drive the demand for a novel mechanism of action and ideally oral therapies that can be used to treat these infections. There is interest in the potential of targeted therapies to treat emerging drug-resistant strains, while providing for fewer adverse effects and less collateral damage, such as disruption of normal flora or resistance pressure on off-target bacteria. Furthermore, targeted therapies could provide a clinical advantage when used as a step down from empirical therapy once staphylococcus infection is confirmed.
This paper summarizes the preclinical evaluation studies of AFN-1252 following its identification in an iterative, structure-guided discovery process involving >500 newly designed compounds. These preclinical studies included target validation, potential for resistance development, in vitro and in vivo efficacy, and pharmacology studies that informed the lead candidate selection processes. The desired target product profile included a highly potent, Staphylococcus-specific agent with a low propensity for resistance development to address a significant need for agents with a new mechanism of action that are active against resistant staphylococci.
AFN-1252, a specific inhibitor of bacterial enoyl-ACP reductase, interrupts fatty acid biosynthesis and has the potential to provide a novel oral therapy that specifically targets staphylococci. Its narrow spectrum of activity is due to a combination of factors, including the presence of alternative AFN-1252-insensitive enoyl-ACP reductases in many bacterial pathogens and in some bacterial pathogens, e.g., Streptococcus agalactiae, the ability to utilize host fatty acids as an alternative to de novo synthesis, bypassing the essentiality of the target enzyme and pathway (4, 9, 40).
Macromolecular synthesis studies demonstrated that fatty acid biosynthesis was the primary target of AFN-1252 in bacteria, consistent with inhibition of FabI inside the bacterial cell. The interactions between AFN-1252 and the FabI protein have been observed through X-ray crystallography of a cocrystal complex. AFN-1252 has a unique mode of binding that overlaps the triclosan-binding site in FabI. Spontaneous AFN-1252 resistance-conferring mutations resulting in FabI amino acid substitutions provide genetic confirmation that FabI is the primary target of AFN-1252, and their effect on AFN-1252 activity is consistent with the AFN-1252–FabI complex structure. Mutation M99T maps to a flexible loop adjacent to the active site. We postulate that substitution at T99 directly affects AFN-1252 binding by steric hindrance. Mutation Y147H had the strongest effect on potency, and it is predicted to directly disrupt the aromatic-ring-stacking and hydrophobic interactions of both AFN-1252 and triclosan.
AFN-1252 demonstrated outstanding potency against clinical isolates of S. aureus and coagulase-negative staphylococci, regardless of drug resistance or hospital- or community-associated origin. The MIC90s for the S. aureus and S. lugdunensis strains tested were low at 0.015 and 0.016 μg/ml, and for coagulase-negative non-S. lugdunensis strains, the MIC90 was 0.12 μg/ml. The highest AFN-1252 MIC observed to date was 0.5 μg/ml for a single S. epidermidis isolate. The extreme potency of AFN-1252 bodes well for its clinical utility.
The potential for resistance development is a key determinant of the long-term clinical utility of any novel antibacterial. AFN-1252 demonstrated low and dose-dependent spontaneous resistance frequencies. Serial-passage studies demonstrated that prolonged exposure to subinhibitory concentrations of AFN-1252 did not select highly resistant strains. MIC studies using diverse panels of staphylococci detected very few isolates with an AFN-1252 MIC more than 4-fold higher than the MIC90; indeed, the maximum MIC for S. aureus reported in this paper is 0.12 μg/ml. To date, no FabI sequencing studies have been conducted upon the isolates reported on in Table 4. Seven isolates from an independent in vitro potency study with AFN-1252 MICs of 0.06 to 0.12 μg/ml have been sequenced, of which three were found to have single amino acid substitutions in FabI (8). Taken together, the low spontaneous resistance frequency found, the inability to select for highly resistant clones in serial-passage studies, and the high potency of AFN-1252 all indicate that it may be suitable for monotherapy in clinical use.
The lethality of the inhibition of fatty acid biosynthesis was evaluated in time-kill studies. AFN-1252 demonstrated a time-dependent reduction of the viability of both MSSA and MRSA, causing a ≥2-log10 reduction in S. aureus counts over 24 h, similar to linezolid. Although fatty acids contribute to multiple essential biosynthetic and structural components of the bacterial cell, depletion of cellular reserves may be required before the effect of FabI inhibition on viability is expressed. This hypothesis is supported by data from an in vitro pharmacodynamic model which shows a 3-log reduction in MSSA and MRSA viability over 26 to 28 h (53) and a mouse granuloma pouch model in which 4-log reductions of viable counts were observed following dosing with AFN-1252 at 100 mg/kg once daily for 3 days (44).
Oral bioavailability is a highly desirable attribute for a new antibacterial therapy, and AFN-1252 demonstrated high levels of plasma exposure following oral dosing in preliminary mouse pharmacokinetic studies. There was a linear relationship between dose and exposure, and plasma drug levels above the MIC90s were observed, even with modest doses. The long elimination half-life of 5 to 7 h in the mouse raises the potential for once- or twice-daily dosing in humans. A single oral AFN-1252 dose of 1 mg/kg was efficacious in a mouse model of septicemia, providing 100% protection from an otherwise lethal peritoneal infection with S. aureus Smith. An ED50 of 0.15 mg/kg indicated that AFN-1252 was 12 to 24 times more potent than linezolid in the model.
Demonstration of the in vivo efficacy of a selective FabI inhibitor adds to the recent debate over the relevance and potential of fatty acid biosynthesis as a novel antibacterial drug target, confirming that in staphylococci, FabI activity and fatty acid biosynthesis are essential in vivo and they cannot be effectively complemented by host fatty acids (40).
Here we have confirmed the mode of action of AFN-1252, showing that it is a potent inhibitor of FabI and fatty acid biosynthesis, we have demonstrated its exquisite activity across a wide range of staphylococci, including drug- and multidrug resistant clinical isolates, we have shown that it has a very low propensity for spontaneous resistance development, we have shown that it is orally bioavailable, and we have proven its efficacy in an animal model of staphylococcal infection. In summary, AFN-1252 has achieved the objectives set in the drug discovery and preclinical evaluation processes. AFN-1252 not only continues to be evaluated in preclinical models of infection with both oral and intravenous formulations but has also progressed into phase II clinical studies, where its potential in a number of indications where staphylococcal infections can be problematic will be evaluated.
Supplementary Material
ACKNOWLEDGMENTS
We thank Nancy B. Walsh (Affinium Pharmaceuticals) for technical assistance; Roger R. Hinshaw, Ronda D. Schaadt, and Gary E. Zurenko (Micromyx) for their contribution to the resistance experiments; William J. Weiss and Mark Pulse (University of North Texas Health Sciences Center) for their pharmacology and efficacy contributions; and Donald E. Low and Susan. M. Poutanen (Mount Sinai Hospital, Toronto, Ontario, Canada) for their clinical isolate studies. Micron Research assisted in the preparation of the manuscript.
This work was supported by, and all of us were or are employees of, Affinium Pharmaceuticals.
Footnotes
Published ahead of print 4 September 2012
This paper is dedicated to the memory of our beloved colleague Nicole L. Brufatto-Hepburn.
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1. Anderson DJ, et al. 2009. Clinical and financial outcomes due to methicillin resistant Staphylococcus aureus surgical site infection: a multi-center matched outcomes study. PLoS One 4:e8305 doi:10.1371/journal.pone.0008305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Asturias FJ, et al. 2005. Structure and molecular organization of mammalian fatty acid synthase. Nat. Struct. Mol. Biol. 12:225–232 [DOI] [PubMed] [Google Scholar]
- 3. Baldock CJ, et al. 1996. A mechanism of drug action revealed by structural studies of enoyl reductase. Science 274:2107–2110 [DOI] [PubMed] [Google Scholar]
- 4. Balemans W, et al. 2010. Essentiality of FASII pathway for Staphylococcus aureus. Nature 463:E3, discussion E4. doi:10.1038/nature08667 [DOI] [PubMed] [Google Scholar]
- 5. Banerjee A, et al. 1994. inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science 263:227–230 [DOI] [PubMed] [Google Scholar]
- 6. Bergler H, Fuchsbichler S, Högenauer G, Turnowsky F. 1996. The enoyl-[acyl-carrier-protein] reductase (FabI) of Escherichia coli, which catalyzes a key regulatory step in fatty acid biosynthesis, accepts NADH and NADPH as cofactors and is inhibited by palmitoyl-CoA. Eur. J. Biochem. 242:689–694 [DOI] [PubMed] [Google Scholar]
- 7. Bergler H, et al. 1994. Protein EnvM is the NADH-dependent enoyl-ACP reductase (FabI) of Escherichia coli. J. Biol. Chem. 269:5493–5496 [PubMed] [Google Scholar]
- 8. Brenwald NP, Andrews J, Frais AP. 2007. Activity of AFN-1252 against Staphylococcus aureus showing reduced susceptibility to triclosan, abstr. F1-926. 47th Intersci. Conf. Antimicrob. Agents Chemother. American Society for Microbiology, Washington, DC [Google Scholar]
- 9. Brinster S, et al. 2009. Type II fatty acid synthesis is not a suitable antibiotic target for Gram-positive pathogens. Nature 458:83–86 [DOI] [PubMed] [Google Scholar]
- 10. Campbell JW, Cronan JE., Jr 2001. Bacterial fatty acid biosynthesis: targets for antibacterial drug discovery. Annu. Rev. Microbiol. 55:305–332 [DOI] [PubMed] [Google Scholar]
- 11. Chambers HF. 2005. Community-associated MRSA—resistance and virulence converge. N. Engl. J. Med. 352:1485–1487 [DOI] [PubMed] [Google Scholar]
- 12. Cleeland R, Squires E. 1991. Evaluation of new antimicrobials in vitro and in experimental animal infections, p 752–783 In Lorian V. (ed), Antibiotics in laboratory medicine, 3rd ed The Williams & Wilkins Co., Baltimore, MD [Google Scholar]
- 13. Clinical and Laboratory Standards Institute 2009. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard, 7th ed, vol 29 CLSI document M07-A7. Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 14. Collaborative Computational Project, Number 4 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50:760–763 [DOI] [PubMed] [Google Scholar]
- 15. Cortés A, Cascante M, Cárdenas ML, Cornish-Bowden A. 2001. Relationships between inhibition constants, inhibitor concentrations for 50% inhibition and types of inhibition: new ways of analysing data. Biochem. J. 357:263–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Delano WL. 2002. The PyMOL molecular graphics system. DeLano Scientific, San Carlos, CA [Google Scholar]
- 17. Diep BA, et al. 2008. The arginine catabolic mobile element and staphylococcal chromosomal cassette mec linkage: convergence of virulence and resistance in the usa300 clone of methicillin resistant Staphylococcus aureus. J. Infect. Dis. 197:1523–1530 [DOI] [PubMed] [Google Scholar]
- 18. Edelsberg J, et al. 2009. Trends in US hospital admissions for skin and soft tissue infections. Emerg. Infect. Dis. 15:1516–1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126–2132 [DOI] [PubMed] [Google Scholar]
- 20. Fan F, et al. 2002. Defining and combating the mechanisms of triclosan resistance in clinical isolates of Staphylococcus aureus. Antimicrob. Agents Chemother. 46:3343–3437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Forsyth RA, et al. 2002. A genome-wide strategy for the identification of essential genes in Staphylococcus aureus. Mol. Microbiol. 43:1387–1400 [DOI] [PubMed] [Google Scholar]
- 22. Frimodt-Moller N, Knudesen JD, Espersen F. 1999. The mouse peritonitis/sepsis model, p 127–136 In Zak O, et al. (ed). Handbook of animal models of infection. Academic Press, London, United Kingdom [Google Scholar]
- 23. Hall LE, Klein EG, Slater LN, Flournoy DJ. 1990. Methicillin-resistant Staphylococcus aureus: a descriptive analysis on veterans. J. Okla. State Med. Assoc. 83(2):60–63 [PubMed] [Google Scholar]
- 24. Heath RJ, Rock CO. 1995. Enoyl-acyl carrier protein reductase (fabI) plays a determinant role in completing cycles of fatty acid elongation in Escherichia coli. J. Biol. Chem. 270:26538–26542 [DOI] [PubMed] [Google Scholar]
- 25. Heath RJ, Rock CO. 2000. A triclosan-resistant bacterial enzyme. Nature 406:145. [DOI] [PubMed] [Google Scholar]
- 26. Heath RJ, Su N, Murphy CK, Rock CO. 2000. The enoyl-[acyl-carrier-protein] reductases FabI and FabL from Bacillus subtilis. J. Biol. Chem. 275:40128–40133 [DOI] [PubMed] [Google Scholar]
- 27. Heath RJ, White SW, Rock CO. 2002. Inhibitors of fatty acid synthesis as antimicrobial chemotherapeutics. Appl. Microbiol. Biotechnol. 58:695–703 [DOI] [PubMed] [Google Scholar]
- 28. Heath RJ, Yu YT, Shapiro MA, Olson E, Rock CO. 1998. Broad spectrum antimicrobial biocides target the FabI component of fatty acid synthesis. J. Biol. Chem. 273:30316–30320 [DOI] [PubMed] [Google Scholar]
- 29. Karlowsky JA, Kaplan N, Hafkin B, Hoban DJ, Zhanel GG. 2009. AFN-1252, a FabI inhibitor, demonstrates a Staphylococcus-specific spectrum of activity. Antimicrob. Agents Chemother. 53:3544–3548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Karlowsky JA, et al. 2007. In vitro activity of API-1252, a novel FabI inhibitor, against clinical isolates of Staphylococcus aureus and Staphylococcus epidermidis. Antimicrob. Agents Chemother. 51:1580–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Klevens RM, et al. 2007. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 298:1763–1771 [DOI] [PubMed] [Google Scholar]
- 32. Kodali S, et al. 2005. Determination of selectivity and efficacy of fatty acid synthesis inhibitors. J. Biol. Chem. 280:1669–1677 [DOI] [PubMed] [Google Scholar]
- 33. Lu H, Tonge PJ. 2008. Inhibitors of FabI, an enzyme drug target in the bacterial fatty acid biosynthesis pathway. Acc. Chem. Res. 41:11–20 [DOI] [PubMed] [Google Scholar]
- 34. Massengo-Tiassé RP, Cronan JE. 2008. Vibrio cholerae FabV defines a new class of enoyl-acyl carrier protein reductase. J. Biol. Chem. 283:1308–1316 [DOI] [PubMed] [Google Scholar]
- 35. McDougal LK, et al. 2003. Pulsed-field gel electrophoresis typing of oxacillin-resistant Staphylococcus aureus isolates from the United States: establishing a national database. J. Clin. Microbiol. 41:5113–5120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McMurry LM, Oethinger M, Levy SB. 1998. Triclosan targets lipid synthesis. Nature 394:531–532 [DOI] [PubMed] [Google Scholar]
- 37. Miesel L, Greene J, Black TA. 2003. Genetic strategies for antibacterial drug discovery. Nat. Rev. Genet. 4:442–456 [DOI] [PubMed] [Google Scholar]
- 38. Miller WH, et al. 2002. Discovery of aminopyridine-based inhibitors of bacterial enoyl-ACP reductase (FabI). J. Med. Chem. 45:3246–3256 [DOI] [PubMed] [Google Scholar]
- 39. Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode, p 307–326 In Carter CW, Jr, Sweet RM. (ed), Methods in enzymology, vol 276: macromolecular crystallography, part A. Academic Press, Inc., New York, NY: [DOI] [PubMed] [Google Scholar]
- 40. Parsons JB, Frank MW, Subramanian C, Saenkham P, Rock CO. 2011. Metabolic basis for the differential susceptibility of Gram-positive pathogens to fatty acid synthesis inhibitors. Proc. Natl. Acad. Sci. U. S. A. 108:15378–15383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Payne DJ, et al. 2002. Discovery of a novel and potent class of FabI-directed antibacterial agents. Antimicrob. Agents Chemother. 46:3118–3124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Payne DJ, Warren PV, Holmes DJ, Ji Y, Lonsdale JT. 2001. Bacterial fatty-acid biosynthesis: a genomics-driven target for antibacterial drug discovery. Drug Discov. Today 6:537–544 [DOI] [PubMed] [Google Scholar]
- 43. Priyadarshi A, Kim EE, Hwang KY. 2010. Structural insights into Staphylococcus aureus enoyl-ACP reductase (FabI), in complex with NADP and triclosan. Proteins 78:480–486 [DOI] [PubMed] [Google Scholar]
- 44. Pulse ME, et al. 2011. Effects of AFN-1252 on in vitro and in vivo Staphylococcus aureus virulence gene expression, abstr. B-1763. 51st Intersci. Conf. Antimicrob. Agents Chemother. American Society for Microbiology, Washington, DC [Google Scholar]
- 45. Ramnauth J, et al. 2009. 2,3,4,5-Tetrahydro-1H-pyrido[2,3-b and e][1,4]diazepines as inhibitors of the bacterial enoyl ACP reductase, FabI. Bioorg. Med. Chem. Lett. 19:5359–5362 [DOI] [PubMed] [Google Scholar]
- 46. Sakuma M. 1998. Probit analysis of preference data. Appl. Entomol. Zool. 33:339–347 [Google Scholar]
- 47. Sakuma M. 2005. PriProbit, 1.63 ed M. Sakuma, Kyoto, Japan [Google Scholar]
- 48. Sampson PB, et al. 2009. Spiro-naphthyridinone piperidines as inhibitors of S. aureus and E. coli enoyl-ACP reductase (FabI). Bioorg. Med. Chem. Lett. 19:5355–5358 [DOI] [PubMed] [Google Scholar]
- 49. Saravolatz LD, Pohlod DJ, Arking LM. 1982. Community-acquired methicillin-resistant Staphylococcus aureus infections: a new source for nosocomial outbreaks. Ann. Intern. Med. 97(3):325–329 [DOI] [PubMed] [Google Scholar]
- 50. Schiebel J, et al. 2012. Staphylococcus aureus FabI: inhibition, substrate recognition, and potential implications for in vivo essentiality. Structure. 20:802–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Seefeld MA, et al. 2001. Inhibitors of bacterial enoyl acyl carrier protein reductase (FabI): 2,9-disubstituted 1,2,3,4-tetrahydropyrido[3,4-b]indoles as potential antibacterial agents. Bioorg. Med. Chem. Lett. 11:2241–2244 [DOI] [PubMed] [Google Scholar]
- 52. Slater-Radosti C, et al. 2001. Biochemical and genetic characterization of the action of triclosan on Staphylococcus aureus. J. Antimicrob. Chemother. 48:1–6 [DOI] [PubMed] [Google Scholar]
- 53. Tsuji BT, et al. 2006. Bactericidal activity of API-1252, a novel bacterial enoyl-ACP reductase inhibitor, against Staphylococcus aureus in an in vitro pharmacodynamic model, abstr F1-0758. 46th Intersci. Conf. Antimicrob. Agents Chemother. American Society for Microbiology, Washington, DC [Google Scholar]
- 54. Turnowsky F, Fuchs K, Jeschek C, Högenauer G. 1989. envM genes of Salmonella typhimurium and Escherichia coli. J. Bacteriol. 171:6555–6565 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.