

Abstract
Metallo-β-lactamases catalyze the hydrolysis of a broad range of β-lactam antibiotics and are a concern for the spread of drug resistance. To analyze the determinants of enzyme structure and function, the sequence requirements for the subclass B1 IMP-1 β-lactamase zinc binding residue Cys221 were tested by saturation mutagenesis and evaluated for protein expression, as well as hydrolysis of β-lactam substrates. The results indicated that most substitutions at position 221 destabilized the enzyme. Only the enzymes containing C221D and C221G substitutions were expressed well in Escherichia coli and exhibited catalytic activity toward β-lactam antibiotics. Despite the lack of a metal-chelating group at position 221, the C221G enzyme exhibited high levels of catalytic activity in the presence of exogenous zinc. Molecular modeling suggests the glycine substitution is unique among substitutions in that the complete removal of the cysteine side chain allows space for a water molecule to replace the thiol and coordinate zinc at the Zn2 zinc binding site to restore function. Multiple methods were used to estimate the C221G Zn2 binding constant to be 17 to 43 μM. Studies of enzyme function in vivo in E. coli grown on minimal medium showed that both IMP-1 and the C221G mutant exhibited compromised activity when zinc availability was low. Finally, substitutions at residue 121, which is the IMP-1 equivalent of the subclass B3 zinc-chelating position, failed to rescue C221G function, suggesting the coordination schemes of subclasses B1 and B3 are not interchangeable.
INTRODUCTION
An increasing prevalence of antibiotic-resistant strains is reducing the available options for treating bacterial infections. β-Lactam antibiotics, such as the penicillins and cephalosporins, are among the most often used antimicrobial agents (31). The major contributors to β-lactam antibiotic resistance are β-lactamase enzymes, which act by hydrolyzing the four-member β-lactam ring (15, 62). Mechanistically, this is accomplished either via an active-site serine in the class A, C, and D enzymes or through the use of one or two Zn2+ ions (class B) (4). Class B metallo-β-lactamases (MβLs) are a group of structurally similar enzymes that exhibit a characteristic αβ/βα sandwich fold, with the active site located at the interface between domains. This scaffold supports up to 6 residues at the active site that coordinate either one or two zinc ions that are central to the catalytic mechanism (1, 6, 35). MβLs have the capacity to hydrolyze most clinically available β-lactam drugs, including extended-spectrum cephalosporins and carbapenems (8, 10, 32, 40, 59, 61).
The IMP-1 metallo-β-lactamase has been identified in several nosocomial, Gram-negative, pathogenic bacteria, including Pseudomonas aeruginosa and Serratia marcesens (22, 28). The blaIMP-1 gene is carried on an integron element that facilitates genetic transfer and is likely why IMP-1 is found among multiple bacterial species (28, 58). The combination of a broad β-lactam antibiotic substrate profile and the genetic capacity for rapid spread is a concern for the efficacy of antibiotic therapy. An understanding of the determinants of stability, catalytic activity, and substrate specificity of the IMP-1 enzyme is needed for the design of novel inhibitors or β-lactam antibiotics that avoid hydrolysis by IMP-1 and related enzymes.
Metallo-β-lactamases are divided into three subgroups (B1 to B3) based on primary amino acid sequence homology (16, 17). The diverged amino acid sequences of the subclasses are reflected in somewhat different catalytic properties of the enzymes. For example, the B1 and B3 enzymes are most active with two zinc ions bound in the active site while the binding of the second zinc inhibits catalysis by the B2 metallo-β-lactamases (6, 20, 51). Subclass B1 contains the largest number of known metallo-β-lactamases, including the Bacillus cereus, IMP, VIM, and NDM enzymes. X-ray crystal structures of B1 enzymes have revealed the two zinc binding sites (labeled Zn1 and Zn2). The Zn1 binding site, also known as the 3H site, contains three histidines (His116, His118, and His196), while the ligands for the Zn2, or DCH, site include Asp120, Cys221, and His263. The zinc ligand residues in both the 3H and DCH sites are strictly conserved among B1 enzymes.
In contrast to the subclass B1 β-lactamases, the B3 enzymes contain either an aspartic acid or serine residue at position 221 that are not Zn2 ligands for the enzyme. Instead, position 121, such as His121 in the S. maltophilia L1 enzyme, provides the Zn2 ligand (1, 56). Thus, the identity of the residue at position 221 is conserved within B1 but varies between subclasses.
The goal of this study was to understand the sequence requirements at residue 221 for enzyme structure and function in the IMP-1 β-lactamase of subclass B1. For this purpose, saturation mutagenesis was performed at position 221, and all 19 possible substitutions were evaluated for protein expression levels in Escherichia coli and for resistance to several β-lactam antibiotics. There are strict limitations on substitutions at position 221 in IMP-1 due in large part to the instability of the majority of enzyme variants, suggesting that residue C221 contributes to enzyme stability by binding to Zn2. The only substitutions that were consistent with enzyme function in E. coli were C221D and C221G. The discovery of glycine as a functional residue in vivo was surprising, since it cannot serve as a zinc ligand. Furthermore, the in vivo activity of the wild-type (WT), C221G, and C221D enzymes, but not other expressed mutants, can be enhanced with increasing exogenous zinc concentrations.
Kinetic studies with purified C221G enzyme indicated that the additional zinc was required for catalytic activity and that at high levels of zinc (1 mM), β-lactam hydrolysis is comparable to that of the wild-type enzyme for some substrates. Thermal stability studies of the C221G enzyme indicated that the addition of zinc stabilizes the enzyme but that even at high zinc concentrations the C221G enzyme is not as stable as the wild type. These findings suggest that zinc concentrations in vivo are sufficient for partial occupancy of the Zn2 site and that the binding location of Zn2 in the C221G enzyme provides high levels of catalysis, but protein stability is not fully restored.
To test the plasticity of location of Zn2 ligand residues for a subclass B1 enzyme scaffold and to mimic the Zn2 coordination of subclass B3 enzymes, we tested the effects of mutations at position 121 on IMP-1 function. Mutations were made at position 121 in the context of the C221G mutation to allow the substitutions to fill the void left by removal of the cysteine. However, suppression of the defective C221G in vivo phenotype was not observed. Additionally, only single Ala, Cys, and Thr substitutions at 121 were functional in a wild-type IMP-1 enzyme background. These data suggest that the wild-type Ser at position 121 is required for catalytic function and/or stability of IMP-1.
MATERIALS AND METHODS
Saturation mutagenesis.
Overlap extension PCR was used to introduce substitutions into the IMP-1 gene using the oligonucleotide primers listed in Table S1 in the supplemental material (34). Briefly, the primers pTP123-top and pTP123-bot, which are complementary to sequences 5′ and 3′ of the cloned IMP-1 gene were paired with mutagenic primers for PCR amplification of the blaIMP-1 gene from the pGR32 plasmid. This yielded two DNA products that were then used as the template with the pTP123-top and pTP123-bot primers for an overlap extension PCR step to amplify the entire gene with a randomized codon (21). The PCR products and the parent pGR32 plasmid were digested with restriction enzymes SacI and XbaI to generate cohesive ends. The resulting PCR products and plasmid backbones were ligated and transformed into E. coli XL1-Blue [F′::Tn10 proA+ proB+ lacIq Δ(lacZ)M15-recA1 endA1 gyrA96(Nalr) thi-1 hsdR17 supE44 relA1 lac] (Stratagene) cells. Colonies were chosen and cells were grown in LB broth culture, and purified plasmid DNA was sequenced to verify the gene orientation and the presence of a mutation(s).
Antibiotic susceptibility determination.
E. coli XL1-Blue cells were transformed with the pGR32 plasmid encoding the IMP-1 mutants. An E. coli culture containing each variant was grown overnight at 37°C in LB containing 12.5 μg/ml chloramphenicol to select for E. coli clones containing the plasmid. The cultures were diluted 1:100, grown to an optical density at 600 nm (OD600) of 0.5 to 0.6, and spread onto LB agar. After drying briefly, Etest (AB Biodisk) strips were applied to the lawns and grown for 16 h at 37°C, and the MICs were recorded.
For experiments performed on M9 minimal medium, pGR32 plasmids encoding wild-type and IMP-1 variants were transformed into E. coli RB791 cells (strain W3110 lacIqL8), and single colonies were grown in LB broth containing chloramphenicol. The cultures were diluted 1:100 into 1× M9 salts (42 mM Na2HPO4, 24 mM KH2PO4, 9 mM NaCl, 19 mM NH4Cl, 0.2 mM MgSO4, 0.1 mM CaCl2, and 0.2% dextrose), which were prepared with high-performance liquid chromatography (HPLC) grade water in acid-washed glassware (rinsed twice with 6 N nitric acid and then twice with HPLC grade water). Overnight M9 broth cultures (OD600, ∼0.5) were swabbed onto M9 agar plates, also prepared with HPLC grade water, with 0 to 50 μM ZnSO4 added to the media. MIC values for ampicillin, as determined by Etest, were observed after incubation at 37°C for 48 h. The results represent three independent experiments.
Protein purification.
IMP-1 enzymes were expressed from the pGR32 vector under the transcriptional control of an isopropyl-1-thio-β-d-galactopyranoside (IPTG)-inducible trc promoter in E. coli RB791 (strain W3110 lacIqL8) cells grown in LB (34). Protein expression was achieved by IPTG induction of mid-log-phase cultures and subsequent incubation at 27°C for 16 h. Following centrifugation, the periplasmic fraction of the pellet was released by osmotic shock (39). The wild-type and mutant enzymes contained a C-terminal 8-amino-acid StrepII tag (WSHPQFEK) that allowed purification. The enzymes were fractionated to greater than 90% purity from periplasmic lysates by affinity chromatography with a Strep-Tactin superflow resin according to the manufacturer's instructions, with the exception that all buffer solutions were reconstructed without EDTA (Novagen). Briefly, 50 to 100 ml of lysate was applied to a 5-ml resin bed in a gravity flow column. Following binding, 5 column volumes of wash buffer (150 mM NaCl and 100 mM Tris-HCl at pH 7.0) was applied. Protein was eluted in 150 mM NaCl and 100 mM Tris-HCl containing 2.5 mM desthiobiotin at pH 7.0. Active fractions were confirmed by monitoring hydrolysis of the colorimetric β-lactam substrate nitrocefin, and active fractions were pooled and concentrated using Amicon concentrator units (Millipore). The final yields of purified enzyme were between 3 and 7 mg per liter of culture.
Immunoblot assay.
The effects of amino acid substitutions at position 221 on steady-state expression levels of IMP-1 β-lactamase were determined by immunoblot analysis of whole-cell lysates of E. coli XL1-Blue expressing each of the mutant IMP-1 enzymes (3, 33). Briefly, overnight E. coli cultures transformed with the pGR32 plasmid encoding wild-type or mutant IMP-1 were diluted 1:100 in fresh LB medium containing chloramphenicol and allowed to grow for 2 h. A total of 1.5 ml of each culture, which was normalized by OD600, was pelleted by centrifugation and resuspended in SDS loading buffer (25% SDS, 62.5 mM Tris-HCl [pH 6.8], 25% glycerol, and 0.01% bromophenol blue). Sample proteins were resolved on a 12% SDS-PAGE gel and electrotransferred to a nitrocellulose membrane (Amersham; GE Healthcare, Piscataway, NJ) using a Bio-Rad semidry transfer apparatus in 25 mM Tris containing 200 mM glycine and 10% methanol. The membrane was blocked overnight in a 5% milk solution prior to probing with mouse monoclonal anti-StrepII-horseradish peroxidase (HRP) antibody, which was detected with the Amersham ECL chemiluminescent detection reagent (GE Healthcare) to visualize IMP-1 proteins. The wild-type and mutant enzymes contained a C-terminal StrepII tag sequence that allowed detection using the anti-StrepII-HRP antibody. The data shown are representative of three independent experiments.
Differential scanning fluorescence.
The thermal stability of purified wild-type and C221G mutant enzymes was examined using a differential scanning fluorescence (thermofluor) assay (14). Wild-type or mutant IMP-1 enzymes, purified as described above, were added to achieve a final concentration of 0.15 mg/ml in 50 mM HEPES, pH 8.0, buffer containing a 1:2,000 dilution of Sypro orange (Invitrogen). A total of 30 μl of 1.5-mg/ml protein solution with or without 100 μM zinc sulfate was added to a final volume of 300 μl in each well of a 96-well MicroAmp plate (ABI). The temperature was raised from 25°C to 99°C over 15 min using an ABI 7900 thermocycler. The data were fitted to the single-site binding equation using nonlinear regression with GraphPad Prism version 5.01 software for Windows (46).
Isothermal titration calorimetry.
Thermodynamic measurements of IMP-1 interaction with zinc were carried out using a VP-ITC calorimeter (GE Healthcare). Purified IMP-1 proteins were dialyzed against 100 mM Tris-HCl buffer at pH 7.0 containing 150 mM NaCl and chelating resin to remove zinc contamination. Protein samples were concentrated to 25 μM using Amicon concentrator units (Millipore). The dialysis buffer was supplemented with 1 mM ZnCl2 to be used as the titrant solution. The isothermal calorimetric titration of Zn binding to IMP-1 was carried out at 30°C. The data were processed using the Origin 7.0 software package with an isothermal titration calorimetry (ITC) add-on (OriginLab Corp., Northampton, MA).
X-ray crystallography. (i) Sulfate-bound enzyme.
The IMP-1 C221G protein was purified as described above to greater than 90% homogeneity, as assessed by SDS-PAGE. The purified C221G protein was screened for crystallization against approximately 400 crystallization conditions of the Joint Center for Structural Genomics (JCSG) Core Suite set (Qiagen) of solutions using a Mosquito automated nanoliter liquid handler robot (TTP LabTech). Crystals were grown by the hanging-drop method in 1:1 drops of 20 mg/ml protein in 25 mM Tris-HCl containing 1 mM zinc sulfate at pH 7.0, and mother liquor containing 0.8 M disodium hydrogen phosphate, 0.8 M dipotassium hydrogen phosphate, and 0.1 M HEPES at pH 7.5 with incubation at 25°C. Diffraction data were collected at the ALS synchrotron facility (Berkley, CA). Data processing and analysis were carried out using the software d*TREK (45). The phase problem was solved through molecular replacement with the published wild-type IMP-1 coordinates (Protein Data Bank [PDB] code 1DDK) as the search model using the Phaser program in the CCP4 software suite (47). Iterative cycles of model building into the 2Fo-Fc maps, further validation by computing Fo-Fc maps, and refinement were carried out using COOT and REFMAC5 as implemented in CCP4i (13, 47). Structure refinement was carried out, and the figures were rendered using the PyMOL Molecular Graphics System (11).
(ii) Citrate-bound enzyme.
Small crystals grown under the conditions described above were macroseeded into fresh hanging drops containing a 1:1 ratio of 10 mg/ml protein in 25 mM Tris-HCl with 1 mM zinc chloride at pH 7.0 and the mother liquor, which consisted of 0.1 M trisodium citrate (pH 5.5) in 40% polyethylene glycol (PEG) 600 at 25°C. The seeds developed into larger crystals within 1 day in 2-μl drops equilibrated against 700 μl of reservoir solution. Diffraction data were collected using a Rigaku FR-E+ SuperBright rotating anode at the Baylor College of Medicine Structural Biology Core. These data were processed using d*TREK (45). For initial phasing, the sulfate-bound C221G structure was used as a search model for molecular replacement using Phaser3. Model building and refinement were carried out using COOT and REFMAC5 (13, 45, 47). Figures were rendered using the PyMOL Molecular Graphics System (11).
(iii) Determination of WT and C221G IMP-1 enzyme zinc contents.
Proteins purified as described above were dialyzed into 100 mM Tris-HCl buffer containing 150 mM NaCl and 5 g of unswelled Chelex resin (Bio-Rad; pH 8.0; 72 h). Samples were then concentrated to approximately 2 ml (3 to 5 mg/ml) using Amicon centrifugation units (Millipore). A total of 500 μl of either concentrated protein or buffer was added to 100 μl of concentrated HNO3, 200 μl of internal standard (I.S.), and 2,200 μl of HPLC grade water and allowed to equilibrate before injection into the 7100-CE inductively coupled plasma mass spectrometry (ICP-MS) instrument (Agilent). Total zinc counts and relative zinc content per enzyme were determined (see Table 2).
Table 2.
IMP-1 enzyme zinc content determined by ICP-MS
| Sample | Zinc concn (μg/liter) | Relative zinc content (per enzyme) |
|---|---|---|
| Buffer | <0.1 | NAa |
| WT IMP-1 | 2,260.0 | 2 |
| C221G IMP-1 | 895.6 | 0.8 |
NA, not applicable.
Steady-state kinetics.
Hydrolysis of ampicillin (Δε235 nm = −8,200 M−1 cm−1), nitrocefin (Δε485 nm = 15,000 M−1 cm−1), ceftazidime (Δε260 nm = −9,000 M−1 cm−1), cefotaxime (Δε260 nm = −7,500 M−1 cm−1), and imipenem (Δε300 nm = 9,000 M−1 cm−1) by the wild-type and mutant IMP-1 enzymes was measured using a DU 640 spectrophotometer (Beckman Coulter) (28). All reactions were carried out in a final volume of 300 μl. For all substrates except ampicillin and imipenem, experiments were performed in 50 mM HEPES buffer containing 50 μM ZnSO4 and 20 μg ml−1 bovine serum albumin (BSA) at pH 7.0. Experiments with ampicillin and imipenem were done in 50 mM MOPS (morpholinepropanesulfonic acid) buffer containing 50 μM ZnSO4, as it was found that these substrates had a high background rate of hydrolysis in the HEPES-based buffer. In experiments described as “metal free,” enzyme preparations were treated with chelating resin to remove exogenous metals (as described for the ICP-MS assays), and all buffers were constructed with HPLC grade water. Prior to beginning kinetic measurements, the buffers were assayed for residual zinc, which was found to be approximately 300 nM by the PAR absorbance assay (23, 48). For all substrates tested, enzyme and substrate were preincubated separately at 30°C for 5 min prior to mixing. Enzyme reactions were performed at 30°C, and the kinetic parameters Km and kcat were derived from initial velocity measurements obtained by fitting to the Michaelis-Menten equation using GraphPad Prism version 5.01 software for Windows.
Estimation of zinc binding by enzyme activity measurements.
IMP-1 enzymes purified as described above were dialyzed overnight against 150 mM NaCl in 100 mM Tris-HCl at pH 7.0 containing a packet of Chelex resin, followed by addition of 50% glycerol by volume for storage at −20°C. To confirm that one zinc ion remained per enzyme, proteins were tested with the PAR assay as described above (see Table S2 in the supplemental material) (23, 48). The kinetic profile of the C221G IMP-1 mutant was determined in metal-free 50 mM HEPES buffer containing 20 μg/ml BSA at pH 7.0, with 10 or more nitrocefin dilutions with concentrations between 3 and 100 μM. The C221G IMP-1 enzyme was stored on ice and incubated in the buffer for 5 min at 30° ± 2°C prior to the addition of the nitrocefin. The rate of nitrocefin hydrolysis was measured spectrophotometrically at 482 nm, and initial velocities were plotted to determine the Km and kcat. The profiles were then conducted in the same HEPES buffer supplemented with 10, 25, 30, 50, 75, 100, 125, 150, or 200 μM ZnSO4 in duplicate. The nitrocefin was also diluted fresh into the zinc-containing buffers immediately prior to the experiment to minimize zinc-catalyzed nitrocefin hydrolysis and stored on ice. The rate constants at each of the zinc concentrations were determined with nonlinear fit to a hyperbola using GraphPad Prism version 5.01 software for Windows. The kcat was then plotted relative to the ZnSO4 concentration and fit using the same nonlinear curve equation to evaluate the affinity of the Zn2 site.
Protein structure accession numbers.
The fully refined structures have been deposited in the Protein Data Bank as PDB 4F6H (sulfate bound) and 4F6Z (citrate bound) (http://www.PDB.org).
RESULTS
Saturation mutagenesis of position 221 and identification of functional mutants.
The role of cysteine at IMP-1 position 221 was examined by randomizing the codon from TGT to NNS, where N is any of the four nucleotides and S is either cytosine or guanine, to allow for all possible amino acid substitutions and testing the resultant mutants for function. A collection of clones representing all 19 non-wild-type amino acids was assembled and verified by DNA sequencing. In order to determine which substitutions are consistent with in vivo function against a representative set of β-lactam substrates, each of the position 221 variants was introduced into E. coli and the MICs of several different β-lactam antibiotics, including ampicillin, cefotaxime, ceftazidime, imipenem, meropenem, and aztreonam, were assessed. The MIC results indicated that only 2 of the 19 mutants retained function, and these variants were only partially functional (Table 1). The highest MIC values were observed for the C221D mutant. The C221G variant also retained partial function, which was unexpected, since glycine lacks a side chain capable of coordinating zinc. MIC determinations also revealed differences in the substrate specificity profiles for wild-type IMP-1 and the C221D and C221G variants. The wild-type IMP-1 enzyme preferentially hydrolyzed ampicillin, cefotaxime, and ceftazidime, as reflected by higher MIC values of these antibiotics. Similarly, E. coli containing the C221D mutant exhibited the highest MICs against ampicillin and ceftazidime but was not as effective against cefotaxime. In contrast, the highest MIC values for the C221G mutant were seen with ceftazidime, followed by ampicillin and cefotaxime (Table 1).
Table 1.
Antibiotic susceptibilities of E. coli expressing position 221 IMP-1 variant enzymes (representative mutants are shown)
| Antibiotic substrate | MIC (μg/ml) |
|||||||
|---|---|---|---|---|---|---|---|---|
| E. coli XL1-Blue | Wild type | C221A | C221D | C221G | C221S | C221T | C221V | |
| Ampicillin | 2 | 125 | 2 | 128 | 4 | 3 | 2 | 2 |
| Cefotaxime | 0.094 | 250 | 0.19 | 0.38 | 3 | 0.19 | 0.064 | 0.047 |
| Ceftazidime | 0.38 | 500 | 0.38 | 48 | 64 | 0.38 | 0.19 | 0.19 |
| Imipenem | 0.25 | 2 | 0.38 | 0.5 | 1 | 1 | 1 | 0.25 |
| Meropenem | 0.032 | 1.5 | 0.094 | 1 | 0.38 | 0.094 | 0.094 | 0.064 |
| Aztreonam | 0.125 | 0.094 | 0.094 | 0.064 | 0.094 | 0.125 | 0.094 | 0.094 |
Steady-state protein expression levels of position 221 mutants.
The effects of amino acid substitutions on the steady-state protein levels for position 221 mutants were assessed by monitoring protein expression levels by immunoblotting of whole-cell lysates of E. coli cells expressing IMP-1 variants. Previous studies from our laboratory and others have shown that mutations affecting the stability of a protein result in expression defects because unstable proteins are proteolyzed in E. coli (43, 44). Protein stability can therefore be qualitatively inferred from relative steady-state protein levels using an immunoblot assay (3, 52).
Steady-state IMP-1 protein levels were evaluated from whole-cell extracts of log-phase cultures of E. coli containing wild-type or mutant enzymes. Extracts were fractionated by SDS-PAGE, and immunoblotting with monoclonal anti-StrepII antibodies was performed to visualize the β-lactamase enzyme. The wild-type and mutant enzymes included a C-terminal StrepII tag sequence that allowed detection by the antibody. Of the 19 IMP-1 mutants tested, only enzymes with Ser, Thr, and Val substitutions, in addition to the functional Asp and Gly mutants, were observed at detectable levels on the immunoblots (Fig. 1). While the Asp, Ser, and Thr variants were expressed at nearly wild-type levels, the enzymes with Val and Gly substitutions were present at somewhat lower levels. All other variants were undetectable following overnight exposure of the immunoblot. In general, there is a correlation between the change in side chain volume of the mutants and protein expression levels in E. coli. The volumes of the side chains of the expressed mutants are relatively small, suggesting a size requirement at position 221 for protein expression (5, 64). The exception is the C221A mutant, which is not detectable in the protein lysates by immunoblotting despite a side chain volume within the range of the substituted residues of other variants whose expression is observed. The immunoblotting results, in combination with the in vivo MIC results, indicate that a limited number of amino acid substitutions at position 221 are consistent with function and that many of the substitutions have a negative effect on protein expression within the bacterial cell. These results are consistent with the wild-type X-ray structure, which indicates that the volume of residue 221 is tightly constrained by the neighboring His118 and Lys224 residues, as well as Zn2. Residues larger than valine would be expected to create steric clashes with Zn2 or other neighboring chelating residues and thereby to prevent Zn2 binding.
Fig 1.

In vivo steady-state protein levels of C221X mutants. Shown are steady-state protein levels of wild-type and mutant IMP-1 metallo-β-lactamases as determined by immunoblotting of E. coli whole-cell lysates. The positions correspond to residue 221 variants represented by single-letter amino acid codes, with the exception of the wild type.
Exogenous zinc affects in vivo enzyme function.
The chelating function of cysteine at position 221 has previously been shown to be essential for optimal IMP-1 function and stability (18, 20). Substitutions of chelating residues in dizinc MβLs have been associated with lower affinity at the Zn2 site than the wild type and, in addition, result in catalytic defects in vitro (7, 9, 20, 30). To assess the role of zinc binding in vivo, the MIC values of E. coli encoding the IMP-1 wild type and mutants were determined for growth in M9 minimal medium containing a range of zinc concentrations. To minimize carryover metal contamination from culture media, cells were subcultured twice into M9 minimal medium without added zinc before being spread onto M9 agar with the desired zinc concentration for MIC determinations. Of the variants that exhibited detectable steady-state protein levels by immunoblotting, E. coli containing the wild-type, C221D, and C221G IMP-1 enzymes displayed increasing resistance to ampicillin with increasing exogenous zinc (Fig. 2). Interestingly, the Ser, Thr, and Val mutants that lacked antibiotic resistance activity above background in rich medium also lacked activity on M9 minimal medium regardless of exogenous zinc concentrations. A similar trend in antibiotic resistance profiles for the wild type and the mutants was observed with increasing zinc for cefotaxime, suggesting the increasing activity applies to general enzyme function and is not substrate dependent in vivo (data not shown).
Fig 2.
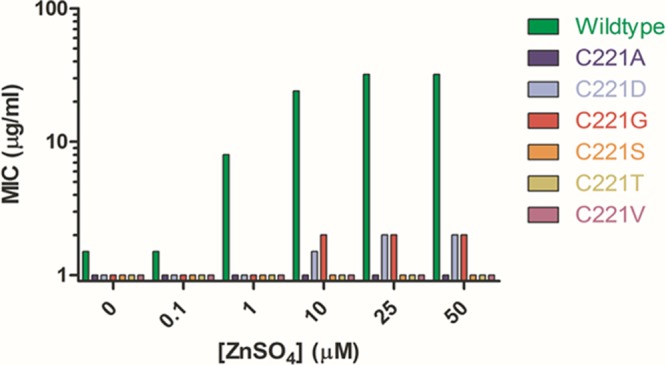
Ampicillin MIC determinations for E. coli grown in M9 minimal medium. Shown are MIC determinations of E. coli expressing IMP-1 221 variants performed on minimal medium containing 0 to 50 μM ZnSO4.
The observed MIC values in minimal medium for the two functional IMP-1 variants appear to plateau between 10 μM and 25 μM ZnSO4, which represents only a fraction of the MIC values observed with rich medium (Table 1). This may reflect the physiological stresses imposed upon the bacteria in the defined versus rich media. Additionally it has been shown that zinc concentrations near 200 μM can decrease growth rates of E. coli (12, 29). Consistent with that finding, we have observed that as little as 50 μM zinc can alter the growth profile of E. coli.
Another interesting finding was the apparent increase in wild-type enzyme activity, as reflected by the antibiotic MIC, with increasing concentrations of environmental zinc. Previous reports on IMP-1 have shown that protein expressed and purified from E. coli grown under rich-medium conditions contains two zinc ions per enzyme molecule and exhibits optimal catalytic function (6, 18, 20, 53). Our current findings from defined minimal medium suggest that in vivo enzyme function is highly dependent on the availability of exogenous zinc in the growth environment of E. coli. The results indicate that rich medium has sufficient zinc for the enzyme to bind both Zn1 and Zn2 sites but that other growth conditions may result in partial occupancy of the zinc sites even with the wild-type enzyme. These findings have implications for antibiotic resistance, since the concentration of zinc in the local environment in which the bacteria are growing during infection may strongly influence the zinc occupancy and thereby the catalytic efficiency of the IMP-1 enzyme.
Zinc contents of wild-type and C221G IMP-1.
Histidine, aspartate, and cysteine residues are known to serve as effective zinc-chelating residues to position zinc atoms in the active sites of metalloenzymes (1, 19, 38). Consistent with our in vivo data, it has been shown in vitro that an Asp substitution at position 221 is associated with partial catalytic function of subclass B1 enzymes (19, 20). It was surprising, however, to find that a glycine substitution at position 221 also provides partial function, since glycine does not have a side chain to serve as a zinc ligand. It was therefore of interest to determine the metal content of the C221G enzyme.
The metallation state was determined using ICP-MS with purified samples of the wild-type and Gly mutant IMP-1 enzymes that had been dialyzed against buffer containing chelating resin to remove exogenous metal (Table 2). As expected, the wild-type enzyme was found to contain the equivalent of two zinc atoms per enzyme molecule, as has been reported previously (20, 53). The C221G protein sample, however, contained approximately half the zinc content of wild-type IMP-1, corresponding to approximately one zinc atom per enzyme molecule. This finding indicates that the C221G substitution results in a significant reduction in affinity for binding a second zinc atom and that, in the absence of exogenous zinc, the C221G enzyme exists as a monozinc enzyme.
Thermal stability of the IMP-1 C221G enzyme.
The immunoblotting results presented above indicate the C221G enzyme is expressed in E. coli but at somewhat lower levels than the wild-type enzyme (Fig. 1). Differential scanning fluorescence (thermofluor) was performed on the wild-type and Gly mutant proteins in the presence and absence of 100 μM ZnSO4 in order to obtain more detailed information on the effect of the glycine substitution at position 221 on protein stability (Fig. 3). The melting temperatures (Tm) of wild-type IMP-1 and the C221G mutant were determined to be 76°C and 62°C, respectively, in the absence of zinc (see Materials and Methods). The C221G enzyme was found to be less stable than the wild-type enzyme in both the presence and absence of zinc, which is consistent with the observed reduced in vivo expression levels of the mutant. The Tm for the wild-type protein exhibited a negligible shift (ΔTm = 1.13°C) in the presence of zinc. In contrast, the Tm for the Gly mutant protein experienced a larger shift (ΔTm = 5.34°C), showing an increase in protein thermal stability. Mass spectrometry data indicated that in the absence of exogenous zinc the enzyme contains an average of one zinc atom per molecule (Table 2). The increased thermal stability of the C221G enzyme in the presence of added zinc is likely due to the binding of a second zinc in the active site. A similar stability profile was observed upon addition of ZnCl2. These results further indicate that binding of the second zinc plays an important role in stabilizing the enzyme.
Fig 3.
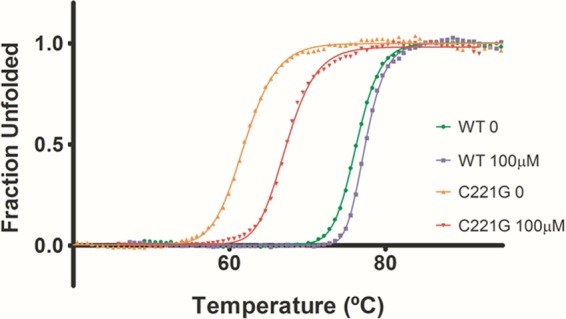
IMP-1 enzyme thermal stability determined by a thermofluor assay. Shown are thermal shift assay results for wild-type and C221G IMP-1 enzymes. Protein samples were diluted in 50 mM HEPES, pH 7.0, and tested in the presence or absence of 100 μM ZnSO4 at pH 7.0.
Kinetic parameter determination.
The catalytic activities of the wild-type and C221G enzymes were assayed for hydrolysis of a series of β-lactam substrates representing penicillins, cephalosporins, and carbapenems (Table 3). Each determination was performed in buffer that had been treated with chelating resin, and assays were performed with or without the addition of zinc. The kinetic parameters for hydrolysis of β-lactam substrates by the wild-type enzyme were similar in the presence or absence of added zinc, although the catalytic efficiency of nitrocefin and ceftazidime hydrolysis was higher in 1 mM zinc. This observation suggests that both active-site zinc atoms are bound, whether or not exogenous zinc is added to the reaction buffer. This is consistent with the mass spectrometry results and with the fact that the wild-type enzyme was purified from E. coli that had been grown in rich medium. The kinetic parameters for the C221G enzyme, however, varied considerably depending on the presence or absence of zinc in the reaction buffer. For all β-lactam substrates tested, the addition of zinc to the reaction mixture was associated with an enhancement of the kcat. The most pronounced difference was seen for the hydrolysis of nitrocefin, where the kcat enhancement from the metal-free condition to buffer containing 1 mM zinc was more than 340-fold. The high zinc concentration places nitrocefin substrate turnover (kcat) by the C221G mutant at a level similar to that observed for wild-type enzyme even without a functional Zn2 zinc ligand. With ampicillin, cefotaxime, and ceftazidime as substrates, the C221G enzyme exhibits little catalytic activity unless zinc is added to the reaction buffer. While the changes seen for these substrates are not as dramatic as for nitrocefin, turnover at high zinc concentrations is comparable to that of the wild-type enzyme for all substrates except ceftazidime (Table 3). Taken together with the ICP-MS results, which indicated the Gly mutant enzyme contains one zinc, these kinetic data suggest that the monozinc enzyme has little catalytic activity alone but that binding of zinc at the Zn2 site leads to an increase in substrate hydrolysis.
Table 3.
Kinetic parameters for hydrolysis of β-lactam antibiotics by wild-type IMP-1 and a varianta
| Variant | Antibiotic substrate |
Km (μM) |
kcat (s−1) |
kcat/Km (μM−1 s−1) |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| No metal | 50 μM Zn | 1 mM Zn | No metal | 50 μM Zn | 1 mM Zn | No metal | 50 μM Zn | 1 mM Zn | ||
| Wild type | Ampicillin | 110 ± 9 | 601 ± 7 | 420 ± 47 | 122 ± 8 | 439 ± 5 | 357 ± 13 | 1 | 1 | 1 |
| Nitrocefin | 79 ± 6 | 302 ± 7 | 9 ± 1 | 790 ± 88 | 489 ± 34 | 580 ± 20 | 10 | 15 | 64 | |
| Cefotaxime | 1 ± 0.1 | 2 ± 0.1 | 1 ± 0.2 | 13 ± 1 | 9 ± 2 | 30 ± 2 | 13 | 5 | 35 | |
| Ceftazidime | 25 ± 5 | 28 ± 7 | 28 ± 8 | 14 ± 1 | 9 ± 1 | 196 ± 14 | 0.6 | 0.3 | 7 | |
| Imipenem | 33 ± 1 | 86 ± 16 | 52 ± 32 | 67 ± 7 | 71 ± 4 | 130 ± 10 | 2 | 1 | 3 | |
| C221G | Ampicillin | NAb | 32 ± 23 | 775 ± 150 | NA | 1 ± 0.1 | 120 ± 10 | NA | 0.01 | 0.2 |
| Nitrocefin | 1 ± 0.2 | 13 ± 5 | 19 ± 6 | 2 ± 0.1 | 275 ± 29 | 692 ± 73 | 1 | 21 | 36 | |
| Cefotaxime | 5 ± 1 | 10 ± 1 | 6 ± 1 | 2 ± 0.3 | 12 ± 1 | 104 ± 6 | 0.3 | 1 | 19 | |
| Ceftazidime | NA | 300 ± 10 | 250 ± 10 | NA | 10 ± 2 | 12 ± 4 | N/A | 0.03 | 0.05 | |
| Imipenem | >1,000 | >1,000 | 663 ± 120 | NDc | ND | 128 ± 25 | NA | NA | 0.2 | |
Kinetic parameters were determined in 50 mM HEPES buffer (made with HPLC grade water) containing 20 μg/ml BSA and either 0 μM, 50 μM, or 1 mM ZnSO4. Variances are representative of the standard deviations from three independent experiments.
NA, not available; no significant hydrolysis above background was detected.
ND, not detected; the Km was too high to determine accurately.
Binding affinity for zinc at Zn2 in C221G IMP-1.
The MIC, mass spectrometry, and enzyme kinetics results suggest the binding affinity of zinc for the Zn2 site is lower in the C221G enzyme. Therefore, it was of interest to determine the binding constant for zinc in the Zn2 site. Zinc binding was determined by ITC using C221G enzyme that had been treated with chelating resin. The ITC results in Fig. 4A revealed a binding constant of 17 μM. The calorimetry data show strong enthalpy-driven binding. Under the titration conditions used (a low c value of ∼1.47), the determination of the binding constant is accurate while the enthalpy could carry a large error (55). The c value is the product of the enzyme concentration and the binding constant (55).
Fig 4.

IMP-1 C221G zinc binding determinations. (A) Isotherm generated by isothermal titration calorimetry showing ZnCl2 binding to chelator-treated C221G IMP-1 protein. (B) Isotherm generated by a thermofluor assay showing ZnCl2 binding to chelator-treated C221G IMP-1 protein. (C) Graph of kcat values for nitrocefin hydrolysis by chelator-treated C221G IMP-1 enzyme in the presence of various ZnSO4 concentrations. A nonlinear regression fit of these data to a hyperbola reveals a Kd (dissociation constant) value of 43 μM for binding of Zn2 and activation of nitrocefin hydrolysis.
The thermofluor protein stability assay that was used for the wild-type and C221G enzymes in Fig. 3 was also employed to estimate a binding constant for the second zinc. For these experiments, the Tm was determined for chelating-resin-treated IMP-1 C221G enzyme with increasing concentrations of ZnCl2. The Tm values versus the ZnCl2 concentration are plotted in Fig. 4B and reveal a binding constant of 30 μM, which is similar to the value obtained by ITC.
A third method for estimating the binding affinity of zinc for chelating-resin-treated IMP-1 C221G was to determine kinetic parameters for nitrocefin hydrolysis with increasing ZnSO4 concentrations. The kcat values for nitrocefin hydrolysis versus the zinc concentration are presented in Fig. 4C. Fitting of the curve indicates a binding constant of 43 μM, which is broadly consistent with the values obtained from the physical measurements.
Structure determination of the IMP-1 C221G enzyme.
The above-mentioned results indicate that the presence of zinc increases both the catalytic activity and the thermal stability of the C221G enzyme. The fact that glycine does not have a side chain and yet activity can be restored in the presence of zinc suggests binding occurs in a similar position as with Zn2 but at lower affinity. One possibility is that a water molecule fills the gap left by the removal of the cysteine side chain and serves as a zinc ligand. This would be consistent with the fact that the expressed C221S mutant does not function and that molecular modeling suggests there is not space for a water molecule when the serine side chain is present (data not shown). It was therefore of interest to determine the X-ray structure of the C221G IMP-1 mutant. To this end, the structure of the C221G mutant enzyme was determined in the presence of excess zinc by X-ray crystallography (Fig. 5).
Fig 5.
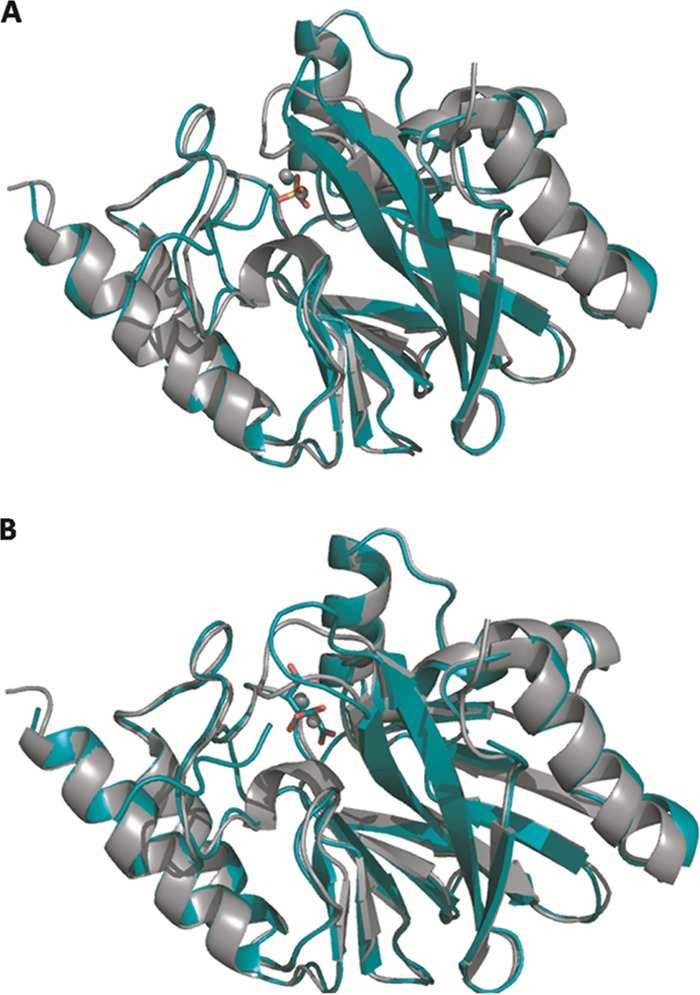
X-ray crystal structures of C221G mutant IMP-1 β-lactamase with bound sulfate and citrate. Shown are cartoon representations of the overlay of wild-type (carbon and zinc atoms in gray) and C221G mutant (carbon and zinc atoms in teal) IMP-1 structures. The C221G mutant is shown bound to either sulfate (A) or citrate (B). Oxygen is shown in red, and sulfur is shown in yellow. The wild-type IMP-1 enzyme structure is from PDB file 1DDK. The figures were rendered using the PyMOL Molecular Graphics System (11).
Crystallization of the C221G enzyme was achieved independently in the presence of ZnSO4 and ZnCl2, and the structures were solved to 1.8 Å and 2.0 Å, respectively (see Materials and Methods). The data collection and refinement statistics for both IMP-1 C221G structures are listed in Table S3 in the supplemental material. While the final concentration of zinc under both crystallization conditions was 500 μM, both C221G mutant structures contained zinc only in the Zn1 site. The structure obtained from a crystal grown with ZnSO4 contains a distinctly tetrahedral density consistent with an SO42− ion in the Zn2 binding site. The presence of a well-coordinated sulfate ion in place of Zn2 has been observed in other Zn2 site mutant MβL structures and may assist in crystal packing (19). The second structure from the ZnCl2 condition also contained a nonspherical density, but it more closely represents the buffer element, citrate, in the Zn2 site. Occupancy of these large ions in the mutant structure appears to affect the conformation of H263 and D120 such that the Zn2 site is shifted away from the Zn1 site (Fig. 6; see Fig. S1 in the supplemental material). This physical shift may contribute to defects observed in the functional assays described above. The absence of Zn2 in these structures could be due to the high concentrations of sulfate (1 mM) and citrate (100 mM) found under the crystallization conditions. LIGPLOT analysis of our mutant IMP-1 structures indicated hydrogen bonds with residues His263 and Ser262 and a potential salt bridge with Lys224 with either sulfate or citrate in the Zn2 site, which may stabilize these ligands in the crystals. Regardless, the presence of these ligands rather than zinc at the Zn2 site does not allow a direct test of the hypothesis that water in the cysteine 221 side chain position serves as a ligand for zinc in the functional C221G mutant enzyme (50, 57).
Fig 6.

Active-site residues of wild-type and C221G IMP-1 enzymes. (A, C, D, and F) Illustrations of wild-type (C and F) and C221G (sulfate-bound [A] and citrate-bound [D]) active-site residue conformations. (B and E) Overlays of the wild-type and C221G structures (sulfate-bound mutant [B] and citrate-bound mutant [E]). Residues are represented as sticks, with carbon atoms shown in gray (wild type) or teal (C221G mutant). Citrate carbon atoms are shown in yellow. Zinc ions are shown as gray (wild-type) or teal (C221G mutant) spheres, and water is shown as small red spheres. Nitrogen is shown in blue, sulfur in yellow, and oxygen in red. The figures were rendered using the PyMOL Molecular Graphics System (11).
The overall structures of both crystal forms were nearly identical to the published wild-type IMP-1 structure, with root mean square deviation (RMSD) values of 0.438 Å for the sulfate-bound and 0.404 Å for the citrate-bound forms. Furthermore, differences in zinc-ligand distances for the three His residues of the Zn1 site fall below the overall RMSD values. A conformational change in active-site loop-containing residues 63 through 69 was found to be the result of an interaction with the protein's uncleaved purification tag and is presumed not to affect function. These data confirm the presence of Zn1 as the single zinc in the Gly mutant enzyme and show that no large-scale structural perturbations are associated with the C221G mutation.
Sequence determinants of Zn2 binding.
The Zn2 binding sites of subclass B1 enzymes differ from those of B3 enzymes in the identities of residues at positions 221 and 121 (1). B3 enzymes include a His that functions as a zinc ligand at position 121 and a Ser or Asp at position 221 that does not interact directly with zinc (2). Subclass B1 enzymes, however, contain a zinc ligand Cys at position 221 and a nonligand Ser at position 121. To understand the plasticity of the Zn2 binding site in an IMP-1 background, we randomly mutated the position 121 codon in the background of the wild-type and C221G IMP-1 genes and measured the in vivo function of sequence-verified mutants by MIC determinations (Table 4).
Table 4.
Antibiotic susceptibilities of E. coli expressing position 121 IMP-1 variants
| Variant | MIC (μg/ml) |
|||
|---|---|---|---|---|
| Wild-type background |
C221G background |
|||
| Ampicillin | Cefotaxime | Ampicillin | Cefotaxime | |
| Wild type | 125 | 250 | ||
| C221G | 6 | 4 | ||
| S121A | >256 | 48 | 3 | 0.75 |
| S121C | 48 | 3 | 1.5 | 0.047 |
| S121D | 1 | 0.023 | 2 | 0.094 |
| S121E | 1.5 | 0.032 | 12 | 4 |
| S121F | 1.5 | 0.032 | 2 | 0.047 |
| S121G | >256 | 32 | 3 | 0.19 |
| S121H | 1.5 | 0.023 | 2 | 0.047 |
| S121I | 1.5 | 0.064 | 1 | 0.023 |
| S121K | 1.5 | 0.032 | 2 | 0.047 |
| S121L | 1.5 | 0.032 | 1.5 | 0.032 |
| S121 M | 1.5 | 0.023 | 1.5 | 0.023 |
| S121N | 6 | 0.38 | 2 | 0.047 |
| S121P | 1 | 0.023 | 1.5 | 0.032 |
| S121Q | 4 | 0.047 | 1.5 | 0.032 |
| S121R | 2 | 0.032 | 2 | 0.047 |
| S121T | 3 | 2 | 48 | 4 |
| S121V | 8 | 1 | 2 | 0.094 |
| S121W | 1.5 | 0.032 | 1.5 | 0.032 |
| S121Y | 1.5 | 0.047 | 1.5 | 0.032 |
The antibiotic MIC values associated with C221G-S121X double mutants suggest that no substitutions at position 121 are able to substantially rescue the in vivo function of the C221G enzyme. Additionally, of the 19 mutants tested, only two were consistent with function at the level of the parent C221G mutant. Therefore, the subclass B1 and B3 enzymes are sufficiently evolutionarily diverged that the 121 and 221 ligand positions are not exchangeable between the subclasses. Interestingly, only five single-position 121 substitutions in the wild-type background were consistent with partial function, and none exhibited wild-type function against ampicillin or cefotaxime. Of the functional single mutants, the Ala and Gly mutants had the highest function while the Cys, Thr, and Val mutants exhibited more compromised functional levels. The negative effects of substitutions seen in the single and C221G double mutant clones point to a previously unreported role for Ser121 in the structure and/or function of IMP-1. It is notable that Ser121 is within range for hydrogen bonds with His116 and Asp120 at the base of the active site. The main-chain amine also appears to hydrogen bond with the main-chain carbonyl of His118. These structural data suggest that Ser121 may directly affect catalysis through positioning of His116 and His118 or through another mechanism affecting protein stability by contributing to the hydrogen-bonding network.
DISCUSSION
Consistent with previous findings, the results of this study indicate that Cys221 of IMP-1 β-lactamase plays a crucial role in maintaining the stability and catalytic efficiency of the enzyme (18–20, 30, 53). Of the 19 mutants tested, only Asp, Gly, Ser, Thr, and Val substitutions resulted in detectable steady-state protein expression levels in E. coli and only the IMP-1 enzymes with Asp and Gly substitutions provided E. coli with resistance to β-lactams. The finding that the C221S mutant exhibits no function above background levels was consistent with previous in vitro observations; however, the identification of a functional Gly mutant was surprising (20).
The C221G mutant provides E. coli with antibiotic resistance levels lower than those observed with wild-type IMP-1 but significantly above background levels on rich medium. We have also shown that binding of exogenous zinc to the monozinc C221G mutant is associated with enhancement of thermal stability, as well as in vivo and in vitro function, despite the absence of a side chain to ligand Zn2. Binding of zinc by the C221G enzyme was measured by multiple techniques to be in the range of a 17 to 43 μM dissociation constant, which is significantly weaker than the recently determined binding constant of 300 nM for cobalt for the Zn2 site in the wild-type enzyme (63). The weaker binding affinity for Zn2 in the C221G enzyme is consistent with the loss of the cysteine side chain, which helps coordinate the zinc ion in the wild-type enzyme.
The pattern of amino acid substitutions at position 221 that are consistent with enzyme function is interesting. Although the enzymes with Val, Ser, and Thr substitutions are expressed in E. coli, only the enzymes with Asp and Gly substitutions exhibit catalytic function. Aspartate is a known zinc ligand and could ligate Zn2. However, molecular modeling suggests there are steric constraints on position 221, and therefore, the mutations at 221 may alter the Zn2 positioning, resulting in the observed reduction in catalytic activity relative to the wild type.
Molecular modeling suggests the Val, Ser, and Thr substitutions can be accommodated in the presence of bound Zn2. However, Ser and Thr are poor zinc ligands and Val has not been shown to coordinate zinc. The lack of zinc coordination by these residues could reduce zinc binding affinity, as well as affect the positioning and/or pKa of the coordinated water between Zn1 and Zn2, which would alter the position and concentration of metal-bound hydroxide ion that is thought to act as a nucleophile in catalysis (41).
The glycine residue in the C221G enzyme cannot directly interact with Zn2, but molecular modeling suggests the gap created by the substitution could be filled with a water molecule, which could serve as a zinc ligand. Modeling also suggests an alanine substitution at 221 would not leave sufficient space for a water molecule between the Ala methyl group and Zn2, which could explain why the C221A mutant is nonfunctional. X-ray crystallography of the C221G IMP-1 enzyme was performed to explore the hypothesis that water fills the void created by the Gly substitution and serves as a surrogate zinc ligand. However, adventitious binding of buffer-supplied sulfate or citrate in the Zn2 site obfuscated the interpretation of these results.
The structure of the sulfate-bound C221G mutant enzyme displays only a small displacement (0.43 Å) between the location of the core of the sulfate ion and the location of the Zn2 ion in the wild-type IMP-1, which is within the overall RMSD for the enzyme. The structural data also indicate that the ions present at the Zn2 site are supported by an extensive network of interactions (Fig. 6; see Fig. S1 in the supplemental material). The Zn2 ligand residue Asp120 appears to stabilize both the sulfate and citrate ions by hydrogen bonding through its carboxyl group. The His263 residue also positions a side chain nitrogen to interact with a sulfate oxygen or with either of two proximal oxygens of the bulkier citrate, while the second-shell Lys224 residue also appears to bind sulfate through its main-chain carbonyl and citrate via a side chain interaction.
It is interesting that in the class A β-lactamases replacement of Ser130 with glycine results in a partially functional enzyme due to a water molecule occupying the position of the missing serine side chain (26, 27, 60). Ser130 is completely conserved among class A enzymes and is suggested to act as a catalytic acid for protonating the nitrogen leaving group in the amide bond and thereby facilitating hydrolysis of the β-lactam ring. Therefore, replacement with glycine would be expected to result in a nonfunctional enzyme. However, the enzyme with an S130G substitution retains significant catalytic activity, and the X-ray structure shows a water molecule in the position of the serine side chain to compensate for loss of the hydroxyl group (60). We postulate that a water molecule plays an analogous role in the C221G mutant, compensating for the loss of the zinc ligand.
The determination of kinetic parameters for nitrocefin hydrolysis at multiple zinc concentrations indicated binding at the Zn2 position in the C221G mutant occurs with a KD (equilibrium dissociation constant) of 43 μM, and zinc concentrations above this value can substantially restore enzyme function. This suggests that, once occupied, the position of Zn2 in the Gly mutant is similar to that of Zn2 in the wild-type enzyme. This result is in contrast to the previously characterized C221S mutant, which is partially occupied at the Zn2 site at high zinc concentrations but does not exhibit an increase in catalytic activity in response to increased available zinc (20).
This study has also shown via in vivo protein expression experiments in defined medium that the function of the IMP-1 enzyme in E. coli is dependent on the concentration of exogenous zinc. This finding suggests that the level of function of MβL enzymes depends critically on the available zinc in the extracellular environment. In the context of a bacterial infection, this observation becomes important, because mechanisms exist within the vertebrate host to limit the concentration of certain metals (24, 25). Scarcity of available zinc could, in turn, limit the catalytic activity of metallo-β-lactamases and reduce the antibiotic resistance potential of the bacteria (25).
It is known that porins such as OmpF in E. coli allow the diffusion of metal ions across the outer membrane of Gram-negative species (12, 42, 54). These studies suggest that the periplasmic space, where MβL enzymes are processed and folded, is continuous with the external milieu with respect to the zinc concentration (12). The concentration of available environmental zinc could then be another selecting factor in the evolution and spread of MβL genes. As we have shown, limited zinc availability affects the function of even the wild-type IMP-1 enzyme in bacteria, and therefore, it will be of interest to identify the metallation state and folded state of enzymes expressed in zinc-limited media (49). Crowder and colleagues have shown that the bioavailability of metal ions has a strong influence on the metal content of the class B3 L1 metallo-β-lactamase when expressed from E. coli grown in minimal medium, and by analogy, the metal content of IMP-1 is likely determined by exogenous metal concentrations (22a).
Last, we have shown that amino acid residues at position 221 (subclass B1 ligand) and position 121 (subclass B3 ligand) are not functionally interchangeable in that no substitutions at position 121 constructed in the background of C221G are able to compensate for the loss of the Cys221 side chain. Consistent with these findings, González et al. concluded that additional sequence determinants beyond positions 121 and 221 likely affect binding at the Zn2 position and therefore enzyme function (19). Structural analysis suggests that a His residue at position 121 of IMP-1 is not possible because of a steric clash with Asp120. Additionally, the wild-type Ser121 is 6.7 Å away from Zn2 and too far to assist in coordination, even if substituted. Ser221 in the L1 enzyme is 4.6 Å away from Zn2 and is also not able to serve as a ligand (Fig. 7). Thus, the B1 and B3 enzymes are evolutionarily diverged to such a point that zinc-chelating residues at these positions are not interchangeable.
Fig 7.
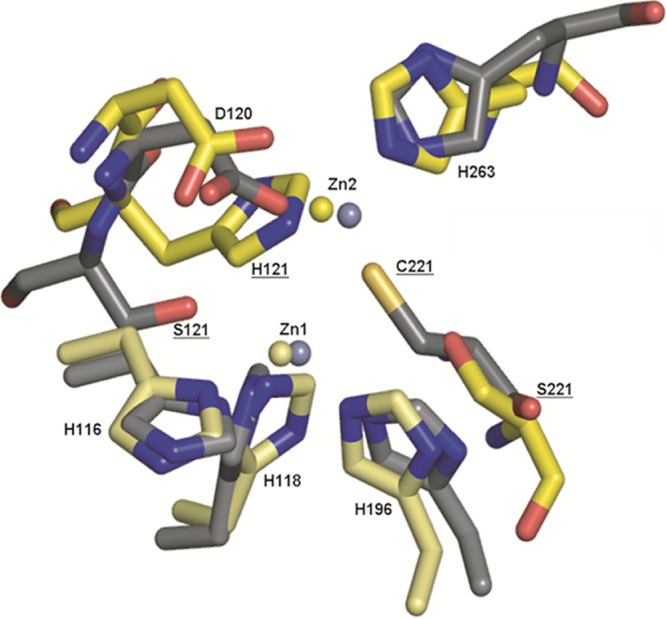
Zinc coordinating active-site residues of IMP-1 and L1 MβLs. Active-site residues of the B1 subgroup IMP-1 (1DDK) and B3 subgroup L1 (1SML) β-lactamase structures are represented as sticks, with oxygen shown in red, nitrogen shown in blue, and sulfur shown in yellow. Carbon atoms are shown in gray (IMP-1) and yellow (L1). Zinc ions are shown in steel gray (IMP-1) and light yellow (L1). The figures were rendered using the PyMOL Molecular Graphics System (11).
Although substitutions at position 121 do not compensate for the loss of Cys221, the mutagenesis results support a role for Ser121 in maintaining the stability and/or catalytic function of IMP-1 and possibly other subclass B1 enzymes. Ser121 is one of a number of hydrophilic residues in the active site that contribute to a hydrogen-bonding network that may properly position other residues for catalysis (36). In a region containing Zn1 and Zn2 ligands, the Ser119 and Ser121 residues on either side of Asp120 engage the catalytically important His116 and His118 residues via main-chain and side chain hydrogen bonds (6). While the side chain of Ser119 faces into the body of the protein, the hydroxyl of Ser121 is directed into the active-site pocket. The positioning of the main chain at position 121 may also explain why this position can be utilized to ligate zinc in the subclass B3 scaffold (56). Although this study shows that 13 of 19 possible substitutions ablate subclass B1 enzyme function, understanding how these substitutions perturb the overall protein structure and the active-site hydrogen bond network will require further structural studies.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH grant AI32956 to T.P. L.B.H. is a fellow of the Pharmacoinformatics Training Program of the W. M. Keck Center for Computational and Structural Biology of the Gulf Coast Consortia (NIH grant T90 DK070109). B.V.V.P. acknowledges support from the Robert Welch Foundation (Q1279).
Footnotes
Published ahead of print 20 August 2012
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1. Bebrone C. 2007. Metallo-beta-lactamases (classification, activity, genetic organization, structure, zinc coordination) and their superfamily. Biochem. Pharmacol. 74:1686–1701 [DOI] [PubMed] [Google Scholar]
- 2. Bebrone C, et al. 2009. The structure of the dizinc subclass B2 metallo-beta-lactamase CphA reveals that the second inhibitory zinc ion binds in the histidine site. Antimicrob. Agents Chemother. 53:4464–4471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brown NG, Horton LB, Huang W, Vongpunsawad S, Palzkill T. 2011. Analysis of the functional contributions of Asn233 in the IMP-1 metallo-{beta}-lactamase. Antimicrob. Agents Chemother. 55:5696–5702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bush K, Jacoby G. 2010. Updated functional classification of beta-lactamases. Antimicrob. Agents Chemother. 54:969–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chothia C. 1976. The nature of the accessible and buried surfaces in proteins. J. Mol. Biol. 105:1–12 [DOI] [PubMed] [Google Scholar]
- 6. Concha NO, et al. 2000. Crystal structure of the IMP-1 metallo beta-lactamase from Pseudomonas aeruginosa and its complex with a mercaptocarboxylate inhibitor: binding determinants of a potent, broad-spectrum inhibitor. Biochemistry 39:4288–4298 [DOI] [PubMed] [Google Scholar]
- 7. Crisp J, et al. 2007. Structural basis for the role of Asp-120 in metallo-beta-lactamases. Biochemistry 46:10664–10674 [DOI] [PubMed] [Google Scholar]
- 8. Crowder MW, Wang Z, Franklin SL, Zovinka EP, Benkovic SJ. 1996. Characterization of the metal-binding sites of the beta-lactamase from Bacteroides fragilis. Biochemistry 35:12126–12132 [DOI] [PubMed] [Google Scholar]
- 9. Crowder MW, Spencer J, Vila AJ. 2006. Metallo-beta-lactamases: novel weaponry for antibiotic resistance in bacteria. Acc. Chem. Res. 39:721–728 [DOI] [PubMed] [Google Scholar]
- 10. Daiyasu H, Osaka K, Ishino Y, Toh H. 2001. Expansion of the zinc metallo-hydrolase family of the beta-lactamase fold. FEBS Lett. 503:1–6 [DOI] [PubMed] [Google Scholar]
- 11. DeLano W. 2002. PyMOL. DeLano Scientific, San Carlos, CA [Google Scholar]
- 12. Easton JA, Thompson P, Crowder MW. 2006. Time-dependent translational response of E. coli to excess Zn(II). J. Biomol. Tech. 17:303–307 [PMC free article] [PubMed] [Google Scholar]
- 13. Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D. 60:2126–2132 [DOI] [PubMed] [Google Scholar]
- 14. Ericsson UB, Hallberg BM, Detitta GT, Dekker N, Nordlund P. 2006. Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal. Biochem. 357:289–298 [DOI] [PubMed] [Google Scholar]
- 15. Fisher JF, Meroueh SO, Mobashery S. 2005. Bacterial resistance to beta-lactam antibiotics: compelling opportunism, compelling opportunity. Chem. Rev. 105:395–424 [DOI] [PubMed] [Google Scholar]
- 16. Frère J-M, Galleni M, Bush K, Dideberg O. 2005. Is it necessary to change the classification of beta-lactamases? J. Antimicrob. Chemother. 55:1051–1053 [DOI] [PubMed] [Google Scholar]
- 17. Galleni M, Lamotte-Brasseur J, Rossolini GM. 2001. Standard numbering scheme for class B β-lactamases. Antimicrob. Agents Chemother. 45:660–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gardonio D, Siemann S. 2009. Biochemical and biophysical research communications chelator-facilitated chemical modification of IMP-1 metallo-β-lactamase and its consequences on metal binding. Biochem. Biophys. Res. Commun. 381:107–111 [DOI] [PubMed] [Google Scholar]
- 19. González JM, et al. 2007. The Zn2 position in metallo-beta-lactamases is critical for activity: a study on chimeric metal sites on a conserved protein scaffold. J. Mol. Biol. 373:1141–1156 [DOI] [PubMed] [Google Scholar]
- 20. Haruta S, et al. 2000. Functional analysis of the active site of a metallo-beta-lactamase proliferating in Japan. Antimicrob. Agents Chemother. 44:2304–2309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77:51–59 [DOI] [PubMed] [Google Scholar]
- 22. Hu Z, Zhao W-H. 2009. Identification of plasmid- and integron-borne blaIMP-1 and blaIMP-10 in clinical isolates of Serratia marcescens. J. Med. Microbiol. 58:217–221 [DOI] [PubMed] [Google Scholar]
- 22a. Hu Z, Gunasekera TS, Spadafora L, Bennett B, Crowder MW. 2008. Metal content of metallo-beta-lactamase L1 is determined by the bioavailability of metal ions. Biochemistry 47:7947–7953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hunt JB, Neece SH, Ginsburg A. 1985. The use of 4-(2-pyridylazo)resorcinol in studies of zinc release from Escherichia coli aspartate transcarbamoylase. Anal. Biochem. 146: 150–157 [DOI] [PubMed] [Google Scholar]
- 24. Kehl-Fie TE, et al. 2011. Nutrient metal sequestration by calprotectin inhibits bacterial superoxide defense, enhancing neutrophil killing of Staphylococcus aureus. Cell Host Microbe 10:158–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kehl-Fie TE, Skaar EP. 2010. Nutritional immunity beyond iron: a role for manganese and zinc. Curr. Opin. Chem. Biol. 14:218–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lamotte-Brasseur J, et al. 1991. Mechanism of acyl transfer by the class A serine beta-lactamase of Streptomyces albus G. Biochem. J. 279:213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lamotte-Brasseur J, Jacob-Dubuisson F, Dive G, Frère JM, Ghuysen JM. 1992. Streptomyces albus G serine beta-lactamase. Probing of the catalytic mechanism via molecular modelling of mutant enzymes. Biochem. J. 282:189–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Laraki N, et al. 1999. Biochemical characterization of the Pseudomonas aeruginosa 101/1477 metallo-beta-lactamase IMP-1 produced by Escherichia coli. Antimicrob. Agents Chemother. 43:902–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee LJ, Barrett JA, Poole RK. 2005. Genome-wide transcriptional response of chemostat-cultured Escherichia coli to zinc. J. Bacteriol. 187:1124–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li Z, Rasmussen B, Herzberg O. 1999. Structural consequences of the active site substitution Cys181→Ser in metallo-beta-lactamase from Bacteroides fragilis. Protein Sci. 8:249–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Livermore DM. 2009. Has the era of untreatable infections arrived? J. Antimicrob. Chemother. 64(Suppl. 1):i29–i36 [DOI] [PubMed] [Google Scholar]
- 32. Maltezou HC. 2009. Metallo-beta-lactamases in Gram-negative bacteria: introducing the era of pan-resistance? Int. J. Antimicrob. Agents 33:405 e1–e7 [DOI] [PubMed] [Google Scholar]
- 33. Marciano DC, et al. 2008. Genetic and structural characterization of an L201P global suppressor substitution in TEM-1 beta-lactamase. J. Mol. Biol. 384:151–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Materon IC, Beharry Z, Huang W, Perez C, Palzkill T. 2004. Analysis of the context dependent sequence requirements of active site residues in the metallo-beta-lactamase IMP-1. J. Mol. Biol. 344:653–663 [DOI] [PubMed] [Google Scholar]
- 35. Materon IC, Palzkill T. 2001. Identification of residues critical for metallo-beta-lactamase function by codon randomization and selection. Protein Sci. 10:2556–2565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McCall KA, Huang C, Fierke CA. 2000. Function and mechanism of zinc metalloenzymes. J. Nutr. 130:1437S–1446S [DOI] [PubMed] [Google Scholar]
- 37. Reference deleted.
- 38. Moran-Barrio J, et al. 2007. The metallo-beta-lactamase GOB is a mono-Zn(II) enzyme with a novel active site. J. Biol. Chem. 282:18286–18293 [DOI] [PubMed] [Google Scholar]
- 39. Neu HC, Heppel LA. 1965. The release of enzymes from Escherichia coli by osmotic shock and during the formation of spheroplasts. J. Biol. Chem. 240:3685–3692 [PubMed] [Google Scholar]
- 40. Oelschlaeger P. 2008. Outsmarting metallo-beta-lactamases by mimicking their natural evolution. J. Inorg. Biochem. 102:2043–2051 [DOI] [PubMed] [Google Scholar]
- 41. Page MI, Badarau A. 2008. The mechanisms of catalysis by metallo beta-lactamases. Bioinorg. Chem. Appl. 2008:576297 doi:10.1155/2008/576297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pagès J-M, James CE, Winterhalter M. 2008. The porin and the permeating antibiotic: a selective diffusion barrier in Gram-negative bacteria. Nat. Rev. Microbiol. 6:893–903 [DOI] [PubMed] [Google Scholar]
- 43. Palzkill T, Le QQ, Venkatachalam KV, LaRocco M, Ocera H. 1994. Evolution of antibiotic resistance: several different amino acid substitutions in an active site loop alter the substrate profile of beta-lactamase. Mol. Microbiol. 12:217–229 [DOI] [PubMed] [Google Scholar]
- 44. Parsell DA, Sauer RT. 1989. The structural stability of a protein is an important determinant of its proteolytic susceptibility in Escherichia coli. J. Biol. Chem. 264:7590–7595 [PubMed] [Google Scholar]
- 45. Pflugrath JW. 1999. The finer things in X-ray diffraction data collection. Acta Crystallogr. D 55:1718–1725 [DOI] [PubMed] [Google Scholar]
- 46. Phillips K, de la Peña AH. 2011. The combined use of the Thermofluor assay and ThermoQ analytical software for the determination of protein stability and buffer optimization as an aid in protein crystallization. Curr. Protoc. Mol. Biol. Chapter 10:Unit10.28 [DOI] [PubMed] [Google Scholar]
- 47. Potterton E, Briggs P, Turkenburg M, Dodson E. 2003. A graphical user interface to the CCP4 program suite. Acta Crystallogr. D 59:1131–1137 [DOI] [PubMed] [Google Scholar]
- 48. Säbel CE, Shepherd JL, Siemann S. 2009. A direct spectrophotometric method for the simultaneous determination of zinc and cobalt in metalloproteins using 4-(2-pyridylazo)resorcinol. Anal. Biochem. 391:74–76 [DOI] [PubMed] [Google Scholar]
- 49. Selevsek N, et al. 2009. Zinc ion-induced domain organization in metallo-beta-lactamases: a flexible “zinc arm” for rapid metal ion transfer? J. Biol. Chem. 284:16419–16431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shanker S, et al. 2011. Structural analysis of histo-blood group antigen binding specificity in a norovirus GII.4 epidemic variant: implications for epochal evolution. J. Virol. 85:8635–8645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sharma N, Toney JH, Fitzgerald PMD. 2005. Expression, purification, crystallization and preliminary X-ray analysis of Aeromonas hydrophilia metallo-beta-lactamase. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 61:180–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sideraki V, Huang W, Palzkill T, Gilbert HF. 2001. A secondary drug resistance mutation of TEM-1 beta-lactamase that suppresses misfolding and aggregation. Proc. Natl. Acad. Sci. U. S. A. 98:283–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Siemann S, et al. 2002. IMP-1 metallo-beta-lactamase: effect of chelators and assessment of metal requirement by electrospray mass spectrometry. Biochim. Biophys. Acta 1571:190–200 [DOI] [PubMed] [Google Scholar]
- 54. Sigdel TK, et al. 2006. Probing the adaptive response of Escherichia coli to extracellular Zn(II). Biometals 19:461–471 [DOI] [PubMed] [Google Scholar]
- 55. Turnbull WB, Daranas AH. 2003. On the value of c: can low affinity systems be studied by isothermal titration calorimetry? J. Am. Chem. Soc. 125:14859–14866 [DOI] [PubMed] [Google Scholar]
- 56. Ullah JH, et al. 1998. The crystal structure of the L1 metallo-beta-lactamase from Stenotrophomonas maltophilia at 1.7 Å resolution. J. Mol. Biol. 284:125–136 [DOI] [PubMed] [Google Scholar]
- 57. Wallace AC, Laskowski RA, Thornton JM. 1995. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 8:127–134 [DOI] [PubMed] [Google Scholar]
- 58. Walsh TR. 2008. Clinically significant carbapenemases: an update. Curr. Opin. Infect. Dis. 21:367–371 [DOI] [PubMed] [Google Scholar]
- 59. Walsh TR, Toleman MA, Poirel L, Nordmann P. 2005. Metallo-beta-lactamases: the quiet before the storm? Clin. Microbiol. Rev. 18:306–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang X, Minasov G, Shoichet BK. 2002. The structural bases of antibiotic resistance in the clinically derived mutant beta-lactamases TEM-30, TEM-32, and TEM-34. J. Biol. Chem. 277:32149–32156 [DOI] [PubMed] [Google Scholar]
- 61. Wang Z, Fast W, Benkovic SJ. 1999. On the mechanism of the metallo-beta-lactamase from Bacteroides fragilis. Biochemistry 38:10013–10023 [DOI] [PubMed] [Google Scholar]
- 62. Wright GD. 2005. Bacterial resistance to antibiotics: enzymatic degradation and modification. Adv. Drug Deliv. Rev. 57:1451–1470 [DOI] [PubMed] [Google Scholar]
- 63. Yamaguchi Y, et al. 2011. A demetallation method for IMP-1 metallo-β-lactamase with restored enzymatic activity upon addition of metal ion(s). Chembiochem 12:1979–1983 [DOI] [PubMed] [Google Scholar]
- 64. Zamyatnin AA. 1972. Protein volume in solution. Progr. Biophys. Mol. Biol. 24:107–123 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.