

Abstract
We prepared monoclonal antibodies against N-(γ-maleimidobutyryloxy)succinimide-conjugated vancomycin (VM). The monoclonal antibody was specific for conjugated or free VM. The monoclonal antibody enabled us to develop an immunocytochemical method for detecting the uptake of VM in the rat kidney and liver. Three hours after a single intravenous (i.v.) injection of VM at the therapeutic dose, the immunocytochemistry revealed that VM accumulated in large amounts in both the S1 and S2 segments and in much smaller amounts in the S3 segment of the proximal tubules as well as in the distal tubules and collecting ducts. The drug was detected in the cytoplasm, cytoplasmic irregular granules, nuclei, and microvilli of the proximal tubule cells. The distal tubules and collecting ducts contained scattered swollen cells in which both the nuclei and cytoplasm were heavily immunostained. Twenty-four hours after injection, most of the swollen cells returned back to normal size and had somewhat decreased immunostaining. Also, significant amounts of VM remained accumulated for as long as 8 days postadministration. In the liver, similar drug accumulation was observed in the Kupffer cells and the endothelial cells of the hepatic sinusoids but not in the hepatocytes, suggesting that vancomycin cannot be eliminated via the liver. Immunoelectron microscopic studies demonstrated that in the collecting ducts, uptake of VM occurred exclusively in the lysosomes and cytoplasm of the principal cells and scarcely in the intercalated cells. Furthermore, double fluorescence staining using rats simultaneously administered with VM and gentamicin strongly suggests that both drugs colocalized in lysosomes in the proximal tubule cells of kidneys.
INTRODUCTION
Vancomycin (VM), one of the most frequently used antimicrobials for the treatment of various serious Gram-positive infections, such as methicillin-resistant Staphylococcus aureus, is a glycopeptide that has been used in clinical practice for almost 5 decades (29, 32). VM sometimes produces several side effects, the most serious of which is nephrotoxicity (3, 39). However, there are few data about its renal uptake and subcellular distribution. Thus, accurate localization of VM in cells and tissues would be useful in developing a better understanding of the nephrotoxicity of the drug. To our knowledge, the only study on VM localization is the paper concerning subcellular localization of VM in proximal tubules of the nephron using the antibody-gold complex technique (1). However, this study was limited to ultrastructural analysis of proximal tubule cells, informing us that the drug localized only in the lysosomes in the cells. Recently, we have developed an original immunocytochemical (ICC) procedure for drugs with an aliphatic primary amino group in their molecule, upgrading the sophistication of the detection sensitivity and applying it to pharmacological and toxicological studies (16–21, 35, 42). These ICC procedures make use of glutaraldehyde (GA)-fixed tissues, which undergo a series of pretreatments to unmask the sites of the accumulation of the drug.
We now report on the development of a monoclonal antibody for VM and an ICC method for the uptake of VM in the kidney and liver of rats administered intravenously (i.v.) with the drug. This method clearly demonstrated the distribution and accumulation in cells of a whole kidney of rats from 1 h to 8 days after the injection, showing that VM accumulates in the cytoplasmic granules of all the segments (S1, S2, and S3) of the proximal tubules, in the tubular lumen side of which numerous small bodies occurred. VM accumulates in the distal convoluted duct and collecting duct cells, and in both cell types, numerous swollen cells, which contained large amounts of VM, being seemingly affected by the drug, occurred. Also, it was found that significant amounts of VM remained accumulated in the sites where it was distributed for as long as 8 days postadministration. This study also includes double immunofluorescence staining for VM and gentamicin (GM) using kidney specimens from rats simultaneously injected with both drugs and immunoelectron microscopy (IEM) for VM in the collecting duct cells as well as VM uptake in the liver.
MATERIALS AND METHODS
Chemicals.
VM hydrochloride, glutaraldehyde (GA; 25% in water), and Triton X-100 were purchased from Nacalai Tesque (Kyoto, Japan). Sodium borohydride (NaBH4) and protease (type XXIV, bacterial) were from Sigma-Aldrich (St. Louis, MO). GM was supplied by Shionogi Co. Ltd. (Osaka, Japan). Anti-GM serum was the same batch as previously used (18).
Preparation of immunogen (VM-GMBS-BSA conjugate).
The immunogen was prepared according to our previous method for anti-daunomycin (DM) serum using a heterobifunctional agent, N-(γ-maleimidobutyryloxy)succinimide (GMBS) (8–10, 17). Briefly, VM HCl (12.5 mg, 8.4 μmol) in 0.1 M phosphate buffer (pH 6.8) and 1.0 ml and 0.7 mg (2.5 μmol) GMBS in 0.5 ml tetrahydrofuran were mixed, constantly stirred, and incubated at room temperature (RT) for 90 min, yielding a GMBS-acylated VM solution. Acetylmercaptosuccinyl bovine serum albumin (AMS-BSA; 15.4 mg, approximately 0.1 μmol) was incubated in 200 μl of 0.1 M hydroxylamine (pH 6.8) at room temperature for 20 min to remove the acetyl group. The resulting mercaptosuccinyl BSA (MS-BSA) was diluted with 3 ml 0.1 M phosphate buffer (pH 7.0), added immediately to GMBS-acylated DM solution, and incubated for 30 min with slow stirring. The conjugate was applied to a 2.5-cm by 45-cm Sephadex G-75 column equilibrated with 20 mM phosphate buffer (pH 7.0) and was eluted with the same buffer. The eluate, monitored at 280 nm, was collected in 3-ml fractions, and the peak fraction was used for immunization. The VM-GA-BSA conjugate was prepared according to our previous method (18).
Preparation of anti-VM monoclonal antibodies (MAbs).
Three 5-week-old, female BALB/c mice were injected intraperitoneally (i.p.) with 100 μg of VM-GMBS-BSA conjugate emulsified in complete Freund's adjuvant (Difco). Subsequently, they received two injections of 50 μg of the conjugate alone at 2-week intervals. Following immunization, antisera were collected, and antibody titers were evaluated with an enzyme-linked immunosorbent assay (ELISA) as described below. The mouse with the best immune response was selected for hybridization. The mouse received a fourth i.p. booster injection and was sacrificed 4 days later.
Cell fusion and cloning.
The spleen cells (2 × 108) from the immunized mouse and 3 × 107 myeloma cells (P3/NS-1) were fused with the help of polyethylene glycol according to our previous method (17). Cells suspended in hypoxanthine-aminopterin-thymidine (HAT) medium were plated out in 96-well tissue culture plates (Corning) at a density of 105 cells per well in which 105 feeder cells had been plated. From 10 to 20 days postfusion, the wells were screened for reactivity using an ELISA method as described below. Limiting dilutions of positive cultures were carried out two or three times to obtain monoclonality, and subisotyping of the MAbs was performed using a mouse monoclonal subisotyping kit (Zymed Lab. Inc., San Francisco, CA). Ascites were raised in BALB/c mice pretreated with Pristene by intraperitoneal injection of 2 × 106 hybridoma cells.
ELISA method.
An ELISA was performed in a manner similar to that for our previous method for anti-DM MAbs (17). In screening clones for the production of antibody against VM-GMBS-BSA, wells in microtiter plates were coated with 10 μg/ml of the VM-GMBS-BSA conjugate for 30 min at RT. The wells were then incubated overnight at 4°C with antiserum (diluted 1:3,000), hybridoma culture supernatant, or ascites fluid (diluted 1:100,000), followed by goat anti-mouse IgG labeled with horseradish peroxidase (HRP) (diluted 1:2,000) for 1 h at RT. The amount of enzyme conjugate bound to each well was measured using o-phenylenediamine as a substrate, and the absorbance at 492 nm was read with an automatic ELISA analyzer (ImmunoMini NJ-2300; Nalje Nunc Int. Co. Ltd., Tokyo, Japan).
Inhibition ELISAs.
Wells in a microtiter plate are coated with 100 μl of the VM-GMBS-BSA conjugate (10 μg/ml) as described above (15). To the coated wells were added 50 μl of a fixed concentration of the anti-VM monoclonal antibody AVM-113 MAb (1:100) and then 50 μl of VM-GMBS-BSA conjugates or free compounds (VM, GM, ampicillin, and peplomycin) at various concentrations, and these were incubated overnight at 4°C, followed by incubation with goat anti-mouse IgG labeled with HRP (1:2,000) for 1 h at RT. The bound HRP activity was measured using the ELISA analyzer.
Binding ELISAs.
According to our previous method (15), the wells in a microtiter plate coated with poly-l-lysine (30 μg/ml) were activated with 2.5% GA in 50 mM borate buffer (pH 10.0) for 1 h. The wells were subsequently incubated with test compounds at various concentrations for 1 h at RT. Excess aldehyde groups were blocked with 0.5% sodium borohydride. The wells were further incubated for 1 h with 1% skim milk to block nonspecific protein binding sites, and then the plates were incubated overnight at 4°C with the primary AVM-113 MAb at 1:50, diluted with phosphate-buffered saline (PBS) containing 0.05% Tween 20 (PBST). The wells were then incubated for 1 h with HRP-labeled goat anti-mouse IgG (diluted 1:2,000). The bound enzyme activity was measured as described above.
Animals.
Normal adult male Wistar rats (Kyudo Exp. Animals; Kumamoto, Japan) with body weights of 200 to 250 g were used in this study. The principles of laboratory animal care and specific national laws were observed. The animals were housed in temperature- and light-controlled rooms (21 ± 1°C and 12 h light/12 h dark) and had free access to standard food and tap water. Twelve Wistar rats were injected i.v. with a single dose of 33.3 mg VM/kg of body weight. At 1, 3, and 24 h and 8 days after the injection, three rats from each group were anesthetized with sodium pentobarbital (60 mg/kg; Abbott Laboratories, North Chicago, IL) and perfused transcardially with PBS (10 mM phosphate buffer [pH 7.2] containing 0.15 M NaCl) at 50 ml/min for 2 min at RT and then with a freshly prepared solution of 2% GA in PBS (pH 7.2) for 6 min. The kidney and liver were quickly excised and postfixed in the same fixative overnight at 4°C with 2.0% GA in PBS. Three rats receiving a saline instead of VM were used as the controls. For the double fluorescence staining for VM and GM, three rats were injected i.v. with a single dose of a mixture of 33.3 mg and 16 mg of VM and GM, respectively, per kg of body weight, and the method mentioned above was followed 12 h after the injection.
ICC.
The ICC method was carried out essentially according to our previous methods (19–21). The postfixed specimens of the liver and kidneys were subsequently embedded in paraffin in a routine way. The samples were cut into 5-μm-thick sections, acidified with 1 N HCl for 30 min (in order to denature the DNA) (25, 46), digested with 0.001% to 0.006% protease (type XXIV, bacterial; Sigma-Aldrich) in 50 mM Tris-HCl buffer (pH 7.4) containing 0.86% NaCl (Tris-buffered saline [TBS]) for different periods of time (15 min to 2 h) at 30°C (in order to facilitate the penetration of antibody to fully detect uptake of VM in different cells and subcellular compartments) (7), and reduced with 1.0% NaBH4 in TBS for 10 min (to prevent unspecific staining by reducing free aldehyde groups of GA bound in tissues) (4, 24). During each process of the treatment, the specimens were washed three times with TBS. Next, the specimens were blocked with a protein solution containing 10% normal goat serum, 1.0% BSA, and 0.1% saponin in TBS for 1 h at RT and were then directly incubated at 4°C overnight with the AVM-113 MAb diluted 1:50 to 1:100 in TBS supplemented with 0.1% Triton X-100 (TBST). The sections were washed with TBST three times, 5 min at a time, and then incubated with HRP-labeled goat anti-mouse IgG (whole IgG; Cappel, West Chester, PA) 1:500 for 2 h at 4°C. After rinsing with TBS, the site of the antigen-antibody reaction was revealed for 10 min with diaminobenzidine (DAB) and H2O2.
Controls for the immunocytochemical method included conventional staining (second-level) controls as well as a peplomycin MAb (20). Absorption controls used conjugates of VM-GMBS-BSA at a concentration of 30 μg/ml and the free compounds VM, amoxicillin (AMPC), and GM at concentrations ranging between 30 μg and 100 μg/ml.
IEM.
The postfixed specimens of the kidneys were cut with a Microslicer (Dosaka EM; Kyoto, Japan) into 50-μm-thick sections which were then applied to a free-floating procedure of the preembedding method (13, 14) in principally the same manner as that used for ICC for paraffin sections except for the HRP-labeled goat anti-mouse IgG/Fab′ (MBL; Nagoya, Japan) used as the second antibody. The color-developed specimens obtained were then fixed with 1.0% osmium tetroxide in 50 mM cacodylate buffer (pH 7.4) for 1 h at RT and dehydrated in a series of graded ethanol solutions. After immersion in propylene oxide (Nacalai Tesque; Tokyo, Japan) (three times for 10 min each), the samples were immersed in a mixture (1:1) of propylene oxide and Epon 812 resin (Taab Lab; Reading, Berks, United Kingdom) overnight and embedded in Epon 812 resin in a routine way. The regions to be studied were cut with a 2-mm-diameter punch, mounted to Epon blocks, and sectioned into ultrathin sections (LKB 8800 Ultrotome III) which were then immediately observed in a 100CX Jeol electron microscope.
Double fluorescence staining for VM and GM.
This was carried out according to our previous method (40, 41). Kidney specimens from rats injected i.v. with a mixture of VM and GM were pretreated in the same manner as described above in the ICC and incubated for 24 h at RT with a mixture of AVM-113 (1:200 diluted in TBS) and rabbit anti-GM serum (1:4,000 diluted in TBS) as the first antibody. After rinsing with TBST two times for 5 min, followed by one 5-min TBS rinse, specimens were incubated in a mixture of Alexa Fluor 488-labeled goat anti-mouse IgG (1:100 diluted in TBS) and Alexa Fluor 555-labeled goat anti-rabbit IgG (1:100 diluted in TBS) without NaN3 at RT for 2 h, mounted in SlowFade Gold (Molecular Probes; Invitrogen, OR), and examined by confocal laser scanning microscopy (Leica; TCS NT).
RESULTS
Generation and specificity of MAbs to VM.
From a fusion experiment, 450 hybridoma lines were produced. Three hybridoma lines (AVM-113, 28, and 74) secreted antibodies that bound to the VM-GMBS-BSA conjugate but did not recognize BSA, as screened by the ELISA system supplemented with BSA (1 mg/ml), which left anti-BSA antibodies out. They continued to secrete antibodies in culture supernatant. Subclones of the hybridoma obtained by limiting dilutions of AVM-113 and 28 were found to produce an antibody of the IgG1 subisotype. The subisotype of AVM-74 was not determined, since no reaction occurred with any of the IgG subtype antibodies available in a mouse MonoAb ID kit (Zymed Lab, Inc., CA).
Microtiter wells coated with VM-GMBS-BSA conjugate (10 μg/ml) were used to test for antibody binding using serial dilutions of the AVM-113 MAb (hybridoma culture supernatants). As shown in Fig. 1a, comparable significant binding activities were observed with the AVM-113 MAb in dilutions of less than 1:1,000. No antibody binding was seen with the type-matched (IgG) MAb of anti-spermine MAb-29, known to be specific to polyamines, spermine, and spermidine (11) (data not shown).
Fig 1.

(a) ELISA measurements of the binding of serially diluted anti-VM MAb (AVM-113) to the solid phase coated with VM-GMBS-BSA or BSA. (b) Reactivity of the AVM-113 MAb as measured by its immunoreactivity in the inhibition ELISA. The curves show the amount (percentage) of bound enzyme activity (B) for various doses of VM-GMBS-BSA, VM-GA-BSA, or free VM as a ratio to that bound using the HRP-labeled second antibody alone (B0). The concentration of VM in the conjugate VM-GMBS-BSA or VM-GA-BSA was photometrically calculated, assuming the molar extinction coefficient of BSA to be 43,000 at 280 nm and the other part (VM-GMBS or VM-GA) of each the conjugates to be ignored. (c) Reactivity of the AVM-113 MAb as determined from its immunoreactivity in the binding ELISA. Activated wells prepared for the binding ELISA were incubated with various concentrations of VM, GM, peplomycin, or AMPC. The wells were reacted with NaBH4 and then with HRP-labeled goat anti-mouse IgG (whole; 1:2,000).
Inhibition ELISA was achieved by the principle of competition between VM or VM conjugates (VM-GMBS-BSA, VM-GA-BSA) (free in solution) and a fixed amount of VM-GMBS-BSA coated on ELISA plates for the limited number of binding sites on the AVM-113 MAb. Calibration curves showing the relationship between the concentrations of the analytes and the percentage of bound MAb were plotted, giving dose-dependent inhibition curves with the conjugates VM-GMBS-BSA and VM-GA-BSA and free VM in the range between 0.1 nM and 1 mM (Fig. 1b). The dose required for 50% inhibition of binding was used as an indication of the strength of inhibition. This dose (the 50% effective concentration [EC50]) was 3.2 nM with VM-GMBS-BSA, 65 nM with VM-GA-BSA, and 800 nM with VM. Almost no inhibition occurred with peplomycin, ampicillin, and GM, even at concentrations of less than 1 mM (data not shown).
The binding ELISA simulates the ICC of tissue sections on the basis of the principle of coupling the amino group of the analytes to the wells of a microtiter plate activated with poly-l-lysine and GA and incubation of the wells by the indirect immunoperoxidase method (15). As shown in Fig. 1c, analysis of the relationship between the concentration of each of the analytes applied to the wells and the bound HRP activity produced a dose-dependent curve, with VM concentrations ranging from 3 μM to 100 μM. Little immunoreaction with peplomycin, GM, or AMPC occurred.
Drug uptake in rat kidney.
In kidney sections taken from rats 3 h postadministration that had been digested with 0.003% for 15 min at 30°C, most noticeable in ICC was the fact that immunostaining for VM occurred in the nuclei and cytoplasm of the collecting duct cells, some of which were heavily stained for VM and swollen while others were very weakly stained, indicating a very heterogenous uptake of VM (Fig. 2a). Also, numerous strongly stained round small bodies occurred in the tubular lumen of all the proximal tubule segments (S1, S2, and S3), and some of the round small bodies adhered to the apical cytoplasm of the tubular cells (Fig. 2b). The best weak staining occurred in the ascending limb cells in ICC tests (see the supplemental material). In an ICC protocol with 1-h preprotease digestion, immunostaining was the strongest in the microvilli, nuclei, and cytoplasm of the S1 and S2 segments of the proximal tubules (the straight proximal segments located in the cortex) (Fig. 2c) but very weak in those of the S3 segment (medullary portions of the straight proximal tubule) (Fig. 2g). Also, the best moderate staining was seen in the podocytes of the glomeruli, in Bowman's capsule squamous epithelium cells, and in the endothelial cells (Fig. 2c). Prolonging the protease digestion to 2 h produced the strongest staining in numerous cytoplasmic irregular granules in all the segments of the proximal tubules (S1, S2, and S3) (Fig. 2d) as well as in the collecting duct cells (see the supplemental material). The best immunostaining also occurred in the nuclei and apical cytoplasm of the distal convolution cells, among which the entirety of some cells was stained very heavily, thus again indicating a heterogenous drug uptake, and they were sometimes disrupted morphologically (Fig. 2e). In the ICC with such strong preprotease digestion, however, immunostaining in the microvilli of the proximal tubules, in cells of the renal corpuscles, and in the thick Henle's loop cells was more or less diminished, and the cell morphology was somewhat disrupted. Almost similar immunostaining patterns were observed for rats 24 h after VM injection (see the supplemental material). However, significant differences from the 3-h postinjection samples were that the number of heavily stained swollen cells in the collecting duct cells markedly decreased (Fig. 2f), as did the heavily stained convoluted distal tubule cells (see the supplemental material). At 8 days postinjection, immunostaining still persisted in essentially the same staining patterns as those of the 3-h and 24-h postinjection specimens (Fig. 2g and h). Some differences were that, on the whole, the intensity of immunostaining in almost all cell types in the kidney only slightly decreased; however, obviously, staining in the microvilli of the proximal tubules became weak, and the number of heavily stained cytoplasmic granules in the proximal tubule cells significantly decreased, especially in the S3 segment, but the granules grew larger in size (Fig. 2i). In control rats receiving a saline instead of VM, no immunoreaction was able to be detected (Fig. 2j). Conventional immunocytochemical staining controls (second-level controls) were all negative (data not shown). The absorption control experiments for the AVM-113 MAb were conducted for all specimens digested with protease under different conditions and showed that an addition of 30 μg/ml of VM-GMBS-BSA or VM-GA-BSA to the antibody abolished all staining (Fig. 2k).
Fig 2.

(a to k) Immunostaining for VM in the kidney of rats administered VM. Rats were i.v. administered VM at 33.3 mg/kg and then sacrificed 1 h, 3 h (a to e, g, and k), 24 h (f), or 8 days (h and i) later. ICC was carried out using a dilution (1:60) of the culture supernatant of the AVM-113 MAb following digestion of sections with protease at 0.003% at 30°C for 15 min (a, b, and f), 1 h (c), or 2 h (d, e, and i to k). Lower (g and h) and higher (a to f and i to k) magnifications are shown. (a) Characteristic immunostaining for VM occurred in the collecting duct cells, in which some cells were heavily stained in the whole cell and swollen but others were stained weakly. (b) Numerous small bodies occurred in the tubular lumen of the S1 and S2 segments of the proximal tubules: some were attached to the apical surface of the tubular cells, and all of them were strongly immunoreactive for VM. (c) Heavily stained microvilli and nuclei of the S1 and S2 proximal tubule cells. Moderate immunostaining was observed in the podocytes of the glomeruli and in Bowman's capsule squamous epithelium cells. (d) The boundary area between the pars radiate and the cortical labyrinth was observed. Note the heavily stained cytoplasmic granules in the S1 and S2 proximal tubule cells and, in contrast, the weakly stained ones in the S3 segment cells. The microvilli were disrupted in parts. (e) Distal convoluted duct cells, showing some where the whole cell was heavily stained and morphologically disrupted, and the apical cytoplasm and nuclei of others moderately stained. (f) Note the collecting duct cells with normal sizes and with slightly decreased immunostaining in comparison with the cells more heavily stained and swollen, shown in panel a. (g and h) Comparable immunostaining occurred in the rat renal cortex 3 h and 8 days postadministration. (i) The cytoplasmic granules grew larger than those in panel d. Most of the microvilli were disrupted by the prolonged protease digestion. (j) No staining in control kidney from normal rats. (k) Staining (in panel d) was completely abolished by the absorption of the MAb with VM-GMBS-BSA (30 μg/ml). G, glomeruli; C, collecting duct; P, proximal tubule; D, distal tubule; S1,2, S1 or S2 segment; S3, S3 segment; m, microvilli; p, podocyte; b, Bowman's capsule; e, endothelial cell. Bars in panels a to f and i = 20 μm. Bars in panels g and h = 100 μm. Bars in panels j and k = 50 μm.
IEM.
IEM was done using kidney specimens at 1 or 3 h postadministration, with the preembedding method using 50-μm Microslicer sections of the specimens nondigested with protease, because ultrastructure of the cell is easily broken by protease digestion. The collecting duct cells are composed of the two cell types of the principal and intercalated cells (30). Immunoreactivity was observed in both the cytoplasm and lysosome of the principal cells (Fig. 3a and b), but only slight immunoreactivity was observed in the apical cytoplasm and almost none was observed in the basal cytoplasm of the intercalated cells, in which immunoreactivity was also visible in the lysosomes (Fig. 3a and b). No immunoreactivity was observed in the absorption controls (Fig. 3c).
Fig 3.
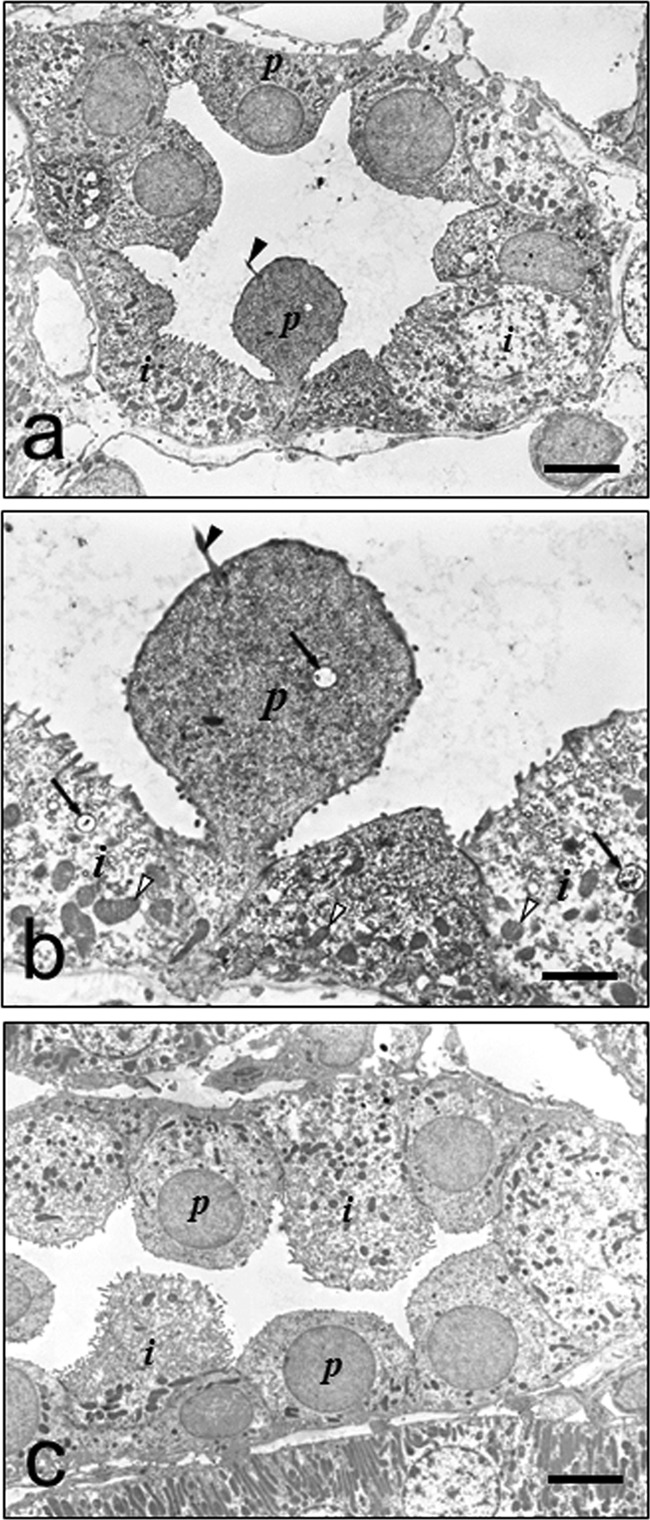
(a to c) Immunoelectron micrographs of collecting duct cells at 3 h after VM injection. (a) Lower magnification. Immunoreactivity occurred mainly in the principal cells that characteristically contain a cilium on the cell surface and scattered mitochondria in the cytoplasm. In contrast, much less immunoreactivity was observed in the cytoplasm of the intercalated cells. (b) Higher magnification. Moderate immunoreactivity occurred in the cytoplasm of the principal cells, and much less occurred in the apical part of the cytoplasm of the intercalated cells. Note that immunoreactivity also occurred in the lysosomes in both cell types (arrows). (c) Immunoreactivity was abolished by preabsorption of the MAb with VM-GA-BSA (30 μg/ml). p, principal cell; i, intercalated cell; arrowhead, cilium; open arrowheads, mitochondria. Bars in panels a and c = 5 μm. Bar in panel b = 2 μm.
Double fluorescence staining for VM and GM.
Kidney specimens 12 h after an i.v. bolus injection of rats with a mixture of VM and GM were subjected to an indirect immunofluorescence method. In the sections that had been digested with 0.003% protease for 15 min at 30°C, strong green fluorescence for VM was observed in both the microvilli and cytoplasmic small granules of the proximal tubule cells (Fig. 4a), whereas red fluorescence for GM was seen in the cytoplasmic granules and at the bottom of the microvilli but not in the microvilli (Fig. 4b). The merged image retained green fluorescence in the microvilli but yielded to yellow fluorescence in the cytoplasmic granules (Fig. 4c). In the sections digested for 60 min, green fluorescence was only in the nuclei of the proximal tubule cells (Fig. 4d to f). In the sections digested for 120 min, green fluorescence in the cytoplasmic granules (Fig. 4g) corresponded well to the red fluorescence in most of the cytoplasmic granules (Fig. 4h), yielding to yellow fluorescence in the merged image (Fig. 4i). Some swollen cells with red fluorescence for GM, possibly corresponding to the distal tubules, were also observed (Fig. 4h and i).
Fig 4.

(a to i) Double fluorescence staining for VM (green) and GM (red) in the proximal tubule cells of the kidney of rats administered with both drugs. Rats were i.v. injected with a mixture of VM (33.3 mg/kg) and GM (16 mg/kg) and then sacrificed 12 h later. This was carried out in essentially the same manner as that used in the ICC except that a mixture of Alexa Fluor 488-labeled goat anti-mouse IgG and Alexa Fluor 555-labeled goat anti-rabbit IgG was used as the second antibody. The first antibody of the culture supernatant of the AVM-113 MAb was used at a dilution of 1:60, and predigestion of specimen sections was performed at 0.004% protease at 30°C for 15 min (a to c), 1 h (d to f), or 2 h (g to i). (a to c) Strong green fluorescence for VM is in both the microvilli and the cytoplasmic granules of the proximal tubule cells, and red fluorescence for GM is weak at the bottom of the microvilli and strong in the cytoplasmic granules. The merged image shows green fluorescence in the microvilli and yellow fluorescence in the cytoplasmic granules. (d to f) Strong green fluorescence is in both the nuclei and the cytoplasmic granules, and red fluorescence is in the cytoplasmic granules. The merged image shows green fluorescence in the nuclei and yellow fluorescence in the cytoplasmic granules. Note that no microvilli were seen in the cells as a result of the prolonged protease digestion. (g to i) Green fluorescence is in most of the cytoplasmic granules, and red fluorescence is in most of the cytoplasmic granules as well as in the three swollen cells. The merged image shows yellow fluorescence in most of the cytoplasmic granules and red fluorescence in the three swollen cells, possibly a part of the distal tubule cells.
Drug uptake in rat liver.
In the liver at 3 h postadministration, ICC without the protease digestion step in the protocol produced no sign of immunoreactivity in any cell types of the liver (data not shown). The protease digestion of the specimens with 0.003% protease for 15 min to 2 h in the protocol produced staining in the endothelial cells of the hepatic sinusoids in Kupffer cells (Fig. 5a) and probably in the macrophages adjacent to the blood vessels in the connective tissues of Glisson's capsule (Fig. 5b). However, no staining was seen in the hepatocytes or on the luminal surface of the bile capillaries and in the interlobular bile ducts (Fig. 5a). Eight days after the administration, essentially the same staining patterns, but with only slightly weakened immunoreactivity, were observed (data not shown).
Fig 5.

(a to d) Immunostaining for VM in the liver of VM-administered rats. ICC was carried out using a dilution (1:60) of the culture supernatant of the AVM-113 MAb following digestion of sections with protease at 0.004% at 30°C for 1 h. (a) Lower magnification. Moderate immunostaining occurred in the sinusoidal capillaries and in Kupffer cells. (b) Higher magnification. Note that there is no staining in the hepatocytes. (c) Immunostaining in the macrophages around blood vessels in the connective tissues. (d) Staining was completely abolished by absorption of the MAb with VM-GMBS-BSA (30 μg/ml). Arrows, sinusoidal capillaries; arrowheads, Kupffer cells. Bars in panels a, c, and d = 20 μm. Bar in panel b = 10 μm.
DISCUSSION
VM is not appreciably absorbed orally and is eliminated primarily via the renal route, with >80 to 90% recovered unchanged in urine within 24 h after administration of a single dose (31, 37). Although VM-associated nephrotoxicity has been studied in humans and animals, its exact mechanism remains to be elucidated (29). Using immunoelectron microscopy and commercial antisera, Beauchamp et al. (2) have reported that VM given at doses of 50 mg/kg/day accumulates in the lysosomes of proximal tubule cells after 10 days of treatment. The study was limited to studies of the proximal tubules. In order to better understand the VM-associated nephrotoxicity, we have prepared MAbs against GMBS-conjugated VM, characterized its specificity, and developed an ICC procedure useful for the study of whole-kidney sections at the light and electron microscopic levels. Our results extend those of Beauchamp et al. (1) by showing that the ICC allowed us to evaluate the sites of the drug uptake in the kidney of a rat which received only a single i.v. injection of VM at 33 mg/kg, the dose/day being equivalent to what humans receive in chemotherapy, and that, as a result, drug uptake is not limited to the proximal tubules.
The AVM-113 MAb prepared in the study was demonstrated to be specific to conjugated or free VM by the inhibition and binding ELISAs. The inhibition results showing that antibody binding was inhibited most effectively by VM-GMBS-BSA followed by VM-GA-BSA and free VM strongly suggest that the AVM-113 MAb recognizes not only the VM molecule but also, in part, the carrier protein conjugation site(s) of GMBS. Furthermore, the fact that the MAb recognized not only the VM-GMBS-BSA conjugate but also the VM-GA-BSA conjugate suggests that it may be useful for ICC studies of drug uptake in GA-fixed tissues.
In ICC studies for small-sized drug molecules, a drug that has been specifically distributed into the cell tissues as a result of its propensity should be fixed in situ without redistribution during fixation. Thus, the fixation must be rapid and effective. Cardiac perfusion fixation of the rats via the left ventricle with 200 to 300 ml of 2% GA at 50 ml/min fulfills these requirements, as the ICC staining revealed a well-localized, specific, and reproducible reaction for VM in the same cell types and cytoplasmic compartments in the kidney and liver of rats. In other ICC studies, GA has proved useful as a fixative for small molecular compounds with an amino group(s) such as the biogenic endogenous amines (5, 12–14, 34) and amino acids (26, 36, 44). Thus, the primary amino group of VM may be the site of fixation for cells and tissues, although VM is a much more complex molecule than all these (10). However, how completely VM, which had been taken up into cells and tissues, was fixed in situ remains unknown because of, at present, no other alternative method that can estimate them.
ICCs for drugs necessitated a series of processes prior to the immunocytochemical reaction (16, 35). Thus, in ICC for VM, we established the optimal conditions for immunostaining for VM in rat kidneys. The protease digestion process was especially critical and significantly differed in the different cells and compartments. These differences may reflect differences in the binding and masking of VM by proteins and other macromolecules in different cellular compartments which may thus constitute different obstacles to the penetration of the antibody into such cell types. Accordingly, to reveal the full compartment of VM localization, results from different digestion procedures needed to be evaluated (7).
In ICC for VM, unmasking of the granular antigens needed strong protease predigestion conditions for kidney sections (18). The cytoplasmic granules got larger in size at 8 days postinjection than those at 3 h and 24 h postinjection. IEM study revealed that VM immunoreactivity occurred in the lysosomes as well as in the cytoplasm of the collecting duct cells (Fig. 3a and b). The double fluorescence staining demonstrated that the two drugs of VM and GM, an aminoglycoside antibiotic, can colocalize in the cytoplasmic granules in the proximal tubule cells. Also, GM has been reported to localize in lysosomes in the proximal tubule cells by the immunogold method and autoradiography (1, 49), as has VM by the immunogold method (2). All these results strongly suggest that the cytoplasmic granules most probably represent lysosomes. These VM staining patterns are similar to those of our other recent ICC for GM (18), with the exception that VM clearly localized in the nuclei of proximal tubule cells but GM scarcely did (18). This was confirmed by the present double fluorescence staining (Fig. 4a to i). Furthermore, VM ICC revealed that significant amounts of VM persisted for as long as 8 days postadministration at the sites where it was distributed, including also the brush border membrane, especially in the S1 and S2 segments of the proximal tubules. Recently, Hodoshima et al. (27) have presumed the relationship between the renal accumulation of the drug and VM nephrotoxicity, determining the levels of VM in the kidney by a fluorescence polarization immunoassay. The occurrence of VM in the microvilli may reflect either ongoing secretion or reabsorption of the drug and/or may reflect the accumulation of the drug. On this issue, Nakamura et al. (33) have suggested that VM excretes in the urine via active tubular secretion, and on the other hand, Sokol has reported that VM crosses the basolateral membrane but does not cross the brush border (43). The immunostaining in the microvilli may also suggest the occurrence of an unknown transporter(s) for VM at the brush border of the proximal tubules in the kidney, as the sites are known to be where a variety of transport systems in drug interactions occur (6, 22, 28, 38, 45, 47, 48). Nakamura et al. (33) have suggested the organic cation transporter (OCT) and P glycoprotein for VM at the basolateral and brush border membranes, respectively, of the renal tubular epithelium, both of which mediate transport of the drug crossing the epithelium in the direction of the lumen side from the blood side. Furthermore, we made observations similar to those in our previous ICCs for AMPC and peplomycin (19–21); small bodies which were extruded into the tubular lumen side from the epithelial tubular cells and which contained VM occurred. Both immunoelectron microscopy and conventional electron microscopy revealed that most of the small bodies were the fragments of the tubular cell cytoplasm or sometimes those including the nuclei but not protein droplets (data not shown). However, whether the occurrence of these small bodies is related to the VM nephrotoxicity remains unclear, as similar phenomena were also sometimes observed in the control rats. Also, in double fluorescence staining, the reason why only GM, but not VM, was taken up into some of the swollen cells of the convoluted distal tubule remains unclear, although it is thought that the combination therapy of VM with aminoglycoside GM can strengthen the nephrotoxic effect (32). All these must be the subjects to be further studied.
Furthermore, a new finding of the occurrence of numerous immunostained swollen cells in the collecting duct cells, as well as in the distal convoluted cells, 3 h after VM injection suggests that the drug somewhat affected the cells in such a short time. Our ICC detected differences in the abilities of neighboring cells to take up VM and/or retain VM. The present IEM study first demonstrated that uptake of VM occurred exclusively in the cytoplasm of the principal cells but scarcely or only very slightly in the intercalated cells, the two cell types which are known to organize both collecting duct cells and convoluted distal tubule cells (30). These phenomena in drug uptake seem to generally occur, as a similar observation has been made with GM or peplomycin in our previous ICCs (18, 20). However, differences, such as that most of the cells seemingly affected by VM disappeared by 24 h after administration, most likely returning back to the normal size of the cells with decreased immunostaining, while those affected by GM or by peplomycin remained morphologically affected during a few experimental days, were also seen (18, 20). This may mean that VM might affect the cells less strongly than GM or peplomycin at the therapeutic dose of the drug administered in the present study.
Although VM distributed in the endothelial cells of the hepatic sinusoids in the liver and then persisted at the sites for as long as 8 days after injection, no VM was able to be detected in the hepatocytes, on the luminal surface of the bile capillaries, or in the interlobular bile duct cells, thus indicating no uptake of VM in the cells under the therapeutic dose conditions. This may possibly mean that there are some strict control systems in the hepatocytes which do not allow VM to be taken up into the cells. The fact that only small amounts of VM distributed in tissue of the liver was in good agreement with the results reported from the previous VM pharmacology (23, 31).
In general, ICC for a drug(s) may have the advantage over autoradiography of being able to elucidate more directly, simply, and precisely the localization of drugs in cells and tissues. ICC can be completed within one or two days, in contrast to autoradiography being time-consuming. Also, its sensitivity may be comparable to that of autoradiography. Contrarily, the weakness of the ICC may be that it necessitates protease digestion of section specimens to elucidate drug distribution, since the protease digestion leads to the disruption of ultrastructure indispensable for IEM, as mentioned in the paper. Also, ICC has drawbacks, as autoradiography does, in exactly quantifying the amount of drug taken up into cells and tissues.
In conclusion, our study clearly demonstrates that the newly developed ICC using MAb is useful for localizing the cellular sites of the accumulation of VM in kidneys from rats receiving as little as 33 mg/kg of VM, a dose corresponding to one therapeutic dose for human patients. The method revealed that VM accumulates for a long time in the proximal and distal tubules as well as in the collecting duct, pinpointing all of these sites as potential targets for VM toxicity. Also, the double fluorescence staining showed that VM can colocalize with GM, possibly in the lysosomes in the proximal tubule cells, and IEM demonstrated that in the distal tubule cells, VM distributed mainly in the principal cells and only very slightly in the intercalated cells.
Supplementary Material
ACKNOWLEDGMENT
We are grateful to K. Tanaka for his technical assistance throughout this study.
Footnotes
Published ahead of print 4 September 2012
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1. Beauchamp D, Gourde P, Bergeron MG. 1991. Subcellular distribution of gentamicin in proximal tubular cells, determined by immunogold labeling. Antimicrob. Agents Chemother. 35:2173–2179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Beauchamp D, Gourde P, Simard M, Bergeron MG. 1992. Subcellular localization of tobramycin and vancomycin given alone and in combination in proximal tubular cells, determined by immunogold labeling. Antimicrob. Agents Chemother. 36:2204–2210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brown J, Brown K, Forrest A. 2012. Vancomycin AUC24/MIC ratio in patients with complicated bacteremia and infective endocarditis due to methicillin-resistant Staphylococcus aureus and its association with attributable mortality during hospitalization. Antimicrob. Agents Chemother. 56:634–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Clancy B, Cauller LJ. 1998. Reduction of background autofluorescence in brain sections following immersion in sodium borohydride. J. Neurosci. Methods 83:97–102 [DOI] [PubMed] [Google Scholar]
- 5. Decavel C, Lescaudron L, Mons N, Calas A. 1987. First visualization of dopaminenergic neurons with a monoclonal antibody to dopamine: a light and electron microscopic study. J. Histochem. Cytochem. 35:1245–1251 [DOI] [PubMed] [Google Scholar]
- 6. Dobson PD, Kell DB. 2008. Carrier-mediated cellular uptake of pharmaceutical drugs: an exception or the rule? Nat. Rev. Drug Discov. 7(3):205–220 [DOI] [PubMed] [Google Scholar]
- 7. Finley JC, Grossman GH, Dimeo P, Petrusz P. 1978. Somatostatin-containing neurons in the rat brain: widespread distribution revealed by immunocytochemistry after pretreatment with pronase. Am. J. Anat. 153:483–488 [DOI] [PubMed] [Google Scholar]
- 8. Fujiwara K, Yasuno M, Kitagawa T. 1981. Novel preparation method of immunogen for hydrophobic hapten, enzyme immunoassay for daunomycin and adriamycin. J. Immunol. Methods 45:195–203 [DOI] [PubMed] [Google Scholar]
- 9. Fujiwara K, Saikusa H, Yasuno M, Kitagawa T. 1982. Enzyme immunoassay for the quantification of mitomycin C using β-galactosidase as a label. Cancer Res. 42:1487–1491 [PubMed] [Google Scholar]
- 10. Fujiwara K, Kitagawa T. 1986. Specificity and use of antisera produced against anticancer drugs, p 251–292 In Ray PK. (ed), Advances in immunity and cancer therapy, vol 2 Springer-Verlag, New York, NY: [DOI] [PubMed] [Google Scholar]
- 11. Fujiwara K, Masuyama Y. 1995. Monoclonal antibody against the glutaraldehyde-conjugated polyamine, spermine. Histochem. Cell Biol. 104:309–316 [DOI] [PubMed] [Google Scholar]
- 12. Fujiwara K, Masuyama Y, Kitagawa T. 1996. Immunocytochemical localization of polyamines in the gastrointestinal tracts of rats and mice. Histochem. Cell Biol. 106:465–471 [DOI] [PubMed] [Google Scholar]
- 13. Fujiwara K, Bai B, Kitagawa T, Tsuru D. 1998. Immunoelectron microscopic study for polyamines. J. Histochem. Cytochem. 46:1321–1328 [DOI] [PubMed] [Google Scholar]
- 14. Fujiwara K, Bai G, Tamura C, Tsuru D. 1999. Immunocytochemical localization of histamine in enterochromaffin-like (ECL) cells in rat oxyntic mucosa: a transmission electron microscopy study using monoclonal antibodies and conventional glutaraldehyde-based fixation. J. Histochem. Cytochem. 47:1031–1038 [DOI] [PubMed] [Google Scholar]
- 15. Fujiwara K, Tanabe T, Yabuuchi M, Ueoka R, Tsuru D. 2001. A monoclonal antibody against the glutaraldehyde-conjugated polyamine, putrescine: application to immunocytochemistry. Histochem. Cell Biol. 115:471–477 [DOI] [PubMed] [Google Scholar]
- 16. Fujiwara K, Takatsu H, Tsukamoto K. 2005. Immunocytochemistry for drugs containing an aliphatic amino group in the molecule, anticancer antibiotic daunomycin as a model. J. Histochem. Cytochem. 53:467–474 [DOI] [PubMed] [Google Scholar]
- 17. Fujiwara K, Shin M, Hougaard DM, Larsson L-I. 2007. Distribution of anticancer antibiotic daunomycin in the rat heart and kidney revealed by immunocytochemistry using monoclonal antibodies. Histochem. Cell Biol. 127:69–77 [DOI] [PubMed] [Google Scholar]
- 18. Fujiwara K, Shin M, Matsunaga H, Saita T, Larsson L-I. 2009. Light-microscopic immunocytochemistry for gentamicin and its use for studying uptake of the drug in kidney. Antimicrob. Agents Chemother. 53:3302–3307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fujiwara K, Shin M, Miyazaki T, Maruta Y. 2011. Immunocytochemistry for amoxicillin and its use for studying uptake of the drug in the intestine, liver, and kidney of rats. Antimicrob. Agents Chemother. 55:62–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fujiwara K, Shin M, Hougaard DH, Saita T. 2011. Distribution study of peplomycin in rat kidney revealed by immunocytochemistry using monoclonal antibody. Histochem. Cell Biol. 135:93–101 [DOI] [PubMed] [Google Scholar]
- 21. Fujiwara K, Shin M, Yoshizaki Y, Miyazaki T, Saita T. 2012. An in vivo role of Mrp2 in the rat hepatocytes by immunocytochemistry for amoxicillin using the transporter-deficient EHBR. J. Mol. Histol. 43:371–378 [DOI] [PubMed] [Google Scholar]
- 22. Gottesman MM, Pastan I. 1993. Biochemistry of multidrug resistance mediated by the multi-drug transporter. Annu. Rev. Biochem. 62:385–427 [DOI] [PubMed] [Google Scholar]
- 23. Gupta A, Biyani M, Khaira A. 2011. Vancomycin nephrotoxicity: myths and facts. Neth. J. Med. 69:379–383 [PubMed] [Google Scholar]
- 24. Haraguchi CM, Yokota S. 2002. Immunofluorescence technique for 100-nm-thick semithin sections of Epon-embedded tissues. Histochem. Cell Biol. 117:81–85 [DOI] [PubMed] [Google Scholar]
- 25. Hayashi A, Koike M, Matsunami M, Hoshino T. 1988. The effects of fixation time and enzymatic digestion on the immunohistochemical demonstration of bromodeoxyuridine in formalin-fixed, paraffin-embedded tissue. J. Histochem. Cytochem. 36:511–514 [DOI] [PubMed] [Google Scholar]
- 26. Hepler JR, et al. 1988. Characterization of antisera to glutamate and aspartate. J. Histochem. Cytochem. 36:13–22 [DOI] [PubMed] [Google Scholar]
- 27. Hodoshima N, Masuda S, Inui K. 2007. Decreased renal accumulation and toxicity of a new VCM formulation in rats with chronic renal failure. Drug Metab. Pharmacokinet. 22:419–427 [DOI] [PubMed] [Google Scholar]
- 28. Inui K, Masuda S, Saito H. 2000. Cellular and molecular aspects of drug transport in the kidney. Kidney Int. 58:944–958 [DOI] [PubMed] [Google Scholar]
- 29. Levine DP. 2006. Vancomycin: a history. Clin. Infect. Dis. 42(Suppl 1):S5–S12 [DOI] [PubMed] [Google Scholar]
- 30. Madsen KM, Tissher CC. 1986. Structural-functional relationship along the distal nephron. Am. J. Physiol. 250:F1–F15 [DOI] [PubMed] [Google Scholar]
- 31. Matzke GR, Zhanel GG, Guay DR. 1986. Clinical pharmacokinetics of vancomycin. Clin. Pharmacokinet. 11:257–282 [DOI] [PubMed] [Google Scholar]
- 32. Moellering RC., Jr 2006. Vancomycin: a 50-year reassessment. Clin. Infect. Dis. 42(Suppl 1):S3–S4 [DOI] [PubMed] [Google Scholar]
- 33. Nakamura T, Takano M, Yasuhira M, Inui K. 1996. In-vivo clearance study of vancomycin in rats. J. Pharm. Pharmacol. 48:1197–1200 [DOI] [PubMed] [Google Scholar]
- 34. Nilsson O, Dahlstrom A, Geffard M, Ahlman H, Ericson LE. 1987. An improved immunocytochemical method for subcellular localization of serotonin in rat enterochromaffin cells. J. Histochem. Cytochem. 35:319–326 [DOI] [PubMed] [Google Scholar]
- 35. Ohara K, Shin M, Larsson L-I, Fujiwara K. 2007. Improved immunocytochemical detection of daunomycin. Histochem. Cell Biol. 127:603–608 [DOI] [PubMed] [Google Scholar]
- 36. Popratiloff A, Valtschanoff JG, Rustioni A, Weinberg RJ. 1996. Colocalization of GABA and glycine in the rat dorsal column nuclei. Brain Res. 706:308–312 [DOI] [PubMed] [Google Scholar]
- 37. Rybak MR. 2006. The pharmacokinetic and pharmacodynamic properties of vancomycin. Clin. Infect. Dis. 42:S35-S-39. [DOI] [PubMed] [Google Scholar]
- 38. Schaub TP, et al. 1997. Expression of the conjugate export pump encoded by the mrp2 gene in the apical membrane of kidney proximal tubules. J. Am. Soc. Nephrol. 8(8):1213–1221 [DOI] [PubMed] [Google Scholar]
- 39. Schilling A, Neuner E, Rehm SJ. 2011. Vancomycin: a 50-something-year-old antibiotic we still don't understand. Cleve Clin. J. Med. 78(7):465–471 [DOI] [PubMed] [Google Scholar]
- 40. Shin M, Larsson L-I, Fujiwara K. 2007. Polyamines in spermatocytes and residual bodies of rat testis. Histochem. Cell Biol. 127:649–655 [DOI] [PubMed] [Google Scholar]
- 41. Shin M, Nakamuta H, Oda-Ueda N, Larsson L-I, Fujiwara K. 2008. Immunocytochemical demonstration of polyamines in nucleoli and nuclei. Histochem. Cell Biol. 129:659–665 [DOI] [PubMed] [Google Scholar]
- 42. Shin M, Larsson L-I, Hougaard DM, Fujiwara K. 2009. Daunomycin accumulation and induction of programmed cell death in rat hair follicles. Cell Tissue Res. 337:429–438 [DOI] [PubMed] [Google Scholar]
- 43. Sokol PP. 1991. Mechanism of vancomycin transport in the kidney: studies in rabbit renal brush border and basolateral membrane vesicles. J. Pharmacol. Exp. Ther. 259:1283–1287 [PubMed] [Google Scholar]
- 44. Spirou GA, Berrebi AS. 1997. Glycine immunoreactivity in the lateral nucleus of the trapezoid body of the cat. J. Comp. Neurol. 383(4):473–488 [DOI] [PubMed] [Google Scholar]
- 45. Sugawara I, et al. 1988. Tissue distribution of p-glycoprotein encoded by a multidrug-resistant gene as revealed by a monoclonal antibody, MRK 16. Cancer Res. 48:1926–1929 [PubMed] [Google Scholar]
- 46. Tang X, Falls DL, Li X, Lane T, Luskin MB. 2007. Antigen-retrieval procedure for bromodeoxyuridine immunolabeling with concurrent labeling of nuclear DNA and antigens damaged by HCl pretreatment. J. Neurosci. 27:5837–5844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Terada T, Inui K. 2008. Physiological and pharmacokinetic roles of H+/organic cation antiporters (METE/SLC47A). Biochem. Pharmacol. 75:1689–1696 [DOI] [PubMed] [Google Scholar]
- 48. Thiebaut F, et al. 1987. Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc. Natl. Acad. Sci. U. S. A. 84:7735–7738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wedeen RP, Batuman V, Cheeks C, Marquet E, Sobel H. 1983. Transport of gentamicin in rat proximal tubule. Lab. Invest. 48:212–223 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.