

Abstract
Toxoplasma gondii is a globally ubiquitous pathogen that can cause severe disease in immunocompromised humans and the developing fetus. Given the proven role of Toxoplasma-secreted kinases in the interaction of Toxoplasma with its host cell, identification of novel kinase inhibitors could precipitate the development of new anti-Toxoplasma drugs and define new pathways important for parasite survival. We selected a small (n = 527) but diverse set of putative kinase inhibitors and screened them for effects on the growth of Toxoplasma in vitro. We identified and validated 14 noncytotoxic compounds, all of which had 50% effective concentrations in the nanomolar to micromolar range. We further characterized eight of these compounds, four inhibitors and four enhancers, by determining their effects on parasite motility, invasion, and the likely cellular target (parasite or host cell). Only two compounds had an effect on parasite motility and invasion. All the inhibitors appeared to target the parasite, and interestingly, two of the enhancers appeared to rather target the host cell, suggesting modulation of host cell pathways beneficial for parasite growth. For the four inhibitors, we also tested their efficacy in a mouse model, where one compound proved potent. Overall, these 14 compounds represent a new and diverse set of small molecules that are likely targeting distinct parasite and host cell pathways. Future work will aim to characterize their molecular targets in both the host and parasite.
INTRODUCTION
Toxoplasma gondii is an obligate intracellular protozoan parasite belonging to the phylum Apicomplexa that causes toxoplasmosis. The parasite is highly promiscuous, infecting all mammalian and avian species (4), including nearly one-third of the world's human population. However, the disease, while asymptomatic in immunocompetent individuals, poses fatal consequences to infected individuals who are immunocompromised, such as HIV-infected/AIDS patients, immunosuppressed organ transplant patients, and the congenitally infected fetus (1, 4, 10). To date there is no cure for toxoplasmosis, since no therapies that target the dormant cyst stages that can persist for the life of the individual are available. In addition, current therapies such as pyrimethamine and clindamycin have significant side effects, including bone marrow suppression and rashes (15, 32).
The parasite kinome has been shown to play a significant role in pathogenesis. Secreted kinases such as ROP18, ROP16, and ROP5 are among the effectors injected into the host during invasion that have a role in virulence and cooption of the host cell for the maintenance of the infection (3, 24, 25, 27, 29, 30). Most of the known secreted parasite kinases are secreted from the rhoptries and are composed of catalytically active and inactive kinases (so-called pseudokinases) (3–5, 24, 25).
Cell-based small-molecule screens have the potential to identify new inhibitors of key parasite processes, including invasion, replication, and egress (7, 13, 20). The advantage of cell-based assays is that the compounds are screened against their potential targets in their native environment, and the success of this approach has been well documented in identifying previously uncharacterized gene products essential for specific parasite processes. For example, the T. gondii gene DJ-1 was found to be crucial for Toxoplasma invasion on the basis of a small-molecule screen (7, 13). Hits from cell-based assays also offer a scaffold for studying parasite mechanisms, which in turn aid in drug development processes using the small molecule as a lead (22). The bottleneck for this approach is target identification, but recent advances in high-throughput sequencing (11) and synthetic chemistry approaches to facilitate identification of the protein targets that interact with the active compound (13) have begun to make this phase more tractable. Successful application of chemical genetics requires a collection of compounds with drug-like properties that are structurally diverse. In addition, small molecules which are amenable to downstream modification for target identification and validation offer a huge advantage (7, 22, 30). These features can be combined with computational chemistry methods aimed at filtering small molecules with undesirable fragments whose features may be problematic for future compound optimization for downstream applications (34).
Our aim was to utilize chemical genetics to probe the Toxoplasma kinome with the hope of identifying novel kinases that are integral to essential pathways, elucidating their mechanism of action, and, ultimately, identifying new drug targets. To this end, we screened a library of 527 structurally diverse kinase inhibitors for growth modulation in vitro. From the screen we identified 14 structurally diverse compounds that altered parasite growth by either inhibiting or enhancing growth in vitro. For a subset of these compounds, we have conducted secondary assays to address specificity for the parasite or the host cell, reversibility, and effect on invasion and motility. In addition, we have investigated the effect of four inhibitors on parasite ultrastructure and their efficacy in an animal model. Our small-molecule approach has identified novel compounds with inhibitory or activating effects on parasite growth, and these chemical probes will open new avenues that will complement both the genomic and proteomic strategies in place for the study of the molecular interactions of Toxoplasma with its host.
MATERIALS AND METHODS
Parasite and host cell maintenance.
Human foreskin fibroblasts (HFFs) were used as the host cell for all assays; cultured in cDMEM, consisting of Dulbecco modified Eagle medium (DMEM; Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS; Atlas Biologicals, Fort Collins, CO), 2 mM l-glutamine (Sigma-Aldrich), and 100 μg/ml penicillin and streptomycin (Mediatech, Manassas, VA); and maintained at 37°C in 5% CO2. Parasites used for assays were harvested from HFF monolayers through syringe release as previously described (14). Gliding motility assays were conducted in HHG medium, consisting of Hanks balanced salt solution (Mediatech, Manassas, VA), 20 mM HEPES (Sigma-Aldrich, St. Louis MO), and 2 mM glutamine.
Parasite strains.
Strains 5A10 (a type III strain) and PB3-10 (a type II strain) were used for the in vitro and in vivo assays; however, for individual compound characterization, we used strain PB3-10 exclusively. Strain 5A10 (described in reference 14) was derived from strain CEPΔHXGPRT, and PB3-10 was derived from strain ME49B7. Both parental strains were transfected with a construct containing green fluorescent protein (GFP) and click beetle luciferase (under the control of the GRA2 [dense granule protein 2] and dihydrofolate reductase promoters, respectively) and sorted using flow cytometry, and GFP-positive clones were isolated using limiting dilution. Strain 5A10 was also transfected with the empty pGRA-HA-HXGPRT vector (27) to complement the missing hypoxanthine-xanthine-guanine phosphoribosyl transferase (HXGPRT) gene. For invasion and motility assays, an RH-derived strain was used. The strain was derived by transfecting RH88 (a type I strain) with a construct containing DsRed, click beetle luciferase, and a phleomycin resistance gene (19), selected for resistance to 100 μM phleomycin, and cloned by limiting dilution.
Compound library.
From a kinase-focused inhibitor library composed of 70,000 compounds, a diverse subset consisting of 527 distinct small molecules was purchased from Life Chemicals (Burlington, Ontario, Canada). The parent library was generated in silico on the basis of (i) similarity to known kinase inhibitors and (ii) virtual docking into known protein kinases. It was further filtered for small molecules with drug-like properties using the Lipinski rule of 5 (17). The subset of 527 compounds was generated using UNITY, a Sybyl-X package (Tripos, St. Louis, MO). UNITY was used to identify 600 of the most diverse hits from the parent library at a Tanimoto score below 0.45. The algorithm was invoked using UNITY's command line dbdiss. The query identified 600 compounds, but only 527 were available for purchase. Each of the purchased compounds (in 96-well plates) was dissolved in 100% dimethyl sulfoxide (DMSO) at a 10 mM concentration, serially diluted to a stock solution of 100 μM in phosphate-buffered saline (PBS)–10% DMSO, and stored at −20°C.
Luciferase-based screen.
The compound library was screened for growth modulation against strains 5A10 and PB3-10 using a luciferase-based assay as described previously (14). Briefly, syringe-released parasites were quantified using a hemacytometer and added to HFF monolayers in 96-well plates at a final parasite concentration of 2 × 104 parasites/well. After 4 h, the plates were washed once with cDMEM, and then test compound (final concentration, 10 μM) or vehicle (cDMEM–1% DMSO) was added. cDMEM–1% DMSO is referred to as “vehicle” for the rest of the report. The plates were then incubated for 72 h to allow parasite replication before harvest. Harvesting was done by washing the plates once with PBS and then adding 50 μl/well of 1× cell culture lysis reagent (Promega, Madison, WI). The plates were then stored at −80° C. Plates were thawed, and the lysate was transferred into a 96-well white-bottomed plate (Greiner Bio-One, Monroe, NC). Seventy microliters per well of luciferase assay reagent (Promega, Madison, WI) was then added. Luciferase activity was measured using a Centro XS3 LB960 luminometer (Berthold Technologies, Oak Ridge, TN).
Host cell viability.
Host cell viability in the presence of the compounds was determined using both a colorimetric assay and microscopy. The colorimetric assay was done as described previously (14) after 48 h compound (10 μM) exposure. Staurosporine was used as a positive control for cytotoxicity, and vehicle was used as the negative control.
Electron microscopy.
Electron microscopy studies were done as previously described (14), with the following minor changes. The monolayers were infected at multiplicities of infection (MOIs) of 5 and 10. After 4 h, monolayers infected at an MOI of 5 were thoroughly washed to remove uninvading parasites and then treated with 10 μM compounds C3 and C5 or vehicle for 40 h. Monolayers infected at an MOI of 10 were grown overnight, washed, and then treated with 10 μM compounds C2 and C1 or vehicle for 24 h. Monolayers were then fixed and processed for electron microscopy (14).
Invasion assays.
Invasion assays were performed as described previously (7). Confluent HFF monolayers on 12-mm coverslips in 24-well plates were infected with 1 × 106 to 2 × 106 RH3-12 tachyzoites per well in the presence of 5 or 10 μM compound or vehicle. Invasion was allowed to proceed for 1 h, and the monolayers were washed once in PBS and fixed with 4% paraformaldehyde in PBS. Cells were not permeabilized. Extracellular (i.e., uninvading) parasites were stained with mouse anti-surface antigen 1 (anti-SAG1; 1:5,000; obtained from mouse serum infected with type II parasites), followed by Alexa Fluor-488-conjugated goat anti-mouse IgG (1:1,000; Invitrogen, Carlsbad, CA). Stained coverslips were briefly stored in 50% glycerol–PBS medium before mounting on glass slides (Fisher Scientific, Pittsburgh, PA) using 3 μl Vectashield mounting medium (Vector Laboratories, Burlingame, CA). Slides were visualized using a Zeiss Axiophot upright microscope equipped for phase-contrast and epifluorescence microscopy and an AxioCam MRC 5 charge-coupled-device camera (Carl Zeiss, Göttingen, Germany). Data were compiled from 4 independent trials each from counting of 6 random fields of view per compound under ×100 magnification. Since RH3-12 also expressed DsRed, parasites that were fluorescent only in the red channel were scored as invading, while those that were red and green were scored as uninvading. Eight compounds (C1, C2, C3, C5, C8, C11, C12, and C13) were assayed to determine their effects on invasion. Compounds C1, C2, C3, and C5 were assayed at 10 μM, while compounds C8, C11, C12, and C13 were screened at 5 μM. Cytochalasin D (1 μm) was also included in the assay as a positive control for inhibition of invasion (7).
Motility assays.
Motility assays were carried out essentially as described previously (12). Twelve-millimeter coverslips in 24-well plates were coated with 100% FBS for 24 h at 4°C and then washed once with PBS. Freshly isolated parasites were filtered through a 5-μm-pore-size filter, pelleted by centrifugation, resuspended in HHG medium containing compound or vehicle (HHG–1% DMSO) at 4 × 107 to 5 × 107 parasites/ml, and then added to coverslips and incubated at 37°C for 30 min. The coverslips were then washed once with PBS, fixed in 2.5% formaldehyde for 20 min, washed once in PBS, and blocked in PBS–5% bovine serum albumin (BSA). SAG1-containing trails were stained using the same antibodies used in the invasion assay described above at 1:1,000 and 1:250 primary and secondary antibodies, respectively. The number of SAG1-positive trails was calculated for 20 random fields of view for each compound, and data were compiled from 3 independent trials. Compounds C8, C11, and C13 were assayed at 5 μM, while compounds C1 to C5 and C12 were assayed at 10 μM.
Reversibility of effect and likely cellular target.
Invading parasites were grown in the presence of four inhibitory compounds for 12 h before compound was removed, the monolayer was washed thoroughly, and fresh cDMEM was added. The plates were then incubated in the absence of the compound for another 12 h before harvest. Parasites were harvested and assayed by luminescence after the 12 h of incubation as well as 12 h after compound removal to assess the ability of the parasites to recover from the treatment. Two independent assays were performed.
To test the effects of compound treatment on extracellular parasites, 1 × 106 parasites/ml freshly lysed parasites were treated for 16 h at 37°C in 5% CO2 with either vehicle or 10 μM compound and then washed 3 times with cDMEM to remove the compound. Treated parasites were then used to infect fresh HFF monolayers for 3 days, prior to assaying for growth by luminescence.
To determine if effects on parasite growth were due to effects on the host cell, uninfected HFF monolayers were treated with either vehicle or 10 μM compound for 24 h and washed thoroughly with cDMEM. Parasites were allowed to invade for 4 h, the monolayer was washed 3 times with cDMEM, and parasite growth was allowed to proceed for 3 days prior to harvest and luminescence assays. In parallel, vehicle-pretreated host cell monolayers were infected with parasites in a similar fashion, but the compound was added at 4 h postinvasion and the parasites were incubated for 3 days. This allowed a comparison of the effects of the compound after pretreatment to the effects of having the compound present for the entire incubation period. In addition, compounds that were effective after compound removal were also classified as having an irreversible effect in these assays.
In vivo studies.
All in vivo studies were carried out in accordance with approved University of Pittsburgh IACUC animal research protocols. Female BALB/c mice 4 to 8 weeks old (Jackson Laboratories, Bar Harbor, ME) were used in all experiments. Freshly isolated tachyzoites of either the PB3-10 or 5A10 strain were pelleted and suspended in PBS at a concentration of 10,000 tachyzoites per 0.2 ml. Mice were injected intraperitoneally (i.p.) with 200 μl of the tachyzoite suspension. The treated groups of mice were also concurrently injected with compound (2.4 mg/kg of body weight) in PBS–10% DMSO, while the untreated group was injected with 10% DMSO–PBS (vehicle) on the day of infection. Thereafter, the treated groups received a compound dose of 4.4 mg/kg/day. In our first study with two mice per group, the mice were infected with strain PB3-10 and treated with compound C1, C2, C3, or C5 or vehicle. From the pilot study, C2 showed a significant reduction in parasite load over that for the controls and was therefore pursued in a follow-up study. In the second study, 6 mice per group were infected with either strain PB3-10 or 5A10 (10,000 tachyzoites) and treated with C2 or vehicle using the same dosing regimen described above. To capture the progression of infection, luminescence readings were acquired daily using an IVIS Lumina II imaging system (Caliper Life Sciences, Alameda, CA) as described previously (6, 28). Before imaging, mice were injected with 200 μl of 15.4 mg/ml d-luciferin in PBS (potassium salt; Caliper Life Sciences). Each mouse was immediately anesthetized in an oxygen-rich induction chamber with 2% isoflurane (TW Medical, Austin, TX). The mice were kept in the induction chamber for 10 min to allow dissemination of d-luciferin and to ensure that they were fully anesthetized before placing them in the imaging chamber. Mice were imaged ventrally for the first 13 days and ventrally and dorsally thereafter. Images were collected for 3 to 5 min, depending on the intensity of the bioluminescent signal. Data acquisition and analysis were done using an IgorPro image analysis package (Caliper Life Sciences, Alameda, CA). Quantitation of emitted light from each mouse was performed by recording the total number of photons per second (total flux).
Statistical analyses.
The 50% effective concentration (EC50) for each compound was determined using the dose-response curve (DRC) (26) package implemented in R (http://www.R-project.org), a language and environment software for statistical computing and graphic analysis, after two independent assays. All other data were analyzed by one-way analysis of variance (ANOVA) via the proc GLM procedure in SAS statistical analysis software (SAS, Cary, NC). Multiple comparisons were carried out using the LSMeans procedure in SAS only if there was a significant (P < 0.05) effect of treatment in the ANOVA, and only preplanned comparisons were conducted to minimize type I error. In all cases, treatments were compared to the vehicle-only control.
RESULTS
Screen identifies 14 compounds that alter parasite growth in vitro.
From the compound screen, we identified small molecules that enhanced, inhibited, or had no effect on parasite growth (Fig. 1). Of the 527 compounds screened, we selected 30 compounds to pursue further on the basis of the level of inhibition or enhancement. Small molecules that reduced parasite growth by >90% or increased growth by over 200% compared to vehicle were categorized as inhibitors and enhancers, respectively. Host cells treated with these compounds were also visually inspected to determine any cytotoxic effects, and those with observable host cell effects were removed. The selected compounds were reassayed to eliminate false positives from the screen (Fig. 2). Of the 30 reassayed compounds, 14 compounds (7 inhibitors and 7 enhancers) showed the same activity profile observed in the initial screen and were pursued further (Fig. 2; see Fig. 3 for structures). These compounds were tested further for cytotoxic effects on the host cell using both a colorimetric assay (Cell Titer Aqueous One Cell Solution assay kit; Promega, Madison, WI) (see Fig. S1 in the supplemental material) and microscopy. On the basis of the results of these assays, none of the 14 compounds were toxic to the host cells at the highest dose used in our assays (10 μM).
Fig 1.

Percent luciferase signal compared to that for the vehicle-treated control for 527 compounds tested in the initial growth screen, sorted from the most effective inhibitors to the most effective enhancers. Small molecules that were not obviously toxic to host cells and that either inhibited parasite growth by >90% or increased parasite growth by >200% were categorized as inhibitors and enhancers, respectively, and were selected for further assays. Thirty compounds satisfied these criteria.
Fig 2.

Parasite growth against compound treatment. Thirty compounds from the primary screen were reassayed at 10 μM to identify false positives. Fourteen (identified with asterisks) out of the 30 small molecules showed levels of inhibition or enhancement reproducible from the initial screen. These compounds were named C1 to C14 (see Table 1, Table S1 in the supplemental material, and Fig. 3 for structures).
Fig 3.

Structures of the 14 small molecules identified in this study. Inhibitors are compounds C1 to C7, and enhancers are compounds C8 to C14.
Compound effects on invasion and motility.
Successful parasite invasion requires the ability of the parasite to glide on, attach to, and penetrate host cells (7, 13, 23). Any defect in any one of these aspects leads to defective invasion and subsequent parasite death. To assess whether the most potent compounds affected invasion by inhibiting or enhancing parasite motility, attachment, or penetration, we did both motility and invasion assays. Both are immunoassays that rely on the ability to detect the parasite surface protein SAG1, which is a component of the parasite plasma membrane and which is also shed on the surface as a slime trail when parasites move (4, 7, 12). On the basis of the results of these assays, the inhibitory compound C2 had no significant effect on parasite motility (Fig. 4A), nor did the enhancing compounds C8, C11, and C13 (Fig. 4A). However, inhibitors C1 and C5 dramatically reduced parasite motility (Fig. 4A; P < 0.001), and parasites treated with both compounds failed to make any detectable trails. Interestingly, compounds C3 and C12 slightly, but significantly (P < 0.05), increased parasite motility.
Fig 4.

(A) Effects of compounds on parasite motility. C1 and C5 completely blocked parasite motility, while C3 and C12 slightly enhanced it. V, vehicle control. (B) Effects of four inhibitors and four enhancers on parasite invasion. Cytochalasin D (Cyt-D) was used as a positive control for invasion inhibition. Effects on invasion were assessed by comparing the mean number of invading or uninvading parasites per field of view to that for the vehicle control. Cytochalasin D and C1 significantly decreased the number of invading parasites, while C1 also decreased the number of uninvading (i.e., attached) parasites, although this difference was not significant (P = 0.08). (C) Effect of pretreatment of extracellular parasites for 16 h on parasite growth after washing out the compound. Compounds C1, C2, C3, and C5 significantly inhibited parasite growth after pretreatment, while C8 significantly enhanced parasite growth. On the basis of the invasion assay results, it is likely that very few, if any, parasites attached to the monolayer after C1 treatment. (D) Effects of host cell pretreatment on parasite growth. Host cells were pretreated with the compounds or vehicle for 24 h and extensively washed prior to parasite invasion and 3 days of growth. Assays where the compound was present for the entire 3-day incubation period were also run in parallel. C8 and C11 resulted in significantly enhanced parasite growth after pretreatment that was comparable to that observed when the compound was present for the entire 3-day growth period. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (compared to the vehicle-treated control for all comparisons).
We also examined the ability of these compounds to block parasite invasion (Fig. 4B). In these assays, only C1 significantly affected the number of invading parasites per field of view (0.5 uninvading parasites per field of view compared to 8.6 in controls; P < 0.05). Cytochalasin D blocks invasion but does not prevent attachment, and under these conditions, 6.5 attached but uninvading parasites were identified per field of view. In contrast, C1-treated parasites averaged only 1.0 attached, uninvading parasites per field of view, suggesting that even very short exposures to C1 either result in the complete disabling of the parasite or, more likely, result in parasite death. However, similar effects on both attachment and invasion have also been observed in parasites treated with 1,2-bis-(2-aminophenoxy)ethanetetraacetic acid-acetoxymethyl ester (BAPTA-AM), a calcium chelator that inhibits attachment and invasion by blocking microneme secretion (8). However, we cannot rule out the possibility that the compound could also be lethal to the parasites. None of the other tested compounds affected parasite invasion, including those with very potent growth-inhibitory activity, such as C2, C3, and C5. Moreover, as described above, C5-treated parasites were defective only in motility and not in invasion. The motility effects of C5 were dramatic: there were few, if any, visible trails. This is a rare uncoupling of parasite motility and invasion which warrants further investigation.
Effective compounds have a variety of effects on parasites and host cells.
The EC50s for the 14 compounds ranged from 0.14 μM to 8.7 μM (see Table 1 and Table S1 in the supplemental material). We performed a variety of assays with four of the most potent inhibitors (C1, C2, C3, and C5) and four of the most potent enhancers (C8, C11, C12, and C13) (Table 1). To begin to determine whether the compounds elicited their effects through a target in the host or parasite, extracellular parasites (16 h) or host cells (24 h) were pretreated with the compounds, and the compound was then thoroughly washed away. All four of the most potent inhibitors reduced parasite growth by over 98% compared to that for vehicle-treated controls, suggesting that the parasite was the target of the compound (Fig. 4C). When the same compounds were used to pretreat host cells prior to parasite invasion, none of the four inhibitors tested negatively affected parasite growth after host cell pretreatment and washout (Fig. 4D).
Table 1.
Effects of the eight compounds described in this studya
| Name | Compound identifier | Effect | % I/E | EC50 (μM) | Likely target | Compound effect on growth | Motility | Invasion |
|---|---|---|---|---|---|---|---|---|
| C1 | F1115-0016 | INH | 0.17 ± 0.1 | 1.36 ± 3.54 | Parasite | IRR | INH | INH |
| C2 | F1041-0001 | INH | 0.19 ± 0.1 | 1.34 ± 0.27 | Parasite | IRR | NE | NE |
| C3 | F3215-0002 | INH | 0.65 ± 0.1 | 0.57 ± 0.06 | Parasite | REV | ENH | NE |
| C5 | F0909-0123 | INH | 1.82 ± 2.2 | 1.12 ± 0.18 | Parasite | IRR | INH | NE |
| C8 | F0406-1005 | ENH | 205.3 ± 7.6 | 0.14 ± 0.02 | Host cell | IRR | NE | NE |
| C11 | F0745-0014 | ENH | 213.8 ± 18.8 | 0.21 ± 0.05 | Host cell | IRR | NE | NE |
| C12 | F0862-0340 | ENH | 214.1 ± 31.0 | 1.19 ± 0.22 | Unknown | REV | ENH | NE |
| C13 | F3220-0514 | ENH | 220.3 ± 1.3 | 0.38 ± 0.26 | Unknown | REV | NE | NE |
Abbreviations: % I/E, percent inhibition or enhancement compared to vehicle control at 10 μM; IRR, irreversible; REV, reversible; INH, inhibitor; ENH, enhancer; NE, no effect.
Interestingly, of the four enhancers, pretreatment of host cells with C8 and C11 significantly increased parasite growth. Specifically, parasite growth in host cells pretreated with C8 was 2.2-fold higher than that for vehicle-treated controls (P < 0.01), which was very similar to the increase in growth observed by incubating the host cells and the parasites for the entire growth period (3.0-fold higher; P < 0.001; Fig. 4D). Similarly, growth in host cells pretreated with C11 was also higher than that in vehicle-treated cells (1.9-fold higher; Fig. 4D; P < 0.01), although this was an effect smaller than that observed when the compound was present for the entire growth period (3.6-fold; Fig. 4D). For these two compounds, it appears that their effects on the host cell that led to increased parasite growth are at least partially irreversible and they act primarily on host cells (or possibly on proteins injected by the parasite into the host cell) to change the growth characteristics of the parasites. Enhancers C12 and C13 had no significant effect on parasite growth after host cell pretreatment (P > 0.05), although growth in C12-treated host cells was 1.6-fold higher than that in vehicle-treated controls.
To determine whether the effects of four inhibitory compounds were reversible, parasite-infected monolayers were treated with each compound for 12 h, and the monolayer was washed 3 times with cDMEM and then incubated for a further 12 h. Among these inhibitors, only the inhibitory effect of C3 was completely reversible, with the parasite growth rate after compound removal being similar to that for the vehicle-treated control (Fig. 5). Parasites treated with C1, C2, and C5 all showed reduced growth after compound removal compared to that for parasites treated with vehicle, suggesting that their inhibitory effects are either completely (C1) or partially (C2, C5) irreversible.
Fig 5.
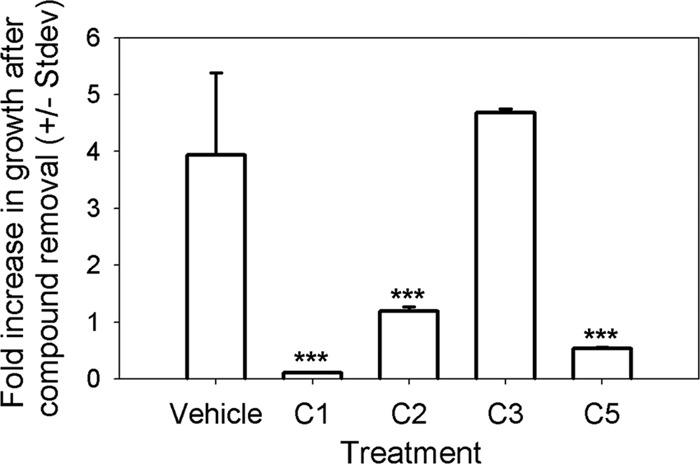
The ability of parasites to recover from treatment with C1, C2 C3, and C5 was assessed by exposing intracellular parasites for 12 h, followed by compound removal for an additional 12 h. Parasites were harvested both after the 12-h exposure and after the 12-h recovery period. Data are expressed as the fold increase in luminescent signal during the 12-h recovery period. C3-treated parasites fully recovered from the treatment, while C1-treated parasites did not. C2- and C5-treated parasites were found to only moderately recover. ***, P < 0.0001 compared to control.
The four most potent inhibitors cause distinct ultrastructural phenotypes.
Parasites exposed to the four most potent inhibitors (C1, C2, C3, and C5) were visualized using electron microscopy to determine changes in parasite ultrastructure. To do this, parasites (PB3-10, an ME49-derived strain) were treated with compounds C3 and C5 for 40 h and with compounds C1 and C2 for 24 h. The shorter exposure time was used for C1 and C2 on the basis of preliminary observations that longer exposure times resulted in nearly complete destruction of parasite-containing vacuoles. After exposure, the monolayers were fixed and processed for electron microscopy. Parasites treated with compound C3 had a characteristic arc-like shape, a well-defined tubulovesicular network, and a parasitophorous vacuolar membrane (PVM), although there was some evidence for disruption of parasite organelles, as they were more poorly defined (Fig. 6A and B). C5-treated parasites contained more apparently empty vacuoles than controls (Fig. 6A and C). Treatment with each of these compounds resulted in growth arrest, based on consistently smaller vacuole sizes compared to those for the controls after 40 h of growth.
Fig 6.
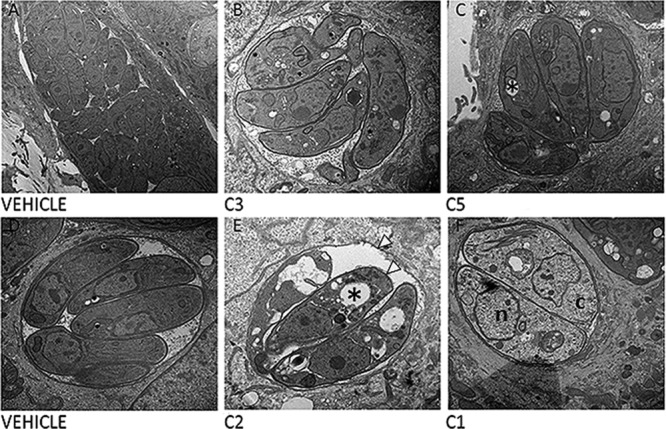
Parasite ultrastructure after exposure to four inhibitory compounds. (A to C) Parasites were treated with either vehicle (A), C3 (B), or C5 (C) for 40 h. (B) C3-treated parasites have a phenotype most similar to the phenotype of parasites treated with the vehicle control, with the exception of growth arrest over the 40-h incubation period. (C) C5-treated parasites have empty (*) vacuoles compared to parasites treated with vehicle. (D to F) In contrast to treatment with C1 and C3, treatment with C2 (E) and C1 (F) showed dramatic effects on parasite morphology compared to treatment with vehicle (D) over a 24-h period. C2-treated parasites have empty vacuoles within them (*) and a ruffled PVM and parasite plasma membrane (white arrowheads), while C1-treated parasites have reduced electron density both in the nucleus (n) and in the cytoplasm (c). Magnifications, ×12,000 (A), ×20,000 (B and D to F), and ×25,000 (C).
In contrast to compounds C3 and C5, parasites treated with compound C1 or C2 had dramatically altered morphology. In both treatments, some of the organelles such as the nucleus and the rhoptries were clearly defined. However, each compound caused distinct changes in parasite ultrastructure. Large numbers of apparently empty vacuoles were found within C2-treated T. gondii parasites, and C2 treatment also resulted in some organellar fragmentation (Fig. 6E) and a ruffled plasma membrane and PVM. On the other hand, C1-treated parasites had an intact PVM and still retained most of their organellar structure but had reduced electron density in both the nucleus and the cytoplasm. Furthermore, individual parasites were swollen and lacked their arc-like shape (Fig. 6F). Overall, these data are consistent with the fact that the effects of treatment with C1 on parasite growth are completely irreversible. Moreover, it is remarkable that C2-treated parasites can at least partially recover from these dramatic changes in compound-induced cell morphology (Fig. 5 and 6E).
Compound C2 is effective during the acute phase of a mouse model of infection.
The most potent inhibitory compounds (C1, C2, C3, and C5) were selected to study their effectiveness against parasite growth in a mouse model. After peritoneal infection with 10,000 tachyzoites of strain PB3-10, mice were treated with the four compounds or vehicle for the life of the mice, as described in the Materials and Methods section. C2 treatment resulted in reduced parasite proliferation during days 1 to 5, which was significantly lower (P < 0.001) by day 4 postinfection (see Fig. S2B in the supplemental material). Despite this decrease in parasite burden, the mice were extremely morbid and were humanely euthanized by day 6 postinfection. Compound C5 was mildly protective, while compounds C1 and C3 were not effective at reducing parasite proliferation (see Fig. S2A and B in the supplemental material).
We performed a 2nd series of experiments to validate the efficacy of C2 in a mouse model. Mice were infected with either PB3-10, a type II strain, or 5A10, a type III strain, which have different virulence phenotypes. For the PB3-10 infections, the C2-treated group exhibited lower parasite proliferation during days 2 to 4 postinfection, which was significantly different from that for vehicle-treated mice on day 4 (P < 0.001; Fig. 7A and B). Treatment with the compound also increased mouse survival from a highly lethal dose of this particular strain (strain PB3-10 and other type II strains have a 50% lethal dose of approximately 100 parasites). The mean time to morbidity was 8 ± 2.1 days for vehicle-treated mice and 10 ± 1.0 days for compound-treated mice (P = 0.04). Taken together, the pilot study and the follow-up study demonstrated that C2 can suppress parasite proliferation in vivo and increase mouse survival, although all mice eventually succumbed to the infection at this dose.
Fig 7.

(A) Effect of C2 treatment in vivo after infection with 10,000 PB3-10 parasites. p/sec/cm∧2/sr, photons per second per cm2 per steradian. (B and C) C2 treatment significantly reduced the parasite growth by day 4 (D4; P < 0.001) and had a significant effect on mouse survival, although all mice eventually became morbid.
With evidence that C2 suppressed tachyzoite growth in vivo, we wanted to study whether the compound would be effective in protecting the mice from developing a chronic infection from an acute one. Infections with 10,000 tachyzoites of most type III strains do not cause morbidity in the mice until the chronic phase of infection, and we used this strain to assess the long-term effects of compound treatment on both the acute phase of infection and the chronic phase. As expected, C2-treated mice showed reduced levels of parasite proliferation and increased survival, although mean survival times were not statistically significant (15 days compared to 19 days for vehicle-treated mice; P = 0.3). During this time, the amount of signal in the abdomen increased for both groups, with the vehicle-treated group recording higher signal levels than the treated group, and then the signal gradually disappeared, such that there was no signal observable ventrally by day 11 (Fig. 8A and B). All mice succumbed to the infection by day 26, and this was correlated with an observable parasite signal in the brain. In fact, even though compound treatment occurred throughout the period of infection, the parasite signal in the brain increased daily in each of the three surviving compound-treated mice (see Fig. S3 in the supplemental material), suggesting that once in the brain the parasites may be protected from the compound by the blood-brain barrier. Regardless, C2 is able to significantly reduce the parasite burden in mice infected with both type II and type III parasite strains.
Fig 8.

(A) Effect of C2 treatment in vivo after infection with 10,000 5A10 parasites. (B and C) C2 treatment again significantly reduced the parasite burden over the course of the experiment (P < 0.001 on day 7) and slightly prolonged mouse survival, although this difference was not significant.
DISCUSSION
In this study, we used a focused screen of compounds that were informatically selected to have the potential to interact with protein kinases to identify those that altered parasite growth in vitro. From these 527 compounds, we identified 14 that either inhibited or enhanced parasite growth. To date, other studies have been carried out using small-molecule libraries (7, 13), but in both of these, the authors were looking for inhibitors of parasite invasion. In our preliminary screen, we exposed parasites to the compounds after invasion and assayed the effects on parasite growth after 72 h in culture. Therefore, our assay encompasses the entire parasite lytic cycle postinvasion, from intracellular replication and egress to reinvasion of new host cells. Therefore, our screen should be able to detect compounds that affect all three of these parasite processes. Using secondary screens for effects on parasite invasion and motility, as well as to determine whether the compound targeted the host cell or the parasite, we found that the most potent inhibitors altered parasite replication rather than invasion or motility and that the most potent enhancer actually targeted the host cell rather than the parasite. As each effective compound develops into its own particular story, our next goal is to identify the molecular targets of these compounds through mutagenesis and/or biochemical strategies.
In this study, we found that the most potent growth enhancer (C8) actually targeted the host cell rather than the parasite, since pretreating host cells but not parasites prior to infection led to growth enhancement that was very similar to that observed while incubating parasites and host cells with the compound. Our data suggest that at least two of these enhancers may have targets within the host cells, while the inhibitors that we identified in this study are likely targeting the parasites directly. Whether this is due to differences in the kinome of Toxoplasma and mammals (the kinome of T. gondii has dramatically expanded; see references 5, 21, 24, and 25) can be determined only after target identification.
In addition, we were curious to see if there were any shared molecular fingerprints between the 14 identified compounds in our study and the 31 invasion modulators identified by Carey et al. (7). When we performed cluster analysis on these 45 compounds, the overall level of similarity across the compound set was quite low (over 95% of the compounds had a maximum similarity of less than 0.30). Moreover, with a few exceptions, compounds from each study tended to cluster together, although, again, with very low similarity (see Fig. S4 in the supplemental material). Therefore, in contrast to the 14 effective compounds identified in this study that primarily alter parasite replication, there does not seem to be any structural correlation with the compounds that alter parasite invasion.
Inhibitors of parasite growth appear either to be lethal or to halt parasite replication.
The effective inhibitors identified in this study varied in terms of their effect on parasites. For example, the target for compounds C1 and C5 is likely the parasite, since pretreatment of extracellular parasites prior to invasion reduced parasite growth to a level similar to that seen after including the compound for the entire growth period. We do acknowledge that T. gondii parasites incubated for 16 h under extracellular conditions will suffer a significant loss in viability and that some of the dramatic inhibitory effects observed after compound pretreatment could be due to synergistic effects between the compound treatment and extracellular exposure. For compounds C1 and C5, this is less likely, since the effects of these compounds were irreversible. Based on light and electron microscopic observations, we hypothesize that the effects of C1 are due to rapid killing of the parasite. This is supported by the fact that (i) parasites do not recover from compound treatment when inside host cells, (ii) parasites treated with C1 were nonmotile in motility assays, and (iii) C1-treated parasites were incapable of invading host cells in the presence of the compound. Since the C1 exposures were very short (∼10 min) prior to the invasion and motility assays, if this compound is lethal to parasites, the effects are most certainly very rapid. However, it is possible that the motility and invasion effects are not due to lethality but, rather, are due to inhibition of microneme secretion (which is well-known to inhibit parasite invasion and motility [7, 13]), although this does not explain the effects on intracellular growth. While C5 did inhibit parasite motility, it had no effect on invasion. The mechanism for this difference is unclear, and future studies will help to address this. Given the strong connection between motility and invasion, it is possible that C5-treated parasites are motile on host cell surfaces but not on the glass surface used in the motility assays. In contrast, the effect of C3 was fully reversible. After 12 h of exposure to the compound, parasites grew at a similar rate after compound removal as those treated with vehicle alone. It is likely that the effects of these compounds are due to the halting of parasite replication, since neither motility nor invasion was altered by these compounds. To date, compounds that block invasion have been shown to also inhibit parasite motility. For example, 4-bromophenacyl bromide (4-BPB) is a potent invasion inhibitor that also inhibits parasite motility by blocking rhoptry secretion (23). In previous studies identifying invasion inhibitors, it was also found that they affected protein secretion and parasite motility. For example, inhibitors 15 and 9 (7) were found to block parasite motility and constitutive secretion of microneme proteins. On the basis of these data, it is likely that the inhibitors identified in our screen may act through mechanisms different from the mechanisms of these previously identified compounds, having more significant effects on parasite growth than on motility and/or invasion.
Enhancers of parasite growth appear to target the host cell.
We identified seven enhancers of parasite growth in this screen. Enhancers of parasite invasion have been identified by others (7, 13), but we are not aware of any studies demonstrating an enhancing effect of parasite growth by small molecules. We were surprised that for two compounds increased parasite growth occurred only when we pretreated the host cells rather than the parasite. This suggests that these compounds may be disrupting host cell pathways or effectors that are responsible for controlling parasite proliferation within the host cell. It is also possible that the compounds present within the host cell are somehow enhancing the ability of secreted parasite effectors to disrupt host cell innate resistance pathways. This observation is in contrast to the findings for previously identified invasion enhancers, which were shown to target the parasite (7) and possibly did so by increasing microneme secretion and parasite gliding motility. Of our growth enhancers, none altered invasion, and only C12 increased parasite motility. This suggests again that the compounds identified in this study likely target cellular pathways different from those identified in the previous studies. Support for this is found in the fact that the most effective enhancer (C8) clearly has its effects on parasite growth by targeting the host cell. In fact, a 24-h host cell pretreatment (followed by extensive washing) was sufficient to increase parasite growth by over 200% of that for the control, which compared quite well with the effects of having the enhancer present for the entire 3-day growth period (>300%). This is particularly exciting, since such a compound can be used to identify host cell pathways that are targeted by the compound and link them in future experiments to their effects on parasite growth. From studies of both viruses (9) and Toxoplasma, it is becoming increasingly clear that there are host cell pathways that, when disrupted, lead to increased parasite proliferation (such as UNC93B1 for Toxoplasma [18]). We are excited by the potential that small molecules may have to identify previously uncharacterized mechanisms of host resistance to intracellular pathogens such as Toxoplasma gondii. While most of the small-molecule screening literature is focused on the identification of inhibitors of key processes, there are a number of studies demonstrating that certain compounds enhance or activate certain cellular pathways. For example, compounds that enhance hepatitis B virus replication in mammalian cells (35) and compounds that activate autophagy (31) and AMP-activated protein kinases (33) have been identified. It will be interesting to identify the host cell pathways altered by compounds such as C8, since these would very likely be involved in host cell resistance to parasitism.
Compound C2 can reduce parasite proliferation in vivo.
For the inhibitory compounds C1, C2, C3, and C5, we also assessed the impact of these compounds in mouse models of toxoplasmosis. C2 was found to be quite effective at decreasing the parasite burden and increasing mouse survival (although all mice eventually succumbed to the infection regardless of drug treatment). This is quite promising, since there are a number of factors that go into whether a compound will be effective in vivo (such as solubility in vivo, access to different tissues, and host metabolic processes). It is possible that some or all of the reduced parasite proliferation in C2-treated mice is due to negative effects on the mice, particularly the host cells that harbor the parasites, but the lack of any cytotoxic effects of C2 on host cells (see Fig. S1 in the supplemental material) provides some support that this may not be the case. Our data also suggest that C2 is very unlikely to cross the blood-brain barrier, since parasites in the drug-treated group proliferated rapidly after their arrival in the brain from day 15 onwards. The mortality observed in this study for type III strain-infected mice was higher than what is typical for this dose of parasites, and this could be due to a number of factors, including the repeated injection of either vehicle or compound and the luciferase imaging, which requires intraperitoneal injection of luciferin and anesthesia for ∼20 min. We think that it is unlikely, however, that the mortality observed in type III strain-infected mice is due solely to negative effects of C2 on the mice, since mortality coincided very well with the increased signal in the mouse brain. Regardless, this compound may represent a starting point to begin developing new anti-Toxoplasma therapies that may be effective in vivo. Other compounds have been found to be more effective than C2 in vivo, including a panel of quinolone derivatives (2) that decreased parasite burdens in the peritoneum, liver, lung, and brain during experimental mouse infections. The doses used in these studies (∼32 mg/kg daily) were much higher than those used in our study due to problems with the solubility of the compounds.
Other compounds that inhibit Toxoplasma growth in vitro have also been found to be effective in a mouse model. Bisphosphonates were found to increase survival against Toxoplasma infection by 80% (16), and 100% survival in mice infected with the highly virulent RH strain was observed when mice were treated with both sulfadiazine and epiroprim, a dihydrofolate reductase inhibitor. Clearly, the protection conferred by C2 in our studies is much less dramatic, but it is possible that we could achieve greater protection after solving the solubility issues, which would then allow us to increase the administered dose.
Supplementary Material
ACKNOWLEDGMENTS
We thank Yaw Adomako-Ankomah, Katelyn Walzer, and Greg Wier for critical reading of the manuscript, George Bondar (Life Chemicals) for helpful discussions about compound libraries, and the directors and staff at the University of Pittsburgh Center for Biologic Imaging for their help with electron microscopy.
This work was supported by NIH scholar development award K22 AI080977 and a Pew Scholarship in the Biomedical Sciences to J.P.B. and University of Pittsburgh Biological Sciences summer and academic year fellowships funded by the Howard Hughes Medical Institute to A.R.S., M.G.F., and E.J.C.
Footnotes
Published ahead of print 20 August 2012
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1. Austeng ME, et al. 2010. Maternal infection with Toxoplasma gondii in pregnancy and the risk of hearing loss in the offspring. Int. J. Audiol. 49:65–68 [DOI] [PubMed] [Google Scholar]
- 2. Bajohr LL, et al. 2010. In vitro and in vivo activities of 1-hydroxy-2-alkyl-4(1H)quinolone derivatives against Toxoplasma gondii. Antimicrob. Agents Chemother. 54:517–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Behnke MS, et al. 2011. Virulence differences in Toxoplasma mediated by amplification of a family of polymorphic pseudokinases. Proc. Natl. Acad. Sci. U. S. A. 108:9631–9636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Black MW, Boothroyd JC. 2000. Lytic cycle of Toxoplasma gondii. Microbiol. Mol. Biol. Rev. 64:607–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boothroyd JC, Dubremetz JF. 2008. Kiss and spit: the dual roles of Toxoplasma rhoptries. Nat. Rev. Microbiol. 6:79–88 [DOI] [PubMed] [Google Scholar]
- 6. Boyle JP, Saeij JP, Boothroyd JC. 2007. Toxoplasma gondii: inconsistent dissemination patterns following oral infection in mice. Exp. Parasitol. 116:302–305 [DOI] [PubMed] [Google Scholar]
- 7. Carey KL, Westwood NJ, Mitchison TJ, Ward GE. 2004. A small-molecule approach to studying invasive mechanisms of Toxoplasma gondii. Proc. Natl. Acad. Sci. U. S. A. 101:7433–7438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Carruthers VB, Giddings OK, Sibley LD. 1999. Secretion of micronemal proteins is associated with Toxoplasma invasion of host cells. Cell. Microbiol. 1:225–235 [DOI] [PubMed] [Google Scholar]
- 9. Coyne CB, et al. 2011. Comparative RNAi screening reveals host factors involved in enterovirus infection of polarized endothelial monolayers. Cell Host Microbe 9:70–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Derouin F, Pelloux H. 2008. Prevention of toxoplasmosis in transplant patients. Clin. Microbiol. Infect. 14:1089–1101 [DOI] [PubMed] [Google Scholar]
- 11. Farrell A, et al. 2012. A DOC2 protein identified by mutational profiling is essential for apicomplexan parasite exocytosis. Science 335:218–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hakansson S, Morisaki H, Heuser J, Sibley LD. 1999. Time-lapse video microscopy of gliding motility in Toxoplasma gondii reveals a novel, biphasic mechanism of cell locomotion. Mol. Biol. Cell 10:3539–3547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hall CI, et al. 2011. Chemical genetic screen identifies Toxoplasma DJ-1 as a regulator of parasite secretion, attachment, and invasion. Proc. Natl. Acad. Sci. U. S. A. 108:10568–10573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kamau E, et al. 2011. A novel benzodioxole-containing inhibitor of Toxoplasma gondii growth alters the parasite cell cycle. Antimicrob. Agents Chemother. 55:5438–5451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kongsaengdao S, Samintarapanya K, Oranratnachai K, Prapakarn W, Apichartpiyakul C. 2008. Randomized controlled trial of pyrimethamine plus sulfadiazine versus trimethoprim plus sulfamethoxazole for treatment of toxoplasmic encephalitis in AIDS patients. J. Int. Assoc. Physicians AIDS Care (Chic.). 7:11–16 [DOI] [PubMed] [Google Scholar]
- 16. Ling Y, et al. 2005. Bisphosphonate inhibitors of Toxoplasma gondii growth: in vitro, QSAR, and in vivo investigations. J. Med. Chem. 48:3130–3140 [DOI] [PubMed] [Google Scholar]
- 17. Lipinski CA. 2000. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 44:235–249 [DOI] [PubMed] [Google Scholar]
- 18. Melo MB, et al. 2010. UNC93B1 mediates host resistance to infection with Toxoplasma gondii. PLoS Pathog. 6:e1001071 doi:10.1371/journal.ppat.1001071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Messina M, Niesman I, Mercier C, Sibley LD. 1995. Stable DNA transformation of Toxoplasma gondii using phleomycin selection. Gene 165:213–217 [DOI] [PubMed] [Google Scholar]
- 20. Muskavitch MA, Barteneva N, Gubbels MJ. 2008. Chemogenomics and parasitology: small molecules and cell-based assays to study infectious processes. Comb. Chem. High Throughput Screen. 11:624–646 [DOI] [PubMed] [Google Scholar]
- 21. Peixoto L, et al. 2010. Integrative genomic approaches highlight a family of parasite-specific kinases that regulate host responses. Cell Host Microbe 8:208–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Puri AW, Bogyo M. 2009. Using small molecules to dissect mechanisms of microbial pathogenesis. ACS Chem. Biol. 4:603–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ravindran S, Lodoen MB, Verhelst SH, Bogyo M, Boothroyd JC. 2009. 4-Bromophenacyl bromide specifically inhibits rhoptry secretion during Toxoplasma invasion. PLoS One 4:e8143 doi:10.1371/journal.pone.0008143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Reese ML, Boyle JP. 2012. Virulence without catalysis: how can a pseudokinase affect host cell signaling? Trends Parasitol. 28:53–57 [DOI] [PubMed] [Google Scholar]
- 25. Reese ML, Zeiner GM, Saeij JP, Boothroyd JC, Boyle JP. 2011. Polymorphic family of injected pseudokinases is paramount in Toxoplasma virulence. Proc. Natl. Acad. Sci. U. S. A. 108:9625–9630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ritz C, Streibig JC. 2005. Bioassay analysis using R. J. Statistical Software 12:5 [Google Scholar]
- 27. Saeij JP, et al. 2006. Polymorphic secreted kinases are key virulence factors in toxoplasmosis. Science 314:1780–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Saeij JP, Boyle JP, Grigg ME, Arrizabalaga G, Boothroyd JC. 2005. Bioluminescence imaging of Toxoplasma gondii infection in living mice reveals dramatic differences between strains. Infect. Immun. 73:695–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Saeij JP, et al. 2007. Toxoplasma co-opts host gene expression by injection of a polymorphic kinase homologue. Nature 445:324–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Taylor S, et al. 2006. A secreted serine-threonine kinase determines virulence in the eukaryotic pathogen Toxoplasma gondii. Science 314:1776–1780 [DOI] [PubMed] [Google Scholar]
- 31. Tian Y, Bustos V, Flajolet M, Greengard P. 2011. A small-molecule enhancer of autophagy decreases levels of Abeta and APP-CTF via Atg5-dependent autophagy pathway. FASEB J. 25:1934–1942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tsai HC, et al. 2002. Treatment of Toxoplasma brain abscess with clindamycin and sulfadiazine in an AIDS patient with concurrent atypical Pneumocystis carinii pneumonia. J. Formos. Med. Assoc. 101:646–649 [PubMed] [Google Scholar]
- 33. Vingtdeux V, Chandakkar P, Zhao H, Davies P, Marambaud P. 2011. Small-molecule activators of AMP-activated protein kinase (AMPK), RSVA314 and RSVA405, inhibit adipogenesis. Mol. Med. 17:1022–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Walters WP, Namchuk M. 2003. Designing screens: how to make your hits a hit. Nat. Rev. Drug Discov. 2:259–266 [DOI] [PubMed] [Google Scholar]
- 35. Yoon SW, et al. 2004. A new compound from Micromonospora sp. SA246, 9-hydroxycrisamicin-A, activates hepatitis B virus replication. Biochem. Biophys. Res. Commun. 319:859–865 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.