

Abstract
Lytic replication of the Kaposi's sarcoma-associated herpesvirus (KSHV) is essential for the maintenance of both the infected state and characteristic angiogenic phenotype of Kaposi's sarcoma and thus represents a desirable therapeutic target. During the peak of herpesvirus lytic replication, viral glycoproteins are mass produced in the endoplasmic reticulum (ER). Normally, this leads to ER stress which, through an unfolded protein response (UPR), triggers phosphorylation of the α subunit of eukaryotic initiation factor 2 (eIF2α), resulting in inhibition of protein synthesis to maintain ER and cellular homeostasis. However, in order to replicate, herpesviruses have acquired the ability to prevent eIF2α phosphorylation. Here we show that clinically achievable nontoxic doses of the glucose analog 2-deoxy-d-glucose (2-DG) stimulate ER stress, thereby shutting down eIF2α and inhibiting KSHV and murine herpesvirus 68 replication and KSHV reactivation from latency. Viral cascade genes that are involved in reactivation, including the master transactivator (RTA) gene, glycoprotein B, K8.1, and angiogenesis-regulating genes are markedly decreased with 2-DG treatment. Overall, our data suggest that activation of UPR by 2-DG elicits an early antiviral response via eIF2α inactivation, which impairs protein synthesis required to drive viral replication and oncogenesis. Thus, induction of ER stress by 2-DG provides a new antiherpesviral strategy that may be applicable to other viruses.
INTRODUCTION
Kaposi's sarcoma-associated herpesvirus (KSHV; HHV8) is the etiologic agent of KS, an AIDS-defining malignancy characterized by intense angiogenesis and the proliferation of spindle-shaped cells (8, 16, 41, 64). Most AIDS-associated KS (AIDS-KS) patients respond favorably to antiretroviral therapy (ART). However, despite the effectiveness of available treatments, KS is not eliminated for at least half of these cases, highlighting the need for novel therapeutic strategies for this serious and deadly form of cancer (14).
Like other herpesviruses, KSHV infection can lead to two different fates: latent infection, in which the viral episome replicates together with the host cell, and a productive cytopathic (also called lytic) infection. During lytic replication, the virus carries out an organized cascade of gene expression spanning the whole viral genome and leading to replication of the viral DNA, infectious virion production, and the death of the host cell (15). Cumulative experimental evidence supports a model of KS oncogenesis in which latently infected KS cell proliferation and angiogenesis are promoted by lytically infected cells (2). This picture of productive viral replication “fueling” the lesion is further supported by the following facts: (i) clinical evidence demonstrating that KS is prevented by antiherpesviral compounds that block lytic replication, such as ganciclovir or foscarnet (42), and that immune reconstitution by ART induces KS regression; (ii) laboratory data showing that the viral angiogenic lytic genes are essential for paracrine maintenance of latent gene-induced tumors (43, 53); (iii) in vitro evidence indicating that continued lytic replication is required for maintaining active latent infection (19); and (iv) epidemiological studies showing that KS incidence is higher in clinical settings, such as immunodeficiency, that permit viral replication to occur (18, 41). Taken together, this cumulative experimental evidence indicates that KSHV lytic replication is required for oncogenesis and the maintenance of KS lesions.
During herpesvirus lytic replication, viral glycoproteins, which are mass produced in the endoplasmic reticulum (ER), increase the demand for protein synthesis and folding, leading to ER stress (9, 21, 26), which is defined as an imbalance between protein load and folding capacity (51). In order for the host to cope with the induced stress and to maintain homeostasis, the cell initiates a compensatory mechanism termed the unfolded protein response (UPR). The signaling pathways evoked in this response involve the reduction of nascent protein translation in the ER as a protection mechanism against further protein load, upregulation of the ER-localized machinery involved in protein folding (i.e., chaperones), and degradation of unfolded proteins (51). The three branches activated during UPR transduction are mediated by the ER resident transmembrane receptors PERK (double-stranded RNA [dsRNA]-activated protein kinase [PKR]-like ER kinase), ATF6 (activating transcription factor 6), and inositol requiring kinase 1 (IRE1). Upon induction of ER stress, activated ATF6 is proteolytically cleaved and translocates to the nucleus, acting as a transcription factor to turn on UPR-related genes such as glucose-regulated protein 78 kDa (GRP78). It has been shown that GRP78 is strongly upregulated upon UPR induction, and its levels serve as a UPR marker (31). At the same time, IRE1 displays endoribonuclease activity by splicing mRNA from the XBP-1 (X-box binding protein 1) transcript for the generation of spliced XBP-1 [XBP-1(s)], which then acts as a transcription factor to turn on other UPR-related genes including chaperones and enzymes involved in protein degradation and ER lipid biosynthesis. The UPR transducer PERK phosphorylates and thereby inactivates the α subunit of eukaryotic initiation factor 2 (eIF2α), which normally is essential for cap-dependent ribosomal protein synthesis. Thus, UPR activation limits the amount of new proteins entering the ER, thereby relieving ER stress. Phosphorylation of eIF2α due to PERK occurs early upon UPR activation prior to the detection of GRP78 and is followed by upregulation of CHOP (C/EBP homologous protein), which induces UPR-mediated apoptosis if signaling is prolonged (20, 29). Taken together, upon ER stress induction, cell homeostasis is maintained by activation of multiple UPR branches acting in concert.
In order to replicate, viruses attenuate host UPR signals after induction of ER stress. Indeed, infection due to hepatitis C virus (HCV) (40) or arenavirus (48) causes a wave of upregulation of UPR markers, which are undetectable at early time points (0 to 2 days postinfection), transiently induced by viral replication (2 to 4 days postinfection), and markedly attenuated thereafter. Similar kinetics have been observed in cells infected with human cytomegalovirus (CMV), which is a herpesvirus (26). The mechanism by which CMV overrides UPR-mediated blockage of protein synthesis is via upregulation of viral genes (TRS1 and IRS1) that have been shown to prevent eIF2α phosphorylation (10). Similarly, in cells infected with herpes simplex virus 1 (HSV-1), it has been reported that viral γ134.5 indirectly dephosphorylates eIF2α (22) and HSV-1 glycoprotein B (gB) directly inhibits cellular PERK (44), both leading to resumption of protein synthesis, facilitating viral replication. Interestingly, only modest activation of the ATF6 and the XBP-1(s) components of the UPR have been detected in latent KSHV-infected primary effusion lymphoma (PEL) cells (28), while dephosphorylation of eIF2α has not been investigated in PEL or other KSHV infections.
It has been shown that 2-deoxy-d-glucose (2-DG) and 2-fluoro-2-deoxy-d-glucose (2-FDG) are powerful agents to block and probe sugar metabolism in cancer cells (33, 34, 39, 61). These compounds also take advantage of the increased glucose uptake that occurs in most tumors, providing a specificity window that increases the selectivity of these analogs (12, 34). As glucose analogs, both 2-DG and 2-FDG are powerful inhibitors of glycolysis, with the latter shown to be a more potent inhibitor of glucose metabolic pathways because of its closer similarity to glucose (34). On the other hand, 2-DG is also known as 2-deoxymannose; because of its mannose-like character, it is better able to compete with mannose in the growing oligosaccharide chain during the initial steps of N-linked glycosylation which occur in the ER (34). Oligosaccharide chains that have incorporated 2-DG cannot form the functional glucose3mannose9 moiety for proper protein glycosylation. Abnormal N-linked glycosylation interferes with protein folding, which induces ER stress, leading to the UPR and resulting in the inhibition of protein synthesis. Thus, we propose that the activity of 2-DG to affect N-linked glycosylation provides a natural window of opportunity to target key processes, i.e., viral replication and expression of viral oncogenes required for KS development. We hypothesize that 2-DG-induced ER stress would counter the ability of KSHV and other herpesviruses to circumvent UPR-mediated blockage of protein synthesis, thereby impairing KSHV gene expression, resulting in inhibition of viral replication and thus oncogenesis.
Here we show that 2-DG induction of ER stress and UPR leads to the inhibition of viral replication in three different KSHV infection in vitro models and in murine gammaherpesvirus 68 (MHV-68) infection. We find that several early members of the lytic viral replication cascade, as well as host angiogenic genes, are significantly decreased with 2-DG treatment. This in vitro study provides the rationale for a new antiviral strategy that may be applicable to inhibiting the replication of KSHV and other viruses.
MATERIALS AND METHODS
Cells and viruses.
293rKSHV.219 are HEK293 cells infected with rKSHV.219 under puromycin selection as described previously (59). Human iSLK.219 is a KSHV-infected cell line derived from endothelium-lineage cells from a KS tumor and also contains a doxycycline-inducible RTA construct (46). 293rKSHV, iSLKrK, and NIH3T12 are maintained in Dulbecco's modified Eagle's medium (DMEM) with 1 g/liter of glucose supplemented with 10% fetal bovine serum (FBS) and penicillin-streptomycin-amphotericin. mECK36, a murine endothelial cell line stably transfected with a KSHV-containing bacterial artificial chromosome (BAC36), is maintained in media further supplemented with 30% FBS, MEM vitamins, insulin, transferrin, selenite, endothelial cell growth supplement, endothelial cell growth factor, and heparin (45). MHV-68 stocks were purchased from the American Type Culture Collection. Cells were grown under 5% CO2 at 37°C.
Cytotoxicity assay.
Cells were seeded onto 48-well plates and cultured overnight. After drug exposure, attached cells and their respective culture media were collected and centrifuged at 400 × g for 5 min. The pellets were then resuspended in Hanks balanced salt solution (HBSS) (Mediatech) and analyzed with a Vi-Cell cell viability analyzer (Beckman Coulter) based on trypan blue exclusion. Results are shown as the percentages of viable cells out of the total cells counted. Data are the averages of triplicate samples ± standard deviations (SD) from one representative experiment out of three independent analyses.
Drugs and antibodies.
2-DG, 2-FDG, mannose, sodium butyrate, and doxycycline were purchased from Sigma-Aldrich. The following rabbit primary antibodies were from Cell Signaling: GRP78, phosphorylated eIF2α, total eIF2α, and GAPDH (glyceraldehyde-3-phosphate dehydrogenase). Mouse anti-β-actin antibody was from Sigma-Aldrich. Mouse anti-K8.1A/B antibody was from Advanced Biotechnologies. Rabbit antiviral interferon regulatory factor 1 antibody (anti-vIRF-1) was from J. Nicholas. Horseradish peroxidase (HRP)-conjugated anti-rabbit and anti-mouse IgG were purchased from Promega.
Lytic induction, TCID50, and plaque assay.
rKSHV.219-infected 293rKSHV cells and BAC36-transfected mECK36 cells were treated at 70% confluence with 3 mM sodium butyrate to induce cells into viral lytic gene expression and replication. rKSHV.219-infected iSLK cells were stimulated with 1 μg/ml doxycycline. At 24 h of treatment, DNA-free RNA was extracted for real-time quantitative reverse transcriptase PCR (qRT-PCR). After 72 h of treatment, viral loads (KSHV DNA copy numbers) were determined by real-time quantitative PCR of cell-free supernatants (virion) and cellular lysates (cell). At 72 h of treatment, cell-free supernatants were collected and used for de novo infection of 293 cells in the presence of 5 μg/μl Polybrene, followed by incubation at 37°C for 90 min and washing with phosphate-buffered saline (PBS). Detection of infected cells and the 50% tissue culture infective dose (TCID50) were determined by counting green fluorescent protein (GFP)-positive cells at 2 days postinfection.
NIH3T12 cells at 50% confluence were infected with MHV-68 at a multiplicity of infection (MOI) of 0.1 in DMEM supplemented with 2% FBS for 2 h. Cells were washed with PBS to remove unbound virus and were maintained in DMEM for 48 h. MHV-68 titers were measured by the collection of supernatants for the infection of uninfected 3T12 cells by serial dilution, followed by the visualization of the cytopathic effect by microscopy and the quantification of plaques after carboxymethylcellulose overlay.
Microscopy.
Cells were seeded onto 8-well Lab-Tek II CC chamber slides (Thomas Scientific) and cultured overnight to reach ∼50% confluence. After 24 h of drug exposure, cells were fixed with 10% neutral buffered formalin solution containing 4% formaldehyde (Sigma-Aldrich), mounted with ProLong Gold antifade reagent with DAPI (4′,6-diamidino-2-phenylindole) (Invitrogen) and visualized with the Laborlux fluorescence microscope (Leitz) equipped with a DFC 340 FX digital camera (Leica). Cells were shown from one representative experiment out of at least three independent analyses.
Flow cytometry.
Cells were washed with PBS and fixed and resuspended with Cytofix fixation buffer (BD Biosciences). Flow cytometric analysis was performed on a Becton, Dickinson LSR analyzer (BD Biosciences).
Real-time qRT-PCR.
SYBR green PCR master mix was used with an ABI Prism 7300 sequence detection software (Applied Biosystems). Primers used were as follows: human β-actin forward, 5′-AGAGCTACGAGCTGCCTGAC-3′, and reverse, 5′-AGCACTGTGTTGGCGTACAG-3′; human GAPDH forward, 5′-GAGTCAACGGATTTGGTCGT-3′, and reverse, 5′-GACAAGCTTCCCGTTCTCAG-3′; mouse GAPDH forward, 5′-AACTTTGGCATTGTGGAAGG-3′, and reverse, 5′-GGATGCAGGGATGATGTTCT-3′; mouse β-actin forward, 5′-GATCTGGCACCACACCTTCT-3′, and reverse, 5′-GGGGTGTTGAAGGTCTCAAA-3′; KSHV RTA/ORF50 (where ORF is the open reading frame) forward, 5′-CAAGGTGTGCCGTGTAGAGA-3′, and reverse, 5′-TCCCAAAGAGGTACCAGGTG-3′; KSHV K8 forward, 5′-GGCCCTAGAGGCCGTCTCCC-3′, and reverse, 5′-GGGAGGTCACGGGACGCTCT; RFP (where RFP is red fluorescent protein) forward, 5′-AGGACGGCTGCTTCATCTAC-3′, and reverse, 5′-TGGTCTTCTTCTGCATCACG-3′; KSHV GPCR/ORF74 (where GPCR is G protein-coupled receptor) forward, 5′-TGTGTGGTGAGGAGGACAAA-3′, and reverse, 5′-GTTACTGCCAGACCCACGTT-3′; KSHV vIL6 forward, 5′-TGCTGGTTCAAGTTGTGGTC-3′, and reverse, 5′-ATGCCGGTACGGTAACAGAG-3′; KSHV vIRF-1/K9 forward, 5′-GGAAGAACAATGCGTGGAATG-3′, and reverse, 5′-CGACTGGCTTGTCGTCAGTA-3′; KSHV K8.1 forward, 5′-CACCACAGAACTGACCGATG-3′, and reverse, 5′-TGGCACACGGTTACTAGCAC-3′; KSHV gB/ORF8 forward, 5′-CTGGGGACTGTCATCCTGTT-3′, and reverse, 5′-ATGCTTCCTCACCAGGTTTG-3′; MHV RTA/ORF50 forward, 5′-CGGTGACAAACCCCTCTAAA-3′, and reverse, 5′-CCCCAATGGTTCATAAGTGG-3′; MHV GPCR/ORF74 forward, 5′-CTGGCCTGGTTTGCAGTTAT-3′, and reverse, 5′-CCCTAGTGGTCCCTCCTAGC-3′; mouse PDGFB forward, 5′-CCTCGGCCTGTGACTAGAAG-3′, and reverse, 5′-CCTTGTCATGGGTGTGCTTA-3′; mouse VEGF-R1 forward, 5′-TATAAGGCAGCGGATTGACC-3′, and reverse, 5′-TCATACACATGCACGGAGGT-3′; mouse VEGF-R3 forward, 5′-GCTGTTGGTTGGAGAGAAGC-3′, and reverse, 5′-TGCTGGAGAGTTCTGTGTGG-3′; mouse Tie1 forward, 5′-CAGGCACAGCAGGTTGTAGA-3′, and reverse, 5′-GTGCCACCATTTTGACACTG-3′; human PDGFA forward, 5′-CAAGACCAGGACGGTCATTT-3′, and reverse, 5′-CTTGACACTGCTCGTGTTGC-3′; human PDGFRβ forward, 5′-GGTGACACTGCACGAGAAGA-3′, and reverse, 5′-CAATGGTGGTTTTGCAGATG-3′; human GRP78 forward, 5′-GGAATTCCTCCTGCTCCTCGT-3′, and reverse, 5′-CAGGTGTCAGGCGATTCTGG-3′; human XBP-1(s) forward, 5′-CTGAGTCCGCAGCAGGTGCA-3′, and reverse, 5′-GGTCCAAGTTGTCCAGAATGCCCAA-3′; and human CHOP forward, 5′-GCGCATGAAGGAGAAAGAAC-3′, and reverse, 5′-TCACCATTCGGTCAATCAGA-3′.
Immunoblotting analysis.
Cells were seeded onto 6-well plates and cultured to reach 70% confluence. Following drug exposure for the indicated times, cells were harvested and lysed with the lysis buffer (Tris-glycine-SDS, phosphatase inhibitor cocktails 2 and 3, and a protease inhibitor cocktail from Sigma-Aldrich). Protein concentrations of each sample were determined using a DC protein assay kit (Bio-Rad) according to the manufacturer's directions, and equal amounts of protein were loaded onto 10% Tris-HCl gels (Bio-Rad). After SDS-PAGE, protein was transferred onto a polyvinylidene difluoride (PVDF) membrane (Millipore), blocked with 5% milk, and probed with the corresponding primary antibodies overnight at 4°C. The membrane was washed and probed with secondary antibodies for 1 h. The membrane was then incubated with ECL Plus (Amersham), and signal was visualized on Blue Lite Autorad Films (ISC BioExpress). All primary antibodies were used at 1:1,000 dilutions, except for vIRF-1 (1:10,000) and β-actin (1:10,000), and the secondary antibodies were used at 1:10,000. Representative blots from at least two independent experiments were shown unless otherwise indicated.
Statistical analysis.
Data were compared using a one-tailed paired Student's t test, and a P value of less than 0.05 was considered significant.
RESULTS
2-DG but not 2-FDG inhibits gammaherpesviral infectivity and replication.
To study the impact of 2-DG on KSHV replication, we determined its effect on the infectivity of supernatants from 293 cells bearing recombinant reporter KSHV that were induced to undergo lytic replication by the addition of butyrate. The reporter virus used, rKSHV.219, encodes green fluorescent protein (GFP) under the cellular control of elongation factor 1 (EF-1; a constitutively expressed marker of infection) (59). Infectivity was monitored by adding the supernatants from lytically induced cells to uninfected control 293 cells and measuring viral titers by monitoring GFP-expressing cells and calculating the 50% tissue culture infective dose (TCID50). Addition of 2-DG during lytic induction produced a dose-dependent decrease in the viral titer (Fig. 1A). To determine whether the decrease in the titer was due either to decreased production of virions or to virions with decreased infectivity, KSHV DNA viral loads in cell extracts and supernatants were determined by qPCR. We found that the 2-DG-induced decrease in the viral titer correlated with the reduced viral DNA copy number (both in supernatants [Fig. 1B] and in cells [Fig. 1C]), indicating that the decrease was more likely due to lowered virion production rather than virions with decreased infectivity. Interestingly, the glucose analog 2-FDG, which is known to preferentially inhibit glycolysis and not N-linked glycosylation (33), did not inhibit KSHV replication. This result was in agreement with our previous findings using 2-DG and 2-FDG in tumor cells, indicating that the inhibitory effect of 2-DG at the employed doses was more likely due to inhibition of N-linked glycosylation than to glycolysis (34). Additionally, to rule out the possibility that lowered viral titer was due to a cytotoxic effect of 2-DG, we treated infected cells with 2-DG up to 3 mM and found no increased cytotoxicity compared to untreated cells (Fig. 1D). Similarly, when 293rKSHV cells were lytically induced with butyrate (Fig. 1E), no increase in cytotoxicity was detected in 2-DG- or 2-FDG-treated cells. Thus, 2-DG inhibits viral replication in the absence of cellular toxicity.
Fig 1.

2-DG inhibits KSHV and MHV-68 replication. (A) rKSHV.219-infected 293 cells were lytically induced with butyrate (3 mM) in the presence of 2-DG at the indicated doses. After 72 h of drug exposure, KSHV titers in the supernatants were measured by TCID50. (B) Infected cells were treated as described for panel A with 2-DG (■) or 2-FDG (●) for 72 h. Bars represent mean percent copy number (triplicates ± SD) of viral DNA levels in cell-free supernatants determined by qPCR (*, P < 0.001). (C) Infected cells were treated as described for panel B, and qPCR of cell lysates was performed to detect intracellular KSHV DNA. GAPDH was used as a loading control (*, P < 0.0001). (D) rKSHV-infected 293 cells were treated with 2-DG (■) or 2-FDG (●) for 72 h, and percent cell viability was determined by trypan blue exclusion of total cells. (E) Infected cells were treated as described for panel D except that they were lytically induced with butyrate (3 mM). (F) NIH3T12 cells were infected with MHV-68 at 0.1 MOI in the presence (black bar) or absence (white bar) of 2-DG (0.4 mM). After 48 h of drug exposure, MHV-68 titers in the supernatants were measured by plaque assay (duplicates ± SD).
To further confirm 2-DG's antiviral activity, we used another gammaherpesvirus, murine γ2-herpesvirus 68 (MHV-68). This virus differs from KSHV in that MHV-68 carries a spontaneous lytic replication in permissive cells (mouse fibroblasts) without the need for chemical stimulation, e.g., butyrate (47). For the MHV-68 replication inhibition assay, infection of 3T12 cells with MHV-68 was carried out in the absence or presence of 2-DG, and the titers of the supernatants were determined by plaque formation assay. Figure 1F shows an 80% decrease of viral titer in 2-DG-treated cells compared to untreated cells. Taken together, these data indicate that 2-DG inhibits lytic replication in two different gammaherpesvirus models.
2-DG inhibits KSHV reactivation by interfering with the N-linked glycosylation pathway.
One possibility to explain the inhibition of virion production and the lowering of viral DNA replication by 2-DG is blockage of the viral gene cascade which drives herpesvirus production. Reactivation of KSHV from the latent to the lytic cycle requires the expression of early viral genes, which include the master regulator RTA (replication and transcription activator). Among several early lytic genes, RTA stimulates PAN (polyadenylated nuclear RNA), which has been reported to be the most upregulated gene during the early phases of reactivation (54). Thus, we used the virus rKSHV.219, which also contains a PAN promoter-driven RFP reporter (59) to assay early viral gene expression and the number of KSHV-infected 293 cells entering the lytic cycle (Fig. 2A). In Fig. 2B (top panels), it can be seen that 2-DG leads to a marked decrease in the number of RFP-expressing 293rKSHV cells when induced with butyrate. As another measure of 2-DG's efficacy in blocking reactivation (which requires early viral gene expression), a cell line (SLK) derived from endothelial-lineage spindle-shaped cells explanted from a KS tumor was used (23). These cells were infected with rKSHV (iSLKrK), in which lytic replication could be induced by doxycycline-inducible expression of RTA (46). Figure 2B (bottom panels) demonstrates a significant decrease in RFP expression in 2-DG-treated iSLKrK cells. Similar to 293rKSHV cells, no increase in cytotoxicity was detected in latent or lytically induced iSLKrK cells in the presence of 2-DG at these concentrations (Fig. 2C). Lytic reactivation was quantified using flow cytometry to count RFP-positive 293rKSHV cells, and 2-DG's inhibitory effect was determined to be dose dependent (Fig. 2D). In contrast, no inhibition by the mostly glycolytic inhibitor 2-FDG was observed, further supporting glycosylation targeting by 2-DG, which occurs through its ability to compete with mannose, as the main mechanism of inhibition (33). Consistent with this proposed mechanism, the addition of mannose reversed 2-DG's inhibitory effect on KSHV reactivation (Fig. 2E).
Fig 2.

2-DG inhibits KSHV reactivation. (A) Schematic diagram of rKSHV.219 GFP and RFP reporters and their use in the detection of the phases of latency and lytic reactivation. (B) rKSHV.219-infected 293 cells were lytically induced with butyrate (3 mM) in the presence or absence of 2-DG (1 mM) for 24 h (top panels). rKSHV.219-infected iSLK cells (iSLKrK) were lytically induced with doxycycline (1 μg/ml) in the presence or absence of 2-DG (1 mM) for 24 h (bottom panels). Viral reactivation was detected by fluorescence microscopy using the RFP reporter driven by the promoter of the early lytic gene PAN. DAPI staining identified the number of cells, and viral infection was detected using the GFP reporter driven by the constitutive promoter of cellular EF-1. (C) iSLKrK cells were treated in the presence (■) or absence (●) of doxycycline as described for panel B, and percent cell viability was determined by trypan blue exclusion. (D) Infected 293 cells were treated as described for panel A, in the presence of 2-DG (■) or 2-FDG (●) at the indicated doses and were collected at 36 h. Flow cytometry of infected cells was performed to detect RFP-expressing (lytic reactivated) cells (duplicates ± SD). (E) Cells were treated as described for panel B except that 2-DG was used at a dose of 4 mM in the presence or absence of mannose (man; 1 mM). Virally infected cells were analyzed as described for panel D.
2-DG inhibits pathogenic lytic gene expression.
In addition to maintaining and sustaining the infected state of KS lesions, the lytic replicative cascade includes expression of key pathogenic viral genes which fuel KS oncogenesis (41, 57). Therefore, we assessed 2-DG's ability to inhibit lytic gene expression upon butyrate induction in the 293 infection system and upon doxycycline induction in infected iSLK cells. Additionally, we used KSHV-containing mouse endothelial cells (mECK36), which we previously have shown to express angiogenic proteins and induce tumorigenesis in mice (45). As depicted in Fig. 3A, B, and C, we found that in the three systems tested, 2-DG inhibited the expression of immediate-early, delayed-early, and late viral lytic genes, including those that encode glycoproteins K8.1 and gB, known to be major components of the virion. In the tested cell lines, inhibition of the expression of the oncogene viral interferon regulatory factor 1 (vIRF-1; the most upregulated lytic gene during mECK36 tumorigenesis) (45) was observed, while inhibition of the angiogenic oncogene viral G protein-coupled receptor (vGPCR) (1) was observed in two of the three cell lines assayed. To further confirm 2-DG's anti-gammaherpesviral activity, murine 3T12 cells infected with MHV-68 were used to measure mRNA expression of the two lytic genes MHV RTA/ORF50 and MHV GPCR/ORF74. Similarly to its effect on KSHV, 2-DG inhibited MHV-68 gene expression by 50% (Fig. 3D). Consistent with lowered viral mRNA, KSHV K8.1 and vIRF-1 were also reduced at the protein level in KSHV-infected 2-DG-treated cells (Fig. 3E and F). Importantly, at least the 2-DG-mediated reduction of expression of the early viral oncogene vIRF-1 was reversed by mannose (Fig. 3F).
Fig 3.

2-DG inhibits lytic gene expression. 293rKSHV (A), mECK36 (B), and iSLKrK (C) cells were lytically induced by butyrate (3 mM) (A and B) or by doxycycline (0.5 μg/ml) (C) in the presence (black bars) or absence (white bars) of 2-DG (1 mM) for 24 h. qRT-PCR was performed to detect levels of immediate-early, delayed-early, and late KSHV lytic gene expression (duplicates ± SD). β-Actin and GAPDH were used as loading controls. (D) NIH3T12 cells were infected with MHV-68 at 0.1 MOI in the presence (black bars) or absence (white bars) of 2-DG (0.4 mM) for 24 h. qRT-PCR was performed to detect levels of MHV RTA and MHV GPCR lytic gene expression (duplicates ± SD). GAPDH and β-actin were used as loading controls. (E) mECK36 cells were treated as described for panel B except that 2-DG was used at a dose of 3 mM. Immunoblotting was performed to detect levels of K8.1 at 24 h after treatment. GAPDH was used as a loading control. (F) iSLKrK cells were treated as described for panel C except that doxycycline was used at a dose of 1 μg/ml and mannose was used at a dose of 3 mM. Immunoblotting was performed to detect levels of vIRF-1 and GRP78 at 24 h after treatment. β-Actin was used as a loading control.
2-DG inhibits host angiogenic gene expression.
According to published results from several labs, viral lytic oncogene expression leads to upregulation of host angiogenic genes, which play a critical role in autocrine and paracrine angiogenesis (1, 41). Our findings as presented above indicate that 2-DG inhibits the lytic cycle of KSHV and the expression of oncogenes such as vIRF-1 and vGPCR (Fig. 3A, B, and C), which could mediate upregulation of vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), and other mediators of the paracrine and autocrine angiogenic phenotype (17). We therefore tested whether 2-DG was able to inhibit the expression of proangiogenic genes that are upregulated during lytic induction in tumorigenic mECK36 cells and in iSLKrK. We found that 2-DG treatment downregulates paracrine and autocrine mediators that are upregulated during KSHV tumorigenesis (41, 45), including PDGFA, PDGFB, PDGF receptor β (PDGFR-β), VEGF receptor 1 (VEGF-R1), VEGF-R3, and the angiopoietin receptor Tie1 (Fig. 4A and B) (39). As KSHV-induced proangiogenic gene expression is essential for in vivo tumor formation, our results suggest that 2-DG alone or in combination could be used to prevent or treat KSHV-induced tumorigenesis.
Fig 4.
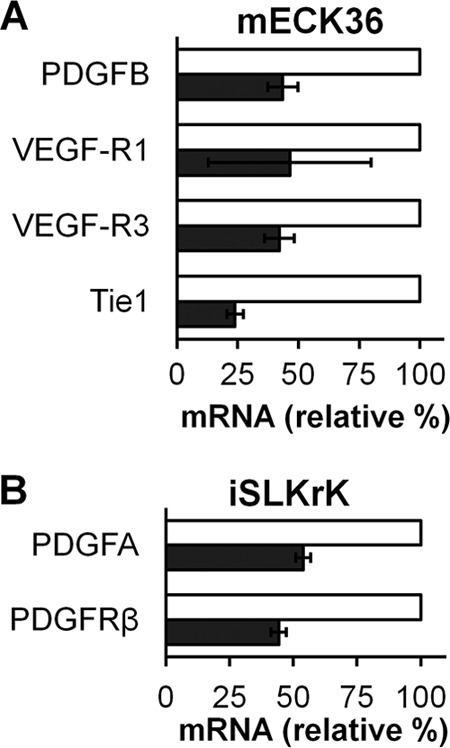
2-DG inhibits angiogenic gene expression. mECK36 (A) and iSLKrK (B) cells were lytically induced by butyrate (3 mM) (A) or by doxycycline (1 μg/ml) (B) in the presence (black bars) or absence (white bars) of 2-DG (3 mM) for 24 h. qRT-PCR was performed to detect levels of gene expression associated with angiogenesis (duplicates ± SD). β-Actin and GAPDH were used as loading controls.
2-DG inhibition correlates with induction of ER stress and a UPR leading to eIF2α inactivation.
We have previously found that inhibition of N-linked glycosylation by 2-DG in cancer cells leads to the accumulation of abortively glycosylated proteins in the ER, leading to ER stress and triggering of the UPR (33), a signaling response that includes the activation of the PERK transducer, phosphorylation of eIF2α, and attenuation of protein synthesis. This led us to hypothesize that activation of ER stress and the UPR by 2-DG would block viral replication and gene expression via PERK-mediated phosphorylation and inactivation of eIF2α. Our results described above show that 2-DG indeed mediates inhibition of KSHV replication predominantly by interfering with the N-linked glycosylation/mannose-dependent pathway. Therefore, we sought to determine if the inhibitory activities of 2-DG depicted in Fig. 1 to 4 were linked to UPR induction. As displayed in Fig. 5A, lytically induced KSHV-infected cells treated with 2-DG show a significant increased phosphorylation (P < 0.01) and thus inactivation of eIF2α at 4 h after induction of butyrate. As predicted for a UPR cascade, eIF2α phosphorylation was followed by upregulation of the UPR marker GRP78 at 8 h (Fig. 5B). In contrast, the responses to 2-FDG of both eIF2α and GRP78 were attenuated. As shown in Fig. 5C, mannose reversed both markers of the UPR. These results further support a mechanism whereby inhibition of N-linked glycosylation by 2-DG leading to ER stress is the pathway responsible for the antiviral effect. All branches of the UPR were activated by 2-DG, as displayed by the qRT-PCR results showing the upregulation of the UPR markers GRP78, XBP-1(s), and CHOP (Fig. 5D). Importantly, we found that lytic induction did not affect the levels of either phosphorylated eIF2α or GRP78 at the time points assayed (4 and 8 h, respectively), indicating KSHV viral replication per se does not activate these UPR markers in 293rKSHV at these early time points. However, 2-DG was found to activate the UPR in both uninduced, latent cells and lytically induced cells (Fig. 5E). Taken together, our results suggest that activation of the UPR by 2-DG elicits an early antiviral response via eIF2α inactivation, which impairs protein synthesis required to drive viral replication and oncogenesis.
Fig 5.

2-DG but not 2-FDG induces ER stress and a UPR in lytically infected cells. (A) 293rKSHV cells were lytically induced with butyrate (3 mM) in the presence of 2-DG (1 mM) or 2-FDG (1 mM). At the indicated times postinduction, cells were harvested and immunoblotting was performed to detect levels of phospho-eIF2α. Densitometric analysis of the immunoblots was performed to quantify phosphorylation levels of eIF2α in butyrate-induced cells in the absence (white bars) or presence (black bars) of 2-DG or 2-FDG (hashed bars). Total eIF2α was used as a control (*, P < 0.01). (B) Induced 293rKSHV cells were treated as described for panel A. At the indicated times postinduction, cells were harvested and immunoblotting was performed to detect levels of GRP78. Densitometric analysis of the immunoblots was performed to quantify GRP78 levels in butyrate-induced cells in the absence (white bars) or presence of 2-DG (black bars) or 2-FDG (hashed bars). GAPDH was used as a loading control (*, P < 0.05). (C) Induced 293rKSHV cells were treated as described for panel A in presence or absence of mannose (man; 0.5 mM). At 4 h postinduction, cells were harvested and immunoblotting was performed to detect levels of phospho-eIF2α and GRP78. Total eIF2α and β-actin were used as loading controls. (D) Infected cells (hashed bars) were induced with butyrate and treated in the absence (white bars) or presence (black bars) of 2-DG as described for panel A except that cells were harvested at 24 h. qRT-PCR was performed to detect RNA fold changes in GRP78, spliced XBP-1, and CHOP (duplicates ± SD). β-Actin and GAPDH were used as loading controls. (E) Infected cells were treated as described for panel A in the presence or absence of butyrate.
DISCUSSION
We provide evidence that induction of ER stress by 2-DG leads to activation of the UPR, which in turn shuts down gammaherpesvirus replication and lytic gene expression in multiple in vitro models. This study supports the therapeutic potential of using glucose/mannose analogs to induce the UPR in KSHV-related malignancies and block viral replication required for its pathogenesis. Our data also suggest the possibility of extending this strategy to other virus-driven cancers. Using 2-DG at clinically relevant concentrations (1 to 3 mM), which have been achieved in two phase I clinical trials (50, 55), we present data which demonstrate the following: (i) 2-DG (50% inhibitory concentration [IC50] ≈ 1 mM) significantly inhibits early viral gene expression and consequently inhibits viral replication; (ii) this inhibition is predominantly due to induction of the UPR by 2-DG, which is reversible through mannose competition and is not mimicked by the glycolytic inhibitor 2-FDG; and (iii) 2-DG-induced UPR inhibits the expression of KSHV-induced proangiogenic genes.
We show that 2-DG downregulates mRNA levels of numerous viral genes required for replication, including RTA, the master regulator of the lytic cascade (Fig. 3A, B, and C). Since expression of this immediate-early gene involves an autotransactivation loop that is essential to amplify and drive the lytic cycle (7, 13), reduction of RTA protein levels would impair the evolution of the lytic cycle. Thus, a potential explanation for our results is that 2-DG-induced eIF2α phosphorylation leads to blockage of RTA protein synthesis, impairment of the RTA transactivation loop, and inhibition of the cascade of KSHV lytic gene transcription.
Recently, 2-DG has been used as a glycolytic inhibitor to assess glycolysis dependency that arises as a consequence of the Warburg effect (i.e., increased aerobic glycolysis and lactic acid production in cancerous cells) in endothelial cells, as a result of latent infection with KSHV (12). Our study shows that, at the doses we used and in the context of KSHV lytic replication, 2-DG is acting predominantly as an inhibitor of N-linked glycosylation leading to ER stress rather than as an inhibitor of glycolysis. This is further supported by our findings that 2-FDG, a more potent inhibitor of glycolysis than 2-DG but a weaker inhibitor of N-linked glycosylation (34), had a negligible inhibitory effect on KSHV replication. Furthermore, we recently reported that 2-DG-induced ER stress (and not inhibition of glycolysis) is the predominant, mannose-reversible pathway by which autophagy is activated in tumor cells at similarly low doses (61).
The results of Fig. 5 show that, as part of the induction of ER stress and the UPR by 2-DG, there is an upregulation of XBP-1(s). However, XBP-1(s) was shown to be able to induce KSHV reactivation in the B cell cancer PEL (35, 63) but not in the cells used in our studies when treated with 2-DG alone at doses as high as 30 mM (H. J. Leung, T. J. Lampidis, and E. A. Mesri, unpublished data). The discrepancy between these results may be explained by the additional activity of XBP-1(s) as a transcription factor that is highly expressed during differentiation of plasma cells of the B lymphoid lineage (5, 27, 28) from which PEL originates, whereas XBP-1(s) function in the adherent cell types (endothelial and epithelial lineages) used in the present study appears to be limited to its UPR activity.
Our data provide evidence that induction of the UPR by 2-DG is a strategy that may be useful to inhibit viral replication in many settings. Previously, 2-DG inhibition of N-linked glycosylation was described in vitro as a means of inhibiting normal HSV-1 glycoprotein assembly and revealed abortive viral DNA-containing particles surrounded by a defective envelope (11, 30). Though the data presented here do not preclude this mechanism, they strongly imply that 2-DG acts primarily through a general repression of the viral replication cascade by inducing ER stress and the UPR, leading to global protein synthesis inhibition. In support of this interpretation, the data in Fig. 1 show that decrease in titer by 2-DG correlates with the reduced viral DNA copy number (both in supernatants and in cells), which indicates that lowered virion production, rather than defective virions, is the mechanism responsible for decreased titers and viral replication.
It has been reported that during peak viral replication, the three arms of the UPR are induced (26, 40, 58). However, viruses overcome certain aspects of the UPR which are detrimental to viral replication, in particular, the circumvention of PERK-mediated phosphorylation of eIF2α that would lead to the shutdown of protein synthesis and impede viral replication (21). Indeed, at early time points upon viral induction with butyrate, we found that UPR markers were not upregulated (Fig. 5). Our strategy was to stimulate activation of the UPR exogenously with 2-DG to block KSHV production. We found that, in KSHV-infected cells, only in the presence of 2-DG was there a sharp increase in the UPR, as reflected by its markers GRP78, XBP-1(s), CHOP, and, most importantly, phosphorylation of eIF2α. It has been reported that HSV-1 regulates the UPR through a mannose-rich viral envelope protein (gB), which directly binds to and thereby inhibits PERK activity, allowing eIF2α to function during viral replication (44). This protein contains multiple N-linked glycosylation sites and shares significant homology in a variety of herpesviruses (6). Although gB has been shown to have divergent functions such as mediating virion egress in KSHV but not in HSV-1, it is functionally conserved relating to virion binding and entry (56). Admittedly, the ability of KSHV gB to block PERK and thereby allow protein synthesis through eIF2α has not been studied. Nevertheless, the marked decrease in gB expression by 2-DG in KSHV-induced cells (as shown in Fig. 3), which correlates with increased eIF2α phosphorylation and significantly lower viral replication, suggests that this protein may have similar activity to that in HSV-1. However, further investigations are warranted to determine whether gB inhibits PERK during KSHV lytic replication.
The inhibition of KSHV replication by inducing ER stress with 2-DG is consistent with the findings for other herpesviruses, such as CMV, in which de novo infection was reduced through calcium-mediated induction of ER stress with thapsigargin, resulting in the UPR-mediated inhibition of replication (25). Interestingly, ritonavir, an HIV protease inhibitor that was reported to prevent AIDS-KS and display anti-KS effects in some animal models (52), has been also shown to induce ER stress and the UPR in other sarcoma cells (32). Thus, this supports the possibility presented here in which 2-DG could be a valid approach for preventing and/or treating KS.
Since KS lesions are characterized by intense angiogenesis and are comprised of cells of endothelial origin, they might be particularly sensitive to the recently identified antiangiogenic effects of 2-DG. In those studies, it was found that the antiangiogenic activity of 2-DG is due to its ability to affect endothelial cell proliferation and microtube formation via interference with the N-linked glycosylation, leading to induction of the ER stress/UPR pathway (39). In addition to the above-described antiangiogenic effects on endothelial cells, we present evidence showing that 2-DG inhibits the expression of several angiogenic genes, which are upregulated during KSHV-mediated tumorigenesis, that are well known to significantly contribute to KS growth and pathogenesis (45). Taken together, our data suggest that 2-DG-mediated inhibition of the KSHV lytic cycle with concomitant inhibition of host angiogenic genes and endothelial cell proliferation could have significant anti-KS effects in vivo. In concurrence with our previous findings that 2-DG inhibition of N-linked glycosylation leading to the UPR could be exploited as an anticancer approach (33), we and others are investigating the use of this glucose analog both as a single agent and in combination in in vitro and in vivo models of human cancer, such as cultured prostate (3), melanoma (36), non-small cell lung cancer (24, 38), osteosarcoma (38), and retinoblastoma (4, 49). The following are among the promising combinations with 2-DG: MBT to inhibit the antiapoptotic activity of Bcl-2, which blocks UPR-induced apoptosis (62), inhibitors of autophagy (61), and rapamycin, which blocks hypoxia-inducible factors (37, 60). Importantly, 2-DG is a relatively nontoxic drug that has displayed minimal adverse effects (subclinical hypoglycemia) in human subjects (50). It has shown both safety and efficacy as an adjuvant drug to cisplatin in a phase I trial for lung cancer (50) and a single agent in a phase I/II trial for prostate cancer (55). Although patent-related issues have somewhat slowed its clinical development, 2-DG is attracting increased interest for its potential anticancer activity. Our results here add to that interest by expanding its potential clinical application as a novel antiviral approach.
As shown in Fig. 1, 2, and 5, doses of 2-DG lead to UPR activation without toxicity in the KSHV-infected cells, as well as in most cancer cells tested in vitro (33). Moreover, at doses much higher than those used here or those required for FDA approval for 2-DG to be used in clinical trials, no detectable toxicity was observed in normal tissues in a variety of animals tested (50). In contrast, at the low and clinically achievable doses used here, 2-DG activation of the UPR has significant anti-KSHV activity. Additionally, most human cancers and KSHV-infected cells display increased uptake of glucose (12), which would further increase 2-DG's selectivity as an anticancer and antiviral agent.
In summary, the in vitro data we present, showing significant inhibitory effects of 2-DG on KSHV replication, gene expression, and pathogenicity, in addition to its virtues as a potent anticancer drug, warrant further examination of this sugar analog in preclinical models of KS, with the eventual goal of clinical implementation. Our data also open the possibility of extending the strategy of 2-DG-induced ER stress to inhibit replication of herpesviruses and potentially other viruses. Furthermore, our approach appears particularly suitable for viral cancers other than KS, such as Epstein-Barr virus (EBV)-induced lymphoma and HCV-induced hepatocellular carcinoma, in which both the combined anticancer and antiviral activities of 2-DG may increase therapeutic efficacy.
ACKNOWLEDGMENTS
We thank Jeffery Vieira (University of Washington, Seattle, Washington) for providing us with recombinant KSHV (rKSHV.219), Gary Hayward (Johns Hopkins University School of Medicine, Baltimore, Maryland) for the anti-vIRF-1 antibody, Lucas Cavallin for the qRT-PCR primers and helpful discussions, Huaping Liu for primary antibodies, George McNamara (Diabetes Research Institute Imaging Core), Jim Phillips (Flow Cytometry Core), the Sheila and David Fuente Graduate Program in Cancer Biology, Sylvester Comprehensive Cancer Center, for support and assistance, and Brittany Ashlock and Qi Ma of the Mesri Lab for invaluable discussions.
This work was supported by Public Health Service grants CA-136387 and CA-037109 from the National Cancer Institute, National Institutes of Health, to E.A.M. and T.J.L., respectively, and an SCCC Developmental Award funded by the Pap Corps (T.J.L.).
We declare no conflicts of interest.
Footnotes
Published ahead of print 27 August 2012
REFERENCES
- 1. Bais C, et al. 1998. G-protein-coupled receptor of Kaposi's sarcoma-associated herpesvirus is a viral oncogene and angiogenesis activator. Nature 391:86–89 [DOI] [PubMed] [Google Scholar]
- 2. Bais C, et al. 2003. Kaposi's sarcoma associated herpesvirus G protein-coupled receptor immortalizes human endothelial cells by activation of the VEGF receptor-2/KDR. Cancer Cell 3:131–143 [DOI] [PubMed] [Google Scholar]
- 3. Ben Sahra I, et al. 2010. Targeting cancer cell metabolism: the combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res. 70:2465–2475 [DOI] [PubMed] [Google Scholar]
- 4. Boutrid H, et al. 2009. Increased hypoxia following vessel targeting in a murine model of retinoblastoma. Invest. Ophthalmol. Vis. Sci. 50:5537–5543 [DOI] [PubMed] [Google Scholar]
- 5. Carbone A, Cesarman E, Spina M, Gloghini A, Schulz TF. 2009. HIV-associated lymphomas and gamma-herpesviruses. Blood 113:1213–1224 [DOI] [PubMed] [Google Scholar]
- 6. Chandran B. 2010. Early events in Kaposi's sarcoma-associated herpesvirus infection of target cells. J. Virol. 84:2188–2199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chang PJ, Miller G. 2004. Autoregulation of DNA binding and protein stability of Kaposi's sarcoma-associated herpesvirus ORF50 protein. J. Virol. 78:10657–10673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chang Y, et al. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869 [DOI] [PubMed] [Google Scholar]
- 9. Cheng G, Feng Z, He B. 2005. Herpes simplex virus 1 infection activates the endoplasmic reticulum resident kinase PERK and mediates eIF-2alpha dephosphorylation by the gamma(1)34.5 protein. J. Virol. 79:1379–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Child SJ, Hakki M, De Niro KL, Geballe AP. 2004. Evasion of cellular antiviral responses by human cytomegalovirus TRS1 and IRS1. J. Virol. 78:197–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Courtney RJ, Steiner SM, Benyesh-Melnick M. 1973. Effects of 2-deoxy-d-glucose on herpes simplex virus replication. Virology 52:447–455 [DOI] [PubMed] [Google Scholar]
- 12. Delgado T, et al. 2010. Induction of the Warburg effect by Kaposi's sarcoma herpesvirus is required for the maintenance of latently infected endothelial cells. Proc. Natl. Acad. Sci. U. S. A. 107:10696–10701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Deng H, Young A, Sun R. 2000. Auto-activation of the rta gene of human herpesvirus-8/Kaposi's sarcoma-associated herpesvirus. J. Gen. Virol. 81:3043–3048 [DOI] [PubMed] [Google Scholar]
- 14. Dittmer DP, Krown SE. 2007. Targeted therapy for Kaposi's sarcoma and Kaposi's sarcoma-associated herpesvirus. Curr. Opin. Oncol. 19:452–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ganem D. 2006. KSHV infection and the pathogenesis of Kaposi's sarcoma. Annu. Rev. Pathol. 1:273–296 [DOI] [PubMed] [Google Scholar]
- 16. Ganem D. 2010. KSHV and the pathogenesis of Kaposi sarcoma: listening to human biology and medicine. J. Clin. Invest. 120:939–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gao SJ, et al. 1997. KSHV ORF K9 (vIRF) is an oncogene which inhibits the interferon signaling pathway. Oncogene 15:1979–1985 [DOI] [PubMed] [Google Scholar]
- 18. Grulich AE, van Leeuwen MT, Falster MO, Vajdic CM. 2007. Incidence of cancers in people with HIV/AIDS compared with immunosuppressed transplant recipients: a meta-analysis. Lancet 370:59–67 [DOI] [PubMed] [Google Scholar]
- 19. Grundhoff A, Ganem D. 2004. Inefficient establishment of KSHV latency suggests an additional role for continued lytic replication in Kaposi sarcoma pathogenesis. J. Clin. Invest. 113:124–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Harding HP, et al. 2000. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6:1099–1108 [DOI] [PubMed] [Google Scholar]
- 21. He B. 2006. Viruses, endoplasmic reticulum stress, and interferon responses. Cell Death Differ. 13:393–403 [DOI] [PubMed] [Google Scholar]
- 22. He B, Gross M, Roizman B. 1997. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. U. S. A. 94:843–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Herndier BG, et al. 1994. Characterization of a human Kaposi's sarcoma cell line that induces angiogenic tumors in animals. AIDS 8:575–581 [DOI] [PubMed] [Google Scholar]
- 24. Inge LJ, Coon KD, Smith MA, Bremner RM. 2009. Expression of LKB1 tumor suppressor in non-small cell lung cancer determines sensitivity to 2-deoxyglucose. J. Thorac. Cardiovasc. Surg. 137:580–586 [DOI] [PubMed] [Google Scholar]
- 25. Isler JA, Maguire TG, Alwine JC. 2005. Production of infectious human cytomegalovirus virions is inhibited by drugs that disrupt calcium homeostasis in the endoplasmic reticulum. J. Virol. 79:15388–15397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Isler JA, Skalet AH, Alwine JC. 2005. Human cytomegalovirus infection activates and regulates the unfolded protein response. J. Virol. 79:6890–6899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Iwakoshi NN, et al. 2003. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat. Immunol. 4:321–329 [DOI] [PubMed] [Google Scholar]
- 28. Jenner RG, et al. 2003. Kaposi's sarcoma-associated herpesvirus-infected primary effusion lymphoma has a plasma cell gene expression profile. Proc. Natl. Acad. Sci. U. S. A. 18:10399–10404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kaufman RJ. 1999. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 13:1211–1233 [DOI] [PubMed] [Google Scholar]
- 30. Klenk HD, Schwarz RT. 1982. Viral glycoprotein metabolism as a target for antiviral substances. Antiviral Res. 2:177–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kozutsumi Y, Segal M, Normington K, Gething MJ, Sambrook J. 1988. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 332:462–464 [DOI] [PubMed] [Google Scholar]
- 32. Kraus M, et al. 2008. Ritonavir induces endoplasmic reticulum stress and sensitizes sarcoma cells toward bortezomib-induced apoptosis. Mol. Cancer Ther. 7:1940–1948 [DOI] [PubMed] [Google Scholar]
- 33. Kurtoglu M, et al. 2007. Under normoxia, 2-deoxy-d-glucose elicits cell death in select tumor types not by inhibition of glycolysis but by interfering with N-linked glycosylation. Mol. Cancer Ther. 6:3049–3058 [DOI] [PubMed] [Google Scholar]
- 34. Kurtoglu M, Maher JC, Lampidis TJ. 2007. Differential toxic mechanisms of 2-deoxy-d-glucose versus 2-fluorodeoxy-d-glucose in hypoxic and normoxic tumor cells. Antioxid. Redox Signal. 9:1383–1390 [DOI] [PubMed] [Google Scholar]
- 35. Lai IY, Farrell PJ, Kellam P. 2011. X-box binding protein 1 induces the expression of the lytic cycle transactivator of Kaposi's sarcoma-associated herpesvirus but not Epstein-Barr virus in co-infected primary effusion lymphoma. J. Gen. Virol. 92:421–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu H, et al. 2009. 2-Deoxy-d-glucose enhances TRAIL-induced apoptosis in human melanoma cells through XBP-1-mediated up-regulation of TRAIL-R2. Mol. Cancer 8:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Maher JC, Wangpaichitr M, Savaraj N, Kurtoglu M, Lampidis TJ. 2007. Hypoxia-inducible factor-1 confers resistance to the glycolytic inhibitor 2-deoxy-d-glucose. Mol. Cancer Ther. 6:732–741 [DOI] [PubMed] [Google Scholar]
- 38. Maschek G, et al. 2004. 2-deoxy-d-glucose increases the efficacy of Adriamycin and paclitaxel in human osteosarcoma and non-small cell lung cancers in vivo. Cancer Res. 64:31–34 [DOI] [PubMed] [Google Scholar]
- 39. Merchan JR, et al. 2010. Antiangiogenic activity of 2-deoxy-d-glucose. PLoS One 5:e13699 doi:10.1371/journal.pone.0013699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Merquiol E, et al. 2011. HCV causes chronic endoplasmic reticulum stress leading to adaptation and interference with the unfolded protein response. PLoS One 6:e24660 doi:10.1371/journal.pone.0024660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mesri EA, Cesarman E, Boshoff C. 2010. Kaposi's sarcoma and its associated herpesvirus. Nat. Rev. Cancer 10:707–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mocroft A, et al. 1996. Anti-herpesvirus treatment and risk of Kaposi's sarcoma in HIV infection. AIDS 10:1101–1105 [PubMed] [Google Scholar]
- 43. Montaner S, et al. 2006. The Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor as a therapeutic target for the treatment of Kaposi's sarcoma. Cancer Res. 66:168–174 [DOI] [PubMed] [Google Scholar]
- 44. Mulvey M, Arias C, Mohr I. 2007. Maintenance of endoplasmic reticulum (ER) homeostasis in herpes simplex virus type 1-infected cells through the association of a viral glycoprotein with PERK, a cellular ER stress sensor. J. Virol. 81:3377–3390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mutlu AD, et al. 2007. In vivo-restricted and reversible malignancy induced by human herpesvirus-8 KSHV: a cell and animal model of virally induced Kaposi's sarcoma. Cancer Cell 11:245–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Myoung J, Ganem D. 2011. Generation of a doxycycline-inducible KSHV producer cell line of endothelial origin: maintenance of tight latency with efficient reactivation upon induction. J. Virol. Methods 174:12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nash AA, Dutia BM, Stewart JP, Davison AJ. 2001. Natural history of murine gamma-herpesvirus infection. Philos. Trans. R. Soc. Lond. B Biol. Sci. 356:569–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pasqual G, Burri DJ, Pasquato A, de la Torre JC, Kunz S. 2011. Role of the host cell's unfolded protein response in arenavirus infection. J. Virol. 85:1662–1670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Piña Y, et al. 2011. Advanced retinoblastoma treatment: targeting hypoxia by inhibition of the mammalian target of rapamycin (mTOR) in LH(BETA)T(AG) retinal tumors. Clin. Ophthalmol. 5:337–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Raez LE, et al. 2007. Responses to the combination of the glycolytic inhibitor 2-deoxy-glucose (2DG) and docetaxel (DC) in patients with lung and head and neck (H/N) carcinomas. J. Clin. Oncol. 25:14025 [Google Scholar]
- 51. Rutkowski DT, Kaufman RJ. 2004. A trip to the ER: coping with stress. Trends Cell Biol. 14:20–28 [DOI] [PubMed] [Google Scholar]
- 52. Sgadari C, Monini P, Barillari G, Ensoli B. 2003. Use of HIV protease inhibitors to block Kaposi's sarcoma and tumour growth. Lancet Oncol. 4:537–547 [DOI] [PubMed] [Google Scholar]
- 53. Sodhi A, et al. 2006. The TSC2/mTOR pathway drives endothelial cell transformation induced by the Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor. Cancer Cell 10:133–143 [DOI] [PubMed] [Google Scholar]
- 54. Song MJ, Deng H, Sun R. 2003. Comparative study of regulation of RTA-responsive genes in Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8. J. Virol. 77:9451–9462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Stein M, et al. 2010. Targeting tumor metabolism with 2-deoxyglucose in patients with castrate-resistant prostate cancer and advanced malignancies. Prostate 70:1388–1394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Subramanian R, Sehgal I, D'Auvergne O, Kousoulas KG. 2010. Kaposi's sarcoma-associated herpesvirus glycoproteins B and K8.1 regulate virion egress and synthesis of vascular endothelial growth factor and viral interleukin-6 in BCBL-1 cells. J. Virol. 84:1704–1714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sun R, et al. 1999. Kinetics of Kaposi's sarcoma-associated herpesvirus gene expression. J. Virol. 73:2232–2242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tirosh B, et al. 2005. Human cytomegalovirus protein US11 provokes an unfolded protein response that may facilitate the degradation of class I major histocompatibility complex products. J. Virol. 79:2768–2779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Vieira J, O'Hearn PM. 2004. Use of the red fluorescent protein as a marker of Kaposi's sarcoma-associated herpesvirus lytic gene expression. Virology 325:225–240 [DOI] [PubMed] [Google Scholar]
- 60. Wangpaichitr M, Savaraj N, Maher J, Kurtoglu M, Lampidis TJ. 2008. Intrinsically lower AKT, mammalian target of rapamycin, and hypoxia-inducible factor activity correlates with increased sensitivity to 2-deoxy-d-glucose under hypoxia in lung cancer cell lines. Mol. Cancer Ther. 7:1506–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Xi H, et al. 2011. 2-Deoxy-d-glucose activates autophagy via endoplasmic reticulum stress rather than ATP depletion. Cancer Chemother. Pharmacol. 67:899–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yamaguchi R, Perkins G. 2012. Finding a panacea among combination cancer therapies. Cancer Res. 72:18–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yu F, et al. 2007. B cell terminal differentiation factor XBP-1 induces reactivation of Kaposi's sarcoma-associated herpesvirus. FEBS Lett. 581:3485–3488 [DOI] [PubMed] [Google Scholar]
- 64. zur Hausen H. 1999. Viruses in human cancers. Eur. J. Cancer. 35:1174–1181 [DOI] [PubMed] [Google Scholar]