

Abstract
The infant gut microbiota undergoes dramatic changes during the first 2 years of life. The acquisition and development of this population can be influenced by numerous factors, and antibiotic treatment has been suggested as one of the most significant. Despite this, however, there have been relatively few studies which have investigated the short-term recovery of the infant gut microbiota following antibiotic treatment. The aim of this study was to use high-throughput sequencing (employing both 16S rRNA and rpoB-specific primers) and quantitative PCR to compare the gut microbiota of nine infants who underwent parenteral antibiotic treatment with ampicillin and gentamicin (within 48 h of birth), 4 and 8 weeks after the conclusion of treatment, relative to that of nine matched healthy controls. The investigation revealed that the gut microbiota of the antibiotic-treated infants had significantly higher proportions of Proteobacteria (P = 0.0049) and significantly lower proportions of Actinobacteria (P = 0.00001) (and the associated genus Bifidobacterium [P = 0.0132]) as well as the genus Lactobacillus (P = 0.0182) than the untreated controls 4 weeks after the cessation of treatment. By week 8, the Proteobacteria levels remained significantly higher in the treated infants (P = 0.0049), but the Actinobacteria, Bifidobacterium, and Lactobacillus levels had recovered and were similar to those in the control samples. Despite this recovery of total Bifidobacterium numbers, rpoB-targeted pyrosequencing revealed that the number of different Bifidobacterium species present in the antibiotic-treated infants was reduced. It is thus apparent that the combined use of ampicillin and gentamicin in early life can have significant effects on the evolution of the infant gut microbiota, the long-term health implications of which remain unknown.
INTRODUCTION
It is becoming increasingly evident that the composition of the human gut microbiota can have a significant impact on health and disease (47, 64, 69, 74). Indeed, several studies have highlighted the role gut microbes play in diverse and important functions in the body, including, for example, vitamin synthesis, immune system development, and toxin metabolism (27, 30). Furthermore, a number of studies have suggested associations between an altered gut microbial composition and Crohn's disease (28), irritable bowel syndrome (42), obesity (46, 51), and other diseases and syndromes. These studies have highlighted the importance of developing and maintaining a “healthy” gut microbiota. Indeed, it was recently established that the fact that the immune system of germfree mice is not exposed to commensal microbes in early life can lead to increased numbers of invariant natural killer T cells, which in turn causes inflammation on exposure to particular microbes, resulting in an increased risk of both colitis and asthma (53). The infant gut microbiota is established early in life, so that although the infant gut is sterile in utero, by the time the infant reaches the age of 2 years, this microbiota resembles that of an adult (3). Consequently, this period of the infant's life represents a unique window of opportunity during which time the gut microbiota may be modified with implications for health outcomes (56). A myriad of factors that affect this composition have been investigated and include mode of delivery (56), feeding choice (i.e., breast versus formula feeding) (8, 45), prematurity (63, 72), and the administration of probiotics (11, 15, 55, 60) and prebiotics (10, 31, 65). It is also thought that exposure to antibiotics can have a significant negative influence on the composition and development of the gut microbiota in early life (7, 23, 35). Antibiotics by their very nature are designed to target and inhibit microorganisms in a variety of ways. The majority of those used clinically have a broad spectrum of activity and, as a consequence, in addition to controlling pathogenic bacteria have the potential to inflict collateral damage on commensal gut bacteria (9), including genera that often have health-promoting roles, such as the bifidobacteria and lactobacilli. Thus far, the most in-depth investigations into the nature and extent of this collateral damage have relied on the use of denaturing gradient gel electrophoresis (DGGE) (23) and have revealed that antibiotic exposure in infancy results in significant decreases in levels of bifidobacteria, lactobacilli, and Bacteroides spp. compared to levels in control infants.
More recently, the impact of antibiotic administration on the gut microbiota has been revealed in even greater depth as a consequence of the use of high-throughput sequencing technologies in both animal and human trials (6, 17, 18, 51, 58, 66). These studies have shown that antibiotics can dramatically alter the gut microbiota, with the effects depending on factors such as the specific antibiotic administered, the spectrum of inhibition, and the duration of treatment (67). While antibiotic administration in adults can have a number of gut microbiota-mediated consequences, such as an increased susceptibility to Clostridium difficile-associated diarrhea (14), there is also evidence to suggest that perturbation of the infant gut microbiota during its rapid developmental phase can have even more significant consequences. For example, an association between antibiotic administration in early life and an increased risk of asthma and allergies, such as atopic eczema, in later life has been noted previously (1, 44, 59). Thus, developing a detailed understanding of the impact of specific antibiotics on the infant gut microbiota is vital in order to begin to understand the mechanism(s) by which these changes increase the risk of disease. It is thus notable that the impact of antibiotics on the composition of the infant gut microbiota has yet to be assessed through high-throughput sequencing technologies. Here, we addressed this issue by using 454 pyrosequencing technology together with quantitative PCR (qPCR) to reveal the short-term (4 to 8 weeks) consequences of the treatment of infants with a combination of ampicillin and gentamicin within the first 48 h of birth.
MATERIALS AND METHODS
Participants.
Approval for this trial was received from the Clinical Research Ethics Committee of the Cork Teaching Hospitals, Cork, Ireland. Details on inclusion criteria, sample collection, and storage have been outlined previously (35). Briefly, 18 infants were recruited, 9 of whom had received parenteral antibiotic treatment with a combination of ampicillin and gentamicin within 48 h of birth and 9 of whom were untreated controls. Infants were excluded if they were born prematurely, required oral antibiotics, were under a “nil by mouth” restriction, required surgery, or had congenital abnormalities. Fecal samples were collected 4 and 8 weeks after the cessation of antibiotic treatment. Of the 18 infants, 8 had been breastfed and 10 formula fed, while 13 were born vaginally and 5 were born by Caesarean section (Table 1).
Table 1.
Details of the infants in the triala
| Infant | Sexb | Mode of delivery | Feeding method | Duration of antibiotic treatment (days) |
|---|---|---|---|---|
| A | M | Caesarean section | Breastfed | 9 |
| B | M | Caesarean section | Breastfed and formula fed | 5 |
| C | M | Caesarean section | Breastfed | 2 |
| D | M | Vaginal delivery | Formula fed | 2 |
| E | F | Caesarean section | Formula fed | 5 |
| F | F | Vaginal delivery | Breastfed | 2 |
| G | F | Vaginal delivery | Breastfed | 2 |
| H | M | Caesarean section | Formula fed | 2 |
| I | M | Vaginal delivery | Formula fed | 2 |
| J | M | Vaginal delivery | Formula fed | |
| K | M | Vaginal delivery | Formula fed | |
| L | F | Vaginal delivery | Formula fed | |
| M | F | Vaginal delivery | Formula fed | |
| N | M | Vaginal delivery | Breastfed | |
| O | F | Vaginal delivery | Breastfed | |
| P | M | Vaginal delivery | Breastfed | |
| Q | M | Vaginal delivery | Formula fed | |
| R | F | Vaginal delivery | Formula fed |
The table was adapted from reference 35.
M, male; F, female.
Generation of 16S rRNA amplicons for high-throughput sequencing.
16S rRNA amplicons were generated as described previously (51). Total bacterial DNA was extracted from fecal samples using the QIAamp DNA stool minikit (Qiagen, West Sussex, United Kingdom) (35). DNA was frozen at −80°C prior to PCR amplification. 16S rRNA bacterial gene amplicons (V4) were generated with a view to high-throughput sequencing using the Roche genome sequencer FLX platform. These amplicons, 239 nucleotides in length, were generated using one forward primer, i.e., F1 (5′ AYTGGGYDTAAAGNG), and a combination of 4 reverse primers, R1 (5′ TACCRGGGTHTCTAATCC), R2 (5′ TACCAGAGTATCTAATTC), R3 (5′ CTACDSRGGTMTCTAATC), and R4 (5′ TACNVGGGTATCTAATCC). These primers also contained an A (F primer) or B (R primers) adapter, and different versions of the F primer (each containing a distinct multiple identifier [MID]) were employed for each sample. PCRs were completed on a G-Storm PCR machine under the following conditions: heated lid at 110°C, 94°C for 2 min followed by 35 cycles of 94°C for 1 min, 52°C for 1 min, and 72°C for 1 min and then by a temperature step of 72°C for 2 min, and holding at 4°C. PCR mixtures had a final volume of 50 μl made up of 25 μl of Biomix red (MyBio, Ireland), 1 μl forward primer (0.15 μM), 1 μl reverse primer (0.15 μM) (mix of 4), template DNA, and sterile PCR-grade water. All samples were assayed in duplicate. PCR products were analyzed by agarose gel electrophoresis (1.5% in 1× TAE [Tris-acetate-EDTA] buffer). Following this, PCR products were cleaned using an Agentcourt AMPure kit (Beckman Coulter Genomics, United Kingdom) as per the manufacturer's protocol. Samples were then quantified using the Quant-iT Picogreen quantification kit (Biosciences, Ireland) and the Nanodrop 3300 fluorospectrometer (Thermo Scientific, Ireland). Equimolar solutions of samples were then pooled for sequencing. These pooled samples were then cleaned and requantified (as before). Emulsion-based clonal amplification was completed as part of the 454 pyrosequencing process. Sequencing took place at the Teagasc 454 sequencing facility on a genome sequencer FLX platform (Roche Diagnostics Ltd., West Sussex, United Kingdom) according to the manufacturer's protocols.
Generation of Bifidobacterium-derived rpoB amplicons for high-throughput sequencing.
A set of PCR primers, which were used previously to facilitate the identification of bifidobacteria (43) and which amplify a 351-bp region from the Bifidobacterium RNA polymerase β-subunit gene, rpoB, were also utilized in this study. Twelve samples from week 4 and 12 from week 8 were selected and amplified using these primers, which had MID tags and 454 adapters attached, allowing pooling of the samples for sequencing while also enabling downstream separation of individual results for analysis (see Table S1 in the supplemental material). PCRs were completed on a G-Storm PCR machine under the following conditions: heated lid at 110°C, 94°C for 2 min followed by 35 cycles of 94°C for 1 min, 60°C for 1 min and 72°C for 1 min and then by a temperature step of 72°C for 2 min, and holding at 4°C. PCR mixtures had a final volume of 50 μl containing 25 μl of Biomix red (MyBio, Ireland), 1 μl forward primer (0.15 μM) (BC1 [5′-TCGATCGGGCACATACGG]), 1 μl reverse primer (0.15 μM) (Rev 1 [5′-CGACCACTTCGGCAACCG]), template DNA, and sterile PCR-grade water. All samples were completed in duplicate. All other steps for sequencing (cleaning, quantifying, pooling, etc.) were completed as outlined above.
Bioinformatic analysis.
Raw 16S rRNA sequencing reads were quality trimmed using a locally installed version of the Ribosomal Database Project (RDP) pyrosequencing pipeline applying the criteria as previously described (54). Trimmed FASTA sequences were then subjected to BLAST searches (5) against a previously published 16S-specific database (70) using default parameters. The BLAST output was then parsed using MEGAN software (version 4.6) (34), which assigns reads to NCBI taxonomies by employing the Lowest Common Ancestor algorithm. Bit scores from within MEGAN were used to filter the results prior to tree construction and summarization. A bit score of 86 was selected, as previously used for 16S ribosomal sequence data (70). Phylum, family, and genus counts for each subject were extracted from MEGAN. Clustering and diversity analysis of the sequence data was performed using the MOTHUR software package (61, 62). For Bifidobacterium analysis, raw rpoB sequencing reads were quality trimmed as described above, with read lengths for the rpoB amplicon above 300 bp being used. Trimmed FASTA sequences were then subjected to BLAST searches (5) against the NCBI nonredundant database using default parameters. The resulting BLAST output was parsed through MEGAN using default parameters (34).
qPCR-based determination of total bacterial and total bifidobacterial numbers.
Absolute quantification of total bacterial numbers (from eight representative infants, infants B, F, G, H, and K to N) and total bifidobacterial numbers (from nine representative infants, infants B, F, G, H, and K to O) was carried out by qPCR using the Roche 480 Lightcycler platform. To determine total bifidobacterial counts, the primers g-Bifid-F (5′-CTCCTGGAAACGGGTGG) and g-Bifid-R (5′-GGTGTTCTTCCCGATATCTACA) were used (49). Bifidobacterium longum ATCC 8809 was used as a reference strain to generate a standard curve for total bifidobacterial quantification (21). B. longum was grown overnight anaerobically at 37°C in modified MRS broth (Difco) (0.05% cysteine) (Sigma-Aldrich). Total bacterial DNA was then isolated using a High Pure PCR template preparation kit (Roche Diagnostics, West Sussex, United Kingdom) as per the manufacturer's instructions and used to establish a standard curve on the Lightcycler 480 platform (Roche Diagnostics, West Sussex, United Kingdom). Total bifidobacterial numbers were quantified using the following program: 95°C for 5 min followed by 50 cycles of 95°C for 10 s, 60°C for 20 s, and 72°C for 20 s, next by melting curve analysis of 95°C for 5 s, 65°C for 1 min and 97°C continuously, and then by cooling at 40°C for 10 s. Reactions took place in a 20-μl volume made up of 3 μl PCR-grade water, 1 μl g-Bifid-F (0.15 μM), 1 μl g-Bifid-R (0.15 μM), 5 μl DNA template, and 10 μl SYBR green (Roche Diagnostics, West Sussex, United Kingdom). To quantify total 16S rRNA bacterial counts, a standard curve was established using 109 to 102 copies 16S rRNA/μl. Values were then converted to copies 16S rRNA/g wet stool using a previously outlined calculation (73). The following program was used to quantify total bacterial numbers: 95°C for 5 min followed by 35 cycles of 95°C for 20 s, 51°C for 20s and 72°C for 20 s followed by melting curve analysis of 95°C for 5 s, 46°C for 1 min, and 97°C continuously and a final cooling at 40°C for 10 s. Samples contained 2 μl of PCR grade water, 1 μl of the forward primer F1 (5′-AYTGGGYDTAAAGNG) (0.15 μM), 1 μl of the reverse primer R1 (5′-TACCRGGGTHTCTAATCC) (0.15 μM), 1 μl template DNA, and 5 μl of SYBR green (Roche Diagnostics, West Sussex United Kingdom), giving a final reaction volume of 10 μl. Samples were run in quadruplicate, while negative controls (where template DNA was replaced with PCR-grade water) and standards were run in triplicate.
Statistical analysis.
Minitab release 15.1.1.0 (Minitab Inc.) was used to perform nonparametric statistical analysis (Mann-Whitney test) to compare two specific subject groups to determine the impact of antibiotic treatment on the microbiota. Statistical significance was defined as a P value of <0.05.
RESULTS
High-throughput sequencing of 16S rRNA amplicons from the fecal samples of antibiotic-treated and control infants.
Eighteen infants, 9 of whom had been treated with a combination of parenteral ampicillin and gentamicin within 48 h of birth and 9 of whom were controls who had received no antibiotic treatment, were recruited (35). Fecal samples were collected 4 and 8 weeks after the cessation of antibiotic treatment, and fecal DNA was extracted and used as a template to generate 16S rRNA amplicons, with a view to determining the composition of the gut microbiota through next-generation sequencing. Diversity, richness, coverage, and evenness estimations were calculated for all data sets (Table 2). The Chao 1 calculation is an estimator of phylotype richness in a data set, and the Shannon index of diversity reflects both the richness and the community evenness (i.e., proportional phylotype abundance). The diversity index was above 3.6 in all samples, indicating an overall high level of biodiversity (Table 2). Good's coverage, a measure of sampling completeness, at the 97% similarity level ranged between 88.6 and 96.1% for the data sets. The lowest value was obtained for the control samples at week 8 and is a reflection of the more diverse nature of the microbiota present (Table 2).
Table 2.
Estimation of diversity within the treated and control groups at week 4 and week 8
| Infant group | Similarity (%) | Chao 1 richness estimation | Shannon's index for diversity | Good's coverage |
|---|---|---|---|---|
| Week 4 | ||||
| Treated | 97 | 243 | 3.6 | 96.1 |
| Control | 97 | 364 | 3.8 | 94.3 |
| Week 8 | ||||
| Treated | 97 | 334 | 3.8 | 93.2 |
| Control | 97 | 490 | 4.6 | 88.6 |
Composition of the gut microbiota of antibiotic-treated and control infants 4 weeks after the conclusion of treatment.
Bioinformatic analysis of 16S rRNA sequence data revealed that there were significant differences in the gut microbiota of antibiotic-treated infants and that of untreated controls, 4 weeks following the cessation of antibiotic treatment. Statistically higher proportions of reads corresponding to the phylum Proteobacteria were detected in the samples from antibiotic-treated infants than in the control samples (P = 0.0049) (Fig. 1). Indeed, the gut microbiota of the antibiotic-treated infants was dominated by Proteobacteria, accounting for 54% of all bacteria present, compared to just 37% in the untreated controls (Fig. 1). While Proteobacteria, Firmicutes, and Actinobacteria were found in all antibiotic-treated infants at week 4, Bacteroidetes were detected in less than half of these infants, and in those where they were detected, levels were notably low (Fig. 1; also, see Fig. S1 in the supplemental material). Actinobacteria levels were also significantly lower in the antibiotic-treated infants than in the controls (3% versus 24%; P = 0.00001).
Fig 1.
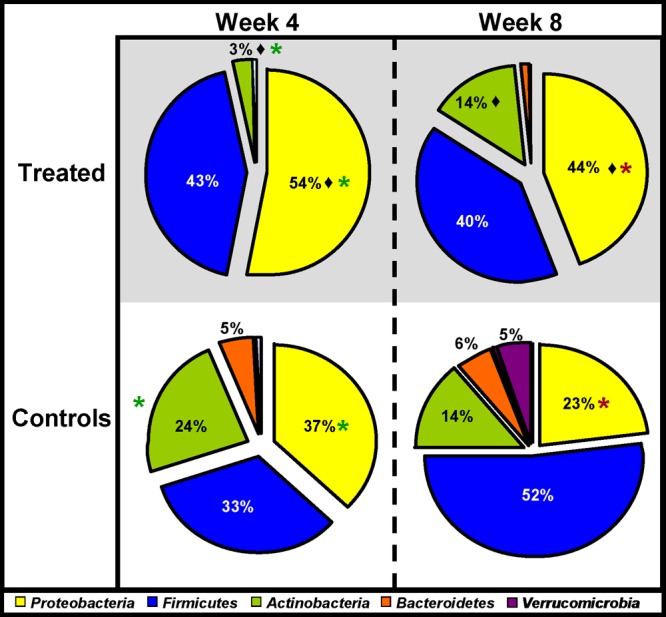
Microbial distributions at the phylum level in the samples from treated and control infants at week 4 and week 8. Statistically significant differences between treated infants and controls at week 4 are indicated by asterisks (P < 0.05). Statistically significant differences between treated infants and controls at week 8 are indicated by asterisks. A statistically significant difference between treated infants at week 4 and week 8 (i.e., the recovery of the treated infants) is indicated by a diamond. Percentages are based on proportions of assignable tags.
At the family level, the antibiotic-treated infants had significantly higher numbers of Enterobacteriaceae (55% versus 37%; P = 0.0073) and Peptostreptococcaceae (23% versus 2%; P = 0.0381) than the control infants at week 4 (Fig. 2). Significantly lower numbers of Bifidobacteriaceae (3% versus 24%; P = 0.0132) were also evident in the antibiotic-treated infants at week 4. In addition, antibiotic treatment resulted in significant differences at the genus level relative to the controls at this time (Fig. 3). Significantly higher levels of Bifidobacterium (25% versus 5%; P = 0.0132) and Lactobacillus (4% versus 1%; P = 0.0088) were present in the untreated controls than in the antibiotic-treated infants. Additionally, the gut microbiota of the antibiotic-treated infants displayed limited diversity, as they were dominated by genera within the family Enterobacteriaceae, with levels of these bacteria being statistically significantly higher in the antibiotic-treated infants than in the controls (P = 0.0073). This pattern was also apparent with respect to proportions of the Firmicutes-associated genus Clostridium (P = 0.0033). Additionally, there was a significantly higher level of enterococci in the treated infants than in the controls at week 4 (P = 0.0172). Despite the fact that the diversity in samples from antibiotic-treated and control infants did not differ significantly (P = 0.5752) (Table 2), the overall numbers of genera detected in the samples from antibiotic-treated infants were notably lower than in the controls, reflecting the restriction in diversity, the dominance of members of the Proteobacteria, and the persistent effects of antibiotic treatment 4 weeks after administration ceased.
Fig 2.

Microbial distributions at the family level in the samples from treated and control infants at week 4 and week 8. Statistically significant differences between treated infants and controls at week 4 are indicated by asterisks (P < 0.05). Statistically significant differences between treated infants and controls at week 8 are indicated by asterisks. A statistically significant difference between treated infants at week 4 and week 8 (i.e., the recovery of the treated infants) is indicated by a diamond. Percentages are based on proportions of assignable reads.
Fig 3.

Microbial distributions at the genus level in the samples from treated and control infants at week 4 and week 8. Statistically significant differences between treated infants and controls at week 4 are indicated by asterisks (P < 0.05). Statistically significant differences between treated infants and controls at week 8 are indicated by asterisks. A statistically significant difference between treated infants at week 4 and at week 8 (i.e., the recovery of the treated infants) is indicated by a diamond. Percentages are based on proportions of assignable reads.
Composition of the gut microbiota of antibiotic-treated and control infants 8 weeks after the conclusion of treatment.
Bioinformatic analysis of the 16S rRNA sequence data revealed that the week 8 samples from the antibiotic-treated infants contained significantly higher proportions of Proteobacteria (44%) than those from controls (23%) (P = 0.0049). Eight weeks after the cessation of antibiotic treatment, Proteobacteria continued to be the dominant phylum present in samples from antibiotic-treated infants despite the fact that the proportions of Proteobacteria reads decreased significantly between week 4 and week 8 (P = 0.0136). During the same period, the proportion of Actinobacteria reads increased significantly (P = 0.0055) in the antibiotic-treated infant samples, to the extent that they no longer differed significantly from those in the control samples (P = 0.1164). Nonetheless, a more diverse gut microbe population was observed in the controls than in the antibiotic-treated infant samples 8 weeks after antibiotic treatment (Shannon's index for diversity was 3.8 and 4.6 in the treated and control infants, respectively) (Fig. 1 and Table 2). Analysis of data from individual infants revealed that the recovery of the infant gut microbiota to one more comparable to that of the controls was also dependent on the duration of treatment (data not shown). For example, the gut microbiota of the infant who underwent the longest antibiotic treatment period (infant A, treated for 9 days) displayed the most limited recovery. This infant's gut microbiota was populated predominantly by Proteobacteria, and this phylum remained dominant at week 8, at which time it accounted for 67% of all of the bacteria detected (see Fig. S1 in the supplemental material).
At the family level at week 8, Enterobacteriaceae remained dominant in the antibiotic-treated infants (45%), despite having significantly decreased in proportion relative to week 4 (P = 0.0136) (Fig. 2). During the same interval, proportions of Enterobacteriaceae decreased in the control infants (37% at week 4 versus 24% at week 8). In the antibiotic-treated group, there was also a significant decrease in levels of Peptostreptococcaceae between week 4 and week 8 (P = 0.0014), whereas a significant increase (P = 0.0182) in the Bifidobacteriaceae levels occurred during this 4-week interval, to the extent that the proportions of this family in the samples from antibiotic-treated and control infants no longer differed significantly by week 8 (P = 0.3927).
At the genus level, the gut microbiota of the antibiotic-treated infants remained predominantly populated with members of the Enterobacteriaceae family, which accounted for half of all genera detected at week 8. The numbers of these bacteria were significantly higher in the antibiotic-treated infants than in the controls at week 8 (P = 0.0061). In contrast, Bifidobacterium numbers were similar in the controls and antibiotic-treated infants at this time (19% versus 15%; P = 0.3927). This was a consequence of the fact that the proportions of Bifidobacterium had increased significantly in the antibiotic-treated infant samples during this 4-week interval (P = 0.0182). Significant differences in the levels of Lactobacillus between the two groups no longer existed at week 8 (P = 0.3253) (Fig. 3), due to a trend toward a significant recovery in Lactobacillus proportions in the antibiotic-treated infants (P = 0.059) during this interval. However, Clostridium proportions remained higher in the treated infants than in the controls at week 8 (7% versus 2%; P = 0.0345), as a consequence of the fact that there was no significant change in the levels of Clostridium in the antibiotic-treated infants between weeks 4 and 8 (P = 0.6132). By week 8, there was no longer a significant difference in the proportions of enterococci in the treated infants and those in the controls (P = 0.1105).
qPCR-based determination of numbers of total bacteria and total bifidobacteria.
To determine the impact of antibiotic treatment on the total numbers of bacteria and of bifidobacteria, absolute quantification was completed using qPCR, with a representative subset of samples. The qPCR results revealed that all infants, i.e., both treated infants and controls, had 107 to 108 copies of the 16S rRNA gene/g wet stool (Table 3) and established that no significant differences existed between total 16S rRNA gene copies (which is representative of total bacterial numbers) when values for antibiotic-treated infant samples were compared to those for controls at week 4 (P = 0.7667) or week 8 (P = 0.7918). However, a statistically significant increase in total 16S rRNA values did occur in the antibiotic-associated samples (P = 0.0005) between weeks 4 and 8. With respect to total bifidobacterial numbers, it was established that counts in both the treated and control infants ranged from 106 to 107 CFU/g wet stool (Table 4). There was no significant difference in the average bifidobacterial numbers in the antibiotic-treated infants relative to the controls at week 4 (P = 0.4273) or at week 8 (P = 0.1548). Furthermore, in the majority of individual infants, the total bifidobacterial numbers did not differ significantly between the two time points (Table 4).
Table 3.
Total bacterial numbers in treated and control samples at week 4 and week 8
| Infant | Copies of 16S rRNA/g wet stool |
Pa | |
|---|---|---|---|
| Wk 4 | Wk 8 | ||
| Treated | |||
| B | 9.79 × 107 | 6.57 × 107 | 0.7728 |
| F | 5.89 × 107 | 3.53 × 108 | 0.0809 |
| G | 3.28 × 107 | 7.79 × 107 | 0.0518 |
| H | 3.52 × 107 | 6.43 × 108 | 0.1489 |
| Average | 4.78 × 107 | 2.48 × 108 | 0.0005 |
| Control | |||
| K | 6.23 × 107 | 7.35 × 107 | 0.1489 |
| L | 2.19 × 107 | 3.61 × 108 | 0.0809 |
| M | 2.37 × 107 | 2.18 × 108 | 0.0518 |
| N | 9.05 × 107 | 5.75 × 106 | 0.0765 |
| Average | 4.96 × 107 | 1.91 × 107 | 0.0289 |
P values are based on Mann-Whitney analysis, with statistical significance being defined as a P value of <0.05. P values indicate whether differences between total bacterial numbers in each infant at week 4 and week 8 are statistically significant.
Table 4.
Total bifidobacterial numbers in the treated and control samples at week 4 and week 8
| Infant | CFU/g stool |
Pa | |
|---|---|---|---|
| Wk 4 | Wk 8 | ||
| Treated | |||
| B | 1.49 × 104 | 1.76 × 107 | 0.0814 |
| F | 7.32 × 106 | 1.10 × 109 | 0.0809 |
| G | 2.51 × 105 | 1.93 × 105 | 0.2472 |
| H | 5.55 × 107 | 4.57 × 108 | 0.0304 |
| Average | 1.58 × 107 | 3.94 × 108 | |
| Control | |||
| K | 6.62 × 107 | 5.98 × 105 | 0.0809 |
| L | 7.95 × 106 | 5.11 × 104 | 0.0809 |
| M | 1.48 × 106 | 2.39 × 108 | 0.0809 |
| N | 4.75 × 106 | 5.47 × 106 | 0.7728 |
| O | 5.05 × 107 | 1.14 × 108 | 0.0369 |
| Average | 2.62 × 107 | 7.18 × 107 | |
P values are based on Mann-Whitney analysis, with statistical significance being defined as a P value of <0.05.
Specific assessment of the composition of the gut Bifidobacterium population in antibiotic-treated and control infants.
Given the health benefits that have been attributed to many strains of Bifidobacterium, a strategy was implemented to specifically assess the impact of antibiotic treatment on gut bifidobacteria. This again relied on the use of high-throughput sequencing but in this instance focused on the sequencing of amplicons corresponding to a region of the Bifidobacterium RNA polymerase β-subunit gene, rpoB, using a set of primers which have been used previously for bifidobacterial species identification (43) but which in this instance contained adapters and MIDs to facilitate the sequencing process. These primers demonstrated excellent specificity, with 99% of the reads at phylum level being assigned to the Actinobacteria. The total number of reads for the antibiotic-treated infants at week 4 was 80,034, averaging 6,670 reads per subject, and the value at week 8 was 36,557, averaging 3,046 reads per subject. While the 16S rRNA data presented above showed that antibiotic treatment decreased the proportion of bifidobacteria present in the gut microbiota of infants, the rpoB data provides further, more detailed insights. More specifically, this analysis revealed that only two species were detected in the majority of cases in the antibiotic-treated infants, namely, B. longum and Bifidobacterium breve. In contrast, the controls showed considerable variation in the composition of individual samples and even between samples from the same individuals at different time points (Fig. 4).
Fig 4.

Individual distributions of bifidobacteria in the treated (B1 to H1 and B2 to H2) and control (J1 to R1 and J2 to R2) samples as detected by using rpoB amplicons for 454 pyrosequencing. Values show the percentage of the different bifidobacterial species present in the individual samples. Samples from treated infants show far less variability both between samples and also between week 4 and week 8 than the controls. The x axis indicates individual infants; the y axis shows the percentage of total bifidobacteria assigned to each species.
DISCUSSION
Antibiotics are of fundamental importance to modern medicine, and their use has been pivotal in the prolongation of human life. Despite this, there are ever-increasing concerns with respect to the negative consequences of antibiotic utilization, including issues revolving around the collateral damage inflicted on the commensal microbiota and the implications thereof (9). Short-term health effects include antibiotic-associated diarrhea, gastrointestinal discomfort, gastritis, and glossitis (24) as well as the possible development of antibiotic-resistant bacterial populations in the gut (39). Furthermore, it has been suggested that a number of long-term health effects are influenced by the development of the gut microbiota (38) and, in turn, the immune system in early life (32, 33, 37, 64), with data suggesting that antibiotic administration contributes to the risk of developing asthma and allergy (13, 25, 40) in addition to heightened risk of obesity (4) later in life. The risks associated with disrupting the gut microbiota may be especially great in young infants, as antibiotic administration can impact the commensal microbiota at a time when this population is in rapid flux and can easily be unbalanced. Despite this concern, there have been no studies to date which have used powerful next-generation sequencing technologies to assess the microbiota of infants who have been administered antibiotics. This study was performed with a view to addressing this issue by employing 454 pyrosequencing, together with qPCR analysis. The results of this relatively small study are important and highlight the apparently major impact that treatment with a combination of ampicillin and gentamicin can have on the gut microbiota of infants. It is evident that the treated infants suffered significant reductions in potentially beneficial bacteria belonging to the phylum Actinobacteria, including Bifidobacterium, as well as some members of the phylum Firmicutes, including Lactobacillus. These appeared to be replaced by members of the Proteobacteria, including members of the family Enterobacteriaceae, which resembles trends previously noted in a terminal restriction fragment length polymorphism-based study of antibiotic-treated infants (68). The dominance of the Proteobacteria, and an overall reduced microbial diversity, continued to be evident even 8 weeks after antibiotic treatment, despite the fact that populations of potentially beneficial bacteria (including Bifidobacterium) recovered somewhat. Given the fact that sequencing data reveal the proportions of different populations present, rather than their absolute number, the question of whether the dominance of Proteobacteria is reflective of an outgrowth of this population or of its numbers' remaining stable among a total bacterial population which is diminished in number exists, and some recent sequencing-based studies have begun to address this issue (51, 58). It is thus important that in this instance, qPCR data establish the fact that there is no significant difference between the total 16S rRNA counts in the treated infants and those in the controls, thereby implying Proteobacteria outgrowth, presumably as a consequence of reduced competition from other more antibiotic-sensitive gut microbes. Others have also documented a corresponding phenomenon of Proteobacteria outgrowth as a consequence of antibiotic administration (22, 51, 58). Notably, the frequency of beta-lactam antibiotic resistance among Enterobacteriaceae, as a consequence of the production of beta-lactamases, has been well established (12, 52, 56, 57). The presence of significantly higher levels of enterococci in the antibiotic-treated infant samples 4 weeks after treatment ended is also consistent with the fact that ampicillin-resistant (41, 71) and gentamicin-resistant (20, 36) Enterococcus strains have been identified on numerous occasions. The ability of the administered antibiotics, and especially ampicillin, to significantly alter the gut microbiota is also reflective of their activity profile. Following parenteral administration, ampicillin is rapidly and widely distributed throughout the body, resulting in high levels in bile (2) and, once excreted, in the gut.
It was notable that while the 16S rRNA sequencing data and the total bacterial qPCR data correlated well, the assessment of the impact of antibiotic administration on relative or total bifidobacterial numbers, as determined by sequencing and qPCR, respectively, was not consistent. More specifically, qPCR analysis at week 4 revealed no significant difference between total bifidobacterial values in the antibiotic-treated infants and those in the controls, while the 16S sequencing data detected significantly lower proportions in infants that had undergone antibiotic treatment. These differences may be accounted for by the fact that only a subset of the 18 infants were included in the rpoB-based qPCR analysis, and as outlined earlier, individual variations occur in response to antibiotic treatment. Furthermore, differences regarding primer specificity between those used for qPCR and for total bacterial 16S rRNA sequencing may also have contributed to this result.
The altered gut microbial composition of antibiotic-treated infants is a concern, given that several members of the genera Bifidobacterium and Lactobacillus have been found to possess health-promoting properties, to the extent that they are frequently employed as probiotic cultures, whereas many Proteobacteria have the potential to become pathogenic given a suitable environment. This study also demonstrated that the collateral damage inflicted on the gut microbiota through the use of broad-spectrum antibiotics is not rapidly repaired, as significant differences between the gut microbiota in the antibiotic-treated and control infants were apparent at 4 and 8 weeks posttreatment. Previous studies, employing temperature gradient gel electrophoresis or DGGE, have also shown that antibiotic treatment causes short- to medium-term effects, in some cases with no bifidobacteria being detected 28 days after treatment ceased (16, 23). However, the fact that some recovery was evident in this and previous trials (16, 35) indicates that the infant microbiota, despite being much less stable than that of an adult, is somewhat resilient. Indeed, on average, bifidobacterial populations recovered to the extent that both sequencing- and qPCR-based analysis revealed that their levels were no longer significantly reduced in the antibiotic-treated infant samples, relative to those of the controls, at week 8. Critically, however, it was apparent that the composition of these Bifidobacterium populations differed from one another. This is consistent with previous studies highlighting the differing susceptibilities of species of bifidobacteria to antibiotics (22, 23, 48). More specifically, in agreement with previous DGGE-based analysis (35), B. longum was found to be more dominant in samples from antibiotic-treated infants. This may be due to the fact that while all bifidobacteria have previously been found to display comparably high levels of sensitivity to gentamicin, strains of B. longum have been found to be more ampicillin resistant than other bifidobacteria (50). The significant impact of antibiotic administration on the Bifidobacterium population at the species level suggests that many other species are similarly affected by antibiotic administration, something which warrants further investigation.
It is important to note that factors other than antibiotic administration may also contribute to the differences in the gut microbial composition of the cohort of infants that were the focus of this investigation. From this perspective, it is notable that the majority of antibiotic-treated infants were delivered by Caesarean section, while the controls were all delivered vaginally. This is particularly relevant, as numerous studies have noted the presence of an altered gut microbiota profile in Caesarean-delivered infants (3, 19, 26). Caesarean-delivered infants have significantly altered profiles compared to vaginally delivered infants, due to a lack of colonization with their mother's vaginal microbiota during delivery, and are instead colonized by skin microbiota (predominantly Staphylococcus and Corynebacterium [19]). However, it has also been demonstrated that while levels of Bifidobacterium were significantly lower in Caesarean-delivered infants than in vaginally delivered infants, Bifidobacterium levels were comparable by 1 month of age (29). This was not the case in our study, in that all antibiotic-treated infants (regardless of delivery mode) had significantly lower levels of Bifidobacterium at 1 month of age and even at 2 months of age possessed a gut microbiota which was different from that of the control group. It is also worth noting that although our microbiota-related data are presented as averages, we also possessed the microbiota-related data (both high-throughput sequencing and qPCR derived) from each infant. Analysis of these data failed to reveal significant differences between the microbial populations of the vaginally delivered and Caesarean delivered subgroups of the antibiotic-treated infants (data not shown). Thus, while we acknowledge that delivery mode may influence the microbial composition of the infants studied, it would seem not to be as significant a factor as antibiotic administration.
Another factor that merits consideration is breastfeeding. While there are a considerable number of publications regarding the benefits of breastfeeding with respect to the development of the infant gut microbiota (8, 56), in this study breastfeeding did not provide any additional protection to the infant gut microbiota against the impact of antibiotic treatment. A failure to observe protection could well be due to the relatively small subgroup of infants in our study who were breastfed. In any case, this is a topic which warrants further investigation.
Regardless of the extent to which factors other than antibiotic administration influence these results, there is a concern that these short-term changes to the microbiota may in turn have long-term health consequences in the form of allergies, asthma, and obesity later in life (4, 13, 25). While follow-up analysis of these infants was outside the scope of this short-term study, we hope to return to this topic in future studies.
In conclusion, this study has shown alterations in the gut microbiota, over 8 weeks, of a group of infants who received parenteral antibiotic treatment within the first 48 h of life. To our knowledge, this is the first study to use high-throughput sequencing of 16S rRNA and/or rpoB amplicons to accurately assess these impacts. While the results may reflect a combination of several environmental effects in early life, it appears that antibiotic administration is the most influential factor. It would thus seem that, where available, the use of narrow-spectrum antibiotics coupled with the use of pre- and probiotics should be considered with a view to minimizing the risk of long-term health effects. While it is evident that the study of the composition of the infant gut microbiota and the effect of antibiotic treatment on this population requires further investigation, it is anticipated that the further application of high-throughput sequencing technologies (including those used in long-term follow-up trials) will shed additional light on the optimal strategies to employ to control infection while minimizing the risks to commensal microbes.
Supplementary Material
ACKNOWLEDGMENTS
We thank Fiona Crispie for high-throughput DNA sequencing services.
Fiona Fouhy is the recipient of an Irish Research Council for Science, Engineering and Technology EMBARK scholarship and is a Teagasc Walsh fellow. This research was supported by the Science Foundation of Ireland-funded Centre for Science Engineering and Technology, the Alimentary Pharmabiotic Centre, and the Irish Government under the National Development Plan (INFANTMET; http://eldermet.ucc.ie). Research in the P.D.C. lab is also supported through the Science Foundation Ireland Investigator award 11/PI/1137.
Footnotes
Published ahead of print 4 September 2012
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1. Abrahamsson TR, et al. 2011. Low diversity of the gut microbiota in infants with atopic eczema. J. Allergy Clin. Immunol. 129:434–440 [DOI] [PubMed] [Google Scholar]
- 2. Acred P, Brown D, Turner D, Wilson M. 1962. Pharmacology and chemotherapy of ampicillin—a new broad-spectrum penicillin. Br. J. Pharmacol. Chemother. 18:356–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Adlerberth I, Wold AE. 2009. Establishment of the gut microbiota in Western infants. Acta Pædiatr. 98:229–238 [DOI] [PubMed] [Google Scholar]
- 4. Ajslev T, Andersen C, Gamborg M, Sørensen T, Jess T. 2011. Childhood overweight after establishment of the gut microbiota: the role of delivery mode, pre-pregnancy weight and early administration of antibiotics. Int. J. Obesity 35:522–529 [DOI] [PubMed] [Google Scholar]
- 5. Altschul SF, et al. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Antonopoulos DA, et al. 2009. Reproducible community dynamics of the gastrointestinal microbiota following antibiotic perturbation. Infect. Immun. 77:2367–2375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bennet R, Eriksson M, Nord CE. 2002. The fecal microflora of 1-3-month-old infants during treatment with eight oral antibiotics. Infection 30:158–160 [DOI] [PubMed] [Google Scholar]
- 8. Bezirtzoglou E, Tsiotsias A, Welling GW. 2011. Microbiota profile in feces of breast- and formula-fed newborns by using fluorescence in situ hybridization (FISH). Anaerobe 17:478–482 [DOI] [PubMed] [Google Scholar]
- 9. Blaser M. 2011. Antibiotic overuse: stop the killing of beneficial bacteria. Nature 476:393–394 [DOI] [PubMed] [Google Scholar]
- 10. Boehm G, et al. 2005. Prebiotic carbohydrates in human milk and formulas. Acta Paediatr. 94:18–21 [DOI] [PubMed] [Google Scholar]
- 11. Bourlioux P, Koletzko B, Guarner F, Braesco V. 2003. The intestine and its microflora are partners for the protection of the host: report on the Danone Symposium “The Intelligent Intestine,” held in Paris, June 14, 2002. Am. J. Clin. Nutr. 78:675–683 [DOI] [PubMed] [Google Scholar]
- 12. Bush K. 2010. Alarming β-lactamase-mediated resistance in multidrug-resistant Enterobacteriaceae. Curr. Opin. Microbiol. 13:558–564 [DOI] [PubMed] [Google Scholar]
- 13. Celedon JC, Fuhlbrigge A, Rifas Shiman S, Weiss ST, Finkelstein JA. 2004. Antibiotic use in the first year of life and asthma in early childhood. Clin. Exp. Allergy 34:1011–1016 [DOI] [PubMed] [Google Scholar]
- 14. Cramer JP, Burchard GD, Lohse AW. 2008. Old dogmas and new perspectives in antibiotic-associated diarrhea. Med. Klinik. (Munich) 103:325–338 [DOI] [PubMed] [Google Scholar]
- 15. Cremonini F, et al. 2002. Meta analysis: the effect of probiotic administration on antibiotic associated diarrhoea. Aliment. Pharmacol. Ther. 16:1461–1467 [DOI] [PubMed] [Google Scholar]
- 16. De La Cochetiere M, et al. 2005. Resilience of the dominant human fecal microbiota upon short-course antibiotic challenge. J. Clin. Microbiol. 43:5588–5592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dethlefsen L, Huse S, Sogin ML, Relman DA. 2008. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 6:e280 doi:10.1371/journal.pbio.0060280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dethlefsen L, Relman DA. 2011. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc. Natl. Acad. Sci. U. S. A. 108:4554–4561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dominguez-Bello MG, et al. 2010. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Natl. Acad. Sci. U. S. A. 107:11971–11976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Donabedian S, et al. 2003. Molecular characterization of gentamicin-resistant enterococci in the United States: evidence of spread from animals to humans through food. J. Clin. Microbiol. 41:1109–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Echarri PP, et al. 2011. Assessment of intestinal microbiota of full-term breast-fed infants from two different geographical locations. Early Hum. Dev. 87:511–513 [DOI] [PubMed] [Google Scholar]
- 22. Fallani M, et al. 2010. Intestinal microbiota of 6-week-old infants across Europe: geographic influence beyond delivery mode, breast-feeding, and antibiotics. J. Pediatr. Gastroenterol. Nutr. 51:77–84 [DOI] [PubMed] [Google Scholar]
- 23. Favier CF, de Vos WM, Akkermans ADL. 2003. Development of bacterial and bifidobacterial communities in feces of newborn babies. Anaerobe 9:219–229 [DOI] [PubMed] [Google Scholar]
- 24. Finegold SM. 1970. Interaction of antimicrobial therapy and intestinal flora. Am. J. Clin. Nutr. 23:1466–1471 [DOI] [PubMed] [Google Scholar]
- 25. Foliaki S, et al. 2009. Antibiotic use in infancy and symptoms of asthma, rhinoconjunctivitis, and eczema in children 6 and 7 years old: international study of asthma and allergies in childhood phase III. J. Allergy Clin. Immunol. 124:982–989 [DOI] [PubMed] [Google Scholar]
- 26. Fouhy F, Ross RP, Fitzgerald G, Stanton C, Cotter PD. 2012. Composition of the early intestinal microbiota: knowledge, knowledge gaps and the use of high-throughput sequencing to address these gaps. Gut Microbes 3:203–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fujimura KE, Slusher NA, Cabana MD, Lynch SV. 2010. Role of the gut microbiota in defining human health. Expert Rev. Anti-infect. Ther. 8:435–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gophna U, Sommerfeld K, Gophna S, Doolittle WF, Veldhuyzen van Zanten SJO. 2006. Differences between tissue-associated intestinal microfloras of patients with Crohn's disease and ulcerative colitis. J. Clin. Microbiol. 44:4136–4141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Grölund MM, Lehtonen OP, Eerola E, Kero P. 1999. Fecal microflora in healthy infants born by different methods of delivery: permanent changes in intestinal flora after cesarean delivery. J. Pediatr. Gastroenterol. Nutr. 28:19–25 [DOI] [PubMed] [Google Scholar]
- 30. Guarner F, Malagelada JR. 2003. Gut flora in health and disease. Lancet 361:512–519 [DOI] [PubMed] [Google Scholar]
- 31. Haarman M, Knol J. 2005. Quantitative real-time PCR assays to identify and quantify fecal Bifidobacterium species in infants receiving a prebiotic infant formula. Applied Environ. Microbiol. 71:2318–2324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hill DA, Artis D. 2010. Intestinal bacteria and the regulation of immune cell homeostasis. Annu. Rev. Immunol. 28:623–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hill DA, et al. 2010. Metagenomic analyses reveal antibiotic-induced temporal and spatial changes in intestinal microbiota with associated alterations in immune cell homeostasis. Mucosal Immunol. 3:148–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huson DH, Auch AF, Qi J, Schuster SC. 2007. MEGAN analysis of metagenomic data. Genome Res. 17:377–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hussey S, et al. 2011. Parenteral antibiotics reduce bifidobacteria colonization and diversity in neonates. Int. J. Microbiol. 2011:130574 doi:10.1155/2011/130574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huycke M, Spiegel C, Gilmore M. 1991. Bacteremia caused by hemolytic, high-level gentamicin-resistant Enterococcus faecalis. Antimicrob. Agents Chemother. 35:1626–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ivanov II, et al. 2008. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe 4:337–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jakobsson HE, et al. 2010. Short-term antibiotic treatment has differing long-term impacts on the human throat and gut microbiome. PLoS One 5:e9836 doi:10.1371/journal.pone.0009836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jernberg C, Löfmark S, Edlund C, Jansson JK. 2010. Long-term impacts of antibiotic exposure on the human intestinal microbiota. Microbiology 156:3216–3223 [DOI] [PubMed] [Google Scholar]
- 40. Johnson CC, et al. 2005. Antibiotic exposure in early infancy and risk for childhood atopy. J. Allergy Clin. Immunol. 115:1218–1224 [DOI] [PubMed] [Google Scholar]
- 41. Jureen R, et al. 2003. Molecular characterization of ampicillin-resistant Enterococcus faecium isolates from hospitalized patients in Norway. J. Clin. Microbiol. 41:2330–2336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kassinen A, et al. 2007. The fecal microbiota of irritable bowel syndrome patients differs significantly from that of healthy subjects. Gastroenterology 133:24–33 [DOI] [PubMed] [Google Scholar]
- 43. Kim BJ, Kim HY, Yun YJ, Kook YH. 2010. Differentiation of Bifidobacterium species using partial RNA polymerase β-subunit (rpoB) gene sequences. Int. J. Syst. Evol. Microbiol. 60:2697–2704 [DOI] [PubMed] [Google Scholar]
- 44. Kozyrskyj AL, Ernst P, Becker AB. 2007. Increased risk of childhood asthma from antibiotic use in early life. Chest 131:1753–1759 [DOI] [PubMed] [Google Scholar]
- 45. Le Huërou-Luron I, Blat S, Boudry G. 2010. Breast- v. formula-feeding: impacts on the digestive tract and immediate and long-term health effects. Nutr. Res. Rev. 23:23–36 [DOI] [PubMed] [Google Scholar]
- 46. Ley RE. 2010. Obesity and the human microbiome. Curr. Opin. Gastroenterol. 26:5–11 [DOI] [PubMed] [Google Scholar]
- 47. Mai V, Draganov PV. 2009. Recent advances and remaining gaps in our knowledge of associations between gut microbiota and human health. World J. Gastroenterol. 15:81–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mangin I, Suau A, Gotteland M, Brunser O, Pochart P. 2010. Amoxicillin treatment modifies the composition of Bifidobacterium species in infant intestinal microbiota. Anaerobe 16:433–438 [DOI] [PubMed] [Google Scholar]
- 49. Matsuki T, et al. 2004. Quantitative PCR with 16S rRNA-gene-targeted species-specific primers for analysis of human intestinal bifidobacteria. Appl. Environ. Microbiol. 70:167–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mättö J, et al. 2007. Susceptibility of human and probiotic Bifidobacterium spp. to selected antibiotics as determined by the Etest method. Int. Dairy J. 17:1123–1131 [Google Scholar]
- 51. Murphy E, et al. 2010. Composition and energy harvesting capacity of the gut microbiota: relationship to diet, obesity and time in mouse models. Gut 59:1635–1642 [DOI] [PubMed] [Google Scholar]
- 52. Nordmann P. 1998. Trends in β-lactam resistance among Enterobacteriaceae. Clin. Infect. Dis. 27:S100–S106 [DOI] [PubMed] [Google Scholar]
- 53. Olszak T, et al. 2012. Microbial exposure during early life has persistent effects on natural killer T cell function. Science 336:489–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. O'Sullivan Ó, et al. 2011. Correlation of rRNA gene amplicon pyrosequencing and bacterial culture for microbial compositional analysis of faecal samples from elderly Irish subjects. J. Appl. Microbiol. 111:467–473 [DOI] [PubMed] [Google Scholar]
- 55. Parracho H, McCartney AL, Gibson GR. 2007. Probiotics and prebiotics in infant nutrition. Proc. Nutr. Soc. 66:405–411 [DOI] [PubMed] [Google Scholar]
- 56. Penders J, et al. 2006. Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics 118:511–521 [DOI] [PubMed] [Google Scholar]
- 57. Qin X, et al. 2009. Prevalence and mechanisms of broad-spectrum β-lactam resistance in Enterobacteriaceae: a children's hospital experience. Antimicrob. Agents Chemother. 53:3909–3914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rea MC, et al. 2011. Effect of broad-and narrow-spectrum antimicrobials on Clostridium difficile and microbial diversity in a model of the distal colon. Proc. Natl. Acad. Sci. U. S. A. 108:4639–4645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Robinson CJ, Young VB. 2010. Antibiotic administration alters the community structure of the gastrointestinal microbiota. Gut Microbes 1:279–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sazawal S, et al. 2010. Prebiotic and probiotic fortified milk in prevention of morbidities among children: community-based, randomized, double-blind, controlled trial. PLoS One 5:e12164 doi:10.1371/journal.pone.0012164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Schloss P, Handelsman J. 2008. A statistical toolbox for metagenomics: assessing functional diversity in microbial communities. BMC Bioinformatics 9:34–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Schloss PD, et al. 2009. Introducing MOTHUR: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75:7537–7541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schwiertz A, Gruhl B, Löbnitz M, Michel P. 2003. Development of the intestinal bacterial composition in hospitalized preterm infants in comparison with breast-fed, full-term infants. Pediatr. Res. 54:393–399 [DOI] [PubMed] [Google Scholar]
- 64. Sekirov I, Russell SL, Antunes LCM, Finlay BB. 2010. Gut microbiota in health and disease. Physiol. Rev. 90:859–904 [DOI] [PubMed] [Google Scholar]
- 65. Sherman PM, et al. 2009. Potential roles and clinical utility of prebiotics in newborns, infants, and children: proceedings from a global prebiotic summit meeting, New York City, June 27–28, 2008. J. Pediatr. 155:S61–S70 [DOI] [PubMed] [Google Scholar]
- 66. Suchodolski J, et al. 2009. The effect of the macrolide antibiotic tylosin on microbial diversity in the canine small intestine as demonstrated by massive parallel 16S rRNA gene sequencing. BMC Microbiol. 9:210–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sullivan Å, Edlund C, Nord CE. 2001. Effect of antimicrobial agents on the ecological balance of human microflora. Lancet Infect. Dis. 1:101–114 [DOI] [PubMed] [Google Scholar]
- 68. Tanaka S, et al. 2009. Influence of antibiotic exposure in the early postnatal period on the development of intestinal microbiota. FEMS Immunol. Med. Microbiol. 56:80–87 [DOI] [PubMed] [Google Scholar]
- 69. Turnbaugh PJ, Gordon JI. 2009. The core gut microbiome, energy balance and obesity. J. Physiol. 587:4153–4158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Urich T, et al. 2008. Simultaneous assessment of soil microbial community structure and function through analysis of the meta-transcriptome. PLoS One 3:e2527 doi:10.1371/journal.pone.0002527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Weisser M, et al. 2012. Dynamics of ampicillin-resistant Enterococcus faecium clones colonizing hospitalized patients: data from a prospective observational study. BMC Infect. Dis. 12:68–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Westerbeek EAM, et al. 2006. The intestinal bacterial colonisation in preterm infants: a review of the literature. Clin. Nutr. 25:361–368 [DOI] [PubMed] [Google Scholar]
- 73. Zhang H, et al. 2009. Human gut microbiota in obesity and after gastric bypass. Proc. Natl. Acad. Sci. U. S. A. 106:2365–2370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zoetendal E, Rajilić-Stojanović M, De Vos W. 2008. High-throughput diversity and functionality analysis of the gastrointestinal tract microbiota. Gut 57:1605–1615 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.