

Abstract
We have developed a series of hybrid P450 BM3 enzymes to perform the light-activated hydroxylation of lauric acid. These enzymes contain a Ru(II)-diimine photosensitizer covalently attached to single cysteine residues of mutant P450 BM3 heme domains. The library of hybrid enzymes includes four non-native single cysteine mutants (K97C, Q397C, Q109C and L407C). In addition, mutations around the heme active site, F87A and I401P, were inserted in the Q397C mutant. Two heteroleptic Ru(II) complexes, Ru(bpy)2phenA (1) and Ru(phen)2phenA (2) (bpy=bipyridine, phen=1,10-phenanthroline, and phenA=5-acetamido-1,10-phenanthroline), are used as photosensitizers. Upon visible light irradiation, the hybrid enzymes display various total turnover numbers in the hydroxylation of lauric acid, up to 140 for the L407C-1 mutant, a 16-fold increase compared to the F87A/Q397C-1 mutant. CO binding studies confirm the ability of the photogenerated Ru(I) compound to reduce the fraction of ferric high spin species present in the mutants upon substrate binding.
Keywords: Hybrid P450BM3 enzymes, Light-initiated hydroxylation, Ru(II)-diimime photosensitizers, Flash quench technique, Ferric heme reduction
1. Introduction
Cytochromes P450 are heme-thiolate enzymes capable of catalyzing the selective oxidation of unactivated C-H bonds in a variety of organic substrates by utilizing two reducing equivalents and molecular dioxygen [1]. The consensus mechanism involves two successive one-electron transfer (ET) steps from the electron provider reductase [2] to the heme center and the formation of several well characterized intermediates (Fig. 1) [3]. The catalytic cycle is often initiated by binding of substrate, resulting in a displacement of the water molecule bound to the resting ferric species and a shift from low to high spin (Fig. 1, Step 1) [4]. This gating step is important in bacterial P450 enzymes; however, it might not be necessary in all human P450s [5]. The binding of the substrate is also associated with a change in the heme redox potential, which promotes the first electron reduction and rapid binding of dioxygen to the reduced ferrous heme. The resulting oxyferrous species is then further reduced by a second electron and protonated (Step 5) to generate a hydroperoxo intermediate, Compound 0. Protonation of the distal oxygen in the Fe(III)-OOH species (Step 6) results in O-O bond cleavage and the formation of a high-valent Fe(IV)-oxo porphyrin radical species, Compound I. Compound I is responsible for the oxygen atom insertion into substrate C-H bond via a rebound mechanism [6,7]. The sequential delivery of electrons by the reductase is well orchestrated in order to achieve high catalytic efficiency and minimize the formation of reactive oxygen species via uncoupled pathways (autooxidation and peroxide shunts).
Fig. 1.

P450 catalytic cycle showing the individual steps and intermediates involved in the mechanism as well as the uncoupling reactions (autooxidation and peroxide shunts) and the formation of Fe(II)-CO adduct.
Due to their great synthetic potential, the P450 enzymes have been of interest for biotechnological applications [8–10]. However, the dependence on the reductase domain and the use of NAD(P)H to initiate the ET cascade remain challenging in their development as biocatalysts [11]. Alternative approaches to deliver electrons have emerged [12], which include artificial fusion P450 proteins [13], regeneration system [14], surrogate oxygen atom donors [15], direct chemical reduction [16,17], and electrochemical reductions [18] as well as light-activated approaches [19,20].
We recently reported an alternative light-activated approach to perform P450 reactions using hybrid P450 BM3 enzymes [21,22]. The hybrid enzymes consist of a Ru(II)-diimine photosensitizer, Ru(bpy)2phenA (1) (bpy=bipyridine and phenA=5-acetamido-1, 10-phenanthroline), covalently attached to non-native single cysteine residues (K97C and Q397C) of the archetypal P450 BM3 heme domain [23]. Under flash quench reductive conditions [24], the photogenerated Ru(I) is sought to provide the necessary electrons to the heme domain in successive one-electron reduction steps. Upon light activation, these hybrid enzymes showed catalytic hydroxylation of lauric acid with total turnover numbers of up to 80.
We have since expanded the library of hybrid enzymes in order to improve their activity, stability and total turnover numbers as well as to gain insights into their mechanism. New non-native single cysteine mutations at position 109 and 407 were generated along with additional mutations, F87A and I401P, around the heme active site of the Q397C mutant. The first set of mutations was engineered in order to modify the distance between the photosensitizer and the heme center. The second set was introduced to alter substrate recognition properties of the heme domain (F87A) [25] as well as the heme redox potential and catalytic activity (I401P) [26]. Aside from the previously used Ru(bpy)2phenA (1), we synthesized and covalently attached another heteroleptic Ru(II) photosensitizer, Ru(phen)2PhenA (2) (phen=1,10-phenanthroline) in order to increase the Eo(Ru(II)/Ru(I)) potential [27]. Overall, the current library comprises seven labeled mutants as follows: K97C-1, Q109C-1, L407C-1, Q397C-1, F87A/Q397C-1, Q397C/I401P-1, and Q397C-2. These hybrid enzymes display different activities and total turnover numbers in the hydroxylation of lauric acid upon light activation. Substrate and CO binding experiments were performed to determine the effects of mutations and covalent attachments of the photosensitizers. In addition, the ability of the photosensitizer to reduce the ferric heme species was investigated in the presence of CO atmosphere under flash quench reductive conditions.
2. Experimental
2.1. Material and reagents
Site-directed mutagenesis on the P450 BM3 heme domain plasmid was performed using the Stratagene QuikChange Mutagenesis kit. Oligonucleotides were purchased from Eurofins MWG Operon. All other reagents used in this work were of analytical grade and purchased from Thermo Fisher Scientific. Accurate mass spectrometry data were obtained from an Agilent 6520 Quadrupole Time-of-Flight LC/MS instrument. Fatty acid analyses were performed on an Agilent 6890N gas chromatography (GC) system equipped with a flame ionization detector and an HP-5MS capillary column. UV-visible (UV–vis) spectra were recorded on a Cary 60 UV–vis spectrophotometer.
2.2. Site-directed mutagenesis and protein expression
The following primers were used for the mutagenesis reactions to create the desired non-native single cysteine mutants: F87A 5′-AGAGACGGGTTAGCGACAAGCTGGAC-3′; Q109C 5′-CTTACTTC CAAGCTTCAGTTGCCAGGCAATGAAAGGCTATC-3′; L407C 5′-CGGTCAGCAGTTCGCTTGCCATGAAGCAACGCTGG-3′. I401P 5′-GTCAGCGTGCGTGTCCGGGTCAGCAGTTCGC-3′. The underlined regions indicate the section of the oligonucleotides modified from the wild-type (WT) sequence to produce the codon change required for the amino acid substitutions. Mutated heme domain plasmids were amplified by PCR using the mentioned primers. PCR was performed using PfuTurbo DNA polymerase (Stratagene). Conditions were as follows: 10 ng template DNA, 125 ng primers, 100 μM dNTPs, 2.5 units of PfuTurbo polymerase in a 50 μL total reaction volume. PCR was performed using an Eppendorf Mastercycler Thermocycler for 18 cycles of 95 °C (50 s), 60 °C (50 s), and 68 °C (7 min) for denaturation, annealing, and extension, respectively. Plasmids were transformed into E. coli XL1 Blue and selected on LB agar/ampicillin plates. DNA sequencing confirmed the correct generation of the desired mutations. Expression and purification of mutant heme domains were carried out as described previously [28]. The E. coli BL21 (DE3) competent cells were used to express the mutant heme domains. Cells were cultured in Terrific Broth (TB) medium enriched with mineral supplements at 37 °C under vigorous agitation. At an OD600 of 0.9, gene expression was induced by the addition of Isopropyl β-D-1-thiogalactopy-ranoside (1 mM) and δ-aminolevulinic acid (1 mM), and the culture was continued for an additional 27 h at 30 °C before harvesting cells. Bacterial cells expressing the heme domains were lysed by sonication on ice, and the solution was centrifuged (17,000 rpm, 60 min, 8 °C). The heme domains containing a His6 tag were first separated using a 5 mL HisTrap HP column and then on a Superdex 200 size exclusion column connected to an ÄKTA Purifier. The integrity and purity of the purified proteins were confirmed by UV–vis spectroscopy, CO-binding, SDS-PAGE and mass spectrometry. After mass spectral deconvolution, the experimental mass values for the purified proteins agree with calculated mass values indicated in parentheses: 53547.3 (53547.2) for K97C, 53547.3 (53547.2) for Q397C and Q109C, 53562.3 (53562.2) for L407C, 53471.2 (53471.1) for F87A/Q397C, and 53531.3 (53531.2) for Q397C/I401P.
2.3. Ru(II)-diimine photosensitizer synthesis
Ru(bpy)2Cl2 and Ru(phen)2Cl2 were prepared following reported procedures [29]. The corresponding 5-acetamido-1,10-phenanthroline (phenIA) derivatives were prepared using literature method [30] in 70 and 65% yields, respectively. The experimentally determined masses for the products were m/z=388.514 [M2+] for Ru(bpy)2phenIA (calculated: 388.514) and m/z=412.514 [M2+]for Ru(phen)2phenIA (calculated: 412.514).
2.4. Labeling reaction and hybrid enzyme purification
The labeling reaction for covalent attachment of Ru(II) photosensitizers to non-native single cysteine residues was performed following reported procedures [21].
Upon completion, the reaction mixture was concentrated using centrifugal filtration, and excess label was removed using a desalting column connected to an ÄKTA Purifier. The labeled heme domain was then separated from unlabeled protein by anionic exchange using a HiTrap Q column with a stepwise elution gradient (Solvent A: 10 mM Tris pH=8.2; Solvent B: 10 mM Tris with 300 mM NaCl, pH 8.2). The purified hybrid enzyme was characterized using UV–vis and luminescence spectroscopy as well as mass spectrometry. After mass spectral deconvolution, the hybrid enzymes have experimental mass values as follows, consistent with calculated values in parentheses: 54196.3 (54196.3) for K97C-1, 54196.3 (54196.4) for Q397C-1 and Q109C-1, 54211.4 (54211.3) for L407C-1, 54120.2 (54120.3) for F87A/Q397C-1, and 54180.3 (54180.3) for Q397C/I401P-1, and 54244.4 (54244.3) for Q397C-2.
2.5. Enzyme activity in photochemical experiments
In a typical experiment, the reaction was initiated by exposing an aerated solution containing 3 μM of hybrid enzymes with 1.5 mM lauric acid and 100 mM sodium diethyldithiocarbamate (DTC) to visible light irradiation from an Orion 1000 W Xenon arc lamp. The solution was maintained at 20 °C using a water bath.
The activity of the enzyme was determined following the general procedure highlighted in our previous study [21]. Briefly, 500 μL aliquots were sampled from the reaction mixture at different time intervals. After addition of 12-hydroxy-dodecanoic acid as internal standard, the products were extracted with dichloromethane. The solvent was evaporated and the residue was silylated using N,O-Bis(trimethylsilyl)trifluoroacetamide (BSTFA) and Pyridine. After a two-fold dilution with dichloromethane, 1 μL of the solution was injected into the GC/MS apparatus.
2.6. Substrate binding titrations
The dissociation constants, Kd, for lauric and palmitic acids were determined for all the hybrid mutants by optical titration with DMSO solutions of the respective fatty acids, keeping the maximum concentration of DMSO below 2.5% (v/v). All hybrid enzyme solutions were at 2.5 μM in 100 mM Tris at pH 8.2 and room temperature. Lauric acid was added to a final concentration of 600 μM with aliquots of 10 mM and 40 mM stock solutions, whereas a final palmitic acid concentration of 100 μM was achieved through additions of 1 mM and 10 mM palmitic acid solutions. The difference spectra between 350 and 650 nm were recorded 30 s after each addition, using the substrate-free hybrid enzyme solution as the reference spectrum. The peak-to-trough difference between 390 and 420 nm was plotted against substrate concentrations. Substrate dissociation constants were extracted from fitting the data of triplicate experiments to a rectangular hyperbolic function.
2.7. CO binding study
A 2.5 μM solution of each hybrid enzyme in 100 mM Tris at pH 8.2 was used. After addition of 150 μM of substrate and 50 mM sodium diethyldithiocarbamate (DTC), the solution was degassed for 30 min with N2 and then bubbled with CO for 1 min. The solution was then exposed to light irradiation from the Orion 1000 W Xenon arc lamp. After 2 min of exposure, the UV–vis difference spectrum from 350 to 500 nm was obtained using the CO-saturated solution before irradiation as the reference spectrum. All solutions were at room temperature, and the enzyme solution was kept in a 20 °C water bath during light irradiation.
2.8. Percent high spin (%HS) determination
The high spin fraction for each hybrid enzyme solution was obtained from the UV–vis spectrum recorded after addition of substrates. The percent high spin (%HS) was calculated from the absorbance at 391 nm and 417 nm using Eq. (1), adapted from ref. [31].
| (1) |
A391 and A417 are absorbance at 391 nm and 417 nm, respectively. The molar extinction coefficient (ε) values determined for the hybrid enzymes are: ; and .
3. Results and discussion
Building on previous work with the Q397C-1 and K97C-1 mutants [21], we have expanded the series of hybrid enzymes to include new mutations and another photosensitizer. These mutants were generated in order to improve the catalytic activity of the hybrid enzymes upon light activation as well as to gain insights into their mechanism.
3.1. Library of hybrid enzymes
Depending on the type of mutants and photosensitizers, the library of mutants can be divided in two series: one consists of non-native single cysteine mutants (Q397C, L407C, K97C, Q109C) labeled with Ru(bpy)2PhenA (1) photosensitizer, and the second includes the non-native single cysteine mutant Q397C with additional mutations around the active site (F87A or I401P) as well as different photosensitizers (1 and 2) containing bipyridine or phenanthroline as ancillary ligands.
In the first series, the position of the covalent attachment relative to the heme domain was modified as illustrated in Fig. 2. The positions of the mutated cysteine residue are selectively varied on the distal face of the heme where the Cys400 is coordinating to the iron center. The K97C and Q109C mutations are located on the C-helix and the L407C mutation on the L-helix. The Q397C mutation is on the highly conserved region close to the thiolate ligating Cys400 residue. The distances from the Cβ carbon of the side chain residue to the Fe center, d(Fe-Cβ) could be estimated from the K97C-1 crystal structure [22] to be 10.0, 13.9, 16.2 and 17.9 Å for the four mutants L407C, Q397C, Q109C and K97C, respectively.
Fig. 2.
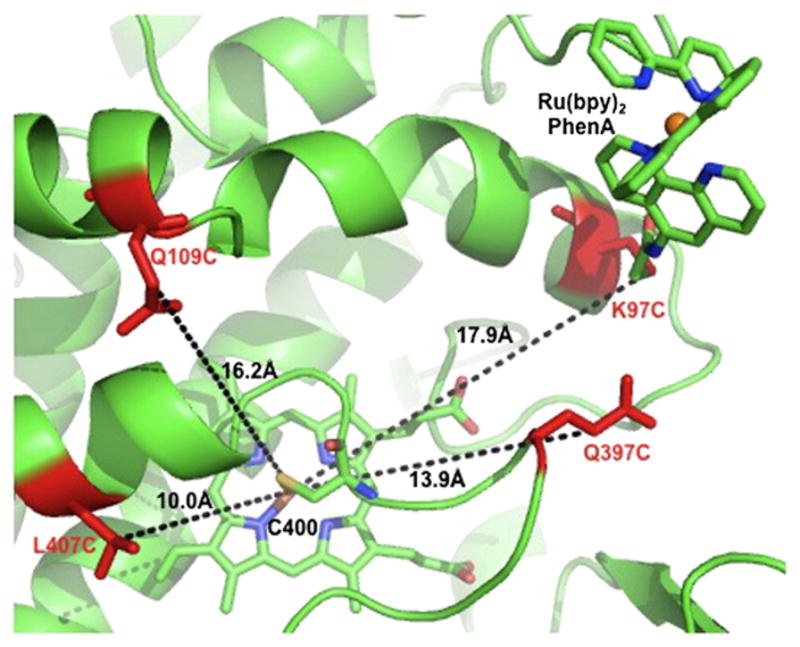
Cartoon representation of the mutated residues Q109C, K97C, Q397C and L407C (red) in the series of hybrid enzymes adapted from the K97C-1 crystal structure (PDB ID: 3NPL). The estimated distances from the Cβ carbon of the side chain residue to the Fe center are indicated.
In the Q397C hybrid enzyme series, it is possible to investigate the influence of the redox potential difference (ΔE) between the electron donor, the photogenerated Ru(I), and the electron acceptor, the ferric heme species. The potential difference in the hybrid enzymes can be tuned by (1) changing the ancilliary ligands on the Ru(II) photosensitizer; (2) changing the potential of the heme by site-directed mutagenesis (I401P mutant) and (3) by binding different type-I substrates, such as lauric or palmitic acids, to the active site.
3.2. Enzyme activity and stability in photochemical experiments
We have studied the ability of the hybrid enzymes to hydroxylate lauric acid upon visible light activation using the photocatalytic cycle highlighted in Scheme 1. Upon light excitation into the metal-to-ligand charge transfer transition, the Ru(II) excited state, Ru(II)*, can be quenched in a bimolecular process by the irreversible sacrificial electron donor, sodium diethyldithiocarbamate (DTC), to form the highly reductive Ru(I) intermediate. Under this flash quench reductive condition, the photogenerated Ru(I) species in the hybrid enzymes is sought to provide the two necessary electrons to the heme domain in successive one-electron transfer steps.
Scheme 1.

Proposed photocatalytic cycle for the hydroxylation of lauric acid by the hybrid enzymes upon light activation and in the presence of a reductive quencher, diethyldithiocarbamate (DTC). The structures of the Ru(II) photosensitizers (1 and 2) used in this study are shown.
The catalytic activity of the hybrid enzymes was determined following procedures described in Section 2.5. The results are summarized in Fig. 3A, which shows progress curves as turnover numbers (concentration of hydroxylated products/enzyme concentration) vs. time. The concentrations and relative distribution of the hydroxylated products were determined by gas chromatography after derivatization (Fig. 3B). Their identity was confirmed from their unique mass fragmentation patterns (Fig. 3C).
Fig. 3.

(A) Progress curves for the series of hybrid enzymes in the light-activated hydroxylation of lauric acid, displaying turnover numbers (concentration of product/enzyme concentration) vs. time. Representative chromatograms (B) and mass spectrometry fragmentation patterns (C) for the trimethylsilylated (TMS) derivatives of the hydroxylated products (ω1, ω2, ω3, ω4, ω5) are shown with their respective structures. 12-Hydroxydodecanoic acid was used as internal standard (I.S., 10 nmol).
In the series of non-native single cysteine mutants containing the Ru(bpy)2phenA (1) photosensitizer, the L407C-1 mutant has the highest turnover numbers (TON) of 140 after 150 min, followed by Q109C-1 (95), Q397C-1 (80), and K97C-1 (60). In the Q397C series, the Q397C-1 shows higher activity than the Q397C-2 and the Q397C/I401P-1 mutants. The F87A/Q397C-1 shows only total TON of 9, 16 times less than that of the L407C-1 mutant. Hydroxylation of lauric acid at the γ- and δ-positions, characteristic of the F87A mutation [25], is observed in the F87A/Q397C mutant (Fig. 3B and C). The apparent specific activities for the hybrid enzymes range from 0.16 min−1 for F87A/Q397C-1 to 0.93 min−1 for L407C-1. It is also worth noting that the initial reaction rates are also different in all the mutants as indicated by the difference in TON after 15 min, with reaction rates varying up to five-fold (Fig. 3A).
In addition, the heme Soret absorption band was monitored at 417 nm in order to probe the stability of the hybrid enzymes during the course of the reaction (Fig. 4). The hybrid enzymes show faster protein degradation compared to the controls using wild-type (WT) heme domain with and without one equivalent of [Ru(bpy)3]2+. Some hybrid enzymes are stable only for 30 min under the current photocatalytic conditions (F87A/Q397C-1 and Q397C/I401P-1), while others, such as the Q109C-1 and L407C-1, are active for more than 3 h.
Fig. 4.

Degradation profiles monitored at 417 nm for the hybrid enzymes during the photochemical reaction and for the wild-type (WT) heme domain in control reactions.
3.3. Substrate binding
Binding of the type-I ligand lauric and palmitic acids to the hybrid enzymes induced the characteristic ferric spin shift with the appearance of a peak at 390 nm and a trough at 420 nm. The peak-to-trough separations in the difference spectra binding titration curves for palmitic acid are shown in Fig. 5A. For three of the mutants, the curves are typical of single site reversible binding of substrate and were fitted using a rectangular hyperbolic function. The determined binding constants for Q109C-1, F87A/Q397C-1 and Q397C-1 are 2.4, 2.7 and 2.8±0.1 μM, respectively. However, different binding curves are obtained for the titration of palmitic acid to the K97C-1 and L407C-1 hybrid mutants. Such binding behavior is also observed with the parent unlabeled K97C and L407C heme domains. It is consistent with a four palmitate binding model involving additional allosteric sites, recently reported for WT P450 BM3 heme domain [32]. The binding curves could then be fitted using an equation developed previously assuming hyperbolic behavior for all sites [33]; however, it was not possible to extract accurate binding constants for these mutants because of a lack of independent variables in the fit.
Fig. 5.
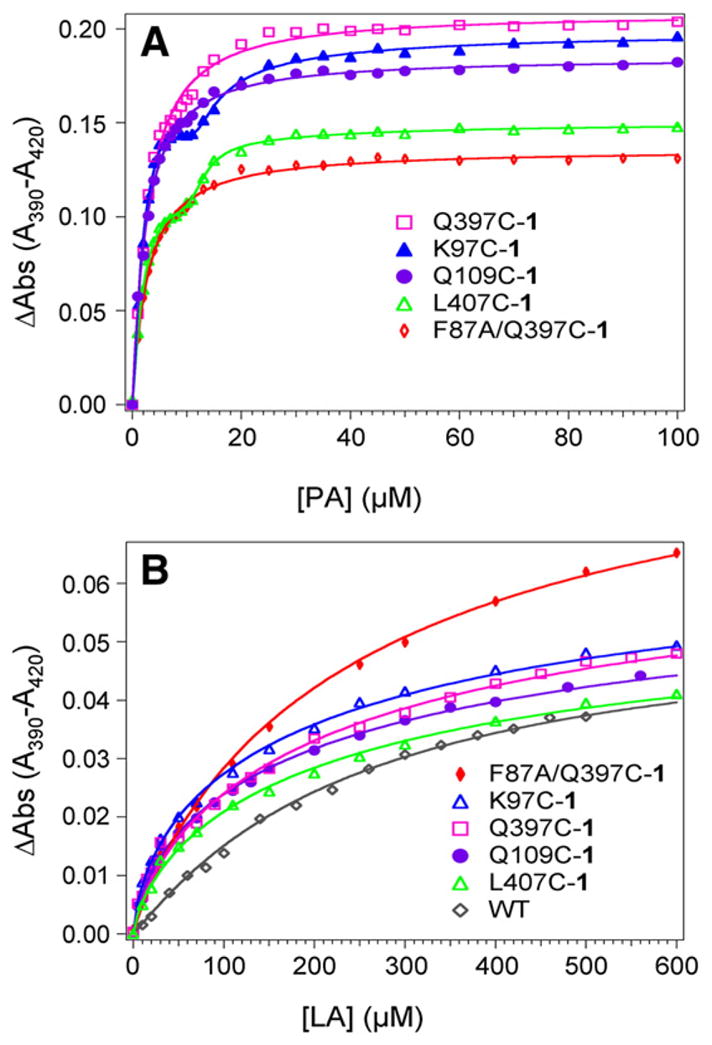
Plots of peak-to-trough separations in the difference spectra upon binding titrations of (A) palmitic acid, PA, and (B) lauric acid, LA, to 2.5 μM hybrid enzymes in 100 mM Tris at 20 °C and pH=8.2.
The binding titration curves for lauric acid (Fig. 5B) were also fitted to rectangular hyperbolic function, giving similar binding constants for all enzymes. The Kd values are 199±18 μM for Q109C-1, 214±17 μM for Q397C-1, 224±22 μM for K97C-1, 258±12 μM for F87A/Q397C-1, and 260±35 μM for L407C-1 compared to 280± 25 μM for WT.
Overall, the mutations and the covalent attachment of the Ru(II) photosensitizer do not significantly alter the binding of substrates to their respective heme domains.
3.4. CO binding study
We probed the ability of the photogenerated Ru(I) species to perform the first reduction step in the hybrid enzymes (Fig. 1, Step 2) by monitoring the formation of the characteristic Fe(II)-CO adduct at 450 nm. Typically, a degassed solution of hybrid enzymes with reductive quencher and substrate was saturated with CO atmosphere and exposed to visible light. The difference spectra in Fig. 6 show significant amounts of Fe(II)-CO formed in the hybrid enzymes upon light irradiation, while no peak at 450 nm was observed for the WT under identical conditions. When one equivalent of [Ru(bpy)3]2+ was added to the solution, only a small amount of Fe(II)-CO adduct was detected.
Fig. 6.
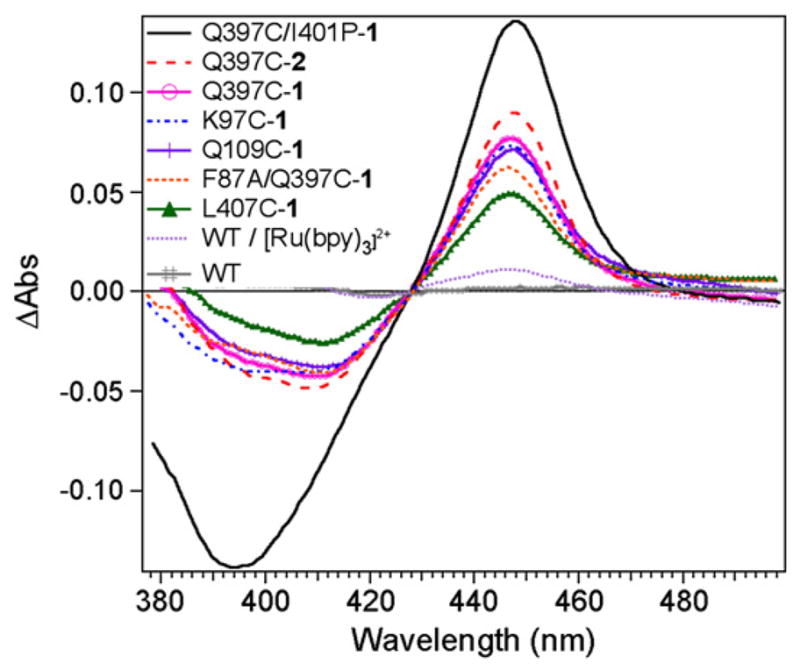
CO difference spectra showing the formation of the Fe(II)-CO adduct with characteristic absorption at 450 nm in the hybrid enzymes under photoreductive conditions and CO atmosphere. (Conditions: 2.5 μM hybrid enzymes with 150 μM palmitic acid and 50 mM DTC).
Palmitic acid is mainly used in this study as it leads to greater amounts of high spin ferric species compared to the lauric acid. While the palmitic dissociation constants for all the mutants are similar (Section 3.3), the spin shift conversion in these mutants is different and reflects small changes in the protein conformation likely due to the mutated residue and/or the presence of the photosensitizer.
In the series of non-native single cysteine mutants, the different amount of Fe(II)-CO adduct correlates well (ρ=0.996) with the percent high spin (%HS) observed in the presence of palmitic acid (Fig. 7). The obtained correlation confirms that the photogenerated Ru(I) species is capable of reducing the high spin ferric fraction.
Fig. 7.

Linear correlation between the concentration of Fe(II)-CO adduct formed upon light excitation and the percent high spin (%HS) observed upon binding of palmitic acid to various non-native single cysteine hybrid mutants (L407C-1, Q109C-1, K97C-1, Q397C-1, and Q397C/I401P-1).
In contrast, no direct correlation is observed between the %HS and the concentration of Fe(II)-CO formed for the Q397C-1, Q397C-2 and Q397C/I401P-1 mutants. For example, the hybrid enzymes Q397C-1 and Q397C-2 have the same %HS but different concentrations of Fe(II)-CO adduct are generated. A linear correlation (ρ=0.989) is obtained when plotting the [Fe(II)-CO] formed vs. the calculated ΔE (Fig. 8). The ΔE was calculated using the heme reduction potentials for the substrate-free WT, the substrate-bound WT and I401P mutant [23], and the E°(Ru(II)/Ru(I)) for [Ru(bpy)2Phen]2+ and [Ru(Phen)3]2+ [27] with respect to NHE. The correlation in the Q397C series reveals that an increase in the redox potential difference, ΔE, between the electron donor Ru(I) and the electron acceptor ferric heme species leads to larger amounts of Fe(II)-CO species formed.
Fig. 8.

Linear correlation between the concentration of Fe(II)-CO adduct formed upon light excitation and the calculated difference in redox potential (ΔE) between the electron donor and acceptor. (SF=substrate-free, LA=lauric acid and PA=palmitic acid).
3.5. Interpretation of the trends observed among the hybrid enzymes
In P450 BM3 and many bacterial P450 enzymes, substrate binding gates the first electron transfer by displacing the water molecule coordinated to the heme Fe(III) center, resulting in a low to high spin conversion and a shift to more positive redox potential. A second electron injection to the oxyferrous species coupled with protonation steps lead to the formation of the hydroperoxo intermediate and ultimately to Compound I (Fig. 1). The timely delivery of the electrons by the reductase and a well-coupled mechanism prevent the formation of reactive oxygen species via the autooxidation and peroxide shunt pathways and lead to high catalytic efficiency.
In the hybrid enzymes, the introduction of mutations and/or covalent attachment of the Ru(II) photosensitizer do not significantly alter the binding of lauric or palmitic acid (Section 3.3). Moreover, the ratio of hydroxylated products in the photocatalytic reaction is maintained relative to that observed with the holoenzyme (Fig. 3B) [23]. The only noticeable difference between the mutants occurs in the percent high spin observed upon substrate binding, especially with palmitic acid (see Section 3.3).
However, the hybrid enzymes display different catalytic activities in the light-initiated hydroxylation of lauric acid, with variation of up to 16-fold in total turnover numbers and up to five-fold in apparent specific activities and initial reaction rates (Section 3.2 and Fig. 3). They also show protein degradation faster than that of the WT heme domain controls under the photocatalyic reaction conditions (Section 3.2). This suggests that the hybrid enzyme degradation could be due to oxidative damages caused by reactive oxygen species (ROS) produced from the autooxidation and peroxide uncoupling pathways as well as from the reduction of dioxygen by the photosensitizer excited states [34]. Attempts to measure the amount of ROS have been limited by the high concentration of diethyldithiocarbamate present.
Overall, the hybrid enzymes showing large turnover numbers and slow degradation rates also have low amounts of Fe(II)-CO formed in the CO binding studies (e.g. L407C-1 and Q109C-1 in Figs. 3, 4 and 6). The observed trends in TON and hybrid enzyme degradation, in conjunction with the results from the CO binding studies, suggest differences in the degrees of uncoupling reactions during the photocatalytic cycle. These variations could arise from differences in stability of the oxyferrous species and/or in rates of electron transfer (Fig. 1, Steps 2 and 5) [35,36]. According to the semi-classical Marcus equation [37], the changes in distance and in redox potential between the Ru(II) photosensitizer and the heme center in the hybrid enzymes should result in different intramolecular electron transfer rates. Experiments to determine the electron transfer rates and stability of the oxyferrous species in the hybrid enzymes are currently ongoing in our laboratory.
4. Conclusion
We have expanded the family of hybrid enzymes by site directed mutagenesis and by modifying the nature of the Ru(II)-diimine photosensitizer, resulting in diverse catalytic activities in the hydroxylation of lauric acid upon light activation. The L407C-1 mutant shows the highest TON of 140 and the slowest degradation rate during the course of the reaction. In addition, upon light activation and in the presence of reductive quencher, the photogenerated Ru(I) species is able to reduce the fraction of high spin ferric heme species. The trends in TON, enzyme degradation and amount of Fe(II)-CO formed suggest differences in stability of the oxyferrous species or in electron transfer rates, which are the focus of ongoing research in our laboratory. These hybrid enzymes can be a valuable alternative approach for biotechnological applications using light to initiate P450 reactions.
Acknowledgments
L.C., N.H. and N.-H. T. would like to thank the National Institute of Health (GM095415) for financial support. This research was also supported by an award from Research Corporation for Science Advancement. L.C. thanks the National Science Foundation (MRI grant 0923573) and San José State University for the use of mass spectrometry facilities in the PROTEIN LAB.
Abbreviations
- %HS
percent of high spin ferric species
- Bpy
2,2′-bipyridine
- BSTFA
N,O-bis(trimethylsilyl)trifluoroacetamide
- DTC
sodium diethyldithiocarbamate
- ET
electron transfer
- GC/MS
gas chromatography–mass spectrometry
- phen
1,10-phenanthroline
- phenA
5-acetamido-1,10-phenanthroline
- TON
turnover numbers (concentration of product/enzyme concentration)
- WT
wild-type P450 BM3 heme domain
References
- 1.Ortiz de Montellano PR. Cytochrome P450: Structure, Mechanism, and Biochemistry. 3. Kluwer Academic/Plenum Publishers; New York: 2005. [Google Scholar]
- 2.Hannemann F, Bichet A, Ewen KM, Bernhardt R. Biochim Biophys Acta. 2007;1770:330–344. doi: 10.1016/j.bbagen.2006.07.017. [DOI] [PubMed] [Google Scholar]
- 3.Denisov IG, Makris TM, Sligar SG, Schlichting I. Chem Rev. 2005;105:2253–2278. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- 4.Hlavica P. Curr Drug Metab. 2007;8:594–611. doi: 10.2174/138920007781368881. [DOI] [PubMed] [Google Scholar]
- 5.Guengerich FP, Johnson WW. Biochemistry. 1997;36:14741–14750. doi: 10.1021/bi9719399. [DOI] [PubMed] [Google Scholar]
- 6.Rittle J, Green MT. Science. 2010;330:933–937. doi: 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]
- 7.Groves JT. J Inorg Biochem. 2006;100:434–447. doi: 10.1016/j.jinorgbio.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 8.Chefson A, Auclair K. Mol Biosyst. 2006;2:462–469. doi: 10.1039/b607001a. [DOI] [PubMed] [Google Scholar]
- 9.Urlacher VB, Eiben S. Trends Biotechnol. 2006;24:324–330. doi: 10.1016/j.tibtech.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 10.Bernhardt R. J Biotechnol. 2006;124:128–145. doi: 10.1016/j.jbiotec.2006.01.026. [DOI] [PubMed] [Google Scholar]
- 11.O’Reilly E, Köhler V, Flitsch SL, Turner NJ. Chem Commun. 2011;47:2490–2501. doi: 10.1039/c0cc03165h. [DOI] [PubMed] [Google Scholar]
- 12.Hlavica P. Biotechnol Adv. 2009;27:103–121. doi: 10.1016/j.biotechadv.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 13.Munro AW, Girvan HM, McLean KJ. Biochim Biophys Acta. 2007;1770:345–359. doi: 10.1016/j.bbagen.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 14.Hollmann F, Witholt B, Schmid A. J Mol Catal B. 2002;19–20:167–176. [Google Scholar]
- 15.Cirino PC, Arnold FH. Angew Chem Int Ed. 2003;42:3299–3301. doi: 10.1002/anie.200351434. [DOI] [PubMed] [Google Scholar]
- 16.Schwaneberg U, Appel D, Schmitt J, Schmid RD. J Biotechnol. 2000;84:249–257. doi: 10.1016/s0168-1656(00)00357-6. [DOI] [PubMed] [Google Scholar]
- 17.Udit AK, Arnold FH, Gray HB. J Inorg Biochem. 2004;98:1547–1550. doi: 10.1016/j.jinorgbio.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 18.Sadeghi SJ, Fantuzzi A, Gilardi G. Biochim Biophys Acta. 2011;1814:237–248. doi: 10.1016/j.bbapap.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 19.Jensen K, Jensen PE, Møller BL. ACS Chem Biol. 2011;6:533–539. doi: 10.1021/cb100393j. [DOI] [PubMed] [Google Scholar]
- 20.Zilly FE, Taglieber A, Schulz F, Hollmann F, Reetz MT. Chem Commun. 2009:7152–7154. doi: 10.1039/b913863c. [DOI] [PubMed] [Google Scholar]
- 21.Tran NH, Huynh N, Bui T, Nguyen Y, Huynh P, Cooper ME, Cheruzel LE. Chem Commun. 2011;47:11936–11938. doi: 10.1039/c1cc15124j. (1938) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ener ME, Lee YT, Winkler JR, Gray HB, Cheruzel L. Proc Natl Acad Sci USA. 2010;107:18783–18786. doi: 10.1073/pnas.1012381107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Whitehouse CJC, Bell SG, Wong LL. Chem Soc Rev. 2012;41:1218–1260. doi: 10.1039/c1cs15192d. [DOI] [PubMed] [Google Scholar]
- 24.Gray HB, Winkler JR. Proc Natl Acad Sci USA. 2005;102:3534–3539. doi: 10.1073/pnas.0408029102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dietrich M, Do TA, Schmid RD, Pleiss J, Urlacher VB. J Biotechnol. 2009;139:115–117. doi: 10.1016/j.jbiotec.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 26.Whitehouse CJC, Yang W, Yorke JA, Tufton HG, Ogilvie LCI, Bell SG, Zhou W, Bartlam M, Rao Z, Wong LL. Dalton Trans. 2011;40:10383–10396. doi: 10.1039/c1dt10098j. [DOI] [PubMed] [Google Scholar]
- 27.Hoffman MZ, Bolletta F, Moggi L, Hug GL. J Phys Chem Ref Data. 1989;18:219–544. [Google Scholar]
- 28.Cirino PC, Arnold FH. Adv Synth Catal. 2002;344:932–937. [Google Scholar]
- 29.Sullivan BP, Salmon DJ, Meyer TJ. Inorg Chem. 1978;17:3334–3341. [Google Scholar]
- 30.Castellano FN, Dattelbaum JD, Lakowicz JR. Anal Biochem. 1998;255:165–170. doi: 10.1006/abio.1997.2468. [DOI] [PubMed] [Google Scholar]
- 31.Fisher MT, Sligar SG. Biochemistry. 1987;26:4797–4803. doi: 10.1021/bi00389a029. [DOI] [PubMed] [Google Scholar]
- 32.Rowlatt B, Yorke JA, Strong AJ, Whitehouse CJC, Bell SG, Wong L-L. Protein Cell. 2011;2:656–671. doi: 10.1007/s13238-011-1082-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Denisov IG, Baas BJ, Grinkova YV, Sligar SG. J Biol Chem. 2007;282:7066–7076. doi: 10.1074/jbc.M609589200. [DOI] [PubMed] [Google Scholar]
- 34.Ruggi A, van Leeuwen FWB, Velders AH. Coord Chem Rev. 2011;255:2542–2554. [Google Scholar]
- 35.Perera R, Sono M, Raner GM, Dawson JH. Biochem Biophys Res Commun. 2005;338:365–371. doi: 10.1016/j.bbrc.2005.08.078. [DOI] [PubMed] [Google Scholar]
- 36.Ost TWB, Clark J, Mowat CG, Miles CS, Walkinshaw MD, Reid GA, Chapman SK, Daff S. J Am Chem Soc. 2003;125:15010–15020. doi: 10.1021/ja035731o. [DOI] [PubMed] [Google Scholar]
- 37.Marcus RA, Sutin N. Biochim Biophys Acta. 1985;811:265–322. [Google Scholar]