

Abstract
To reduce shrinkage stress which arises during the polymerization of crosslinked polymers, allyl sulfide functional groups were incorporated into methacrylate polymerizations to determine their effect on stress relaxation via addition-fragmentation chain transfer (AFCT). Additionally, stoichiometrically balanced thiol and allyl sulfide-containing norbornene monomers were incorporated into the methacrylate resin to maximize the overall functional group conversion and promote AFCT while also enhancing the polymer’s mechanical properties. Shrinkage stress and reaction kinetics for each of the various functional groups were measured by tensometry and Fourier-transform infrared (FTIR) spectroscopy, respectively. The glass transition temperature (Tg) and elastic moduli (E′) were measured using dynamic mechanical analysis. When the allyl sulfide functional group was incorporated into dimethacrylates, the polymerization-induced shrinkage stress was not relieved as compared with analogous propyl sulfide-containing resins. These analogous propyl sulfide containing monomers are incapable of undergoing AFCT while having similar chemical structure and crosslink density to the allyl sulfide containing methacrylates. Here, a monomethacrylate monomer that also contains a cyclic allyl sulfide (PAS) was found to increase the crosslinking density nearly 20 times as compared to an analogous monomethacrylate in which the allyl sulfide was replaced with an ethyl sulfide. Despite the much higher crosslink density, the PAS formulation exhibited no concomitant increase in stress. Thiol-norbornene resins were copolymerized in PAS to promote AFCT as well as to synergistically combine the ring opening benefits associated with the thiol-ene reaction. AFCT resulted in a 63% reduction of polymerization stress and a 45°C enhancement of the glass transition temperature in the allyl sulfide-containing thiol-norbornene-methacrylate system compared with rubbery dimethacrylates. When compared with conventional glassy dimethacrylates, this combined system has less than 10% of the typical shrinkage stress level while having similarly excellent mechanical properties.
Introduction
Polymerization shrinkage stress is a deleterious phenomenon that plagues numerous polymer material applications1,2,3 and results in warping, microcracks, and material failure. Polymerization shrinkage itself results from the decrease in molecular spacing between monomers as the van der Waals interactions are replaced with covalent bonds, bringing the monomer units more closely together. Previously, various approaches have been used to combat polymerization stress including incorporation of materials that exhibit low polymerization shrinkage, such as ring-opening monomers4 or formulations that undergo polymerization induced phase separation.5 Additionally, strategies that in which the gel-point conversion is delayed, such as thiol-ene6 and thiol-yne7,8,9 polymerizations, have been used to enable the material to shrink in the early stages of the reaction without leading to any significant stress accumulation. Under these circumstances, prior to the gel-point, a material is capable of dissipating shrinkage via the viscous flow of the monomer/oligomer species; however, after gelation, molecular rearrangement is reduced and stress accumulates. To address this issue, we have explored a methodology utilizing reversible covalent bond structures to enable the rearrangement of network strands. This approach facilitates an elastic to plastic transition in behavior during the polymerization that provides a route for post-gel-point network stress relaxation through accommodation of the shrinkage. Specifically, we have utilized addition-fragmentation chain transfer (AFCT) to enable non-degradative radical-mediated breaking and reforming of polymer network strands throughout the polymerization.10,11,12 The initial demonstrations of reduced polymerization shrinkage stress using AFCT employed the allyl sulfide functional group in both thiol-ene11 and thiol-yne13 materials. These systems have the additional advantages of possessing delayed gelation as well as negligible oxygen inhibition.6,14 While these systems demonstrated decreased polymerization stress, the crosslinking density of these step-growth polymers was necessarily lower than their chain growth counterparts, leading to a lower crosslink density, glass transition temperature, and modulus, which precluded their use in many applications.
Methacrylate-based materials are widely used in a variety of applications, such as electrical materials15, optical materials, and dental restorative materials16, as they are typically lightweight, possess a high modulus, exhibit good impact-strength, and are transparent. Unfortunately, the rapid polymerization of multifunctional methacrylate (or acrylate) monomers generates a large amount of stress, on the order of megapascals17, owing to the typical chain-growth polymerization characteristics, particularly early gelation and the ultimate formation of a glassy, highly crosslinked polymer.18,19 To achieve enhanced mechanical properties as well as the stress relaxation capabilities of the AFCT mechanism, thiol-ene resins containing allyl sulfide moieties were incorporated into dimethacrylate resins20 to induce stress relaxation. As expected, the increased methacrylate content enhances the mechanical properties such as the glass transition temperature and elastic modulus.21,22 While stress relaxation was observed, the effect was significantly reduced or even eliminated for materials possessing a super-ambient glass transition temperature due to a limited allyl sulfide concentration which was incorporated only into the thiol-ene component of the resin.
In contrast, here, we incorporate the allyl sulfide functional group directly into the methacrylate monomer to investigate its effect on stress relaxation. In addition, tetrafunctional thiol and allyl sulfide-containing difunctional norbornene monomers were formulated into the methacrylate resin to enhance further the mechanical properties and the capability for undergoing AFCT.
Experimental section
Materials
Chemical structures of materials utilized in this study are shown in Figure 1.
Figure 1.

Materials used: (1) SAS, (2) SPS, (3) PAS, (4) PES, (5) NAS, (6) NPS, (7) PETMP, and (8) HCPK.
Synthesis of 2-Methylenepropane-1,3-bis(2-[2-methacryloyloxyethyl succinyl]ethyl sulfide) (SAS, Succinate Allyl Sulfide)
To synthesize the desired molecular precursor, 2-methylenepropane-1,3-bis(2-hydroxyethyl sulfide), 2-mercaptoethanol (25.60 g, 0.328 mol, Alfa Aesar) was dissolved in 100 ml ethanol in a 500 ml 2-neck round-bottom flask equipped with a magnetic stirring bar, condenser and a dropping funnel. With vigorous stirring, sodium metal (8.20 g, 0.356 mol, Alfa Aesar) was added slowly in small pieces to minimize the reaction exotherm. After complete addition of the sodium metal, the mixture was stirred under a nitrogen blanket until the flask temperature returned to ambient. A solution of 3-chloro-2-chloromethyl-1-propene (20 g, 0.16 mol, Secant Chemicals Inc.) in 50 mL ethanol was added drop-wise to give a white cloudy mixture that transitioned to a white solid by the time all the dichloropropene component had been added. The flask contents were then refluxed for 45 minutes and cooled to ambient temperature. The white solid was removed by vacuum filtration and the filter cake was washed with excess ethanol. The solvent was removed in a rotary evaporator followed by drying using a vacuum pump to give a colorless liquid. If required, vacuum distillation (6–7 torr) at 135–150 °C can be used for purification of the crude product.
The synthesized 2-methylenepropane-1,3-bis(2-hydroxyethyl sulfide) (10.6 g, 0.051 mol) was dissolved in 100 ml ethylacetate in a 500 ml 3-neck round bottom flask equipped with a dropping funnel on one arm, a magnetic stirring bar and a dry air blanket. To the solution was added mono-methacryloyloxyethylsuccinic acid (25 g, 0.108 mol, Sigma Aldrich), followed by N,N-dimethylamino pyridine (DMAP, 1.35 g, 0.011 mol, Alfa Aesar). The mixture was cooled in an ice bath with continuous stirring. In a separate flask, dicyclohexyl carbodiimide (DCC, Alfa Aesar) was dissolved in 150 ml ethylacetate. The DCC solution was placed in the dropping funnel and added drop-wise to the flask as the flask contents were kept cold (0 – 10 °C). After complete addition of the DCC solution, the flask contents were continuously stirred in the ice bath for 30 minute then at ambient temperature overnight. The following day, 50 ml of hexanes were added and all solids formed in the reaction flask were removed and washed by vacuum filtration. The filtrate was then extracted with a mixture of 100 ml 1 N HCl, 150 mL 10% aqueous sodium bicarbonate and 100 ml water. The organic layer was dried (Na2SO4) then concentrated in a rotary evaporator followed by drying to give a light yellow liquid in 90% yield. The structure was confirmed by 1H NMR spectroscopy (Varian INOVA 500) (see Supporting Information).
Synthesis of 2-Methylpropane-1,3-bis(2-[2-methacryloyloxyethyl succinyl]ethyl sulfide) (SPS, Succinate Propyl Sulfide)
To synthesize 2-methylpropane-1,3-bis(2-hydroxyethyl sulfide), crude 2-methylpropane-1,3-bis(2-hydroxyethyl sulfide) was prepared from 2-methyl-1-bromo-3-chloropropane (9.57g, 0.169 mol, Sigma Aldrich), mercaptoethanol (26.44 g, 0.339 mol, TCI America) and sodium metal (8.6 g, 0.374 mol, Lancaster Synthesis Inc.) in a method similar to the one described for methylenepropane-1,3-bis(2-hydroxyethyl sulfide) above. Vacuum distillation at 6–8 torr and 140–160 °C gave a colorless liquid with 77.5% recovery.
The mono-HEMA succinic acid intermediate was made in situ then used in the second step in a one-pot fashion. 2-Hydroxyethyl 2-methylprop-2-enoate (HEMA, 10 g, 0.084 mol, Sigma Aldrich) and succinic anhydride (9.35 g, 0.094 mol, Sigma Aldrich) were charged into a round bottom flask equipped with a magnetic stirring bar, a dry air blanket and dropping funnel. DMAP (1.15 g, 0.009 mol) and butylated hydroxytoluene (BHT, 0.035 g, 0.16 mmol) were added to the flask. The mixture was heated with stirring at 90 °C for 5 hours to give a colorless, viscous liquid, and the flask was left to cool to ambient temperature. Ethyl acetate (approximately 50 ml) was subsequently added and the mixture was cooled in an ice bath to 0–5°C. Charged into the dropping funnel was a solution of DCC (17.3 g, 0.089 mol, Alfa Aesar) in 50 ml ethylacetate. The DCC solution was carefully and slowly added to the vigorously stirred cold solution over 30 minutes, and the mixture was stirred at 0–5°C for 30 minutes then at ambient temperature overnight. The following day, the reaction was worked up similarly to SAS above, and the structure was confirmed by 1H NMR spectroscopy (Varian INOVA 500) (see Supporting Information).
Synthesis of 2-(methacryloyloxyethyl) 7-methylene-1,5-dithiocan-3-yl phthalate (PAS, Phthalate Allyl Sulfide) and 2-(methacryloyloxyethyl) 7-methyl-1,5-dithiocan-3-yl phthalate (PES, Phthalate Ethyl Sulfide)
PAS and PES were synthesized following the procedure described in the literature.23 Mono-2-methacryloyloxyethyl phthalate (9.4g, 31 mmol, Sigma Aldrich) and C-8 alcohol (5.38g, 31 mmol), DMAP (400 mg), DCC (6.95g, 34 mmol, Alfa Aesar), and methylene chloride (50ml) were utilized. The structures of products were confirmed by 1H NMR spectroscopy (Varian INOVA 500) (see Supporting Information).
Synthesis of 2-Methylene-propane-1,3-di(norbornene sulfide) (NAS, Norbornene Allyl Sulfide)
5-bromomethyl norbornene was synthesized from 1,3-dicyclopentadiene (16g, 0.12mol, ACROS), allyl bromide (35g, 0.29mol, Sigma Aldrich), and hydroquinone (81mg, 0.74mmol, Aldrich) by adding them in a 100 ml pressure vessel followed by heating at 170 °C for 12 hours. The crude oil was purified with normal distillation at 175 °C. 3-mercapto-2-(mercaptomethyl)-1-propene was synthesized according to the method described in the literature.11,24,25 Then, a slightly diluted solution of 19 g (172.4 mmol) of 5-bromomethyl norbornene and 10.3 g (86.2 mmol) of 2-methyl-1,3-dimercaptopropene in methanol was prepared and added to a refluxing solution of sodium (5g, 215.5 mmol) in 300ml of methanol under argon protection. The following day, methanol was dried with vacuum and the dried crude oil was purified using liquid-liquid water-ether solution. The product extracted in the ether phase was dried and vacuum distilled at 230°C and 0.3 mm Hg to give a waxy/hazy solid. The structure was confirmed by 1H NMR spectroscopy (Varian INOVA 500) (see Supporting Information).
Synthesis of 2-Methyl-propane-1,3-di(norbornene sulfide) (NPS, Norbornene Propyl Sulfide)
NPS was synthesized from 5-bromomethyl norbornene with 1,3-dimercapto-2-methylpropane with the same synthetic procedure of NAS. 3-dimercapto-2-methylpropane was synthesized following the procedure in the literature.11 A viscous/colorless liquid (NPS) was given after vacuum purification at 200°C and 0.3 mmHg, and its structure was confirmed by 1H NMR spectroscopy (Varian INOVA 500) (see Supporting Information).
Tetrathiol (pentaerythritol tetrakis(3-mercaptopropionate) (PETMP, Evans Chemetics), 1-hydroxycyclohexylphenylketone (HCPK, Ciba Specialty Chemicals), and sodium metal (Sigma Aldrich) were provided or purchased, and no further purification was performed. All resin mixtures were produced based on a desired functional group ratio. For SAS-PETMP, SPS-PETMP resins, the functional group ratio (methacylate : thiol) was 7 to 3. NAS-PETMP-PAS and NPS-PETMP-PAS were stoichiometically balanced, implying that the ratio of norbornene to thiol to methacrylate was 1 : 1 : 1. Throughout this study, all resins includes 1 wt% of HCPK as a UV-activated photoinitiator.
Methods and Equipment
The shrinkage stress and functional group conversion were simultaneously observed during polymerization using a tensometer coupled with a FTIR spectrometer (Nicolet 670), which was equipped with near-infrared transmitting optical fiber patch cables and an indium gallium arsenide (InGaAs) detector.6, 26 The tensometer was developed by the Paffenbarger Research Center (American Dental Association Health Foundation) to measure the stress during photopolymerization. Stress evolution is measured by the cantilever beam deflection that is detected by the LVDT (linear variable displacement transducer) displacement. In the tensometer, one glass rod is fixed to the bottom plate and another rod is connected to the cantilever beam. The formulated resin was injected between the two glass rods, which results in a specimen geometry of 6 mm diameter and 1mm thickness. Samples were irradiated with 365 nm filtered UV light at 10 or 50mW/cm2 (Acticure 4000, EXPO) for 5 or 16 minutes. PAS and PES samples were exposed to the higher light intensity because they polymerize much more slowly otherwise than the remainder of the monomers considered here. Evolution of the methacrylate, norbornene and allyl sulfide double bond concentrations were determined by monitoring the infrared absorption peaks centered at 6164 cm−1 (C=C-H stretching, overtone), 6111 cm−1 (C=C-H stretching, overtone), and 6121 cm−1 (C=C-H stretching, overtone), respectively. As the methacrylate peak is overlapped with the norbornene and allyl sulfide peaks, Gaussian fitting was used to deconvolute these peak areas. See the supporting information for additional detail on the infrared spectra and the conversion measurement.
The elastic moduli (E′) and glass transition temperatures (Tgs) of polymerized samples were measured by dynamic mechanical analysis (DMA, TA Instruments Q800). Specimens (25mm * 5mm * 1mm) for DMA were prepared and irradiated under the identical conditions as performed in the tensometer experiment. DMA experiments were performed at a constant strain and frequency of 0.1% and 1 Hz, respectively, scanning the temperature from −50°C to 140°C twice at 1°C/minute; the temperature scan was repeated to ensure the absence of dark polymerization at temperatures greater than the Tg.27–28
Results and Discussion
The allyl sulfide functional group was incorporated into two different monomers containing methacrylate functionalities: a symmetric dimethacrylate with a central allyl sufide functional group (succinate-based allyl sulfide, SAS) and an asymmetric monomethacrylate with an allyl sulfide containing ring that participates in the polymerization through a ring-opening reaction (phthalate-based allyl sulfide, PAS). Both monomers were designed to undergo radical-mediated AFCT during polymerization while PAS has the ring opening functional group for further stress reduction and the stiff benzene ring structure to promote a higher glass transition temperature than SAS. To isolate the effects of AFCT on stress relaxation, analogous monomers were synthesized that possess a nearly identical molecular structure; however, each “control” monomer does not contain an allyl sulfide and is thus incapable of undergoing AFCT. These monomers were created such that, to the greatest extent possible, differences in the polymerization stress development could be attributed to the plasticity enabled by the AFCT mechanism for stress relaxation. Specifically, the allyl sulfides in SAS and PAS were replaced by propyl sulfide (SPS) or ethyl sulfide (PES), respectively.
The comparison between the conversion and stress evolution in the methacrylate monomers is shown in Figure 2. When the allyl sulfide was incorporated into the dimethacrylate-based monomer (SAS), the stress evolution is nearly identical when compared with SPS, indicating that the AFCT mechanism is not promoting plasticization under these circumstances. A possible explanation for this behavior is that the allyl sulfide exhibits reduced reactivity towards carbon-centered radicals such as those involved in the methacrylate polymerization. Further, the reaction of a carbon-centered radical with the allyl sulfide is not a reversible reaction as the product contains an allylic group that is distinct from the initial allyl sulfide (Scheme 1 (B)). Ultimately, both of these explanations would lead to reduced capability for undergoing chain cleavage and reformation, as is necessary to promote stress relaxation. These explanations would also result in a low conversion of the allyl sulfide in SAS (Table 1) and lead to SAS and SPS exhibiting similar mechanical properties (Figure 3), as observed. To explore this possibility further, a tetra-functional thiol (PETMP) was formulated into these resins to introduce thiyl radicals, which have been shown to have excellent reactivity towards allyl sulfides and thus promote AFCT in a fully reversible manner, as shown in scheme 1 (A).11 Interestingly, the addition of PETMP resulted in an enhanced shrinkage stress for the allyl sulfide-containing resins compared with the propyl sulfide-containing resins (Figure 2(B)) although stresses of both systems were reduced compared with those of SAS or SPS due to the addition of PETMP. This outcome is attributed to the enhanced crosslinking density of the SAS-PETMP resin that results from the higher conversion of the allyl sulfide functional group (33%), which, under these circumstances, leads to additional crosslinking in the allyl sulfide containing system.
Figure 2.

(A) Methacrylate conversion and (B) shrinkage stress versus time for PAS(□) (allyl sulfide-based monomethacrylate), PES(■) (ethyl sulfide-based monomethacrylate), SAS(○) (allyl sulfide-based dimethacrylate), SPS(●) (propyl sulfide-based dimethacrylate), SAS-PETMP(△), and SPS-PETMP(▲). Samples were formulated with 1 wt % HCPK as the photoinitiator. All resins except PAS and PES (50mW/cm2) were irradiated with 365 nm light at 1 mW/cm2 for 16 min.
Scheme 1.

(A) Reversible AFCT is mediated by reaction with the thiyl radical while (B) irreversible allyl sulfide AFCT is mediated by reaction with a carbon-centered radical.
Table 1.
Summary of the final functional group conversions, Tg, and elastic moduli. Rubbery moduli are measured at the same temperature for both the allyl sulfide-based resin and its analogous non-allyl sulfide containing resin. When the Tgs are different for these formulations, the rubbery modulus is taken at the higher of the two glass transition temperatures plus 40°C.
| Vinyl ether monomer used in the ternary system (thiol:vinyl ether:methacrylate ratio) | Final functional group conversion [%]
|
Stress (MPa) | Tg (°C) | Rubbery elastic modulus at T = Tg + 40°C (MPa) | Elastic modulus at T = 25°C (MPa) | ||
|---|---|---|---|---|---|---|---|
| Methacrylate | Norbornene | Allyl sulfide | |||||
| SAS | 84 ± 0.8 | - | 10 ± 1.5 | 0.55 ± 0.004 | 24 ± 1 | 19 ± 0.2 | 86 ± 7 |
| SPS | 92 ± 0.5 | - | - | 0.57 ± 0.004 | 20 ± 1 | 18 ± 0.1 | 55 ± 2 |
|
| |||||||
| PAS | 77 ± 0.2 | - | 16 ± 0.2 | 0.36 ± 0.01 | 102 ± 0.7 | 35 ± 0.3 | 2490 ± 500 |
| PES | 82 ± 0.7 | - | - | 0.33 ± 0.01 | Below 80 ± 4 | 2 ± 0.8 | 1870 ± 300 |
|
| |||||||
| NAS-PETMP-PAS | 100 ± 0.1 | 64 ± 0.5 | 21 ± 0.9 | 0.21 ± 0.01 | 65 ± 1 | 19 ± 0.5 | 2140 ± 400 |
| NPS-PETMP-PAS | 100 ± 0.1 | 70 ± 0.8 | 5 ± 0.2 | 0.28 ± 0.01 | 65 ± 2 | 14 ± 0.4 | 1900 ± 400 |
Figure 3.
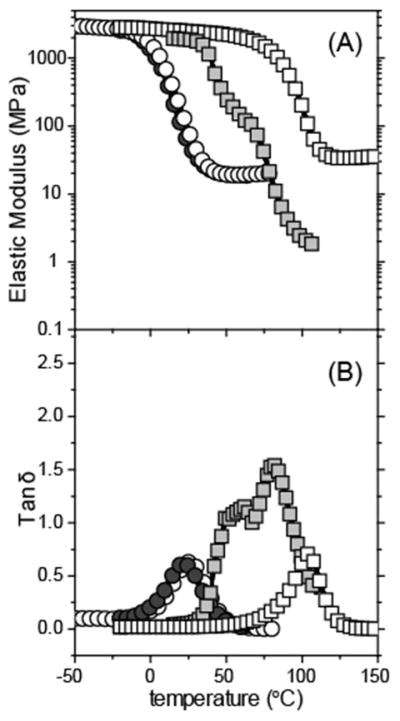
(A) Elastic modulus and (B) tand versus temperature for PAS(□) (allyl sulfide-based monomethacrylate), PES(■) (ethyl sulfide-based monomethacrylate), SAS(○) (allyl sulfide-based dimethacrylate), and SPS(●) (propyl sulfide-based dimethacrylate). Samples were formulated with 1 wt % HCPK. SAS and SPS were irradiated with 365 nm light at 1 mW/cm2 for 16 min. PAS and PES were irradiated with 365 nm light at 50 mW/cm2 for 16 min.
The shrinkage stress of the phthalate allyl sulfide-based methacrylate (PAS) resin is lower than in the succinate allyl sulfide-based methacrylate resin (SAS). However, as shown in Figure 2(B), the phthalate ethyl sulfide methacrylate (PES) demonstrated similar stress evolution behavior to the the allyl sulfide-containing material (PAS) which serves as a molecular control. Despite serving as a molecular control, the PES by its nature is significantly different both in how it polymerizes and in enabling AFCT. While the molecular structures of the two monomers (PAS and PES) are nearly identical, the allyl sulfide functional group in this monomer uniquely acts both to promote AFCT and as an additional polymerizable functional group through a ring opening reaction. This ring opening reaction occurs during the first allyl sulfide AFCT reaction of the monomer and leads to crosslinking in this material. Thus, the PES monomer is not capable of AFCT but also not capable of crosslinking, leading to dramatic differences in the crosslink density, modulus, and inherent shrinkage stress that will arise. These effects are clearly seen in the material properties for these two resins (see Figure 3) as well, where PES does not exhibit a clearly defined rubbery modulus but instead the behavior more closely resembles that of a very loosely crosslinked polymer melt. This outcome suggests that, as expected, the ethyl sulfide ring in the PES structure does not undergo AFCT but also does not significantly react to form crosslinks. Nevertheless, it should be noted that the two materials have nearly identical polymerization stresses, indicating that the additional crosslinking and higher modulus resulting from the crosslinking of the allyl sulfide monomer are counterbalanced by AFCT. Overall, this combination leads to enhanced mechanical properties and behavior with no concomitant stress increase.
As discussed above, the addition of thiol-ene monomers into a methacrylate resin can have the effect of enhancing stress reduction by promoting allyl sulfide-mediated AFCT in the methacrylate resins.20 As our goal is to have a low-stress material possessing a super-ambient Tg, a stoichiometrically balanced mixture of thiol-ene monomers (ene functional group : thiol functional group = 1:1) was incorporated into the phthalate-based methacrylate (PAS), since its Tg is significantly higher than those of the SAS and SPS resins. Further, to maximize the amount of allyl sulfide and thus the AFCT reaction in the crosslinked network, a dinorbornene (i.e., ‘diene’) monomer was synthesized. The dinorbornene allyl sulfide (NAS) monomer was compared with a propyl sulfide-containing norbornene (NPS) to isolate the stress relaxation effect of the allyl sulfide functional group. Thus, by utilizing PAS with thiol-NAS and thiol-NPS, the effect of the allyl sulfide’s concentration in this ternary resin is elucidated. In each of the two combinations presented here, the norbornene, thiol, and methacrylate functional group were stoichiometrically balanced (norbornene : thiol : methacrylate = 1:1:1), though it is not necessary to maintain a 1:1 ratio of methacrylate to thiol-ene formulation.
The reaction kinetics in these resins exhibited similar behavior whether the allyl sulfide or propyl sulfide-based norbornene was included, where the methacrylate functional group proceeds to nearly complete conversion at a more rapid rate than the thiol-ene component (Figure 4(A)). A relatively low final norbornene conversion was observed, likely resulting from the reduced molecular mobility associated with vitrification (Table 1) that arises at the end of these polymerizations. The allyl sulfide conversion was limited to at most 20% conversion (Table 1), indicative of a largely reversible AFCT reaction as desired to minimize the stress that develops. In particular, the allyl sulfide functional group was only negligibly consumed when present only in the methacrylate monomer (i.e., the NPS-PETMP-PAS resin). As the rubbery modulus of the PAS is much greater than PES (i.e., without the ring opening component), the allyl sulfide is being incorporated into the network via ring-opening, but it is not being consumed by irreversible side reactions. Since the AFCT mechanism results in ring-opening while also preserving the allyl sulfide functional group (see scheme 1A), we conclude that stress relieving AFCT must occur subsequent to the ring opening reaction in order to promote plasticity and stress relaxation during the polymerization. This outcome is further evidenced by NAS-PETMP-PAS having lower polymerization stress than NPS-PETMP-PAS, which has a reduced allyl sulfide content in the formulation (i.e., NPS versus NAS). Since these two ternary resins form very similar polymer networks as demonstrated in Figure 5, the reduced stress in the NAS-PETMP-PAS system results from stress relaxation by AFCT. In addition, NAS-PETMP-PAS has a higher crosslink density (Table 1), which in the absence of AFCT would lead to larger polymerization stress. The higher stress is not observed because of the AFCT-induced rearrangement of the network.
Figure 4.

(A) Methacrylate conversion and (B) shrinkage stress versus time for stoichiometric mixtures of NAS-PETMP-PAS(○) (functional group ratio norbornene : thiol : methacrylate = 1:1:1) and NPS-PETMP-PAS(●) (functional group ratio norbornene : thiol : methacrylate = 1:1:1). Samples were formulated with 1 wt % HCPK, and irradiated with 365 nm light at 50 mW/cm2 for 5 min.
Figure 5.

(A) Elastic modulus and (B) tan δ versus temperature for stoichiometrically balanced mixtures of NAS-PETMP-PAS(○) (functional group ratio norbornene : thiol : methacrylate = 1:1:1) and NPS-PETMP-PAS(●) (functional group ratio norbornene : thiol : methacrylate = 1:1:1). Samples were formulated with 1 wt % HCPK, and irradiated with 365 nm at 50 mW/cm2 for 5 min.
Amazingly, despite the formation of glassy, crosslinked polymers that would be expected to limit the amount of relaxation, this approach to stress reduction was highly successful at alleviating stress relative to almost any other approach. In particular, the allyl sulfide-incorporated norbornene-PETMP-PAS systems demonstrated 63% reduced stress as compared to the dimethacrylate resin (SPS) while achieving higher functional group conversions and exhibiting glass transition temperatures that were as much as 40°C higher than their high stress counterparts. Comparisons with conventional high Tg dimethacrylates are even more astonishing as the NAS-PETMP-PAS system demonstrated more than a factor of 10 lower stress level as compared with that previously reported for conventional dimethacrylates (2.9MPa)6 while simultaneously achieving excellent mechanical properties. Thus, this resin system is an outstanding candidate for applications in which the stress level is critical such as electronic materials, optical materials, and dental materials.
Conclusions
Stress relaxation during the photopolymerization of monomers containing allyl sulfide functional groups was evaluated in methacrylate and thiol-norbornene-methacrylate systems. The allyl sulfide’s capability to undergo AFCT resulted in a decrease in polymerization stress when compared with analogous materials incapable of undergoing AFCT while also having similar crosslink densities. Although AFCT did not relieve stress when the allyl sulfide functionality was incorporated directly into dimethacylates, the ring opening allyl sulfide-based methacrylate (PAS) increased the crosslinking density nearly 20 times and resulted in a Tg increase of up to 20°C without a concomitant increase in stress as compared with the ethyl sulfide-based methacrylate (PES). PAS was then copolymerized in a thiol-norbornene resin (NAS-PETMP) to facilitate AFCT as well as to incorporate the benefits associated with the thiol-ene reaction. The allyl sulfide functional group in the NAS-PETMP-PAS system resulted in a factor of 3 lower stress as compared to the dimethacrylate resin (SPS) while achieving a higher Tg by up to 40°C and excellent conversion. PAS and NAS-PETMP-PAS systems demonstrate more than a factor of 10 lower stress as compared with conventional dimethacrylates6 while also exhibiting excellent mechanical properties.
Supplementary Material
Acknowledgments
This investigation was supported by NIDCR 2 R01 DE-010959-11 from the National Institutes of Health and NSF 0933828.
References
- 1.Drury CJ, Mutsaers CMJ, Hart CM, Matters M, de Leeuw DM. Applied Physics Letters. 1998;73:108–110. [Google Scholar]
- 2.Bowman CN, Cramer NB, Stansbury JW. J Dent Res. 2011;90:402–416. doi: 10.1177/0022034510381263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yoffe AD. Advances in Physics. 2001;50:1–208. [Google Scholar]
- 4.Alcoutlabi M, McKenna GB, Simon SL. Journal of Applied Polymer Science. 2003;88:227–244. [Google Scholar]
- 5.Lu H, Trujillo-Lemon M, Ge J, Stansbury JW. Compend Contin Educ Dent. 2010;31(Spec No 2):1–4. [PubMed] [Google Scholar]
- 6.Lu H, Carioscia JA, Stansbury JW, Bowman CN. Dent Mater. 2005;21:1129–1136. doi: 10.1016/j.dental.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 7.Chan JW, Shin J, Hoyle CE, Bowman CN, Lowe AB. Macromolecules. 2010;43:4937–4942. [Google Scholar]
- 8.Fairbanks BD, Scott TF, Kloxin CJ, Anseth KS, Bowman CN. Macromolecules. 2009;42:211–217. doi: 10.1021/ma801903w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lowe AB, Hoyle CE, Bowman CN. Journal of Materials Chemistry. 2010;20:4745–4750. [Google Scholar]
- 10.Kloxin CJ, Scott TF, Adzima BJ, Bowman CN. Macromolecules. 2010;43:2643–2653. doi: 10.1021/ma902596s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kloxin CJ, Scott TF, Bowman CN. Macromolecules. 2009;42:2551–2556. doi: 10.1021/ma802771b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scott TF, Schneider AD, Cook WD, Bowman CN. Science. 2005;308:1615–1617. doi: 10.1126/science.1110505. [DOI] [PubMed] [Google Scholar]
- 13.Park HY, Kloxin CJ, Scott TF, Bowman CN. Macromelecules. 2010;43:10188–10190. doi: 10.1021/ma1020209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carioscia JA, Lu H, Stanbury JW, Bowman CN. Dent Mater. 2005;21:1137–1143. doi: 10.1016/j.dental.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 15.Kloosterboer JG. Advances in Polymer Science. 1988;84:1–61. [Google Scholar]
- 16.Peutzfeldt A. European Journal of Oral Sciences. 1997;105:97–116. doi: 10.1111/j.1600-0722.1997.tb00188.x. [DOI] [PubMed] [Google Scholar]
- 17.Kleverlaan CJ, Feilzer AJ. Dent Mater. 2005;21:1150–1157. doi: 10.1016/j.dental.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 18.Labella R, Lambrechts P, Van Meerbeek B, Vanherle G. Dent Mater. 1999;15:128–137. doi: 10.1016/s0109-5641(99)00022-6. [DOI] [PubMed] [Google Scholar]
- 19.Walls AWG, Mccabe JF, Murray JJ. Journal of Dentistry. 1988;16:177–181. doi: 10.1016/0300-5712(88)90032-2. [DOI] [PubMed] [Google Scholar]
- 20.Park HY, Kloxin CJ, Scott TF, Bowman CN. Dent Mater. 2010;26:1010–1016. doi: 10.1016/j.dental.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee TY, Carioscia J, Smith Z, Bowman CN. Macromolecules. 2007;40:1473–1479. [Google Scholar]
- 22.Lee TY, Smith Z, Reddy SK, Cramer NB, Bowman CN. Macromolecules. 2007;40:1466–1472. [Google Scholar]
- 23.Abuelyaman Ahmed S, SBM, Lewandowski Kevin M, Plaut David J. Dental compositions containing hybrid monomers. 3M Innovative Properties Company; US: 2011. [Google Scholar]
- 24.Evans RA, Rizzardo E. Macromolecules. 2000;33:6722–6731. [Google Scholar]
- 25.Evans RA, Rizzardo E. J Polym Sci Pol Chem. 2001;39:202–215. [Google Scholar]
- 26.Lu H, Stansbury JW, Dickens SH, Eichmiller FC, Bowman CN. Journal of Materials Science-Materials in Medicine. 2004;15:1097–1103. doi: 10.1023/B:JMSM.0000046391.07274.e6. [DOI] [PubMed] [Google Scholar]
- 27.Ferrillo RG, Achorn PJ. Journal of Applied Polymer Science. 1997;64:191–195. [Google Scholar]
- 28.Li G, Lee-Sullivan P, Thring RW. Journal of Thermal Analysis and Calorimetry. 2000;60:377–390. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.