

Abstract
In this issue, Soufir et al. report a founder mutation in the XPC DNA repair gene in 74% of families with xeroderma pigmentosum (XP) in the Maghreb region (Algeria, Morocco, and Tunisia) of northern Africa. These patients have a high frequency of skin cancer. The presence of this founder mutation provides an opportunity for genetic counseling and early diagnosis of XP.
Genetics of xeroderma pigmentosum
Xeroderma pigmentosum (XP) is a rare autosomal recessive genodermatosis with a markedly elevated risk of developing sunlight-induced cancers of the skin and eyes (Kraemer et al., 2007). XP is caused by mutations in DNA repair genes that protect cells from UV-induced DNA damage. There are seven XP complementation groups (XP-A to XP-G) representing nucleotide excision repair genes that are mutated in patients with X P. An additional “variant” form (XPV) has a defect in polymerase eta that bypasses DNA photoproducts (Kraemer et al., 2007).
XP is found in all races and throughout the world, with a frequency that ranges from about 1 per million in Europe and the United States (Kleijer et al., 2008) to 1 per 22,000 in Japan (Hirai et al., 2006). In this issue, Soufir et al. (2010) report a large genetic analysis of 86 patients with XP in 66 unrelated families from the Maghreb region (Algeria, Morocco, and Tunisia) of North Africa. They reported that 85% of families had mutations in the XPC gene and 12% had mutations in the XPA gene. Importantly, 74% of families had the same homozygous two-base deletion (c.1643_1644delTG) in the XPC gene that leads to premature termination of the XPC protein. This is in accord with reports from the same region of this homozygous mutation in 100% of 14 XP families from different parts of Tunisia (Ben et al., 2009), in two other African patients with XP (Khan et al., 2006), and in a Sudanese XP family (Mahindra et al., 2008).
Mutation hotspot or founder mutation?
Why was the same mutation present in so many patients? In theory, the mutations could have arisen independently. The localized DNA sequence in the XPC gene might be genetically unstable and produce a mutation “hotspot” (Figure 1a and Table 1). Indeed, a mutation hotspot is present in the XPD gene. Mutations leading to p.R683W are present in unrelated patients with XP from many parts of the world (Lehmann, 2001).
Figure 1. Haplotype analysis distinguishes a mutation hotspot from a founder mutation.
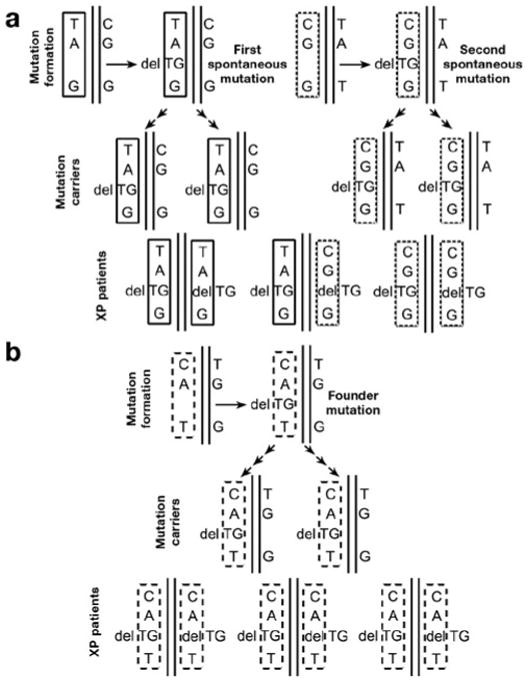
(a) Mutation hotspot. (Top row) The same mutation (delTG) arises independently in two individuals with different single-nucleotide polymorphism (SNP) haplotypes in the region of the mutation: TA (delTG) G (solid rectangle) and CG (delTG) G (dotted rectangle). Over generations these individuals pass the delTG mutation and adjacent regions to their progeny, who are asymptomatic carriers with one mutated allele and one normal allele (center row). Eventually, carriers of the delTG mutation have children with the same mutation in both alleles (bottom row). All of these affected individuals (XP patients) are homozygous for the delTG mutation. However, different XP patients have different haplotypes (TA (delTG) G, CG (delTG) G, or both). (b) Founder mutation. (Top row) The delTG mutation arises in one individual with a SNP haplotype in the region of the mutation: CA (delTG) T (dashed rectangle). This individual passes the delTG mutation and adjacent regions to many generations of progeny who are asymptomatic carriers with one mutated allele and one normal allele (center row). Eventually, two carriers of the delTG mutation have a child with the same mutation in both alleles (bottom row). All of the affected individuals are homozygous for the delTG mutation and have the same haplotype (CA (delTG) T).
Table 1. Primer on founder mutations.
| Founder mutation | A mutation that arose in a common ancestor; the same mutation is found in related individuals who share the same haplotype; it may have arisen many generations ago; mutation carriers often live in same isolated geographic region; it can be of low or high frequency in the population |
| Mutation hotspot | An area within a gene that is prone to frequent spontaneous mutations such that the same mutation is found arising in unrelated individuals; often found scattered in different parts of the world; may have arisen recently; represents genetic DNA instability at the site of mutation |
| Public health measures | Screen for frequency of the founder mutation in the general population; newborn nursery screen if high frequency exists |
| Genetic counseling | Counseling of concerned individuals regarding the risks for genetic disorders; premarital founder mutation screening in members of a defined group; prenatal diagnosis may be possible in families in which both parents are carriers of the founder mutation |
| XP founder mutations identifed | Japan: XPA, XP variant; North Africa: XPC; Israel: XPD |
An alternative explanation is that the African patients with XP had a common ancestor who carried this mutation— a “founder” mutation. In this case the two-base deletion mutation (delTG) in the XPC gene might have arisen spontaneously in one allele in an individual many generations ago (Figure 1b). Because the other allele is normal, this person would have been a normal-appearing heterozygous carrier. Social customs in some parts of the world encourage marriage within the local community or among cousins. Similarly, geographic isolation, such as living on an island (as in Japan) or in an isolated area (the Maghreb region is bounded by the Sahara Desert, the Atlas Mountains, and the Mediterranean Sea) would promote intermarriage. These factors could eventually produce individuals with the same mutation in both alleles, i.e., individuals with clinical XP with a homozygous two-base deletion, as seen in most of the African patients with XPC reported by Soufir et al. (2010). The DNA surrounding the mutation (the haplotype or pattern of single-nucleotide polymorphisms (SNPs) or microsatellites) would also have been passed from generation to generation. Thus, individuals with a founder mutation would share the same haplotype (Figure 1b and Table 1). In contrast, individuals with hotspot mutations would have different haplotypes (Figure 1a).
XP founder mutations
The observation that most North African patients with XP in a single geographic area have the same mutation suggests the presence of a founder mutation. Finding that they share the same haplotype would provide direct evidence of a common ancestor. The size of the common haplotype region of the chromosome can be related to the number of generations that link the patients to their “most recent common ancestor.” A large region of haplotype commonality suggests a recent ancestor. Changes in DNA, such as chromosome crossing over during multiple generations, may result in reductions in the size of the common region. Thus, smaller regions of identical haplotype suggest a more distant ancestor. Soufir et al. (2010) used this elegant technique to show that the most recent common ancestor of the Maghrebi patients with XPC with delTG lived about 50 generations ago. If a generation is 25 years, then this XPC founder mutation arose in the Maghreb region about 1,250 years ago. Similarly, a founder mutation in the XPA gene of Japanese patients with XP was described as arising about 120 generations (or about 2,400 years) ago (Imoto et al., 2008).
Carriers of XP founder mutations in the general population
The frequency of the founder mutation in the population may vary. Evidence of a common ancestor was found linking XP families in Italy and Turkey with a common ancestor 300–500 years ago (Gozukara et al., 2001). However, the mutation Soufir et al. (2010) found in the XPC gene (c.1840C>T) was identified in only two families. On the other hand, the XPA founder mutation in Japan was estimated to be present in 1% of the general population, or about 1 million people (Hirai et al., 2006). Four other founder mutations were reported in Japanese patients with variant XP (Masaki et al., 2008). An XPD founder mutation was also reported in Kurdish Iraqi Jews in Israel, but the population frequency is not known. The finding of the homozygous XPC delTG founder mutation in many families in North Africa suggests that it is not rare. For recessive disorders, normal-appearing heterozygous carriers are much more common than homo zygotes in the general population. A survey of the Maghreb population for the frequency of this founder mutation should be performed.
High frequency of a founder mutation for a serious disease in the general population could have an impact on medical care and genetic counseling. In the United States, genetic screening is performed on newborns for diseases that meet several criteria: it can be identified at a time (24–48 hours after birth) at which it would not ordinarily be detected clinically; a test with appropriate sensitivity and specificity is available for it; and there are demonstrated benefits of early detection, timely intervention, and efficacy (Watson et al., 2006). Neonatal testing is currently performed for several dozen genetic disorders, including phenylketonuria, cystic fibrosis, and sickle cell disease. In the United States, most of the XPC mutations identified to date have been found in one or a small number of families without a founder mutation (Khan et al., 2006). High frequency of an XP found er mutation in other parts of the world makes genetic screening by DNA analysis feasible. Early diagnosis and institution of sun protection measures are essential for preventing skin cancer and preserving vision in patients with XP, and early detection can save lives. Thus, it is reasonable to consider newborn screening for XP in selected populations with a high frequency of a founder mutation. Similarly, premarital screening for the presence of the founder mutation may be a part of genetic counseling. The founder mutation also may be used for prenatal diagnosis in pregnancies where the parents are carriers.
Clinical phenotype of XP patients with founder mutation
Most African patients with XPC with the delTG mutation in both alleles have similar clinical features consisting of photosensitivity, pigmentary changes, and early onset of skin cancer without neurological involvement (Mahindra et al., 2008; Ben et al., 2009; Soufir et al., 2010). However, two patients were reported to be homozygous for the delTG but with neurological involvement (Soufir et al., 2010). This is similar to XP-C patients reported with homozygous mutations in the initiation codon with and without neurological involvement (Khan et al., 2009). These data suggest that neurological involvement may be caused by consanguinity with homozygosity of other unknown genes or by modifying genes or prenatal exposure to teratogens or infectious agents. The data also point to possible problems in trying to predict phenotype when only the genotype is known.
Acknowledgments
This work was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health. We thank Xiaolong (Alan) Zhou for assistance with the illustration.
This is a commentary on article Soufir N, Ged C, Bourillon A, Austerlitz F, Chemin C, Stary A, Armier J, Pham D, Khadir K, Roume J, Hadj-Rabia S, Bouadjar B, Taieb A, de Verneuil H, Benchiki H, Grandchamp B, Sarasin A. A prevalent mutation with founder effect in xeroderma pigmentosum group C from north Africa. J Invest Dermatol. 2010;130(6):1537-42.
Footnotes
Conflict of Interest: The authors state no conflict of interest.
References
- Ben RM, Messaoud O, Talmoudi F, et al. High frequency of the V548A fs X572 XPC mutation in Tunisia: implication for molecular diagnosis. J Hum Genet. 2009;54:426–9. doi: 10.1038/jhg.2009.50. [DOI] [PubMed] [Google Scholar]
- Gozukara EM, Khan SG, Metin A, et al. A stop codon in xeroderma pigmentosum group C families in Turkey and Italy: molecular genetic evidence for a common ancestor. J Invest Dermatol. 2001;117:197–204. doi: 10.1046/j.1523-1747.2001.01424.x. [DOI] [PubMed] [Google Scholar]
- Hirai Y, Kodama Y, Moriwaki S, et al. Heterozygous individuals bearing a founder mutation in the XPA DNA repair gene comprise nearly 1% of the Japanese population. Mutat Res. 2006;601:171–8. doi: 10.1016/j.mrfmmm.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Imoto K, Nadem C, Moriwaki S, et al. Ancient origin of a Japanese xeroderma pigmentosum founder mutation. J Invest Dermatol. 2008;128:S28. doi: 10.1016/j.jdermsci.2012.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SG, Oh KS, Emmert S, et al. XPC initiation codon mutation in xeroderma pigmentosum patients with and without neurological symptoms. DNA Repair (Amst) 2009;8:114–25. doi: 10.1016/j.dnarep.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SG, Oh KS, Shahlavi T, et al. Reduced XPC DNA repair gene mRNA levels in clinically normal parents of xeroderma pigmentosum patients. Carcinogenesis. 2006;27:84–94. doi: 10.1093/carcin/bgi204. [DOI] [PubMed] [Google Scholar]
- Kleijer WJ, Laugel V, Berneburg M, et al. Incidence of DNA repair deficiency disorders in western Europe: xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. DNA Repair (Amst) 2008;7:744–50. doi: 10.1016/j.dnarep.2008.01.014. [DOI] [PubMed] [Google Scholar]
- Kraemer KH, Patronas NJ, Schiffmann R, et al. Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: a complex genotype–phenotype relationship. Neuroscience. 2007;145:1388–96. doi: 10.1016/j.neuroscience.2006.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann AR. The xeroderma pigmentosum group D (XPD) gene: one gene, two functions, three diseases. Genes Dev. 2001;15:15–23. doi: 10.1101/gad.859501. [DOI] [PubMed] [Google Scholar]
- Mahindra P, DiGiovanna JJ, Tamura D, et al. Skin cancers, blindness, and anterior tongue mass in African brothers. J Am Acad Dermatol. 2008;59:881–6. doi: 10.1016/j.jaad.2008.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masaki T, Ono R, Tanioka M, et al. Four types of possible founder mutations are responsible for 87% of Japanese patients with xeroderma pigmentosum variant type. J Dermatol Sci. 2008;52:144–8. doi: 10.1016/j.jdermsci.2008.07.001. [DOI] [PubMed] [Google Scholar]
- Soufir N, Ged C, Bourillon A, et al. A prevalent mutation with founder effect in xeroderma pigmentosum group C from North Africa. J Invest Dermatol. 2010;130:1537–42. doi: 10.1038/jid.2009.409. [DOI] [PubMed] [Google Scholar]
- Watson MS, Mann MY, Lloyd-Puryear M, et al. Newborn screening: toward a uniform screening panel and system. Genet Med. 2006;8(Suppl 1):1S–252S. doi: 10.1097/01.gim.0000223891.82390.ad. [DOI] [PMC free article] [PubMed] [Google Scholar]