

Abstract
Rapidly increasing fields, such as systems biology, require the development and implementation of new technologies, enabling high-throughput and high-fidelity measurements of large systems. Microfluidics promises to fulfill many of these requirements, such as performing high-throughput screening experiments on-chip, encompassing biochemical, biophysical, and cell-based assays1. Since the early days of microfluidics devices, this field has drastically evolved, leading to the development of microfluidic large-scale integration2,3. This technology allows for the integration of thousands of micromechanical valves on a single device with a postage-sized footprint (Figure 1). We have developed a high-throughput microfluidic platform for generating in vitro expression of protein arrays (Figure 2) named PING (Protein Interaction Network Generator). These arrays can serve as a template for many experiments such as protein-protein 4, protein-RNA5 or protein-DNA6 interactions.
The device consist of thousands of reaction chambers, which are individually programmed using a microarrayer. Aligning of these printed microarrays to microfluidics devices programs each chamber with a single spot eliminating potential contamination or cross-reactivity Moreover, generating microarrays using standard microarray spotting techniques is also very modular, allowing for the arraying of proteins7, DNA8, small molecules, and even colloidal suspensions. The potential impact of microfluidics on biological sciences is significant. A number of microfluidics based assays have already provided novel insights into the structure and function of biological systems, and the field of microfluidics will continue to impact biology.
Keywords: Bioengineering, Issue 66, Genetics, Chemistry, Molecular Biology, In vitro protein expression, microfluidics, protein microarray, systems biology, high-throughput, screening
Protocol
1. Device Fabrication
Purchased DTPA-D SU-8 control mold and SPR220-7 flow mold from the Stanford Microfluidics Foundry (www.stanford.edu/group/foundry).
Expose the silicone molds to chlorotrimethylsilane (TMCS) vapor for 10 min to promote elastomer release after the baking steps9.
Prepare a mixture of silicone based elastomer and curing agent (mix well) in two different ratios 5:1 and 20:1 for the control and flow molds, respectively. The different ratios are necessary for proper bonding of multiple layers.
Pour the 5:1 PDMS on the control layer (approximately 5 mm height) . Degas the control layer and bake it for 30 min at 80 °C. Spin coat (Laurell, USA) the 20:1 PDMS mixture on the flow layer at 2,600 rpm for 60 sec and then bake it at 80 °C for 30 min. The spin coater velocity is optimized for valve actuation at a pressure of 15 psi. Faster spinning will result in a thinner layer with lower activation pressure and vice versa. The devices we use have a limit of 25 psi in the control and 10 psi in the flow, above which detachment from the substrate may occur.
Separate the control layer from the mold. Do it slowly and be careful not to peel the SU8 pattern. Then cut the device around its perimeter and punch holes to access the control channels.
Align the flow and control layers manually under a stereoscope. Start by aligning the upper left corner; the button valve (control layer) must be in the middle of the reaction chamber (flow layer). Next, align the first row and gently release the control layer row by row. Make sure that all the button valves are in the middle of the reaction chambers and that the address and input valves cross the flow channels at the correct position. The process can be repeated locally until all rows are aligned. At the end, release any tension in the PDMS by lifting it carefully from the sides and corners. Do not lift too much or misalignment may occur.
Bake it for 2 hr at 80 °C.
Cut around the perimeter of the device and peel the two layer device (chip) from the flow mold. Punch holes to access the flow channels (Figure 1).
2. DNA Arraying and Device Alignment
Produce synthetic genes by assembly PCR. The synthetic genes are compose of T7 promoter, ribosome binding site (RBS), ORF with two epitope tags (one at each end), and T7 terminator 4. The genes can vary in length from 100 bp up to, at least, 5,000 bp.
Prepare the synthetic genes for arraying: prepare a mixture of poly ethylene glycole (1.25%) and D-trehalose dihydrate (125 mg/ml) and dispense 2 μl per reaction into 384 wells plate. This solution will reduce irreversible binding of the DNA to the glass as well as for visualization during alignment 10.
Transfer the synthetic genes to the 384 wells plate. Usually DNA concentrations can range from 10 ng/μl to 100 ng/μl final concentration. Add dH2O to a final volume of 20 μl (depends on microarrayer and pin type).
Spot a series of synthetic genes onto epoxy coated glass substrates using a microarrayer. We use MicroGrid 610 (Bio Robotics) with SMT-S75 silicone pins (Parallel Synthesis, USA). Contact printing of the DNA, with these specific pins, results in spots with diameter of about 100 μm on the glass surface. Each pin load is enough for approximately 100 spots. Make sure the column and row pitches correspond to the specific device used. The device we use contains 16 columns and 40 rows with a pitch of 680 μm by 320 μm, respectively.
Manually align the microfluidic device to the gene array under a stereoscope. The DNA spot should be in the middle of the DNA chambers. Start the alignment by locating the first row of spots below the first row of DNA chambers. Then, bring into line the rest of the rows finishing with fine tuning of the whole device. Once the array is aligned care should be taken to alleviate any stress in the PDMS, by lifting it locally. If there is tension left in the PDMS it may not bond well to the microarray.
Finally, bond the device to the glass slide by overnight incubation on a hot plate at 80 °C.
3. Priming and Activating the Device
The valves in the control layer are actuated from the computer using LabVIEW. The LabVIEW script controls a series of solenoid micro valves via an electronic control box (purchased from Stanford Microfluidics Foundry). The solenoid valve manifold is connected to compressed air, which control the air flow and pressure into the control valves on the device.
Connect the device to the solenoid valves manifold using a flexible plastic tubing with internal diameter of 0.02" (Tygon) and stainless steel pin (New England Small Tubes Corporation).
Fill the tubes with DDW and insert the pin into the access holes of the control layer. Make sure that each tube is connected to its corresponding control channel.
Run the LabVIEW application on the computer to activate the control channels. By activating the valve from the LabVIEW script, we apply air pressure, which in turn will push the water into the PDMS control channels. Set the air pressure to 5 psi. It is recommended to fill the 'sandwich' and the address valves first, in order to identify cross-talks between control channels of defective devices. In case of cross-talks, actuation of the 'sandwich' control line will lead to the actuation of either the neck or button control lines. This is similar to a short in an electrical circuit. A device with cross-talk is defective and cannot be used.
After all the control channels and valves are full of DDW, the valves are primed and ready to block the flow channels underneath them. Increase the air pressure to 15 psi. Valve activation is controlled from a LabVIEW program through a set of on/off switches connected to individual solenoid valves. Each switch controls a specific control line through its corresponding solenoid valve. Turning on the switch button (in the script) will apply air pressure in the corresponding tube. The air pressure will push the DDW in the control line and will result in expansion of the microfluidic valves, blocking the channel underneath. Turning off a control valve, will release the air pressure and subsequently release the blocking of the respective flow channel.
To ensure that all the valves are open, connect a tube to one of the flow inputs and flow air into the device at 4-5 psi. This will release any sticky valves from the glass slide.
Finally, the device is primed and ready. Close all the input and 'neck' valves, to start the surface chemistry.
4. Surface Chemistry
In order to facilitate the self-assembly of a protein array on the surface and prevent non-specific adsorption within the microfluidic device, the surface is chemically modified:
To flow a component through the device, connect a new tube with the required solution to one of the flow channels in the device. Connect the free side of the tube to the manual manifold and open the air pressure flow (5 psi).
Load 40 μl of Biotinylated-BSA (1 μg/μl) in a new tube and flow approximately half of it for 20 min through the device, the BSA will bind to the epoxy surface.
Use Hepes (50 mM) for washing unreacted substrate between each of the different surface chemistry steps.
Flow 25 μl Strepavidin (0.5 μg/μl) for 20 min on top of the biotinylated-BSA.
Wash with Hepes for 5 min.
Close the 'button' valve and flow the rest of the biotinylated-BSA (as described above), passivating the surface surrounding the button.
Wash with Hepes for 5 min.
Release the 'button' valve and flow 30 μl of penta-His-biotin (0.16 μg/μl) for 20 min. The antibody will bind to the exposed strepavidin; specifically to the area under the 'button' creating an anti His-tag array.
5. Protein Expression
Express the proteins on the device using rabbit reticulocyte quick coupled transcription and translation reaction. Protein expression from the spotted synthetic genes on the device creates an array of proteins ready to use. For example, one such use is for a protein binding screen.
Open the 'neck' valves and flow rabbit reticulocyte quick coupled transcription and translation reaction through the device into the DNA chamber. Next, close the 'sandwich' valves and separate each gene from its environment. Incubate the device on a hot plate for 2.5 hr at 30 °C. Expressed proteins will diffuse from the DNA chamber to the reaction chamber spontaneously (neck valve is open) and bind to the anti-His antibody under the 'button' valve immobilizing the protein through its C-terminus tag.
Label the proteins with C-myc Cy3 antibody. The antibody will bind to its corresponding epitope located at the protein N-terminus.
Determine protein expression levels with a microarray scanner (LS Reloaded, Tecan) using a 532 nm laser and 575 nm emission filter.
6. Representative Results
1. Device Fabrication
A graphic design for the device was created by AutoCAD based on our experimental needs. The design was printed on a transparency film by a high-resolution image setter. This transparency serves as a photomask in contact photolithography. A surface micromachining technique was used to create 3-D templates on a silicon wafer determined by the patterns inscribed on the masks used.
The microfluidic device was fabricated on silicone mold casting silicone elastomer polydimethylsiloxane (PDMS, SYLGARD 184, Dow Corning, USA) (Figure 1). Each device consists of two aligned PDMS layers, the flow and the control layer. The mold was first exposed to chlorotrimethylsilane (TMCS, Aldrich) vapor for 10 min to promote elastomer release after the baking steps. A mixture of silicone based elastomer and curing agent was prepared in two different ratio 5:1 and 20:1 for the control and flow molds, respectively. The control layer was degassed and baked for 30 min at 80 °C. The flow layer was initially spin coated (Laurell, USA) at 2,600 rpm for 60 sec and baked at 80 °C for 30 min. The control layer was separated from its mold and control channel access holes were punched. Next, the flow and control layers were aligned manually under a stereoscope and baked for 2 hr at 80 °C. The two layer device was peeled from the flow mold and flow channels access holes were punched.
2. Device Description
The device capacity can range from 500 up to 10,000 unit cells (Figure 2). The device consists of two layers; flow layer (grey) and control layer (color). The control layer contain a variety of valves with different function. Each unit cell, in the flow layer, is composed of one DNA and one reaction chamber and is controlled by three types of micromechanical valves; 'neck', 'button', and 'sandwich' (Figure 2A). A spotted 'synthetic gene' deposited within the DNA chamber is blocked from the reaction chamber by the 'neck' valve (green). Trapping and mechanical washing of surface bound proteins in the reaction chamber is performed by the 'button' valve (blue). The 'sandwich' valve enables each reaction to occur in its own unit cell (red). The address valves can split the device up to 8 independent sections, for different condition assay. In addition, the control layer contain input valves that enable the flow of selected fluid into the flow layer. The PDMS control channels are full with DDW. When a valve is actuated the pressure increase (15 psi) results in the expansion of the PDMS. In places where the membrane is thin enough (i.e. crossing of control and flow lines) this is sufficient to completely block the flow line. Average unit cell height is 10 μm, and average unit cell volume is less than 1 nl.
3. Proteins Expression and Detection
Proteins were expressed on the device using rabbit reticulocyte quick coupled transcription and translation reaction (Promega). The expression of the proteins created an array of proteins ready for use in a binding screen (Figure 3). An expression mix (12.5 μl) was loaded into the device and then flooded into the DNA chambers by opening the 'neck' valves. Next, the 'sandwich' valves were closed leaving each unit cell separated from its neighboring cells and the device was incubated on a hot plate for 2.5 hr at 30 °C. Expressed proteins diffused through the gene chamber into the reaction chamber, where their C-terminus His tag could bind the anti-His antibody (Qiagen) localized under the 'button' valve immobilizing the protein to the surface. Proteins were labeled with a C-myc Cy3 (Sigma), which bound to its corresponding epitope located at the protein N-terminus and labeled it. Protein expression levels were determined with a microarray scanner (LS Reloaded, Tecan) using a 532 nm laser and 575 nm filter. The resulting protein array consists of varying levels of protein expression. Usually, about 20% of a gene library fail to express to detectable levels. No correlation with protein size was observed 3. Background levels were determined using chambers that were not spotted with DNA. Thus, signals from the corresponding protein chambers are due to either noise or non-specific adsorption of the labeling antibodies.
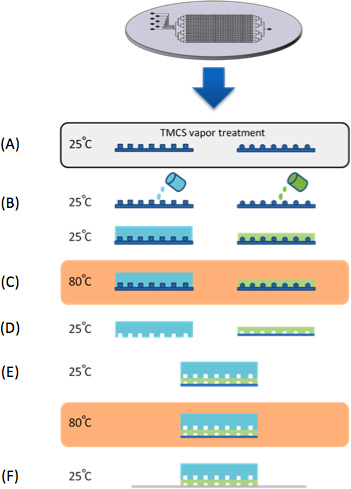 Figure 1. Chip Fabrication. (A) Molds were treated with TMCS vapors for 10 min. (B) PDMS is casted on control and flow silicon molds. (C) Both control and flow molds are baked in 80 °C for 30 min. (D) The control layer peeled of the mold, cut to size and control inlets are punched. (E) The control layer is aligned to the flow layer under a stereoscope and then baked in 80 °C for 2 hr. In parallel to the PDMS device fabrication, a series of synthetic genes are microarrayed onto epoxy coated glass substrates (CEL Associates). (F) The device is then aligned to the DNA microarray trapping the 'synthetic genes' within the DNA chambers.
Figure 1. Chip Fabrication. (A) Molds were treated with TMCS vapors for 10 min. (B) PDMS is casted on control and flow silicon molds. (C) Both control and flow molds are baked in 80 °C for 30 min. (D) The control layer peeled of the mold, cut to size and control inlets are punched. (E) The control layer is aligned to the flow layer under a stereoscope and then baked in 80 °C for 2 hr. In parallel to the PDMS device fabrication, a series of synthetic genes are microarrayed onto epoxy coated glass substrates (CEL Associates). (F) The device is then aligned to the DNA microarray trapping the 'synthetic genes' within the DNA chambers.
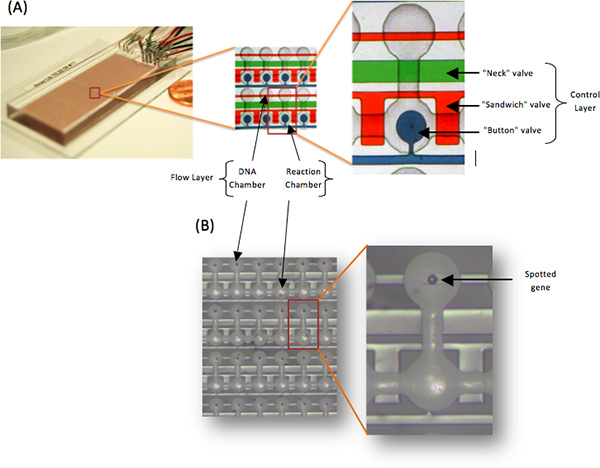 Figure 2. A Photo of the PING Device. (A) The device consist of two aligned PDMS layers (control and flow). The flow layer contains parallel DNA and reaction chambers (grey) controlled by micromechanical valves ("button", "sandwich" and "neck") in the control layer. (B) The layers are aligned to a printed microarray, which programs each DNA chamber with a single spot. This eliminates any potential contamination.
Figure 2. A Photo of the PING Device. (A) The device consist of two aligned PDMS layers (control and flow). The flow layer contains parallel DNA and reaction chambers (grey) controlled by micromechanical valves ("button", "sandwich" and "neck") in the control layer. (B) The layers are aligned to a printed microarray, which programs each DNA chamber with a single spot. This eliminates any potential contamination.
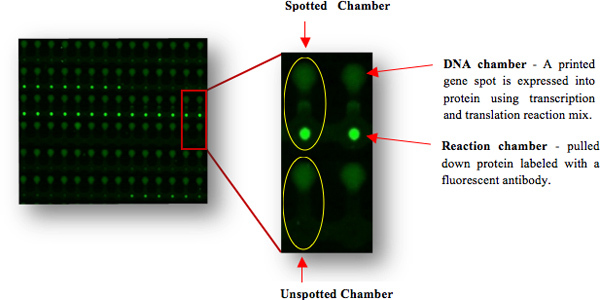 Figure 3. Fluorescent Images of a Protein Array Created with a Microfluidic Chip. The printed spot genes were expressed to proteins within the device (in the DNA chamber) and were pulled down to the surface (below the "button" valve in the reaction chamber) through their C-terminus His tag. Protein expression was detected using their C-myc N-terminus tag and a specific fluorescent antibody. Different signal intensities indicate different protein expression levels. Un-spotted chambers served as controls for background levels.
Figure 3. Fluorescent Images of a Protein Array Created with a Microfluidic Chip. The printed spot genes were expressed to proteins within the device (in the DNA chamber) and were pulled down to the surface (below the "button" valve in the reaction chamber) through their C-terminus His tag. Protein expression was detected using their C-myc N-terminus tag and a specific fluorescent antibody. Different signal intensities indicate different protein expression levels. Un-spotted chambers served as controls for background levels.
Discussion
In this paper we present a method for generation protein arrays in high-throughput using a microfluidic platform. The array generation is based on microarray printing of DNA templates and in vitro protein expression from the DNA within the microfluidic device.
Our novel microfluidic platform has several important advantages over currently used methods, which make it a promising and general tool for proteomics. One advantage is with membrane-bound proteins. In vitro protein synthesis using mammalian reticulocyte lysates in the presence of microsomal membranes11 provides "natural like" conditions required for membrane proteins. Furthermore, microfluidics allows for protein expression in very low volumes and protein purification is not necessary. These are the most common bottlenecks in conventional methodologies. In fact, further optimization of proteins can be achieved by matching the in vitro cell lysate to the proteins, for example E. coli lysate for bacterial proteins.
Microfluidics allows protein expression in very low volumes. In our specific example the chamber volume is 1 nl. There are two advantages to the low volumes. The obvious one is lower consumption of reagents and ability to work with rare materials (i.e. membrane proteins). Another significant advantage is that concentrating the proteins on the surface with antibodies in such a small volume allows us to achieve relatively high concentrations of proteins for interaction assays, which will increase the assay sensitivity.
The button valves have dual role. First they are used for patterning an antibody under the button in each chamber. This allows us to pull down the proteins only under the buttons, while the rest of the chamber is passivated. Second, the buttons allow for mechanical washing and trapping of the proteins by displacing a volume of liquid underneath when actuated. This washing and trapping increases the overall sensitivity of the platform and allows detection of weak and transient molecular interactions 4.
Finally, the microfluidic device can easily be automated and is capable of making many parallel measurements. Thereby saving on costs as well as time. Our estimate is that the cost per protein experiment using PING is about 2 cents per protein, with current devices ranging up to 10,000 experiments per device 4. This estimate includes fabrication and materials. Thus, PING has the potential to be a robust tool for protein array in general with specific advantages such as for membrane proteins.
Disclosures
No conflicts of interest declared.
Acknowledgments
This work was supported by Marie Curie international reintegration grant.
References
- Maerkl SJ. Integration column: Microfluidic high-throughput screening. Integrative biology quantitative biosciences from nano to macro. 2009;1:19–29. doi: 10.1039/b819762h. [DOI] [PubMed] [Google Scholar]
- Hong JW, Quake SR. Integrated nanoliter systems. Nature. 2003;21:1179–1183. doi: 10.1038/nbt871. [DOI] [PubMed] [Google Scholar]
- Unger M. A Monolithic Microfabricated Valves and Pumps by Multilayer Soft Lithography. Science. 2000;288:113–116. doi: 10.1126/science.288.5463.113. [DOI] [PubMed] [Google Scholar]
- Gerber D, Maerkl SJ, Quake SR. An in vitro microfluidic approach to generating protein-interaction networks. Nature. 2009;6:71–74. doi: 10.1038/nmeth.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einav S. Discovery of a hepatitis C target and its pharmacological inhibitors by microfluidic affinity analysis. Nature. 2008;26:1019–1027. doi: 10.1038/nbt.1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fordyce PM. De novo identification and biophysical characterization of transcription-factor binding sites with microfluidic affinity analysis. Nature Biotechnology. 2010;28:962–967. doi: 10.1038/nbt.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H. Global analysis of protein activities using proteome chips. Science (New York, N.Y.) 2001;293:2101–2105. doi: 10.1126/science.1062191. [DOI] [PubMed] [Google Scholar]
- Ramachandran N. Self-assembling protein microarrays. Science (New York, N.Y.) 2004;305:86–90. doi: 10.1126/science.1097639. [DOI] [PubMed] [Google Scholar]
- Zhong JF. A microfluidic processor for gene expression profiling of single human embryonic stem cells. Lab on a chip. 2008;8:68–74. doi: 10.1039/b712116d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusnezow W, Hoheisel JD. Solid supports for microarray immunoassays. Journal of molecular recognition JMR. 2003;16:165–176. doi: 10.1002/jmr.625. [DOI] [PubMed] [Google Scholar]
- Lundin M, Monne M, Widell A, Von Heijne G, Persson MAA. Topology of the membrane-associated hepatitis C virus protein NS4B. Journal of virology. 2003;77:5428. doi: 10.1128/JVI.77.9.5428-5438.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]