

Abstract
Synapse strength refers to the amplitude of postsynaptic responses to presynaptic neurotransmitter release events, and has a major impact on overall neural circuit function. Synapse strength critically depends on the abundance of neurotransmitter receptors clustered at synaptic sites on the postsynaptic membrane. Receptor levels are established developmentally, and can be altered by receptor trafficking between surface-localized, subsynaptic, and intracellular pools, representing important mechanisms of synaptic plasticity and neuromodulation. Rigorous methods to quantify synaptically-localized neurotransmitter receptor abundance are essential to study synaptic development and plasticity. Fluorescence microscopy is an optimal approach because it preserves spatial information, distinguishing synaptic from non-synaptic pools, and discriminating among receptor populations localized to different types of synapses. The genetic model organism Caenorhabditis elegans is particularly well suited for these studies due to the small size and relative simplicity of its nervous system, its transparency, and the availability of powerful genetic techniques, allowing examination of native synapses in intact animals.
Here we present a method for quantifying fluorescently-labeled synaptic neurotransmitter receptors in C. elegans. Its key feature is the automated identification and analysis of individual synapses in three dimensions in multi-plane confocal microscope output files, tabulating position, volume, fluorescence intensity, and total fluorescence for each synapse. This approach has two principal advantages over manual analysis of z-plane projections of confocal data. First, because every plane of the confocal data set is included, no data are lost through z-plane projection, typically based on pixel intensity averages or maxima. Second, identification of synapses is automated, but can be inspected by the experimenter as the data analysis proceeds, allowing fast and accurate extraction of data from large numbers of synapses. Hundreds to thousands of synapses per sample can easily be obtained, producing large data sets to maximize statistical power. Considerations for preparing C. elegans for analysis, and performing confocal imaging to minimize variability between animals within treatment groups are also discussed. Although developed to analyze C. elegans postsynaptic receptors, this method is generally useful for any type of synaptically-localized protein, or indeed, any fluorescence signal that is localized to discrete clusters, puncta, or organelles.
The procedure is performed in three steps: 1) preparation of samples, 2) confocal imaging, and 3) image analysis. Steps 1 and 2 are specific to C. elegans, while step 3 is generally applicable to any punctate fluorescence signal in confocal micrographs.
Keywords: Neuroscience, Issue 66, Developmental Biology, Neurotransmitter receptors, quantification, confocal microscopy, immunostaining, fluorescence, Volocity, UNC-49 GABA receptors, C. elegans
Protocol
1. Preparation of Worms for Imaging
This segment of the protocol is based on published C. elegans culture techniques1,2, and is outlined in Figure 1.
Grow worms on high peptone NGM agar plates (10 cm) seeded with NA22 E. coli bacteria until nearly starved. If immunostaining, one plate will reliably produce enough worms for 1 individual stain, taking into account losses along the way, and the strict criteria used to select worms for imaging (see Part 2).
Harvest worms by pouring a few ml of ddH20 onto plate, swirling briefly, and pouring liquid into a 15 ml conical tube (repeat if necessary to transfer most of the worms to the tube). Pellet in clinical centrifuge for 3 min, 1000x g. Wash pellet 1-3x with ddH2O until supernatant is clear to remove residual bacteria. Use polypropylene transfer pipettes to remove supernatants, as worms stick to glass (from this step onward it is critical to minimize time between steps to avoid losses in egg viability).
After the last wash, resuspend worm pellet in 5 ml alkaline hypochlorite solution to lyse worms and release eggs. Rock gently, and examine tubes under a dissecting microscope about once per minute. When about 50% of worms are lysed (they will appear bent and broken open), terminate lysis by filling the tube to the top with Egg Buffer, inverting several times, and pelleting for 3 min, 1000x g. Lysis time must not exceed 5 min.
Remove supernatant with transfer pipette, and wash pellet 3x with Egg Buffer.
To separate eggs from debris, float them on a cushion of 30% sucrose in ddH2O: after the final wash from step 1.4, carefully remove supernatant and resuspend pellet in 5 ml ddH20. Add 5 ml sterile 60% sucrose in ddH2O, and mix well. Spin in clinical centrifuge 6 min, 1000x g. Eggs will collect at the meniscus (they will have a cloudy appearance), while debris will pellet.
Using a plastic transfer pipette, suck up eggs in a minimal volume and transfer to unseeded NGM agar plates. Any eggs adhering to side of tube can be rinsed down gently and transferred as well. One 10 cm unseeded plate per strain analyzed is sufficient.
Incubate the unseeded plates with the eggs overnight (~16 hr) at 20 °C. Vent the plate for the first few hours to dry it by leaving the lid slightly off for a few hours, but be sure to cover overnight.
After hatching, transfer the L1 larvae to a 15 ml conical tube in S Basal (add a few ml S Basal to plate, swirl, pour into tube). Pellet in clinical centrifuge (3 min, 1000x g), resuspend in a minimal volume of S Basal, and transfer to 10 cm NGM agar plates seeded with NA22 bacteria. Use 2 plates here for each original plate from step 1.1. Incubate until worms reach desired age.
Monitor the cultures once or twice per day. If they are in danger of starving, transfer to fresh NA22 plates. For wild-type N2 worms, starvation is preceded by the formation of a line or wave of worms that moves across the plate as animals migrate en masse away from food-depleted zones. Once this forms, the food will be completely depleted within a few hours.
Worms are now ready for immunostaining or live imaging. Fixing and staining worms in suspension is optimal because large numbers of intact worms can be imaged easily. Staining procedures based on the Finney and Ruvkun protocol3-6 are preferable to freeze-crack procedures because large numbers of worms are needed for imaging.
2. Confocal Imaging
Mount worms on slides for confocal imaging. Adjust density of worms to a few hundred per microliter. Make a pad of 2% agarose in S-basal surrounded by a thin ring of Vaseline to prevent evaporation during imaging (applied via a 3 ml syringe fitted with a cut-off 25 gauge needle). Pipette a few microliters of the worm suspension onto a cover slip (use anaesthetic if imaging live worms), and lower the agarose pad onto the cover slip, spreading the worms evenly over the pad.
Select worms for imaging. Choose worms where the synapses of interest are oriented toward the objective lens, with radial displacement of no more than ±45° (where 0° means oriented directly toward the objective). Perform this assessment quickly (a few seconds) to avoid photobleaching. The scale of the initial cultures is optimized to ensure sufficient worms will satisfy these geometric criteria.
Complete the confocal imaging of the specimen. Relatively flat samples that can be encompassed within about 14 0.4 μm thick sections produce the best results. High speed, low resolution images (512 x 512 pixels) are sufficient for rapid accurate data collection. Avoid saturating the detector with excessive signal strengths, as this will cause underestimation of fluorescence signals.
3. Automated Identification and Analysis of Individual Synaptic Clusters
Open multi-TIF output files from the confocal microscope using Volocity 4.0 (or higher) software (PerkinElmer).
Crop away areas of the image that do not contain the structure of interest (Image -> Extended Focus -> Select area of interest using 'Freehand ROI' tool -> Actions -> Crop to Selection; Figure 2A). Note, Extended Focus produces a z-plane projection on the computer screen to aid your analysis, but does not affect the underlying data; the same is true for brightness and contrast adjustments, if necessary).
Measure the length of the area analyzed using the Line Tool. The length of the line will be calculated and added to the data table (Figure 2B).
Identify objects using the 'Objects by Intensity' filter (Measurements mode). Specify a threshold such that synaptic clusters are highlighted and non-synaptic background is not (Figure 2C). Inclusion of an independent synaptic marker labeled with another color may help unambiguously identify synapses. Typical synapses range from a few to a few hundred voxels. Set the threshold once using a control sample, and use it for all subsequent samples. Thresholding each sample separately is unacceptable because it introduces the potential for experimenter bias. Objects are selected and tabulated automatically (Figure 2D).
Arrange and Export the data. Eliminate objects of 2 or fewer voxels, since these are usually background specks. These can be eliminated by further filtering, or during subsequent analysis. To facilitate their removal downstream, re-sort the data file by 'object size' so they will all group together. Export the data in CSV format, which can be opened by Microsoft Excel or other spreadsheet programs. Each image produces one output file.
If Volocity software is unavailable, alternative methods to analyze Z-plane projections of confocal data may be utilized (e.g. ref. 7, or manual analysis using ImageJ), although 3-dimensional information is lost (see Discussion).
4. Representative Results
The quantification method presented should be able to distinguish between cluster populations of different brightness and different volumes. Representative images and corresponding quantitative data presented in Figure 3 demonstrate examples of differentiation on the basis of these parameters. As a general rule the results should conform to what is evident to the eye. In the case of UNC-49 GABA receptor immunostaining the ventral nerve cord of C. elegans, all animals typically appeared very similar in size and maturity, and total synaptic fluorescence values for a group of five worms (normalized to the length of nerve cord analyzed) showed Standard Error values of about 10% of the mean 4. Genetic mutation and other experimental treatments (e.g. drug exposure) may alter not only the volume and intensity values and frequency distributions of synaptic clusters, but possibly the developmental time-course and synchrony of the worms, leading to higher variability. However the quantitative results should always reflect what can be appreciated visually and if not, inspection of the images and the objects identified by Volocity should reveal where the error occurred and suggest corrective action such as re-thresholding or removal of artefactual objects.
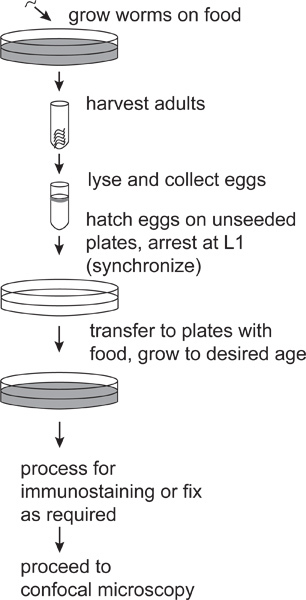 Figure 1. Preparation of synchronous C. elegans cultures. Synchronous cultures are obtained from this procedure because C. elegans development arrests and the L1 larval stage in the absence of food, and resumes when food is introduced.
Figure 1. Preparation of synchronous C. elegans cultures. Synchronous cultures are obtained from this procedure because C. elegans development arrests and the L1 larval stage in the absence of food, and resumes when food is introduced.
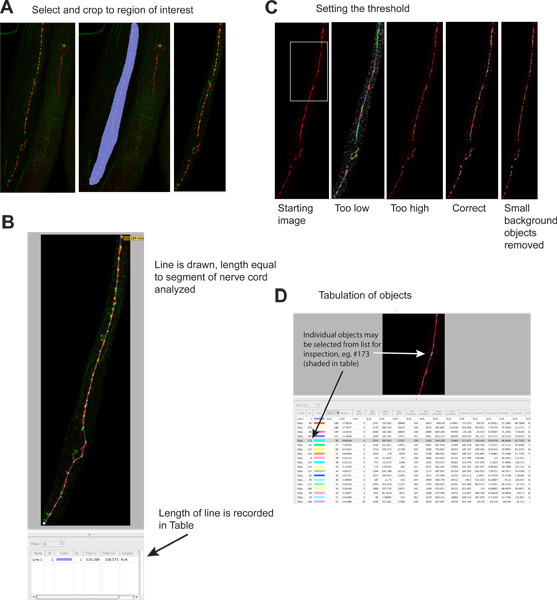 Figure 2. Quantitation of synaptic puncta.
Figure 2. Quantitation of synaptic puncta.
Extraneous regions of the image are cropped to reduce file sizes and increase specificity. Left hand panel shows starting image, middle panel shows selected region, right panel shows image after cropping. Red signal is UNC-49 GABA receptor immunofluorescence, green signal is synaptobrevin-GFP (a presynaptic marker) expressed in presynaptic GABA neurons 4. Click here to view larger figure.
A line is drawn the length of the nerve cord segment to be analyzed, and its length is automatically recorded in the results table. This information is necessary to normalize synaptic data due to the length of analyzable nerve cord, which can vary several fold if worms become fragmented during staining. Click here to view larger figure.
A threshold is applied to identify individual synaptic clusters. Left panel shows image before thresholding; examples of different thresholding levels are shown in separate panels (green signal omitted). Each identified object is depicted on the image as a colored region. Note the correspondence between the visually evident synaptic clusters in the left-most panel and the colored regions on the right-most panel. Click here to view larger figure.
Each identified object is listed separately in the results table. Individual objects may be selected, and are highlighted on the image for inspection if desired. Note that table contains information for the red ('rhodamine') and green ('EGFP') channels; the green channel information is potentially misleading since objects were identified based strictly on red fluorescence. This analysis may also be performed on single color images. Click here to view larger figure.
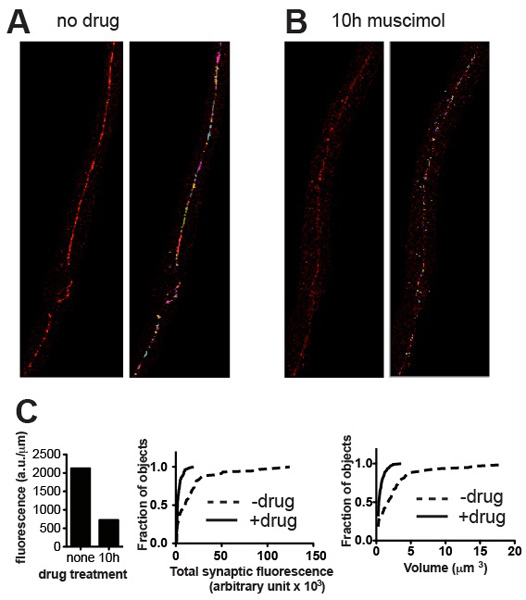 Figure 3. Representative results. Representative micrographs from wild-type C. elegans stained for UNC-49 GABA receptors before (A) and after (B) treatment with muscimol, a GABA receptor agonist that causes receptors to become downregulated after long exposure. Panels show cropped images before (left) and after (right) thresholding and removal of small background specks. (C) Plots of quantitative synaptic parameters for the specimens shown in (A) and (B): total fluorescence normalized to nerve cord length (left), and cumulative probability histograms of individual synapse fluorescence content (middle) and synapse volume (right), demonstrating statistically significant reduction in synaptic content and volume induced by agonist exposure (n = 60 synapses for untreated, n = 115 synapses for muscimol-treated; p < 0.001, Kolmogorov-Smirnov test http://www.physics.csbsju.edu/stats/KS-test.html).
Figure 3. Representative results. Representative micrographs from wild-type C. elegans stained for UNC-49 GABA receptors before (A) and after (B) treatment with muscimol, a GABA receptor agonist that causes receptors to become downregulated after long exposure. Panels show cropped images before (left) and after (right) thresholding and removal of small background specks. (C) Plots of quantitative synaptic parameters for the specimens shown in (A) and (B): total fluorescence normalized to nerve cord length (left), and cumulative probability histograms of individual synapse fluorescence content (middle) and synapse volume (right), demonstrating statistically significant reduction in synaptic content and volume induced by agonist exposure (n = 60 synapses for untreated, n = 115 synapses for muscimol-treated; p < 0.001, Kolmogorov-Smirnov test http://www.physics.csbsju.edu/stats/KS-test.html).
Discussion
The method presented here is designed to extract quantitative multi-parameter data for large populations of synapses in C. elegans, while maximizing consistency within treatment groups. Three features contribute to these objectives. First, immunostaining is performed on synchronous worm populations to ensure that all animals are the same age. This step is critical because developmental regulation of expression levels may obscure effects of the experimental treatment (e.g. UNC-49 GABA receptor immunofluorescence varies several-fold during development (K. Davis et al., in preparation)). Additionally, permeabilization and fixation can be highly variable, making quantitative comparisons difficult. Using synchronized cultures minimizes this inherent variability, improving the reliability of the technique. Other ways to address this variability include using a second antibody to an unrelated target that can be independently visualized as an internal control for permeabilization, or imaging GFP-tagged proteins in live worms, which avoids the need for permeabilization altogether. However this latter approach introduces new problems because the endogenous protein is no longer being measured directly, so potential artefacts due to inconsistencies in transgene expression/copy number, structural alteration due to the fusion of GFP, or non-physiological transgene expression levels need to be considered. No single method is perfect; ideally, corroborating data can be obtained using multiple approaches. In this protocol, the size of the starter cultures has been optimized to result in sufficient numbers of fixed and stained worms for confocal analysis. Large numbers of worms are required because the geometric criteria used to select worms for imaging are strict, which is the second feature of the protocol to minimize variability. The same anatomical structure must be imaged in each animal (either the most biologically relevant site, or an arbitrarily chosen site in the case of broad tissue distribution of the protein of interest), and that structure must be oriented toward the objective lens with only slight deviation to either side. This criterion improves consistency by eliminating variability in the amount of intervening tissue that can scatter excitation and emission light, affecting signal strength. It is important that this is the ONLY criterion used to select worms, as it is easy to bias the results based on expectation of a certain outcome. Indeed experiments are best performed blind where possible. The third feature that contributes to the power of this technique is the automated identification and quantitative characterization of synaptic clusters in three dimensions using Volocity. This analysis is simple and rapid, capable of identifying and tabulating hundreds to thousands of individual synapses for each experimental condition. More importantly, Volocity includes data from all of the planes of multi-plane confocal data stacks, rather than discarding data to generate the 2-dimensional z-projections used in earlier protocols. Downstream of this analysis, experimental groups may be compared in terms of total synaptic fluorescence per animal, the spatial distribution of synapses, and frequency distributions of volumes, total fluorescence, and maximal fluorescence for individual synapses. These parameters can reveal key transitions indicating synaptic development events, the roles of developmental and regulatory proteins, and synaptic plasticity.
Quantitation of fluorescence signals using Volocity has applications in neurobiology beyond analysis of permeabilized fixed C. elegans samples, being useful for quantification of any fluorescent signal localized to high-contrast puncta. Therefore it is suitable for analysis of fixed tissue or cells from any organism. However, fixation and permeabilization leads to visualization of total synaptic protein populations regardless of whether they are surface-expressed, whereas for receptors at least, only the surface-expressed fraction contributes to synaptic strength. Volocity-based analysis can also be used to quantify surface-expressed populations, provided non-permeabilizing fixation conditions are used. Volocity analysis will also be useful to study protein dynamics in live cells. Many protein domains shuttle between vesicular and extracellular environments with different pH during neuronal signaling events (e.g. the carboxy terminus of synaptobrevin during synaptic vesicle release 8-10, or the amino terminal domains of neurotransmitter receptors during trafficking to the cell surface 11). Fusing pH sensitive ecliptic GFP tags to these regions can produce in vivo reporters of these transitions that can be studied quantitatively using a Volocity-based approach. Similarly, GFP-tagging of neuropeptide genes results in GFP-labeled dense-core vesicles that de-stain as these vesicles fuse with the plasma membrane 12; Volocity analysis may be useful in this context as an assay for specific neuropeptide release in vivo. The advantage of Volocity analysis in all of these settings is its ability to rapidly identify and quantify large numbers of synapses, enabling subtle differences in their individual properties or population distributions to be demonstrated with statistical rigor.
Disclosures
No conflicts of interest declared.
Acknowledgments
The authors would like to thank A. Benham for assisting with the development of the protocol. This work was funded by NIH grant NS06747 to B. A. B.
References
- Christensen M. A primary culture system for functional analysis of C. elegans neurons and muscle cells. Neuron. 2002;33:503–514. doi: 10.1016/s0896-6273(02)00591-3. [DOI] [PubMed] [Google Scholar]
- Lewis JA, Fleming JT. Basic culture methods. Methods Cell Biol. 1995;48:3–29. [PubMed] [Google Scholar]
- Bettinger JC, Lee K, Rougvie AE. Stage-specific accumulation of the terminal differentiation factor LIN-29 during Caenorhabditis elegans development. Development. 1996;122:2517–2527. doi: 10.1242/dev.122.8.2517. [DOI] [PubMed] [Google Scholar]
- Davis KM. Regulated lysosomal trafficking as a mechanism for regulating GABAA receptor abundance at synapses in Caenorhabditis elegans. Mol. Cell Neurosci. 2010;44:307–317. doi: 10.1016/j.mcn.2010.04.002. [DOI] [PubMed] [Google Scholar]
- Finney M, Ruvkun G. The unc-86 gene product couples cell lineage and cell identity in C. elegans. Cell. 1990;63:895–905. doi: 10.1016/0092-8674(90)90493-x. [DOI] [PubMed] [Google Scholar]
- Rowland AM. Presynaptic terminals independently regulate synaptic clustering and autophagy of GABAA receptors in Caenorhabditis elegans. J. Neurosci. 2006;26:1711–1720. doi: 10.1523/JNEUROSCI.2279-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbea M. Ubiquitin and AP180 regulate the abundance of GLR-1 glutamate receptors at postsynaptic elements in C. elegans. Neuron. 2002;35:107–120. doi: 10.1016/s0896-6273(02)00749-3. [DOI] [PubMed] [Google Scholar]
- Miesenbock G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- Oda S, Tomioka M, Iino Y. Neuronal plasticity regulated by the insulin-like signaling pathway underlies salt chemotaxis learning in Caenorhabditis elegans. J. Neurophysiol. 2011;106:301–308. doi: 10.1152/jn.01029.2010. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan S. The use of pHluorins for optical measurements of presynaptic activity. Biophys. J. 2000;79:2199–2208. doi: 10.1016/S0006-3495(00)76468-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald NA. Generation and functional characterization of fluorescent, N-terminally tagged CB1 receptor chimeras for live-cell imaging. Mol. Cell Neurosci. 2007;35:237–248. doi: 10.1016/j.mcn.2007.02.016. [DOI] [PubMed] [Google Scholar]
- Shakiryanova D. Synaptic neuropeptide release induced by octopamine without Ca2+ entry into the nerve terminal. Proc. Natl. Acad. Sci. U. S. A. 2011;108:4477–4481. doi: 10.1073/pnas.1017837108. [DOI] [PMC free article] [PubMed] [Google Scholar]