

Abstract
Epithelial ovarian cancers (EOCs) are the leading cause of death from gynecological malignancy in Western societies. Despite advances in surgical treatments and improved platinum-based chemotherapies, there has been little improvement in EOC survival rates for more than four decades 1,2. Whilst stage I tumors have 5-year survival rates >85%, survival rates for stage III/IV disease are <40%. Thus, the high rates of mortality for EOC could be significantly decreased if tumors were detected at earlier, more treatable, stages 3-5. At present, the molecular genetic and biological basis of early stage disease development is poorly understood. More specifically, little is known about the role of the microenvironment during tumor initiation; but known risk factors for EOCs (e.g. age and parity) suggest that the microenvironment plays a key role in the early genesis of EOCs. We therefore developed three-dimensional heterotypic models of both the normal ovary and of early stage ovarian cancers. For the normal ovary, we co-cultured normal ovarian surface epithelial (IOSE) and normal stromal fibroblast (INOF) cells, immortalized by retrovrial transduction of the catalytic subunit of human telomerase holoenzyme (hTERT) to extend the lifespan of these cells in culture. To model the earliest stages of ovarian epithelial cell transformation, overexpression of the CMYC oncogene in IOSE cells, again co-cultured with INOF cells. These heterotypic models were used to investigate the effects of aging and senescence on the transformation and invasion of epithelial cells. Here we describe the methodological steps in development of these three-dimensional model; these methodologies aren't specific to the development of normal ovary and ovarian cancer tissues, and could be used to study other tissue types where stromal and epithelial cell interactions are a fundamental aspect of the tissue maintenance and disease development.
Keywords: Cancer Biology, Issue 66, Medicine, Tissue Engineering, three-dimensional cultures, stromal-epithelial interactions, epithelial ovarian cancer, ovarian surface epithelium, ovarian fibroblasts, tumor initiation
Protocol
Figure 1 illustrates an overview of the workflow described below.
1. Isolation of Normal Ovarian Fibroblasts and Extension of in vitro Lifespan by Overexpression of the Catalytic Subunit of the hTERT Holoenzyme
Ovarian tissues can be collected with informed patient consent and approval of the Institutional Review Board (for US institutions). Normal ovarian tissues can be collected following total abdominal hysterectomy or total laparoscopic hysterectomy with bilateral salpingo-oophorectomy. In this study, tissues were examined by a pathologist and a portion of the ovarian stroma biopsied for cell culture. Tissue samples of approximately 2-5 mm2 were harvested from the central region of the ovary to attempt to reduce the chance of also sampling ovarian epithelia.
Transport fresh ovarian tissue to the cell culture laboratory in immortalized normal ovarian fibroblast (INOF) growth medium, supplemented with extra antibiotics and antimycotics. INOF growth medium (INOF-GM) comprises: Medium 199 and MCDB105 mixed in a 1:1 ratio, supplemented with 15% fetal bovine serum, 2 mM L-glutamine, 50 μg/ml gentamicin and 250 ng/ml amphotericin B.
Working inside a biosafety cabinet: Remove any epithelial cells that remain loosely attached to the tissue, by washing the sample with 5 ml PBS. Aspirate and repeat twice more.
Transfer the sample plus PBS into a clean 10 cm diameter (P100) culture dish. Use sterile forceps to handle the tissue and place into a second, dry P100 clean culture dish. Use sterile scalpel to cut tissue 0.5 cm2 pieces. Place each piece of tissue into a separate P100 dish and further cut into pieces <1 mm2 in size. Add 20 ml INOF-GM (plus antibiotics and antimycotics), and incubate at 37 °C, 5% CO2. Monitor cell growth using phase microscopy. Cells can be seen adhering to the dish within 24 hr.
Re-feed after one week, removing any non-adherent tissue by aspiration. Thereafter, refeed cultures twice a week. Cells will form colonies, usually within 1-3 weeks. Once these colonies are large enough to subculture (once they reach around 500 μm), isolate the clones using trypsin-coated cloning disks. Confirm cells are 100% fibroblastic and free of other cellular contaminants by plating a sample of the cell line onto glass coverslips and performing immuno-fluorescent staining for a fibroblastic marker (FSP), an epithelial marker (pan-cytokeratin) and an endothelial cell marker (von Willenbrand Factor VIII).
Passage primary normal ovarian fibroblast (NOF) cell isolates from a single P100 dish into 3 P100 dishes one day prior to retroviral transduction of hTERT. Pre-made viral supernatants can be purchased, or produced in-house using standard protocols for co-transfecting HEK293T cells (see www.stanford.edu/group/nolan/). Retroviral supernatants can be produced using the pBABE-hygro-hTERT vector (Addgene). Remove NOFs from the incubator and then aspirate the cell culture medium. Add 3 ml of viral supernatant to each dish: one dish receives hTERT-retrovirus, the 2nd dish receives GFP-retrovirus and the third dish receives no virus. Overlay viral supernatants with INOF-GM containing polybrene to give a final concentration of 8 μg/ml. Re-feed cells the following day with fresh medium. Two days after infection, begin selection with low dose (10 U/ml) hygromycin B. After seven days this dose can be increased to 20 U/ml; selection should be complete within 10-14 days.
Clones expressing hTERT or GFP can be isolated by ring cloning once they reach around 500 μm. Cell lines require additional characterization to confirm the successful expression of hTERT and retention of primary cell line features. This involves: (a) confirm telomerase activity by PCR-ELISA and telomere length, by Southern blot or PCR 6,7; (b) confirming the expression of critical markers (e.g. immunostaining with an anti-pan-cytokeratin antibody to identify epithelial cells, and with an anti-fibroblast surface protein antibody for fibroblasts) are similar between primary and hTERT-immortalized cells; (c) confirming the cells do not show properties of neoplastic transformation (e.g using anchorage independent growth assays); (d) confirming extension of in vitro lifespan observed in cells expressing hTERT but not GFP. hTERT-expressing immortalized normal ovarian fibroblasts (INOFs) should bypass replicative senescence but not be neoplastically transformed; additionally the INOF cells should maintain the expression of fibroblast surface protein but not epithelial markers such as cytokeratin (Figure 2).
2. Coating of Tissue Culture Dishes with PolyHEMA for Non-adherent Cell Culture
To prepare the polyHEMA solution, weigh out 1.5 g polyHEMA, and place into a sterile bottle. Add 95 ml molecular biology grade absolute ethanol and 5 ml cell culture grade distilled water. The polyHEMA hydrogel will dissolve in the water content of the solution, and the ethanol will sterilize the preparation.
Place the polyHEMA solution in a waterbath at 65 °C until fully dissolved. This may take up to 8 hr. Note that the polyHEMA solution cannot be stored for long periods of time and so should always be prepared fresh. TROUBLESHOOTING: Solutions of polyHEMA require long incubation times at 65 °C. Regularly invert the polyHEMA solution to mix. Viscosity observed at the bottom of the glass bottle indicates the polyHEMA is not fully dissolved and further incubation at 65 °C is required.
Working inside a laminar flow hood, coat tissue culture dishes with the polyHEMA solution. Any size of tissue culture dish, flask or multiwell plate can be coated with polyHEMA. Recommended volumes of polyHEMA solution required for each vessel are given in Table 2.
Allow cell culture dishes to dry inside the laminar flow hood. This may take 2-3 days. Gentle rocking on a rocking platform can be used to ensure even coating of larger-sized dishes or flasks. If the polyHEMA solution dries too fast the surface may not be smooth. To avoid this, ensure the lid of the dish/flask remains on throughout the drying process.
Apply a second coat of polyHEMA and allow to dry as described above. TROUBLESHOOTING: Holes in the polyHEMA coating can occur, particularly when plates dry too quickly. Double-coating the plates with polyHEMA helps to cover gaps or cracks in the coating are covered before use. Plates usually take 2-3 days for each polyHEMA coat to dry and so should be prepared at least a week before they will be needed for cell culture.
PolyHEMA-coated plates can be stored at room temperature until use.
NOTE: 1% agarose solutions, dissolved in 1X PBS and autoclaved to sterilize, can also be used to coat cell culture dishes to create a non-adherent surface for short-term 3D culturing (less than one week). Agarose coating of multi-well plates creates a concave surface that promotes the formation of a single spheroid per well for many cell lines. However, for longer 3D cultures polyHEMA coating is recommended as agarose coating often disintegrates after longer culture periods.
3. Generating Three-dimensional (3D) Heterotypic Spheroids and Re-feeding of 3D Cultures
Recover and expand INOF and epithelial cell cultures in vitro to prepare enough cells for establishing 3D heterotypic spheroids. For 'mass cultures' we typically seed 2-4x106 INOFs per P100 dish and 0.5-1x106 epithelial cells to give a stromal:epithelial ratio of 4:1. INOF cells are grown in INOF growth medium (INOF-GM) without gentamicin or amphotericin B. If desired, fibroblast cells can be pretreated prior to 3D culture (e.g. for induction of senescence, cells can be treated with sub-lethal doses of hydrogen peroxide).
Wash a dry polyHEMA plate with 5 ml phosphate-buffered saline (PBS) for 5 min. Aspirate the PBS and replace with 15 ml INOF-GM.
Meanwhile, trypsinize immortalized normal ovarian fibroblast cells to detach them from the culture dish, after 3-5 min neutralize the trypsin with an equal volume of culture media and pellet the cells by centrifugation at 450 x g for 5 min.
Discard the supernatant. Create a single-cell suspension by resuspending the cell pellet in 5 ml INOF-GM, mix well by pipetting up and down or vortexing gently. Determine cell concentration by mixing 10 μl cell suspension and 10 μl trypan blue and enumerating using a hemocytometer. Trypan blue exclusion can be used to identify live cells.
Add 4x106 INOF cells to the medium already dispensed into the polyHEMA-coated plate. Make up the culture media volume to 20 ml using an appropriate volume of fresh INOF-GM and incubate at 37 °C, 5% CO2. Cells begin to aggregate into multicellular spheroids within 24 hr. Spheroid formation can be monitored by phase microscopy. Accurate counting of spheroid numbers under mass culture conditions is difficult, but we estimate that we obtain 50-100 spheroids when 4x106 stromal cells are plated. Spheroid sizes typically range from 80-250 μm.
Re-feed the spheroid cultures every 2-3 days by resting the plates at an angle on a pipette placed within the laminar flow hood. Allow the spheroids to settle. This may take up to 20 min. Carefully remove the exhausted media with a 5 ml pipette, until only approximately 4 ml remain in the dish. Take care not to aspirate the spheroids. Re-feed with 16 ml fresh INOF-GM and return to the incubator.
On day 7, add epithelial cells to fibroblast spheroids to create heterotypic cultures as follows. Prepare suspensions of ovarian epithelial cells (phenotypically normal or transformed) by trypsinization as described in section 3.4-3.5. Determine cell concentration using a hemocytometer. Wash once in INOF-GM to prevent any growth factors from being carried over from the epithelial growth media. In our laboratory, normal ovarian surface epithelial cell (OSEC) lines were collected with informed patient consent from ovaries removed by total abdominal hysterectomy or total laparoscopic hysterectomy with bilateral salpingo-oophorectomy. Ovaries were brushed with a sterile cytobrush to collect the epithelium. Primary cell lines were established and transduced with hTERT using the approach described above. Details of OSEC culture in 2D, 3D and OSEC immortalization can be found in references 10 and 11.
To form heterotypic spheroids, first re-feed cultures of fibroblast spheroids alone. Then inoculate fibroblast spheroid cultures with 1x106 epithelial cells at a ratio of 4:1 fibroblasts to epithelial cells. Culture the heterotypic spheroids in INOF-GM.
Re-feed heterotypic cultures every 2-3 days for an additional 14 days.
4. Processing for Imaging and Molecular Analyses
- Many different techniques can be used to analyze the heterotypic spheroids. For imaging, it is recommended that spheroids are harvested from the culture dish with a 25 ml pipette. Pipetting slowly will minimize chances of shear forces disrupting the architecture of the spheroid. Spheroids can then be washed in PBS and pelleted by gently centrifugation at 100 x g for 10 min; higher centrifugal forces may damage the spheroids. Alternatively, cultures can be transferred to a 50 ml falcon tube and allowed to settle, and the growth medium then removed.
- For immunohistochemistry, fix spheroids with neutral buffered formalin for 30 min at room temperature. Pellet the spheroids and replace the formalin with 70% ethanol. Process into paraffin using standard protocols used for human tissues. Paraffin-embedded spheroids can be sectioned, and stained with either hematoxylin and eosin or specific molecular markers using standard immunohistochemistry (Figure 3).
- Alternatively, spheroids washed in PBS can be fixed in 4% paraformaldehyde (prepared in PBS), embedded in Optimal Cutting Temperature OCT medium and snap frozen for frozen sectioning and immunofluorescent staining.
For molecular analyses, the spheroids can be harvested, pelleted and washed twice in PBS before lysing cells for DNA, RNA or protein extraction.
For analysis by flow cytometry, spheroids can be washed in PBS and dissociated by trypsin or accutase digestion at 37 °C. To do this, submerge the Falcon tube containing spheroids in a waterbath to facilitate trypsinization. Complete dissociation of spheroids into a single cell suspension can be promoted by pipetting the solution up and down with a 1 ml pipette tip. Take care not to over-trypsinize the cells.
Neutralize the reaction with an equal volume of culture medium and remove cell clumps by passing the suspension through a 40-70 μm cell strainer. Pellet cells, wash with PBS, enumerate and resuspend the desired number of cells in FACS buffer. Stain cells according to manufacturers instructions.
5. Representative Results
This approach can be used for a broad range of applications, and for the study of normal ovarian biology as well as the study of ovarian cancer initiation. A major advantage of this approach over 3D cultures created using commercially available extracellular matrix gel is that the normal ovarian fibroblasts produce the extracellular matrix that makes up the core of the spheroids. In our experience, the matrix material produced by normal ovarian fibroblasts is heterogeneous and more closely resembles the extracellular matrix of the ovary than any commercially available matrix gel. In our laboratory we have utilized epithelial cells that have been partially transformed in vitro using defined genetic elements (e.g.CMYC overexpression) and co-cultured these cells with normal and senescent ovarian fibroblasts to mimic early-stage ovarian cancer in a post-menopausal ovary. Our data suggest that the aging microenvironment promotes neoplastic features (e.g. proliferation) of partially transformed epithelial cells (Figure 3), whereas a normal microenvironment inhibits neoplastic progression 8. Our lab and others have also used similar approaches to model normal ovarian epithelial cells 9, fallopian tube epithelial cells, step-wise models of neoplastic progression10, and ovarian and endometrial cancer cell lines 11-13 as homotypic spheroid cultures. We have observed that histological features of normal and malignant cells that are not detectable when the cells are cultured as monolayers are restored when the cells are transitioned into spheroid culture. For example, histological features of clear cell ovarian cancers can be detected in 3D cultures of clear cell ovarian cancer cell lines (Figure 4), but not in monolayer cultures of the same cells. Moreover, 3D cultured cells more closely reflect the tissue specific protein expression seen in primary tissues compared to the same cells cultured in traditional 2D monolayer cultures 9-11. These approaches to modeling normal and malignant ovarian tissues also represent valuable tools for the discovery and functional analysis of novel candidate ovarian cancer tumor suppressor genes and oncogenes and other molecular biomarkers that may be associated with disease development 10,14.
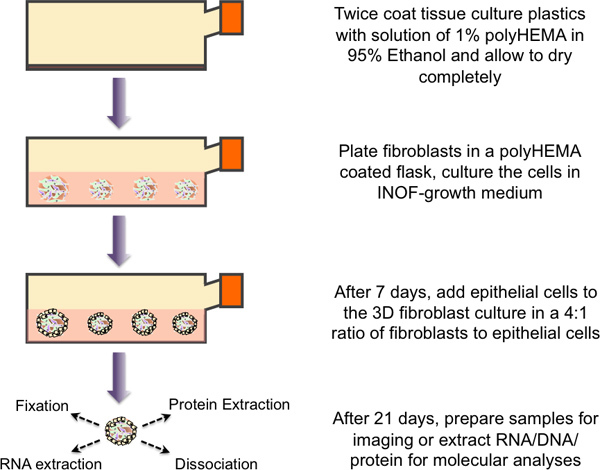 Figure 1. Schematic overview of the protocol for 3D heterotypic culturing. Tissue culture flasks are twice coated with a 1% polyHEMA solution and allowed to dry. PolyHEMA coated plastics are washed with 1x PBS immediately prior to use. Immortalized normal ovarian fibroblast cells (4x106 cells) are plated into the flask with 20 ml growth medium. Spheroid formation and growth monitored over 7 days. At this point, fibroblast spheroid cultures are inoculated with 1x106 epithelial cells, and the two cell types are co-cultured for 14 days before the multicellular spheroids are harvested and processed for downstream applications (molecular analyses, imaging, etc).
Figure 1. Schematic overview of the protocol for 3D heterotypic culturing. Tissue culture flasks are twice coated with a 1% polyHEMA solution and allowed to dry. PolyHEMA coated plastics are washed with 1x PBS immediately prior to use. Immortalized normal ovarian fibroblast cells (4x106 cells) are plated into the flask with 20 ml growth medium. Spheroid formation and growth monitored over 7 days. At this point, fibroblast spheroid cultures are inoculated with 1x106 epithelial cells, and the two cell types are co-cultured for 14 days before the multicellular spheroids are harvested and processed for downstream applications (molecular analyses, imaging, etc).
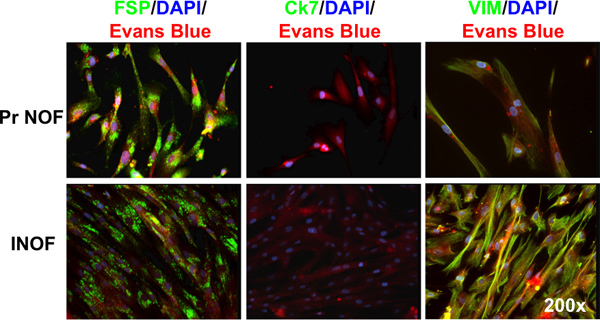 Figure 2. Immunofluorescence staining of hTERT immortalized normal ovarian fibroblasts. Both primary normal ovarian fibroblasts (Pr NOFs) and immortalized normal ovarian fibroblasts (INOFs) express fibroblast specific protein (FSP), do not express cytokeratin-7 (Ck-7) and express vimentin (VIM). Markers of interest are shown in green, DNA is stained with DAPI and shown in blue, cytoplasm is counterstained with Evan's blue and shown in red.
Figure 2. Immunofluorescence staining of hTERT immortalized normal ovarian fibroblasts. Both primary normal ovarian fibroblasts (Pr NOFs) and immortalized normal ovarian fibroblasts (INOFs) express fibroblast specific protein (FSP), do not express cytokeratin-7 (Ck-7) and express vimentin (VIM). Markers of interest are shown in green, DNA is stained with DAPI and shown in blue, cytoplasm is counterstained with Evan's blue and shown in red.
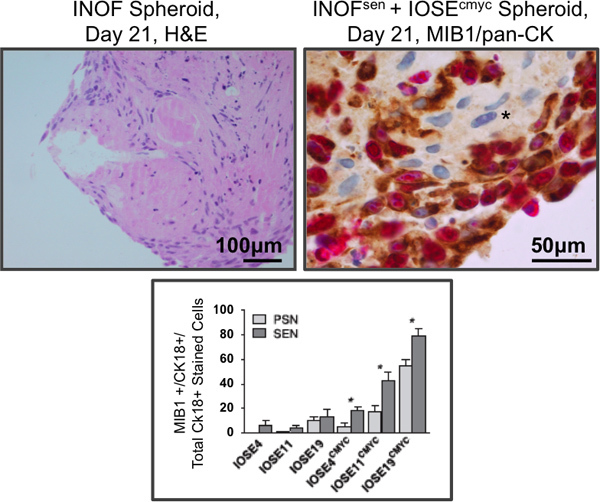 Figure 3. Immunohistochemistry of co-cultured stromal and epithelial cells. Left panel, normal ovarian fibroblast spheroid monoculture, stained by hematoxylin and eosin. Normal ovarian fibroblast cells cultured alone in 3D form spheroids consisting of cells with a spindled-morphology and abundant extracellular matrix. Right panel: normal ovarian fibroblasts were exposed to hydrogen peroxide to induce senescence. Partially transformed ovarian epithelial cells (IOSEcmyc) were added to 7-day senescent fibroblast spheroids. After an additional 14 days of co-culture, spheroids were fixed, paraffin-embedded, sectioned and dual stained for a proliferation marker (MIB1, pink) and, to distinguish the two cell types, an epithelial marker (pan-cytokeratin, brown). Eosin was used as a counterstain, the unstained fibroblasts at the core of the spheroid are clearly visible (asterisks). Lower panel: quantification of epithelial cell proliferation in dual stained co-cultures. Immortalized ovarian surface epithelial (IOSE) cell lines from three different patients were used in this study (IOSE4, IOSE11 and IOSE19). Senescent fibroblasts (SEN) promote proliferation of co-cultured IOSEcmyc cells significantly more than the same cells prior to senescence induction (pre-senescent, PSN). Parental IOSE cells without CMYC overexpression show no difference in proliferation when co-cultured with PSN or SEN fibroblasts. These data suggest that in our in vitro co-culture model, aging processes in the ovarian stroma may promote the development of OCs 11. *P≤0.05, two-tailed paired T-Test.
Figure 3. Immunohistochemistry of co-cultured stromal and epithelial cells. Left panel, normal ovarian fibroblast spheroid monoculture, stained by hematoxylin and eosin. Normal ovarian fibroblast cells cultured alone in 3D form spheroids consisting of cells with a spindled-morphology and abundant extracellular matrix. Right panel: normal ovarian fibroblasts were exposed to hydrogen peroxide to induce senescence. Partially transformed ovarian epithelial cells (IOSEcmyc) were added to 7-day senescent fibroblast spheroids. After an additional 14 days of co-culture, spheroids were fixed, paraffin-embedded, sectioned and dual stained for a proliferation marker (MIB1, pink) and, to distinguish the two cell types, an epithelial marker (pan-cytokeratin, brown). Eosin was used as a counterstain, the unstained fibroblasts at the core of the spheroid are clearly visible (asterisks). Lower panel: quantification of epithelial cell proliferation in dual stained co-cultures. Immortalized ovarian surface epithelial (IOSE) cell lines from three different patients were used in this study (IOSE4, IOSE11 and IOSE19). Senescent fibroblasts (SEN) promote proliferation of co-cultured IOSEcmyc cells significantly more than the same cells prior to senescence induction (pre-senescent, PSN). Parental IOSE cells without CMYC overexpression show no difference in proliferation when co-cultured with PSN or SEN fibroblasts. These data suggest that in our in vitro co-culture model, aging processes in the ovarian stroma may promote the development of OCs 11. *P≤0.05, two-tailed paired T-Test.
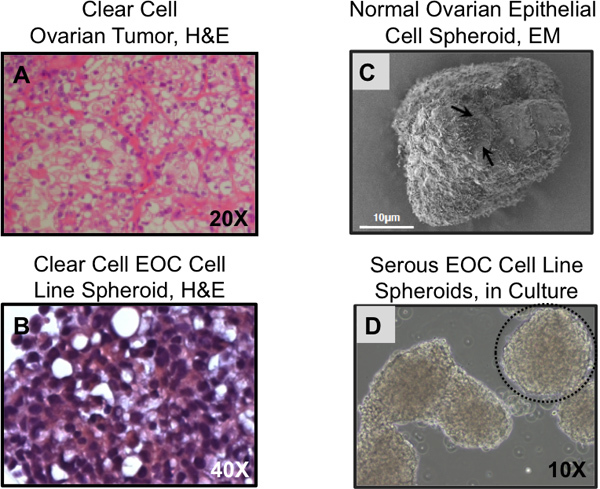 Figure 4. Histological features of 3D cultured cells reflect primary tissues. (A) Clear cell ovarian carcinomas display characteristic clear cell structures, these are absent when clear cell EOC lines are grown on plastic but similar structures are detected when the same cells are grown as 3D spheroids (B). (C) Scanning electron micrograph of normal ovarian epithelial cells grown as spheroids for 14 days. Surface microvilli are visible (arrows). (D) Serous ovarian cancer cell line spheroids, in culture. (A,B) Hematoxylin and eosin staining of sectioned paraffin embedded tumor tissue or 3D spheroids; (C) scanning electron microscopy; (D) phase contrast microscopy. Spheroids can be spherical (dashed circle) or irregular in shape, as in the case of the structure seen in the left side of the image.
Figure 4. Histological features of 3D cultured cells reflect primary tissues. (A) Clear cell ovarian carcinomas display characteristic clear cell structures, these are absent when clear cell EOC lines are grown on plastic but similar structures are detected when the same cells are grown as 3D spheroids (B). (C) Scanning electron micrograph of normal ovarian epithelial cells grown as spheroids for 14 days. Surface microvilli are visible (arrows). (D) Serous ovarian cancer cell line spheroids, in culture. (A,B) Hematoxylin and eosin staining of sectioned paraffin embedded tumor tissue or 3D spheroids; (C) scanning electron microscopy; (D) phase contrast microscopy. Spheroids can be spherical (dashed circle) or irregular in shape, as in the case of the structure seen in the left side of the image.
| Dish/Plate Size | Volume of PolyHEMA (to be applied twice) | Culture Media Final Volume |
| 6-well plate | 3 ml | 5-7 ml |
| 12-well plate | 1.5 ml | 2 ml |
| 24-well plate | 500 μl | 1.5 ml |
| 48-well plate | 250 μl | 500 μl |
| 96-well plate | 50 μl | 200 μl |
| P100 culture dish | 4-5 ml | 20 ml |
| P60 culture dish | 2-3 ml | 5-7 ml |
| F25 flask | 2-3 ml | 7 ml |
| F75 flask | 4-5 ml | 20-30 ml |
| F175 flask | 15 ml | 30-50 ml |
Table 2. Recommended volumes for polyHEMA coating and 3D culturing.
Discussion
The biology of early-stage epithelial ovarian cancer (EOC) is poorly understood. Perhaps one of the main impediments in this field for many years has been the lack of understanding of the tissue specific origins of the disease, and of the significance of the role of the microenvironment in EOC development. Over the past few years, is has become clear that EOC is a heterogeneous disease with multiple distinct histophathological subtypes, probably with different cellular origins for different subtypes. For example, the current data suggest that high-grade serous EOCs may originate from both fallopian tube secretory epithelial cells and ovarian surface epithelial cells; at least a proportion of endometrioid and clear cell EOCs are thought to arise from ovarian endometriosis (endometrioma); and mucinous EOCs may arise from paraovarian or paratubal transitional-type epithelium 15,16. However, there have been only limited in vitro and in vivo models of disease progression for any of these histotypes. In vitro heterotypic modeling of EOC to study the tumor stroma-epithelial interactions and the microenvironment associated with developing ovarian carcinomas is an even less developed area of research. The tumor stroma is likely to be both a source of tumor biomarkers as well as a novel target for therapy. The development and use of heterotypic models of EOC may therefore be key to identifying novel biomarkers associated with early stage disease which could be used as part of a program of screening and early disease detection; or for the identification of novel, druggable therapeutic targets to improve treatment regimens for patients diagnosed with EOC.
More fundamentally, the models we have described could be used to gain a deeper understanding of the underlying phenotypic and molecular changes that occur during EOC initiation and early disease development. It is now clear that different EOC subtypes have distinct molecular profiles, clinical characteristics and underlying biology. By generating a library of EOC precursor cells and modeling the development of each cancer subtype, including the microenvironmental influences, it may be possible to gain novel insights into the development of EOC histotypes through models that accurately reflect disease development in the human condition. The methodologies we describe can be applied to a broad range of studies and applications, including the study of other solid tumors. We have tested spheroid formation ability in over fifty different normal and transformed ovarian cell lines to date, and observed that the vast majority of cell lines are able to form spheroids when plated at high cell densities on polyHEMA-coated plates. We therefore anticipate that cell culture models from other mammalian organs and also other species could also be cultured in 3D using this approach.
These approaches may represent an advance specifically for studying the role of the microenvironment during tumor initiation, for example in studies of the interactions of discrete somatic genetic aberrations in tumor epithelial cells existing concurrently with specific microenvironmental variables. We have shown that phenotypic changes in stromal cells can affect epithelial cell phenotype 8. By altering the cell types included in the model, one could investigate whether different stromal cells differentially influence epithelial cell proliferation and also expression of other specific markers e.g. markers associated with invasion or metastasis. Moreover, these models can also be used in the study of benign disease and normal ovarian physiology. For example, such 3D models could be used in fertility studies to evaluate the role of the ovarian stroma during oocyte maturation.
For studying the contribution of the microenvironment to EOC development and the role of ovarian cancer-associated fibroblasts in malignant EOCs, these models can also be adapted by co-culturing ovarian cancer cells with fibroblasts isolated from tumor specimens. More complex cultures could also be generated by introducing additional, relevant cell types into the heterotypic structure. For example, the model of stromal and epithelial cell interactions we developed could by supplemented with the introduction of granulosa cells, immune or endothelial cells. Another possibility would be to co-culture other precursor cell types in the ovarian stroma model, such as epithelial cells from the fallopian tube and endometrium. It is also possible to model additional paracrine interactions that are proposed to be important during EOC initiation. For example, steroid hormones could be introduced into the heterotypic model; or expression of paracrine acting genes could be perturbed exclusively in the stromal compartment and the effect on epithelial cells measured.
The majority of ovarian cancers are identified at a late stage, after the disease has disseminated widely throughout the peritoneum. Ovarian cancers diagnosed when localized to the ovaries (Stage 1) can be effectively treated with surgery and in over 90% of cases patients are cured of their disease. It is clear that strategies for earlier detection of ovarian cancers could significantly reduce ovarian cancer morbidity and mortality. By gaining a deeper understanding of early-stage disease development using organotypic modeling approaches such as those described here, it is anticipated that new opportunities for disease detection in at-risk populations will be revealed and ultimately translated into clinical practice.
Disclosures
No conflicts of interest declared.
Acknowledgments
This research was performed at the Keck School of Medicine, University of California, USA, and University College London, UK. KL is funded by National Institute of Health grant 5 U19 CA148112-02. BG was funded by a project grant from the Eve Appeal gynecological oncology charity (UK). Some of this work undertaken at UCLH/UCL was partly funding from the Department of Health's NIHR Biomedical Research Centre funding scheme.
References
- Jelovac D, Armstrong DK. Recent progress in the diagnosis and treatment of ovarian cancer. CA Cancer J. Clin. 2011. [DOI] [PMC free article] [PubMed]
- Office for National Statistics. Cancer Statistics registrations: registrations of cancer diagnosed in 2008. England: 2011. [Google Scholar]
- Köbel M, Kalloger SE, Santos JL. Tumor type and substage predict survival in stage I and II ovarian carcinoma: insights and implications. Gynecol. Oncol. 2010;116:50–56. doi: 10.1016/j.ygyno.2009.09.029. [DOI] [PubMed] [Google Scholar]
- Köbel M, Kalloger SE, Boyd N. Ovarian carcinoma subtypes are different diseases: implications for biomarker studies. PLoS Med. 2008;5:e232. doi: 10.1371/journal.pmed.0050232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LH, Morris CR, Yasmeen S. Ovarian cancer: can we make the clinical diagnosis earlier. Cancer. 2005;104:1398–1407. doi: 10.1002/cncr.21310. [DOI] [PubMed] [Google Scholar]
- Aviv A, Hunt SC, Lin J, Cao X, Kimura M, Blackburn E. Impartial comparative analysis of measurement of leukocyte telomere length/DNA content by Southern blots and qPCR. Nucleic Acids Res. 2011;39:e134. doi: 10.1093/nar/gkr634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim NW, Wu F. Advances in quantification and characterization of telomerase activity by the telomeric repeat amplification protocol (TRAP. Nucleic Acids Res. 1997;25:2595–2597. doi: 10.1093/nar/25.13.2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrenson K, Grun B, Benjamin E. Senescent fibroblasts promote neoplastic transformation of partially transformed ovarian epithelial cells in a three-dimensional model of early stage ovarian cancer. Neoplasia. 2010;12:317–325. doi: 10.1593/neo.91948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrenson K, Benjamin E, Turmaine M. In vitro three-dimensional modelling of human ovarian surface epithelial cells. Cell Prolif. 2009;42:385–393. doi: 10.1111/j.1365-2184.2009.00604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrenson K, Sproul D, Grun B. Modelling genetic and clinical heterogeneity in epithelial ovarian cancers. Carcinogenesis. 2011;32:1540–1549. doi: 10.1093/carcin/bgr140. [DOI] [PubMed] [Google Scholar]
- Grun B, Benjamin E, Sinclair J. Three-dimensional in vitro cell biology models of ovarian and endometrial cancer. Cell Prolif. 2009;42:219–228. doi: 10.1111/j.1365-2184.2008.00579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zietarska M, Maugard CM, Filali-Mouhim A, Alam-Fahmy M, Tonin PN, Provencher DM, Mes-Masson AM. Molecular description of a 3D in vitro model for the study of epithelial ovarian cancer (EOC. Mol. Carcinog. 2007;46:872–885. doi: 10.1002/mc.20315. [DOI] [PubMed] [Google Scholar]
- Shield K, Ackland ML, Ahmed N, Rice GE. Multicellular spheroids in ovarian cancer metastases: Biology and pathology. Gynecol Oncol. 2009;113:143–148. doi: 10.1016/j.ygyno.2008.11.032. [DOI] [PubMed] [Google Scholar]
- Dafou D, Grun B, Sinclair J. Microcell-mediated chromosome transfer identifies EPB41L3 as a functional suppressor of epithelial ovarian cancers. Neoplasia. 2010;12:579–589. doi: 10.1593/neo.10340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levanon K, Crum C, Drapkin R. New insights into the pathogenesis of serous ovarian cancer and its clinical impact. J. Clin. Oncol. 2008;26:5284–5293. doi: 10.1200/JCO.2008.18.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurman RJ, Shih IeM. Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer--shifting the paradigm. Hum. Pathol. 2011;42:918–931. doi: 10.1016/j.humpath.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]