

Abstract
The acid-base behavior of amino acids is an important subject of study due to their prominent role in enzyme catalysis, substrate binding and protein structure. Due to interactions with the protein environment, their pKas can be shifted from their solution values and, if a protein has two stable conformations, it is possible for a residue to have different “microscopic”, conformation-dependent pKa values. In those cases, interpretation of experimental measurements of the pKa is complicated by the coupling between pH, protonation state and protein conformation. We explored these issues using Nitrophorin 4 (NP4), a protein that releases NO in a pH sensitive manner. At pH 5.5 NP4 is in a closed conformation where NO is tightly bound, while at pH 7.5 Asp30 becomes deprotonated, causing the conformation to change to an open state from which NO can easily escape. Using constant pH molecular dynamics we found two distinct microscopic Asp30 pKas: 8.5 in the closed structure and 4.3 in the open structure. Using a four-state model, we then related the obtained microscopic values to the experimentally observed “apparent” pKa, obtaining a value of 6.5, in excellent agreement with experimental data. This value must be interpreted as the pH at which the closed to open population transition takes place. More generally, our results show that it is possible to relate microscopic structure dependent pKa values to experimentally observed ensemble dependent apparent pKas and that the insight gained in the relatively simple case of NP4 can be useful in several more complex cases involving a pH dependent transition, of great biochemical interest.
Author Summary
The interaction of an amino acid with its protein environment can result in an acid-base behavior that is very different from what would be observed in solution. This environment can be greatly altered when the protein changes conformation. As a result, the amino acid will have two different “microscopic” pKa values. Nitrophorin 4 is a good case study to explore this behavior, because it undergoes a pH-dependent conformational change that is well characterized experimentally. Using computer simulation tools, we found that the key titratable Aspartic acid 30, has two very different microscopic pKas: 4.3 and 8.5, which are significantly different to the observed transition pKa in solution. However, using a simple model, we were able to understand how this causes the conformational change to take place at pH∼6.5, as measured experimentally. The insight gained in this relatively simple case can be useful in other more complex cases where the apparent pKa is also a result of the interplay of different conformations where some amino acids experience very different environments.
Introduction
Ionizable amino acid residues have been shown to play important roles in the binding of proteins to other molecules and in enzyme mechanisms. They also have a large influence on protein structure, stability and solubility [1], [2]. The types of interactions these side chains will have with their environment depend on their protonation state. Because of this, their pKa values and the factors that influence them are a subject of intense biochemical interest. Strongly altered pKa values are often seen in the active sites of enzymes, to enhance the ability of ionizable residues of acting as nucleophiles, electrophiles or general bases and acids [3]. As a consequence of the change in protonation of these residues, the stability of proteins is pH-dependent [4]. Changes in intracellular pH have been shown to regulate essential processes like cell proliferation and apoptosis. However, our understanding of how changes in environmental pH affect proteins is limited [5].
The pKa values of ionizable residues in folded proteins can be strongly influenced by the local environment. The three main factors affecting these values are charge-charge interactions [6], charge-dipole interactions, which include hydrogen bonding and the interaction with macroscopic helix dipoles [3], [7], and the Born effect (dehydration) [8], [9]. The effect of charge-charge and charge-dipole interactions on the pKa value becomes stronger when the dielectric constant decreases, as in the case of the hydrophobic interior of proteins [10].
Through these effects, the environment can alter the pKa of a residue to varying extents. For example, while the average pKa value measured in proteins for Asp is 3.5±1.2, values ranging from 0.5 to 9.2 have been reported for this residue [1]. Usually, these values are measured using nuclear magnetic resonance (NMR) by fitting chemical shift vs. pH curves. In some cases they are obtained indirectly by measuring other properties, such as equilibrium constants [11] or midpoint potentials [12] as a function of pH. However, changing the pH can modify the protonation state of a residue, which can couple with a conformational change that causes the environment of this residue to change [2], [13], possibly altering its pKa value. Tanford has shown that, if the equilibrium between two conformations of a protein is pH dependent, then at least one titratable group must have a different pKa in the two conformations [14]. In this manuscript, we will refer to these as “microscopic” pKas. Experimentally, usually only one apparent macroscopic pKa is measured for each residue, which cannot be directly assigned to any individual microscopic conformation.
The coupling of protonation and conformational equilibria is one particular example of the concept of thermodynamic linkage, which was first introduced by Wyman to describe the Bohr effect [15]. Other work in this area includes the study of protein denaturation by pH [16], the development of methods that can accurately treat the coupling between multiple titration sites [17], [18], and the improvement of pKa calculations by taking into account the coupling between conformational flexibility and ionization states [19], [20]. In particular, García-Moreno has studied the linkage between local conformational changes and proton binding in SNase both experimentally [21] and computationally, using an ensemble-based description [22].
Nitrophorin 4 (NP4) is a system displaying this complexity and we will use it as a case study to explore the relationship between the microscopic and macroscopic “apparent” pKas. We will show how two distinct microscopic pKas can give rise to a macroscopic pKa in the physiologically relevant range of 6.5–7.5. Scenarios like NP4 occur in several systems of high biochemical interest where the insight obtained for the relatively simple case of NP4 can be applied. Some of these will be discussed in more detail in the discussion section of this manuscript.
Nitrophorins (NPs) are nitric oxide (NO) carrier heme proteins found in the saliva of some blood-sucking insects [23]. The most extensively studied NPs are those found in the salivary glands of the kissing bug, Rhodnius prolixus, which can act as a vector of the parasite Trypanosoma cruzi, the causative agent of Chagas' Disease (American Trypanosomiasis) [24]. Up to seven of these NPs have been cloned [25] and spectroscopically, kinetically [11] and structurally [26]–[29] characterized. Among these, NP4 is the most extensively studied. In order to fulfill its biological role, NP4 must retain NO in the insect's saliva and release it in the victim's tissues. This selectivity is achieved by binding NO in a pH sensitive manner. The protein binds NO tightly inside the salivary glands of the bug, where the pH is approximately 5. However, when it is injected into the victim's tissues, which have a pH of approximately 7.4, NP4 undergoes a conformational change that reduces substantially its affinity for NO [11], [30].
NP4 is a good case for studying pH-dependent conformational changes using computational tools because there are NO-bound and unbound crystallographic structures available of both the low pH and the high pH state [31] (referred to as “closed” and “open” structures, respectively) which show that this conformational change involves mainly loops AB and GH, which cap the heme side of the β barrel to which NO is bound [31], [32] (Figure 1 and Figure S1). Furthermore, the pH-dependent conformational change that leads to NO release has been studied both theoretically and experimentally [31]–[34]. In this context, although it has been shown that the strength of the heme-NO bond is pH-independent [11], results from our group show that NO is only able to escape in the open conformation because in the low pH state the kinetic escape barrier is very high [33].
Figure 1. Structure of the AB and GH loops in the open and closed conformations.
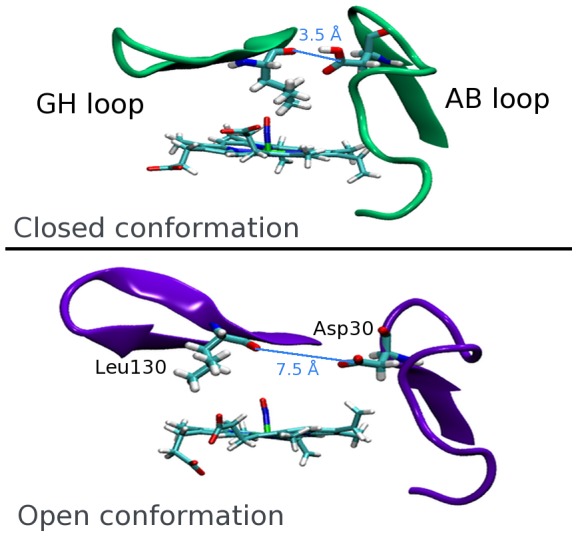
The position of Asp30 and Leu130, and the distance between these residues is also shown. The open conformation is predominant at pH 7.5, while the closed structure is predominant at pH 5.5.
Previous studies have also shown that the protonation of Asp30, which is conserved in the four major NPs of adult insects [12] plays an important role in the conformational change. In the closed conformation this residue is protonated and interacting with Leu130 through a hydrogen bond, keeping loops AB and GH close together. This interaction is lost in the open structure, in which Asp30 is ionized and becomes solvent exposed [32], [35] (Figure 1). In earlier work we have shown that setting the protonation state of this key residue is enough to maintain a closed or open conformation during extensive molecular dynamics (MD) simulations [35]. Although much has been learned about the way NPs work, the fundamental question of what is the value for the Asp30 pKa, and how it is related to the conformational change remains unanswered.
The first attempt to measure a pKa value in NP4 was derived from NO affinity studies as a function of pH. The observed NO dissociation equilibrium constant (KdNO) shows a simple sigmoidal behavior when the pH is changed from 5.0 to 8.0, allowing the determination of an observed apparent pKa from the NO binding data of 6.5 [11]. Later, studies tried to focus on particular residues using a combined site directed mutagenesis (changing the titratable residues for alanines) and electrochemical approach. The resulting apparent pKa value of Asp30 in NP4 was estimated to be 5.5±0.4 [12].
In this work we have used constant pH MD (CpHMD) [36] to study the titration of Asp30 in NP4 in order to determine its microscopic pKas and relate them to the NP4 conformational transition that governs NO affinity. Traditionally, the treatment of pH in MD has been limited to setting a fixed protonation state for each titratable residue, but there are several problems related to this approach: (a) the pKa of a given residue may be unknown, (b) if the pKa of one residue is similar to the solvent pH, a single protonation state is insufficient to appropriately describe the ensemble of protonation states that will be observed at that pH, (c) it is not possible to study the coupling between pKa, protonation state and conformation. A significant advantage of the CpHMD method is that by using MD, the method allows the sampling of different conformations. This is very important because pKa calculations tend to be highly sensitive to details of the environment around the residue that is being titrated. Methods that incorporate little to no conformational flexibility suffer from problems like, for example, exaggerating the magnitude of electrostatic effects, which results in extreme pKas [4], [19]–. CpHMD makes it possible to study the coupling between the solvent pH, the microscopic pKas and the conformation. Given that the pH-dependent conformational change of NP4 is relatively large, an implicit solvation model will be used to ensure that the relevant conformational space will be sampled during the MD simulation. Other possible strategies to deal with this issue include driving the conformational change using Umbrella sampling or an overcharging strategy [38], [39].
The CpHMD [36] method allows titratable residues to be in any discrete protonation state during the computational simulation. Every 10 MD steps, a Monte Carlo (MC) sampling scheme is used to determine the protonation state based on the energy differences between the possible protonation states [36], [40]–[41]. The pKa of the amino acid residue can be estimated based on the fraction of time it spent protonated at a given pH, assuming ergodicity and Henderson-Hasselbach titration curves [36]. A slightly different approach for performing constant pH MD simulations allows protonation parameters to vary continuously between protonation states [42]–[44].
Our results show that the microscopic pKa of Asp30 in the closed conformation (pKa = 8.5) is significantly different from that in the open conformation (pKa = 4.3). Furthermore, we also show that the protein conformation and, therefore, the pKa of Asp30 are closely coupled to the solvent's pH, and we trace how the pKa changes as the conformational change takes place. With this data, we performed an analysis similar in spirit to Tanford's [14] but with a focus on how the microscopic pKa values of this key residue determine the macroscopic, observed pKa. We were able to set limits for the free energy values for the conformational closed-to-open transition for both protonation states of Asp30 and calculate the pH at which the closed to open transition takes place. The computed value, which is close to 6.5, is in excellent agreement with the apparent pKa obtained for the experimental titration of NO affinity. We determined that the apparent pKa corresponds to the pH at which the conformational change takes place. The analysis performed and the conclusions drawn for the case of NP4 can be applied to several more complex cases of great biochemical interest.
Results
The results are organized as follows. First, we will describe simulations of the open and closed structure at the solvent pH values at which they are stable. From these we were able to estimate the microscopic pKa of Asp30 in both conformations. We will then describe the results from simulations in which each structure was placed at a solvent pH at which it is not stable, focusing on how the conformational change is coupled to the microscopic pKa of Asp30. Finally, in the discussion we will show how the microscopic pKas obtained for each conformation are related to the macroscopic pKa.
Stable simulations: Closed conformation at pH 5.5 and open conformation at pH 7.5
As expected, CpHMD simulations of the closed conformation at pH = 5.5 and of the open conformation at pH = 7.5 show that both structures remain stable during the simulation timescale. In our previous studies we were able to determine some key interactions that characterize each protein conformational state (open or closed) and that remain stable during fixed Asp30 protonation state simulations [35]. Distance CAsp30-OLeu130 shows whether the hydrogen bond that keeps the AB and GH loops close together is formed. Distances CBVal36-CGLeu130 and CGAsp35-OAsp129 can be monitored to know whether the NO escape route is open (Figure S2). As can be seen from Table 1, the results for the CpHMD simulations show that the interactions are maintained to similar values as those obtained previously in ref. 35.
Table 1. Average distances for selected characteristic interactions in the open and closed conformations of NP4.
| Structure | dCAsp30-OLeu130 | dCBVal36-CGLeu130 | dCGAsp35-OAsp129 |
| Closed, fixed (*) | 3.6±0.3 | 5.9±0.7 | 5±1 |
| Closed, pH 5.5 | 3.5±0.3 | 6.4±0.8 | 5±1 |
| Open, fixed (*) | 7.2±0.9 | 13±1 | 12±2 |
| Open, pH 7.5 | 8±1 | 14±2 | 14±3 |
Distances and their respective standard deviations are shown in Å.
Data from structures labeled “fixed” corresponds to simulations where the protonation state of Asp30 is fixed, while in the rest of the cases the protonation of Asp30 is allowed to change.
A detailed analysis of Asp30 shows that in the simulations of the closed state at a solvent pH of 5.5, Asp30 remains protonated during most of the simulation, resulting in a pKa value of 8.5. The simulations of the open state at solvent pH = 7.5 show that Asp30 remains ionized most of the time, resulting in a pKa value of 4.3. Therefore, the pKa of Asp30 in the closed conformation is significantly different from that in the open conformation. The obtained pKa values can be rationalized looking at the Asp30 interactions along the MD. In the open state the residue is solvent exposed, with a solvent-accessible surface area (SASA) value of 40±2 Å2 and, therefore, has a pKa value close to 4.0, the pKa for an isolated Asp [3]. In the closed state, the residue is almost completely buried (the SASA value is reduced to 9±4 Å2) and involved in a hydrogen bond. As a consequence, the resulting pKa is 4.5 units higher than the intrinsic Asp pKa. Although this is a large pKa shift, even higher pKas have been reported for Asp in the literature [1].
Transition inducing simulations: Closed conformation at pH 7.5 and open conformation at pH 5.5
We now turn our attention to CpHMD simulations that start from the closed conformation and are run at pH = 7.5 and that of the open conformation at pH = 5.5, i.e. corresponding to conditions where the starting structure is not optimal. As expected, results show that these structures are not stable under these conditions (see RMSD values in Figure S3). In both cases the CpHMD scheme changes the protonation state of Asp30 during the dynamics and this triggers the onset of the conformational change, as evidenced by the shift in the monitored distances towards the typical values of the opposite structure, i.e. the stable structure at the simulation pH. It is important, then, to highlight that using this method we were not only able to measure the pKas, but also to observe the conformational change that takes place upon a change in protonation of Asp30. As consequence of the onset of the structural transition, the microscopic pKa changes along the simulation, a fact that is also accompanied by a change in the SASA of the residue (Figure S4). Figure 2 illustrates this point, by showing the time evolution of relevant distances and of the pKa as a function of time when the closed conformation is placed in pH 7.5 solvent. A close look at the Asp30 interactions shows that ionization of this residue causes the hydrogen bond between Asp30 and Leu130 to break after 9 ns of simulation time. This change is coupled to the opening of the NO escape route, as shown by the increase in the distance Asp35-Asp129, which reaches values that are close to those expected in the open conformation. The RMSD plots of the AB and GH loops with respect to the closed and open structure (Figure S3), also show that the position of these loops changes to resemble that of the open conformation.
Figure 2. Time evolution of relevant parameters when closed NP4 is placed at a solvent pH of 7.5.
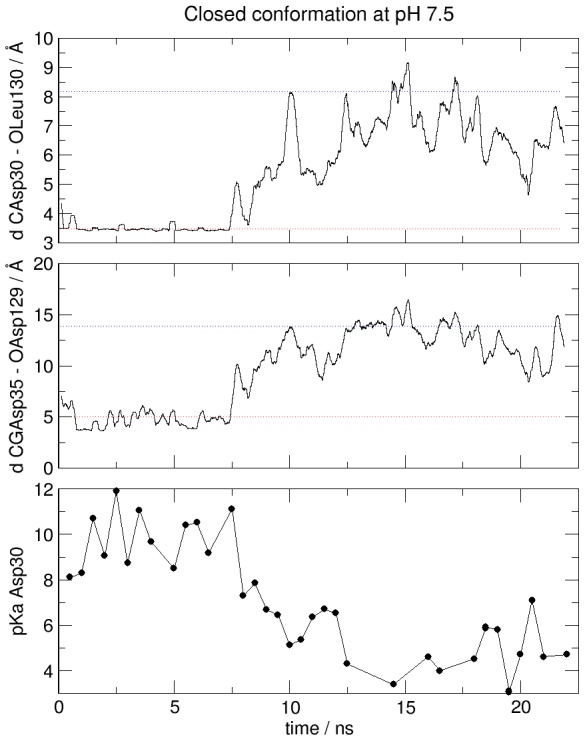
Top: Running average of distance Asp30-Leu130; Middle: Running average of distance Asp35-Asp129; Bottom: Asp30 microscopic pKa. The average values of these distances in the stable simulations of the closed (red) and the open (blue) structures are also shown.
On the other hand, for the simulations which start from the open conformation but are placed at pH 5.5, as Asp30 becomes protonated, its side chain is rapidly buried inside the protein (SASA is reduced), bringing it close to Leu130 and allowing the hydrogen bond between these residues to form, closing the NO escape route. Also in this case it is possible to see that as the conformational change occurs, Asp30 pKa increases to reach values typical of the closed conformation (for additional detail, see Figure S5 and text S1).
In summary, our results show that the microscopic pKa of Asp30 in the open conformation (4.3) is significantly different from that in the closed conformation (8.5). These pKa values enable the protein to be stable in its closed conformation at a low pH, and the open conformation to be stable at a high pH, as required to fulfill its biological role. Transition-inducing CpHMD simulations enabled us to show that when a conformation is placed at a pH at which it is not stable, a conformational change takes place and, as this happens, the pKa of Asp30 changes in time.
Discussion
The relationship between the microscopic and apparent macroscopic pKa values
We have shown that there are two distinct microscopic pKas, 4.3 and 8.5, with neither corresponding to the experimentally observed estimated value assigned to Asp30 of 5.5±0.4, as derived from electrochemical measurements [12], or the apparent pKa of 6.5 derived from the pH dependence of the NO binding equilibrium constant [11]. The obvious question now is: Where does the observed pKa come from? To relate the computed microscopic pKa values to the experimental observations, and to understand how the NP4 population ensemble changes with the solvent pH, we postulate a simple four state system (similar to Tanford's [14]). We analyzed the relative populations of the different NP4 states according to the chemical equilibria shown in Figure 3.
Figure 3. Thermodynamic cycle relating the relevant chemical species and their equilibrium constants.
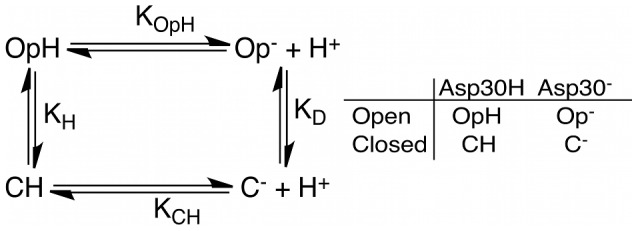
According to Figure 3, the equilibrium between the open state with Asp30 protonated (OpH) and deprotonated (Op−) is described by the computed pKa in the open conformation (pKOpH) value of 4.3. The corresponding value in the case of the closed conformation is pKCH = 8.5. The values of KH = (OpH)/(CH) and KD = (Op−)/(C−) correspond to the equilibrium constants determined by the free energy difference between the open and closed conformations and with Asp30 neutral or charged, respectively. Although these values are unknown, reasonable bounds can be estimated. In order to function efficiently, at low pH NP4 must be predominantly in the closed state, but since Asp30 is mostly protonated, for this to be true, KH≪1. On the other hand, at a high pH, the open structure must be predominant. Since at this pH Asp30 is found deprotonated, it follows that KD≫1. However, as shown by Figure 3, both KH and KD are not independent. Using the thermodynamic cycle, both equilibrium constants can be related by equation 1:
| (1) |
Using Equation 1, KH for example can be calculated if all other constants are known, leaving KD as the only unknown parameter. Using the cycle above, mass balance equations and the values of pKCH and pKOpH obtained from our calculations, the equilibrium concentrations of all the relevant chemical species can be calculated at any pH value for any given value of KD (see text S2 for complete derivation). Figure 4 is an example of this, in which KD was chosen to be 100 and, as a consequence, KH equals 6.31×10−3.
Figure 4. Mole fractions of the relevant chemical species as a function of pH.
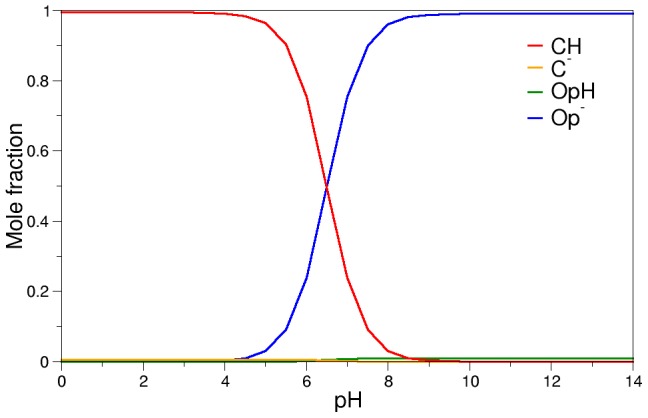
KD = 100, KH = 6.31×10−3.
Figure 4 shows that at a pH of less than 5, the closed conformation in which Asp30 is protonated is predominant. Raising the pH results in a dramatic increase of the concentration of the open conformation with Asp30 ionized, which is the predominant chemical species at pH higher than 8. These results are consistent with the kinetic and crystallographic evidence available. It is important to note that using the pKOpH and pKCH values obtained in the present work, the diagram will show the same behavior only if 10<KD<1500. For values outside this range, either the concentration of OpH or of C− become significant (>10%) at extreme pH values, contradicting the known experimental data. It is also possible to calculate what is the KD value that maximizes the efficiency of the protein to fulfill its biological role. Figure 5 shows how the concentration of CH at pH 5.5 and Op− at pH 7.5 varies as a function of KD. NP4 will be most efficient by simultaneously maximizing the concentration of the closed form at pH 5.5 and the concentration of the open form at a high pH, to ensure that NO is released in the victim's tissues and not in the salivary glands of the insect. This is achieved when KD is approximately 100 Larger values of KD result in an increase of the population of OpH at a low pH, which would cause release of NO in the insect's salivary glands while lower values would cause an increase in the population of C− at high pH, which would reduce the amount of NO released into the victim's tissues. It is important to note that using this KD value also yields results which are in agreement with the observation that at pH 5.5 and 7.5 there is a mixture of the open and closed conformations [45], [46]: the population of the open conformation at pH 5.5 and of the closed conformation at pH 7.5 are approximately 10%. Studies of CO rebinding upon laser photolysis show that the population of the closed conformer at neutral pH is significant [47], and estimated to be 23% [48]. This is also in good agreement with our results: for KD = 100 the closed population is 24%.
Figure 5. Concentration of CH at pH 5.5 and Op− at pH 7.5 as a function of KD.
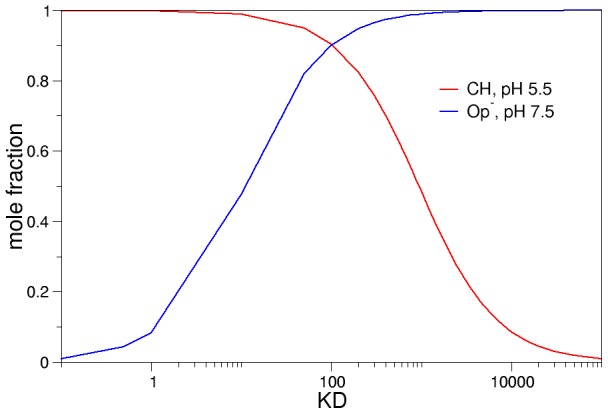
(CH) at pH 5.5 shown in red, while (Op−) at pH 7.5 is shown in blue. The concentrations are given as mole fractions.
As the experiments designed to measure the pKa typically report on the overall protein conformation, we can calculate the observed signal (i.e. spectroscopic signature, redox potential or any other conformation-dependent measurable property) according to Equation 2.
| (2) |
In this equation, α and β are parameters that correspond to the limiting values of the measured signal, in the closed and open conformations respectively. Equation 2 can be applied, for example, in the case where the measured property is the NO dissociation equilibrium constant, KdNO, in NP4. Using the computed pKa values, KD = 100 and obtaining α and β from the limits of the KdNO vs. pH plot taken from ref. 11 we can plot the signal as a function of pH, as shown in Figure 6. This plot has the expected sigmoidal shape and is in excellent agreement with the experimental data. The apparent pKa can be calculated as the inflection point of this curve according to Equation 3 (see Text S2 for more detail), yielding a value of 6.5, also in excellent agreement with the reported data [11].
| (3) |
From Equation 3, it follows that the pKa is always bounded by the values of the pKa in the open and closed conformations, as shown in Figure 7. Moreover, varying KD in the range of values discussed above does not significantly change the population transition pKa, but only the [OpH]/[CH] and [Op−]/[C−] ratios at high or low pH values, underscoring the reliability of the obtained results and at the same time providing bounds for the transition free energies.
Figure 6. Calculated signal as a function of pH, and comparison to experimental results.
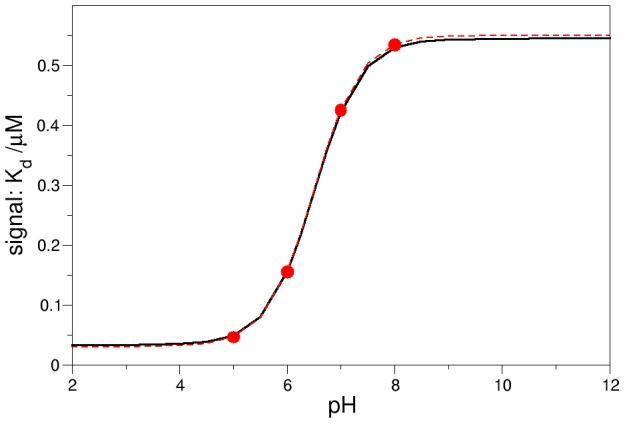
Black: Signal, as defined in Equation 2, as a function of pH for KD = 100, KH = 6.31×10−3, α = 0.03, β = 0.55 (α and β are obtained from ref. 11). Red: NO dissociation equilibrium constant (KdNO) vs pH and fit to the equation of a titration curve with an apparent pKa of 6.5, as obtained from ref. 11.
Figure 7. Apparent pKa, calculated using Equation 3, as a function of KD.
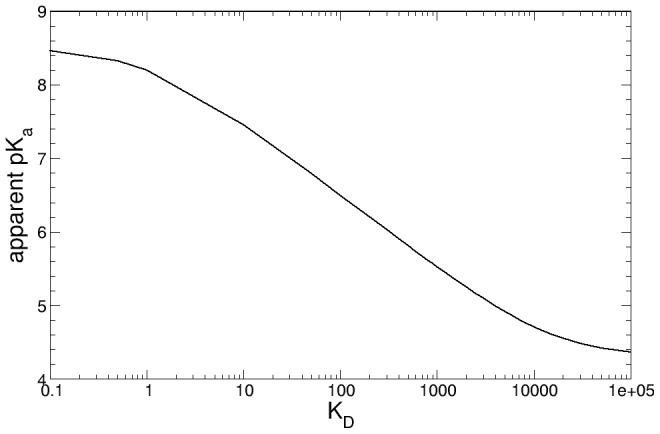
Finally, we analyzed the dependence of the apparent pKa on the estimated KD value, as shown in Figure 7. The results show that for a KD in the range 10<KD<1500 the apparent pKa varies only between 5.4 and 7.5, showing that our results allow us to predict the experimental apparent pKa within one pKa unit, having only made broad assumptions regarding the value of KD. However, using the arguments presented in Figure 5, we can see that a KD of approximately 100 gives an apparent pKa value in very good agreement with the experimental results based on NO equilibrium [11]. A higher KD (KD≈1000), is necessary to obtain a lower apparent pKa of 5.5, consistent with the estimate for Asp30 [12].
At this point it is very important to note that although the microscopic structure dependent pKa values are assigned to a particular residue, Asp30, the apparent pKa which is related to the experiment is not necessarily the apparent pKa value for this residue, but the pKa of the structural transition, which as will be discussed below is what is usually measured. Moreover, although in the present work only Asp30 was set as titratable, the choice is justified by the facts that a) differentially setting this residue protonation state is enough to maintain each conformation during the MD simulations [35] b) the resulting apparent pKa derived from the computed values is in excellent agreement with the experimental available data. Moreover, the fact that changing the protonation state of Asp30 is enough to describe with reasonable accuracy the system behavior, although it does not rule out the possibility that other titratable residues contribute to the observed process, strongly suggests, in agreement with experimental data, that it plays a primary role.
The type of analysis presented here to relate the microscopic pKa values to the apparent pKa can be applied to other proteins that undergo pH-dependent conformational changes and may provide useful insight for understanding more complex cases, where several titratable residues interact.
Relationship to other systems
Our results may explain why titrations of several aspartic and glutamic acid residues in NP1, NP2 and NP4 (including Asp30) yield values in the 5.5–7.5 range [12]. Specifically, NP2 undergoes a pH-dependent conformational change that is very similar to NP4, with Asp29 playing a key role similar to that of Asp30 [49]. The experimentally measured pKa for NP2Asp29 is 5.4±0.4 [12]. The arguments presented here can be extended to that case and a small change in each microscopic pKa or the KD would result in the observed macroscopic value. It is likely that they can also be extended to the recently crystallized NP7, in which the key aspartic acid residue is also conserved [50]. Also, β-lactoglobulin displays a pH-dependence similar to that observed in NPs. The protein undergoes a conformational change, known as the Tanford transition, at pH∼7.5 [51] that is coupled to a change in the protonation state of Glu89 [52]–[53]. Tanford used a setup similar to ours to show that the microscopic pKa of this residue must be different in the two conformations [14].
Several proton translocation pathways in active transporters have functionally important carboxylates with pKa values close to physiological pH for at least some stage of the transport cycle [54]. For example, in the multidrug transporter AcrB, protonation and deprotonation events trigger conformational changes that allow drug transport. Two essential Asp residues (Asp407 and 408) are responsible for proton translocation. In one of the conformations, these are considered to be deprotonated and interacting with a positively charged Lys side-chain. However, when the carboxylates become protonated, Lys turns away from them and towards a Thr residue [55]. In the context of our analysis, the experimentally measured pKa value of 7.4 for Asp408 can be interpreted as arising from two very different microscopic pKas coupled by a conformational change. Another case where carboxylates play a key role is the Nha Na+/H+ antiporter of E. coli. In the pH = 4 structure multiconformation continuum electrostatics calculations predict that Asp 163 and 164, which are part of the Na+ binding site, have a pKa of >15. However, a structural change that lowers the pKa of these residues is required for the antiporter to be active at its normal pH range of 6.5–8.5 [56]. Finally, the type of analysis presented in this work could be useful to interpret the pKa value of ∼7 measured for Glu65 [57]–[59], the residue thought to interact with the key Arg (known as stator charge) at the a/c interface of F1F0 ATP synthase. The current model for torque generation in this system involves Arg forming an ion pair with a deprotonated Glu65. Upon competition with the coupling ion, Arg is displaced and Glu65 becomes protonated, triggering a conformational change that buries this residue in a hydrophobic region. Based on the environment of Glu65 in each state, we expect there will be two very different microscopic pKas. However, the measured pKa of 7 is interpreted in some of the work on this topic as arising from a hydrophobic environment and a local structure around this residue that has been optimized to raise the pKa to the physiological range [58]–[59]. We want to emphasize that in cases such as this it is crucial to think of the measured pKa as arising from the coupling of two structures and not from a single static structure.
Can microscopic pKas be observed experimentally?
Our results also suggest the possibility that the microscopic pKas cannot directly be measured experimentally, but can certainly be computed via a thermodynamics model like ours. Typically, the experiments involve the acquisition of data (e.g. NMR, IR, enzymatic rates, equilibrium binding constants) as a function of solvent pH over the range of interest and subsequent fitting of the data to a titration curve [60]. These are structural probes and, as such, they do not report directly on protonation states. In our system, the following paradox applies: to be able to accurately measure the pKa of Asp30 in the closed structure it would be necessary to make measurements at a solvent pH close to the residue's pKa of 8.5. However, at this pH the protein is found in its open conformation, so the experiment seems impossible to perform. A similar reasoning applies to the measurement of the pKa in the open conformation. Only in some particular cases it is possible to obtain the value of the microscopic pKas of one residue in two different conformations using pH-jump experiments [61]–[64]. Using a thermodynamic cycle similar to the one shown in Figure 3 it is possible to obtain a relationship between the relaxation time of the experiment and the pKas. Knowledge of microscopic pKas can be used to improve the analysis of NMR spectra tracking a pH-induced conformational change [2]. It is also important to keep in mind that pH-dependent conformational changes may be taking place when comparing data from pKa calculations to experimental results [44].
Conclusion
Our results show that there are two distinct microscopic Asp30 pKa values: 8.5 in the closed conformation and 4.3 in the open conformation. These values are reasonable once the environment of Asp30 in each state is considered: in the closed structure it is buried and forming a hydrogen bond with Leu130 that is key to keeping the AB and GH loops close together, while in the open structure this bond is broken and the residue becomes exposed to the solvent.
We were also able to confirm that the closed structure is stable at a pH of 5.5, while the open structure is stable at pH 7.5. However, we found that the closed structure, when studied at a high pH, spontaneously undergoes a conformational change to the open structure. Similar results were obtained when the open structure was placed at a low pH. We found that these transitions are coupled to a change in the protonation state of Asp30 and that they can be traced by analyzing the change in the distances between amino acids significantly involved in the process and in the microscopic pKa of Asp30. These results show that the microscopic pKa of Asp30 is highly coupled to the conformation of NP4, which in turn is pH-dependent.
Finally, we were able to reconcile the two microscopic pKas obtained with the apparent pKa measured experimentally. Our results show that the apparent pKa that governs the structural transition is 6.5, in excellent agreement with the pKa obtained from NO affinity studies. The apparent pKa corresponds to the pH at which the conformational change from the closed to the open state takes place. It is important to highlight that while the microscopic pKas were significantly different from the apparent pKa, the coupling between the conformation and the solvent pH gives rise to an apparent pKa that is within the physiological range. This is a remarkable property because it allows for amino acids that do not normally titrate in this pH range to act as conformational change triggers: if an amino acid has an atypical pKa in one of the conformations then the apparent pKa will be intermediate and may fall in this range.
These conclusions can be extended to other systems of great biochemical interest, where the environment of the amino acid of interest changes significantly with pH. In these systems, where there is a coupling between protonation and conformational states, no possible conclusion can be derived from looking at individual structures and the apparent pKa should be interpreted as the pH at which the conformational change takes place.
Methods
The initial structures for the CpHMD simulations were built starting from the crystal structures of NO bound NP4 at pH 5.6 (PDB code 1X8O) and at pH 7.4 (PDB code 1X8N) [31]. In these structures, the conformations of the AB and GH loops are characteristic of the closed and open states, respectively. The protonation states of all residues except Asp30 were assigned as suggested from previous experimental [31] and theoretical [33]–[34] studies, and were fixed throughout the simulations. Asp30 was kept as the only titratable residue. This choice is justified by previous studies, which show that CH and Op− are stable conformations while C− and OpH are unstable and begin to undergo the conformational change to the structure that is more stable given the selected protonation state of Asp30 [35] and also, as shown in the results, by the good agreement with the available experimental data.
All simulations were performed at 300 K, maintained using Langevin dynamics with a collision frequency of 2.5 ps−1 [65]. The SHAKE algorithm was used to keep bonds involving H atoms at their equilibrium length [66]. Newton's equations were integrated with a time step of 2 fs. The Amber ff99SB force field parameters were used for all residues [67] except the heme, for which parameters developed and tested by our group on previous works were used [68]. The parameters for Asp30 were taken from the original CpHMD article [36]. All simulations were performed with the SANDER module of the AMBER 11 suite [69]. Frames were collected at 1 ps intervals. All simulations were performed using the Generalized Born implicit solvent model [70]. The salt concentration was set at 0.1 M and the cutoff for non-bonded interactions and Born radii was 30 Å.
The CpHMD method [36] was used to allow Asp30 to adapt its protonation state to the specific conformation. This method involves periodic MC sampling of protonation states during the standard generalized Born simulation. At each MC step, a new protonation state is chosen randomly for Asp30 and the free energy of the transition is computed. This value, which depends on both the environment of the titrated residue and the solvent pH, is used as the basis for applying the Metropolis criterion [71] to determine whether the transition will be accepted. If the transition is accepted, the MD simulation continues with Asp30 in the new protonation state. Otherwise, the MD continues with no changes in the protonation state. In this work, a MC step was performed every 20 fs. The pKa value of Asp30 is then computed based on the population of the protonated and deprotonated states using Henderson-Hasselbach's equation.
Four types of CpHMD simulations were performed. Two of these started from the closed conformation, but the solvent pH was set at 5.5 in one case and 7.5 in the other. The other two types were analogous, but started from the open conformation. Also, simulations of the closed structure with a protonated Asp30 and of the open structure with Asp30 ionized were performed for comparative purposes. Over 150 ns of CpHMD simulations were obtained.
Supporting Information
AB and GH loops in the context of the complete protein. A) Closed conformation. B) Open conformation. The AB and GH loops are shown in a different color than the rest of the protein. Leu130 and Asp30 are shown. Asp30 is displayed protonated in the closed structure and deprotonated in the open conformation.
(TIF)
Val36, Asp35 and Asp129 in the closed and open conformations. A) Closed conformation. B) Open conformation. Asp 30 is shown in gray for reference. The AB and GH loops are colored according to the conformation. Average values for dCBVal36-CGLeu130 and dCGAsp35-OAsp129 are also shown.
(TIF)
RMSD of the AB and GH loops. Running average of the time evolution of the RMSD of the AB and GH loops with respect to the closed (red) and open (black) structures. A) Initially closed structure, simulation at pH 5.5, B) Initially closed structure, simulation at pH 7.5, C) Initially open structure, simulation at pH 5.5, D) Initially open structure, simulation at pH 7.5. In case C the transition to the closed structure takes place almost immediately, but after ∼8 ns the simulation becomes trapped in an intermediate structure (see Text S1 for further detail).
(TIF)
Time evolution of the Solvent Accessible Surface Area (SASA) of Asp30. Running average of the time evolution of the SASA of Asp30. A) Initially closed structure, simulation at pH 5.5, B) Initially closed structure, simulation at pH 7.5, C) Initially open structure, simulation at pH 5.5, D) Initially open structure, simulation at pH 7.5. In case C the transition to the closed structure takes place almost immediately, but after ∼8 ns the simulation becomes trapped in an intermediate structure (see Text S1 for further detail).
(TIF)
Time evolution of relevant parameters when open NP4 is placed at a solvent pH of 5.5. Top: Running average of distance Asp30-Leu130; Middle: Running average of distance Asp35-Asp129; Bottom: Asp30 microscopic pKa. The average values of these distances in the stable simulations of the closed (red) and the open (blue) structures are also shown.
(TIF)
Additional data for transition inducing simulations. Further details on the simulation of the open conformation at pH 5.5.
(PDF)
Derivations of the equations presented in the main text. Details of the calculation of the equilibrium concentration of all chemical species and the apparent pKa.
(PDF)
Funding Statement
This work is supported by the National Institute of Health (http://www.nih.gov/) under contract 1R01AI073674. This work was partially supported by grants PICT-2010-0416 (http://www.agencia.gov.ar/), UBACYT 2010-2012 (http://www.uba.ar/) and a grant from the Bunge & Born Foundation (http://www.fundacionbyb.org/) to MAM. Computer resources and support were provided by the Large Allocations Resource Committee through grant TG-MCA05S010 and the University of Florida High-Performance Computing Center. Publication of this article was funded in part by the University of Florida Open Access Publishing Fund. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Grimsley GR, Scholtz JM, Pace CN (2009) A summary of the measured pK values of the ionizable groups in folded proteins. Protein Sci 18: 247–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kukić P, Farrell D, Søndergaard CR, Bjarnadottir U, Bradley J, et al. (2010) Improving the analysis of NMR spectra tracking pH-induced conformational changes: removing artefacts of the electric field on the NMR chemical shift. Proteins 78: 971–984. [DOI] [PubMed] [Google Scholar]
- 3. Harris TK, Turner GJ (2002) Structural Basis of Perturbed pKa Values of Catalytic Groups in Enzyme Active Sites. IUBMB Life 53: 85–98. [DOI] [PubMed] [Google Scholar]
- 4. Gunner MR, Mao J, Song Y, Kim J (2006) Factors influencing the energetics of electron and proton transfers in proteins. What can be learned from calculations. Biochim Biophys Acta 1757: 942–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Srivastava J, Barber DL, Jacobson MP (2007) Intracellular pH sensors: design principles and functional significance. Physiology 22: 30–39. [DOI] [PubMed] [Google Scholar]
- 6. McIntosh LP, Hand G, Johnson PE, Joshi MD, Körner M, et al. (1996) The pKa of the general acid/base carboxyl group of a glycosidase cycles during catalysis: a 13C-NMR study of Bacillus circulans xylanase. Biochemistry 35: 9958–9966. [DOI] [PubMed] [Google Scholar]
- 7. Chivers PT, Prehoda KE, Volkman BF, Kim BM, Markley JL, et al. (1997) Microscopic pKa values of Escherichia coli thioredoxin. Biochemistry 36: 14985–14991. [DOI] [PubMed] [Google Scholar]
- 8. Dwyer JJ, Gittis AG, Karp DA, Lattman EE, Spencer DS, et al. (2000) High apparent dielectric constants inthe interior of a protein reflect water penetration. Biophys J 79: 1610–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stites WE, Gittis AG, Lattman EE, Shortle D (1991) In a staphylococcal nuclease mutant the side-chain of a lysine replacing valine 66 is fully buried in the hydrophobic core. J Mol Biol 221: 7–14. [DOI] [PubMed] [Google Scholar]
- 10. Schutz CN, Warshel A (2001) What Are the Dielectric “Constants” of Proteins and How To Validate Electrostatic Models? Proteins 44: 400–417. [DOI] [PubMed] [Google Scholar]
- 11. Andersen JF, Ding XD, Balfour CA, Shokhireva TK, Champagne DE, et al. (2000) Kinetics and equilibria in ligand binding by nitrophorins 1–4: evidence for stabilization of a nitric oxide-ferriheme complex through a ligand-induced conformational trap. Biochemistry 39: 10118–10131. [DOI] [PubMed] [Google Scholar]
- 12. Berry RE, Shokhirev MN, Ho AYW, Yang F, Shokhireva TK, et al. (2009) Effect of mutation of carboxyl side-chain amino acids near the heme on the midpoint potentials and ligand binding constants of nitrophorin 2 and its NO, histamine, and imidazole complexes. J Am Chem Soc 131: 2313–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chakrabarty S, Namslauer I, Brzezinski P, Warshel A (2011) Exploration of the cytochrome c oxidase pathway puzzle and examination of the origin of elusive mutational effects. Biochim Biophys Acta 1807: 413–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tanford C (1961) Ionization-linked Changes in Protein Conformation. I. Theory. J Am Chem Soc 83: 1628–1634. [Google Scholar]
- 15. Wyman J (1948) Heme Proteins. Adv Protein Chem 4: 407–531. [DOI] [PubMed] [Google Scholar]
- 16. Yang A, Honig B (1993) On the pH dependence of protein stability. J Mol Biol 231: 459–474. [DOI] [PubMed] [Google Scholar]
- 17. Antosiewicz J, McCammon JA, Gilson MK (1994) Prediction of pH-dependent properties of proteins. J Mol Biol 238: 415–436. [DOI] [PubMed] [Google Scholar]
- 18. Bashford D, Karplus M (1991) Multiple-site titration curves of proteins: an analysis of exact and approximate methods for their calculation. J Phys Chem 95: 9556–9561. [Google Scholar]
- 19. Georgescu RE, Alexov EG, Gunner MR (2002) Combining conformational flexibility and continuum electrostatics for calculating pK(a)s in proteins. Biophys J 83: 1731–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Alexov EG, Gunner MR (1997) Incorporating protein conformational flexibility into the calculation of pH-dependent protein properties. Biophys J 72: 2075–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Karp DA, Gittis AG, Stahley MR, Fitch CA, Stites WE, et al. (2007) High apparent dielectric constant inside a protein reflects structural reorganization coupled to the ionization of an internal Asp. Biophys J 92: 2041–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Whitten ST, García-Moreno E B, Hilser VJ (2005) Local conformational fluctuations can modulate the coupling between proton binding and global structural transitions in proteins. Proc Natl Acad Sci U S A 102: 4282–4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Montfort WR, Weichsel A, Andersen JF (2000) Nitrophorins and related antihemostatic lipocalins from Rhodnius prolixus and other blood-sucking arthropods. Biochim Biophys Acta 1482: 110–118. [DOI] [PubMed] [Google Scholar]
- 24. Kirchhoff LV (1993) American trypanosomiasis (Chagas' disease)–a tropical disease now in the United States. N Engl J Med 329: 639–644. [DOI] [PubMed] [Google Scholar]
- 25. Champagne DE, Nussenzveig RH, Ribeiro JMC (1995) Purification, partial characterization, and cloning of nitric oxide-carrying heme proteins (nitrophorins) from salivary glands of the blood-sucking insect Rhodnius prolixus . J Biol Chem 270: 8691–5. [DOI] [PubMed] [Google Scholar]
- 26. Andersen JF, Champagne DE, Weichsel A, Ribeiro JMC, Balfour CA, et al. (1997) Nitric oxide binding and crystallization of recombinant nitrophorin I, a nitric oxide transport protein from the blood-sucking bug Rhodnius prolixus . Biochemistry 36: 4423–4428. [DOI] [PubMed] [Google Scholar]
- 27. Andersen JF, Montfort WR (2000) The crystal structure of nitrophorin 2. A trifunctional antihemostatic protein from the saliva of Rhodnius prolixus . J Biol Chem 275: 30496–30503. [DOI] [PubMed] [Google Scholar]
- 28. Andersen JF, Weichsel A, Balfour CA, Champagne DE, Montfort WR (1998) The crystal structure of nitrophorin 4 at 1.5 A resolution: transport of nitric oxide by a lipocalin-based heme protein. Structure 6: 1315–1327. [DOI] [PubMed] [Google Scholar]
- 29. Weichsel A, Andersen JF, Champagne DE, Walker FA, Montfort WR (1998) Crystal structures of a nitric oxide transport protein from a blood-sucking insect. Nat Struct Mol Biol 5: 304–309. [DOI] [PubMed] [Google Scholar]
- 30. Weichsel A, Andersen JF, Roberts SA, Montfort WR (2000) Nitric oxide binding to nitrophorin 4 induces complete distal pocket burial. Nat Struct Mol Biol 7: 551–554. [DOI] [PubMed] [Google Scholar]
- 31. Kondrashov DA, Roberts SA, Weichsel A, Montfort WR (2004) Protein functional cycle viewed at atomic resolution: conformational change and mobility in nitrophorin 4 as a function of pH and NO binding. Biochemistry 43: 13637–13647. [DOI] [PubMed] [Google Scholar]
- 32. Maes EM, Weichsel A, Andersen JF, Shepley D, Montfort WR (2004) Role of binding site loops in controlling nitric oxide release: structure and kinetics of mutant forms of nitrophorin 4. Biochemistry 43: 6679–6690. [DOI] [PubMed] [Google Scholar]
- 33. Martí MA, González Lebrero MC, Roitberg AE, Estrin DA (2008) Bond or cage effect: how nitrophorins transport and release nitric oxide. J Am Chem Soc 130: 1611–1618. [DOI] [PubMed] [Google Scholar]
- 34. Menyhárd DK, Keserü GM (2005) Protonation state of Asp30 exerts crucial influence over surface loop rearrangements responsible for NO release in nitrophorin 4. FEBS Lett 579: 5392–5398. [DOI] [PubMed] [Google Scholar]
- 35. Martí MA, Estrin DA, Roitberg AE (2009) Molecular basis for the pH dependent structural transition of Nitrophorin 4. J Phys Chem B113: 2135–2142. [DOI] [PubMed] [Google Scholar]
- 36. Mongan J, Case DA, McCammon JA (2004) Constant pH molecular dynamics in generalized Born implicit solvent. J Comput Chem 25: 2038–2048. [DOI] [PubMed] [Google Scholar]
- 37. Baran KL, Chimenti MS, Schlessman JL, Fitch C, Herbst KJ, et al. (2008) Electrostatic effects in a network of polar and ionizable groups in staphylococcal nuclease. J Mol Biol 379: 1045–1062. [DOI] [PubMed] [Google Scholar]
- 38. Kato M, Warshel A (2006) Using a charging coordinate in studies of ionization induced partial unfolding. J Phys Chem B 110: 11566–11570. [DOI] [PubMed] [Google Scholar]
- 39. Damjanović A, Wu X, García-Moreno EB, Brooks BR (2008) Backbone relaxation coupled to the ionization of internal groups in proteins: a self-guided Langevin dynamics study. Biophys J 95: 4091–4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Baptista AM, Teixeira VH, Soares CM (2002) Constant-pH molecular dynamics using stochastic titration. J Chem Phys 117: 4184–4200. [Google Scholar]
- 41. Walczak AM, Antosiewicz JM (2002) Langevin Dynamics of Proteins at Constant pH. Phys Rev E Stat Nonlin Soft Matter Phys 66: 051911/1–051911/8. [DOI] [PubMed] [Google Scholar]
- 42. Lee MS, Salsbury FR, Brooks CLI (2004) Constant-pH molecular dynamics using continuous titration coordinates. Proteins 56: 738–752. [DOI] [PubMed] [Google Scholar]
- 43. Khandogin J, Brooks CL (2005) Constant pH molecular dynamics with proton tautomerism. Biophys J 89: 141–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shi C, Wallace JA, Shen JK (2012) Thermodynamic Coupling of Protonation and Conformational Equilibria in Proteins: Theory and Simulation. Biophys J 102: 1590–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Maes EM, Roberts SA, Weichsel A, Montfort WR (2005) Ultrahigh resolution structures of nitrophorin 4: heme distortion in ferrous CO and NO complexes. Biochemistry 44: 12690–12699. [DOI] [PubMed] [Google Scholar]
- 46. Nienhaus K, Maes EM, Weichsel A, Montfort WR, Nienhaus GU (2004) Structural dynamics controls nitric oxide affinity in nitrophorin 4. J Biol Chem 279: 39401–39407. [DOI] [PubMed] [Google Scholar]
- 47. Abbruzzetti S, He C, Ogata H, Bruno S, Viappiani C, et al. (2012) Heterogeneous kinetics of the carbon monoxide association and dissociation reaction of nitrophorin 4 and 7 coincide with structural heterogeneity of the gate-loop. J Am Chem Soc 134: 9986–9998. [DOI] [PubMed] [Google Scholar]
- 48. Benabbas A, Ye X, Kubo M, Zhang Z, Maes EM, et al. (2010) Ultrafast dynamics of diatomic ligand binding to nitrophorin 4. J Am Chem Soc 132: 2811–2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Swails JM, Meng Y, Walker FA, Martí MA, Estrin DA, et al. (2009) pH-dependent mechanism of nitric oxide release in nitrophorins 2 and 4. J Phys Chem B 113: 1192–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ogata H, Knipp M (2012) Crystallization and preliminary X-ray crystallographic analysis of the membrane-binding haemprotein nitrophorin 7 from Rhodnius prolixus . Acta Cryst Sect F Struct Biol Cryst Commun 68: 37–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tanford C, Bunville LG, Nozaki Y (1959) Reversible transformation of β-lactoglobulin at pH 7.5. J Am Chem Soc 81: 4032–4036. [Google Scholar]
- 52. Tanford C, Taggart VG (1961) Ionization-linked Changes in Protein Conformation. II. The N→R transition in β-lactoglobulin. J Am Chem Soc 83: 1634–1638. [Google Scholar]
- 53. Qin BY, Bewley MC, Creamer LK, Baker HM, Baker EN, et al. (1998) Structural basis of the Tanford transition of bovine beta-lactoglobulin. Biochemistry 37: 14014–14023. [DOI] [PubMed] [Google Scholar]
- 54. Seeger MA, vonBallmoos C, Verrey F, Pos KM (2009) Crucial role of Asp408 in the proton translocation pathway of multidrug transporter AcrB: evidence from site-directed mutagenesis and carbodiimide labeling. Biochemistry 48: 5801–5812. [DOI] [PubMed] [Google Scholar]
- 55. Murakami S, Nakashima R, Yamashita E, Matsumoto T, Yamaguchi A (2006) Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. Nature 443: 173–179. [DOI] [PubMed] [Google Scholar]
- 56. Olkhova E, Hunte C, Screpanti E, Padan E, Michel H (2006) Multiconformation continuum electrostatics analysis of the NhaA Na+/H+ antiporter of Escherichia coli with functional implications. Proc Natl Acad Sci U S A 103: 2629–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kluge C, Dimroth P (1993) Kinetics of Inactivation of the F1F0 ATPase of Propionigenium modestum by Dicyclohexylcarbodiimide in Relationship to H+ and Na+ Concentration: Probing the Binding Site for the Coupling Ions. Biochemistry 32: 10378–10386. [DOI] [PubMed] [Google Scholar]
- 58. Assadi-Porter FM, Fillingame RH (1995) Proton-Translocating Carboxyl of Subunit c of F1F0- H+ ATP Synthas: The Unique Environment Suggested by the pKa determined by 1H NMR. Biochemistry 34: 16186–16193. [DOI] [PubMed] [Google Scholar]
- 59. Rivera-Torres IO, Krueger-Koplin RD, Hicks DB, Cahill SM, Krulwich TA, et al. (2004) pKa of the essential Glu54 and backbone conformation for subunit c from the H+-coupled F1F0 ATP synthase from an alkaliphilic Bacillus. FEBS Lett 575: 131–5. [DOI] [PubMed] [Google Scholar]
- 60. Webb H, Tynan-Connolly BM, Lee GM, Farrell D, O'Meara F, et al. (2011) Re-measuring HEWL pKa values by NMR spectroscopy: Methods, analysis, accuracy and implications for theoretical pKa calculations. Proteins 79: 685–702. [DOI] [PubMed] [Google Scholar]
- 61. Garel JR, Epely S, Labouesse B (1974) The Acidic Transition of δ-Chymotrypsin. Biochemistry 13: 3117–3123. [DOI] [PubMed] [Google Scholar]
- 62. Garel JR, Labouesse B (1971) Rate of ligand-promoted isomerization of proteins. Relaxation study of the “alkaline-transition” of δ-chymotrypsin. Biochimie 53: 9–16. [DOI] [PubMed] [Google Scholar]
- 63. Mcphie P (1979) The Alkaline Transition of Swine Pepsinogen. Biophys Chem 9: 281–287. [PubMed] [Google Scholar]
- 64. Fersht AR (1972) Conformational Equilibria in alpha and delta Chymotrypsin. J Mol Biol 64: 497–509. [DOI] [PubMed] [Google Scholar]
- 65. Xiang T, Liu F, Grant DM (1991) Generalized Langevin equations for molecular dynamics in solution. J Chem Phys 94: 4463–4471. [Google Scholar]
- 66. Ryckaert JP, Ciccotti G, Berendsen HJC (1977) Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys 23: 327–341. [Google Scholar]
- 67. Hornak V, Abel R, Okur A, Strockbine B, Roitberg AE, et al. (2006) Comparison of Multiple Amber Force Fields and Development of Improved Protein Backbone Parameters. Proteins 725: 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bikiel DE, Boechi L, Capece L, Crespo A, De Biase PM, et al. (2006) Modeling heme proteins using atomistic simulations. Phys Chem Chem Phys 8: 5611–5628. [DOI] [PubMed] [Google Scholar]
- 69.Case DA, Darden TA, Cheatham TE, Simmerling CL, Wang J, et al.. (2010) AMBER 11. University of California, San Francisco (San Francisco).
- 70. Onufriev A, Bashford D, Case DA (2004) Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 55: 383–394. [DOI] [PubMed] [Google Scholar]
- 71. Metropolis N, Rosenbluth AW, Rosenbluth MN, Teller AH, Teller E (1953) Equation-of-state calculations by fast computing machines. J Chem Phys 21: 1087–1092. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
AB and GH loops in the context of the complete protein. A) Closed conformation. B) Open conformation. The AB and GH loops are shown in a different color than the rest of the protein. Leu130 and Asp30 are shown. Asp30 is displayed protonated in the closed structure and deprotonated in the open conformation.
(TIF)
Val36, Asp35 and Asp129 in the closed and open conformations. A) Closed conformation. B) Open conformation. Asp 30 is shown in gray for reference. The AB and GH loops are colored according to the conformation. Average values for dCBVal36-CGLeu130 and dCGAsp35-OAsp129 are also shown.
(TIF)
RMSD of the AB and GH loops. Running average of the time evolution of the RMSD of the AB and GH loops with respect to the closed (red) and open (black) structures. A) Initially closed structure, simulation at pH 5.5, B) Initially closed structure, simulation at pH 7.5, C) Initially open structure, simulation at pH 5.5, D) Initially open structure, simulation at pH 7.5. In case C the transition to the closed structure takes place almost immediately, but after ∼8 ns the simulation becomes trapped in an intermediate structure (see Text S1 for further detail).
(TIF)
Time evolution of the Solvent Accessible Surface Area (SASA) of Asp30. Running average of the time evolution of the SASA of Asp30. A) Initially closed structure, simulation at pH 5.5, B) Initially closed structure, simulation at pH 7.5, C) Initially open structure, simulation at pH 5.5, D) Initially open structure, simulation at pH 7.5. In case C the transition to the closed structure takes place almost immediately, but after ∼8 ns the simulation becomes trapped in an intermediate structure (see Text S1 for further detail).
(TIF)
Time evolution of relevant parameters when open NP4 is placed at a solvent pH of 5.5. Top: Running average of distance Asp30-Leu130; Middle: Running average of distance Asp35-Asp129; Bottom: Asp30 microscopic pKa. The average values of these distances in the stable simulations of the closed (red) and the open (blue) structures are also shown.
(TIF)
Additional data for transition inducing simulations. Further details on the simulation of the open conformation at pH 5.5.
(PDF)
Derivations of the equations presented in the main text. Details of the calculation of the equilibrium concentration of all chemical species and the apparent pKa.
(PDF)