

Abstract
Neuroplastic changes at the spinal synapses between primary nociceptors and second order dorsal horn neurons play key roles in pain and analgesia. NMDA receptor-dependent forms of long-term plasticity have been studied extensively at these synapses, but little is known about possible contributions of the endocannabinoid system. Here, we addressed the role of cannabinoid (CB)1 receptors in activity-dependent plasticity at these synapses. We report that conditional low-frequency stimulation of high-threshold primary sensory nerve fibres paired with depolarisation of the postsynaptic neuron evoked robust long-term depression (LTD) of excitatory synaptic transmission by about 40% in the vast majority (90%) of recordings made in wild-type mice. When recordings were made from global or nociceptor-specific CB1 receptor-deficient mice (CB1−/− mice and sns-CB1−/− mice), the portion of neurons exhibiting LTD was strongly reduced to about 25%. Accordingly, LTD was prevented to a similar extent by the CB1 receptor antagonist AM 251 and mimicked by pharmacological activation of CB1 receptors. In a subset of neurons with EPSCs of particularly high stimulation thresholds, we furthermore found that the absence of CB1 receptors in CB1−/− and sns-CB1−/− mice converted the response to the paired conditioning stimulation protocol from LTD to long-term potentiation (LTP). Our results identify CB1 receptor-dependent LTD as a form of synaptic plasticity previously unknown in spinal nociceptors. They furthermore suggest that prevention of LTP may be a second hitherto unknown function of CB1 receptors in primary nociceptors. Both findings may have important implications for our understanding of endogenous pain control mechanisms and of analgesia evoked by cannabinoid receptor agonists.
Key points
Synaptic plasticity between primary nociceptors and second order dorsal horn neurons serves key roles in pain and analgesia
A contribution of NMDA receptors to long-term potentiation and long-term depression at these synapses has been demonstrated before, but much less is known about a possible role of endocannabinoids and cannabinoid (CB)1 receptors.
Here we show that CB1 receptors residing on the spinal terminals of primary nociceptors critically contribute to an NMDA receptor-independent form of long-term depression at these synapses, which requires simultaneous pre- and postsynaptic activity.
A similar long-lasting depression of nociceptive signal transmission can also be obtained with application of CB1 receptor agonists in the presence of presynaptic stimulation alone.
These findings identify a previously unknown form of long-term depression at spinal nociceptor synapses, which may be important for our understanding of pain-related neural plasticity and analgesic actions of CB1 receptor agonists.
Introduction
Changes in the efficacy of synaptic transmission between primary nociceptive fibres, which convey information about potentially painful stimuli from the periphery of the body to the CNS, and intrinsic dorsal horn neurons play key roles in the generation of persistent pain states and as mechanisms of analgesic drug actions. Transient reductions in synaptic strength contribute to analgesia evoked by centrally acting drugs such as morphine. Longer lasting changes in synaptic efficacy occur in response to intense or prolonged activation of different classes of primary nociceptors. Intense input from nociceptive C fibres to the spinal dorsal horn induces LTP at the synapses between C fibres and certain types of projection neurons in the superficial spinal dorsal horn (Randic et al. 1993; Ikeda et al. 2003, 2006). Many lines of evidence indicate that this LTP makes an important contribution to persistent pain states following inflammation or tissue trauma (Ikeda et al. 2006). Conversely, LTD, which has been reported to occur mainly at Aδ fibre synapses (Sandkühler et al. 1997), is a candidate mechanism of analgesia evoked by conditioning nerve stimulation. While the role of NMDA receptors in these plastic changes has been investigated extensively, little is known about potential NMDA receptor-independent plasticity at spinal nociceptor synapses.
Plenty of evidence indicates that CB1 receptors and endocannabinoids mediate at least some forms of NMDA receptor-independent plasticity in many CNS areas. CB1 receptors and endocannabinoids have particularly been implicated in short-term and long-term depression (STD and LTD) of synaptic transmission (for reviews see Chevaleyre et al. 2006; Heifets & Castillo, 2009; Kano et al. 2009). Both forms require the postsynaptic production of endocannabinoids, mainly of 2-arachidonoyl glycerol (2-AG), that act as retrograde messengers to activate presynaptic CB1 receptors and to subsequently inhibit transmitter release. Recent evidence suggests that such retrograde synaptic signalling depends on diacylglycerol lipase-α (DGL-α) (Tanimura et al. 2010), which is the rate-limiting enzyme of 2-AG production in the CNS (Bisogno et al. 2003). Typical triggers of DGL-α activation are intracellular Ca2+ rises and activation of postsynaptic group I metabotropic glutamate receptors (mGluR1/5) or of other Gq/11 coupled G protein-coupled receptors (GPCRs) (Katona & Freund, 2012). Increases in intracellular free Ca2+ concentrations evoked by prolonged depolarisation initiate the 2-AG production underlying depolarisation-induced suppression of inhibition (DSI) (Ohno-Shosaku et al. 2001; Wilson & Nicoll, 2001) or of excitation (DSE) (Kreitzer & Regehr, 2001; Ohno-Shosaku et al. 2002), both of which are forms of short-term synaptic plasticity. Glutamate release and subsequent activation of mGluR1/5 receptors stimulate a different 2-AG production pathway, which does not require intracellular Ca2+ and which is capable of inducing both short-lasting and long-lasting synaptic depression after conditioning stimulation of glutamatergic synapses. The induction of LTD requires probably prolonged activation of CB1 receptors (Kreitzer & Malenka, 2005) possibly together with concomitant presynaptic activity (Robbe et al. 2002; Chevaleyre & Castillo, 2003; Singla et al. 2007).
It is tempting to speculate that such signalling pathways also contribute to plasticity at dorsal horn nociceptor synapses. Powerful analgesic effects are observed in response to plant-derived or synthetic cannabinoids in many rodent models of inflammatory and neuropathic pain as well as in some clinical trials (Walker & Hohmann, 2005; Lever & Rice, 2006; Pacher et al. 2006). Compelling evidence supports a contribution of endocannabinoids to intrinsic pain control (Lewis et al. 1980; Meng et al. 1998; Hohmann et al. 2005; Agarwal et al. 2007; Petrosino et al. 2007). Most of these actions occur through CB1 receptors at different levels of the neuraxis including the spinal dorsal horn (Cravatt & Lichtman, 2004). At this site, CB1 receptors are densely expressed on the terminals of primary nociceptors (Liang et al. 2004; Hegyi et al. 2009), while the opposing postsynaptic structures contain large amounts of DGL-α (Nyilas et al. 2009). Collectively, these findings indicate that the major molecular prerequisites for endocannabinoid-dependent plasticity are present at spinal nociceptor synapses. We now report that presynaptic CB1 receptors expressed on the spinal terminals of primary nociceptors mediate a previously unidentified form of LTD in primary nociceptor synapses, which exists in parallel to well-known NMDA receptor-dependent LTD. We further provide evidence that primary nociceptor CB1 receptors serve an additional function by hindering the induction of LTP at spinal nociceptor synapses.
Methods
Animals
Mice lacking CB1 receptors either globally (CB1−/−; genetic background C57BL/6N; Marsicano et al. 2002) or specifically in primary nociceptors (sns-CB1−/− mice; mixed genetic background C57BL/6J × C57BL/6N; Agarwal et al. 2007) and their respective littermates were investigated in electrophysiological and morphological experiments. sns-CB1−/− mice were generated by crossing CB1fl/fl mice (Marsicano et al. 2003) with bacterial artificial chromosome (BAC) transgenic sns-cre mice expressing the cre recombinase under the transcriptional control of the sns (Nav1.8) gene, which enables gene recombination specifically in primary nociceptors (Agarwal et al. 2004). The killing of the mice for preparation of sections for morphology or electrophysiology was carried out in accordance with the regulations of the animal welfare committee of the canton of Zurich, and conform to the principles of UK regulations, as described in Drummond (2009).
Morphology
For immunohistochemistry, adult CB1fl/fl and sns-CB1−/− littermates (three of each genotype) were deeply anaesthetised with a mixture of ketamine–xylazine injected intraperitoneally and perfused transcardially with 0.9% NaCl for 2 min followed by 100 ml fixative containing 4% paraformaldehyde (PFA) in 0.1 m phosphate buffer (PB; pH 7.4) for 20 min. After perfusion, the spinal cord was immediately isolated, postfixed in 4% PFA for 2 h and washed in 0.1 m PB. Transverse sections 50 μm thick at the lumbar level were cut using a vibratome (Leica, VTS-1000).
For immunofluorescence staining, free-floating spinal cord sections were first extensively washed in 0.1 m PB. Following washing in 0.05 m Tris-buffered saline (TBS; pH 7.4) containing 0.3% Triton X-100, the sections were blocked in 10% normal donkey serum (Vector Laboratories, Burlingame, CA, USA) for 45 min. Sections were then incubated in a mixture of polyclonal affinity-purified guinea pig anti-CB1 (1:200; Fukudome et al. 2004; generously provided by Dr Masahiko Watanabe, Hokkaido), rat anti-substance P (SP) (1:250, AbD Serotec) and rabbit anti-calcitonin gene-related peptide (CGRP) antibodies (1:40,000, Baffi et al. 1992; or 1:1000, Calbiochem) in TBS at 4°C for 48 h. After multiple washings in TBS, the sections were treated with the mixture of Dylight-594-labelled donkey anti-guinea pig, Dylight-488-labelled donkey anti-rat and DyLight-649-conjugated donkey anti-rabbit fluorescent secondary antibodies (1:400; Jackson ImmunoResearch) at room temperature for 2 h in the dark. After extensive washing in TBS and PB, sections were mounted onto glass slides, covered in Vectashield (Vector Laboratories). Coverslips were sealed with nail polish.
Image acquisition was performed on a Nikon A1R confocal laser-scanning system built on a Ti-E inverted Nikon microscope in a sequential acquisition mode, using a 1.4 NA 60× CFI Plan Apochromat VC (Nikon) oil-immersion objective and NIS elements (Nikon) software. Confocal settings (confocal aperture, laser power, gain, offset, pixel dwell and pixel size) were identical for all scans. Optical sections were acquired at a z-separation of 200 nm. For restoration of 3D image stacks, Huygens deconvolution software (Scientific Volume Imaging) was used.
Co-localization of CB1 immunoreactivity with markers for nociceptive primary afferent terminals was quantified by an experimenter blind to the genotypes, and performed manually on single optical sections taken from the same focal depth of the samples (2 μm from the upper surface of the sections) using Adobe Photoshop CS5. Boutons immunopositive for both SP and CGRP were selected randomly in laminae I and II of the superficial dorsal horn (100 SP+/CGRP+ boutons per animal). The number of SP+/CGRP+ axon terminals containing CB1 immunoreactive puncta per number of all SP+/CGRP+ boutons ratio was calculated for each animal.
Electrophysiology
Mice 12–28 days old of either sex were decapitated under isoflurane anaesthesia. Laminectomy was performed to prepare lumbar spinal cords with dorsal roots attached. The isolated lumbar spinal cords were glued onto a gelatine block with cyanolyt and cut into 300- to 450-μm-thick transverse slices. Slices were kept in oxygenated (95% O2–5% CO2) artificial cerebrospinal fluid containing (in mm): 120 NaCl, 5 Hepes, 26 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 2 CaCl2, 1 MgCl2 and 10 glucose (pH 7.35) at 35°C (for details see Ahmadi et al. 2002).
Slices were transferred to a recording chamber and continuously superfused with extracellular solution (for composition see above) equilibrated with 95% O2–5% CO2 at a flow rate of 1–2 ml min−1. Neurons in the superficial dorsal horn (lamina I and II, ≤150 μm from the dorsal margin) were visually identified using the infrared gradient contrast technique coupled to a video microscopy system (Dodt & Zieglgänsberger, 1994). Whole-cell patch-clamp recordings were performed at room temperature with borosilicate glass recording pipettes (resistance 4–6 MΩ) filled with intracellular solution containing (in mm) 135 CsF, 5 CsCl, 5 EGTA, 10 Na-Hepes, 1 Mg-ATP, 0.1 Na-GTP and 2 QX-314 (305 mosmol l−1, pH 7.35, adjusted with CsOH) using a HEKA EPC 10 amplifier controlled with PatchMaster (HEKA electronics) acquisition software. The partial substitution of Cl−1 by F−1 in the intracellular solution allowed minimising the contamination of EPSCs by inhibitory postsynaptic currents without compromising inhibitory synaptic transmission in general. EPSCs were elicited through electrical stimulation of the dorsal root at a frequency of 3 min−1 (pulse duration 100–500 μs, voltage 1–70 V) using bipolar tungsten or suction electrodes. In the experiments on pharmacologically induced LTD (Fig. 4 and 5) and on NMDA receptor-mediated EPSCs, the internal solution contained (in mm): 130 potassium gluconate, 20 KCl, 2 MgCl2, 0.05 EGTA, 3 Na-ATP, 0.1 Na-GTP, 10 Na-Hepes and 5 QX-314 (305 mosmol l−1, pH 7.35 adjusted with KOH). In these experiments, GABAergic and glycinergic synaptic currents were blocked with bicuculline (10 μm) and strychnine (0.5 μm). NMDA receptor-mediated EPSCs were recorded in the presence of strychnine (0.5 μm), bicuculline (10 μm) and NBQX (10 μm), at a holding potential of −30 mV to partially relieve the NMDA receptors from block by extracellular Mg2+. In all preincubation experiments (i.e. in the experiments on NMDA–EPSC and in the experiments shown in Figs 3 and 7) antagonists were added immediately after reaching the whole-cell configuration and thus about 10–15 min prior to the conditioning low-frequency stimulation. All electrical signals were sampled at 5 kHz and filtered off-line at 2.9 kHz. Data were analysed using the FitMaster software (HEKA electronics). Input and access resistance of each neuron were continuously monitored by delivering short (20 ms) hyperpolarising voltage steps (to −5 or −10 mV) between the synaptic stimulations. Drug containing solutions were applied via perfusion of the recording chamber. All recordings were carried out at room temperature.
Figure 4. CB1 receptor agonists cause a specific inhibition of high-threshold primary afferent EPSCs.
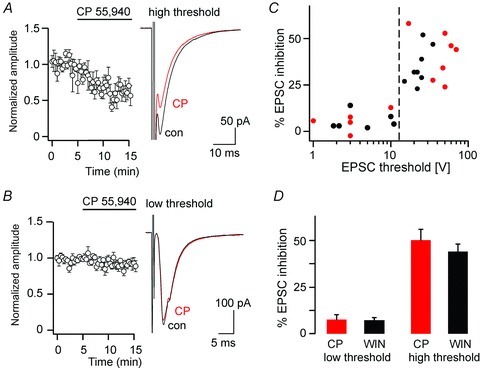
A, inhibition of high-threshold EPSCs (threshold ≥ 15 V) by CP 55,940 (3 μm). Left, time course. Right, averages traces of 10 consecutive EPSCs before application of CP 55,940 (black, control) and after (CP, red). B, same as A but low-threshold EPSCs (threshold < 15 V). C, degree of EPSC inhibition plotted versus the stimulation threshold of the EPSC (black, WIN 55,212-2; red CP 55,940). D, per cent EPSC inhibition by CP 55,940 and WIN 55,212-2 in low-threshold and high-threshold EPSCs. The difference between high-threshold EPCSs (≥15 V) and low-threshold EPSCs (<15 V) was statistically significant (P≤ 0.0001, unpaired Student's t test), while differences between WIN 55,212-2 and CP 55,940 were small and remained below statistical significance (P= 0.95 and P= 0.34, for low-threshold and high-threshold EPSCs).
Figure 5. CP 55,940-mediated inhibition of high-threshold primary afferent EPSCs is absent in CB1−/− and sns-CB1−/− mice.
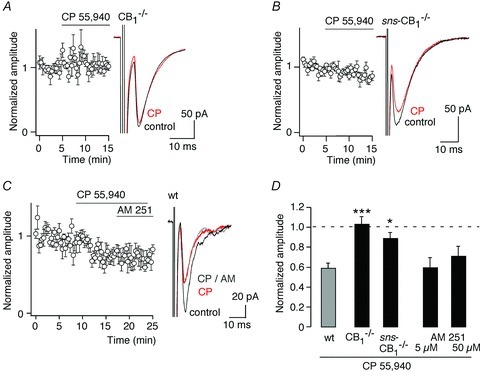
A, lack of inhibition by CP 55,940 (3 μm) of high-threshold EPSCs in CB1−/− mice. Left, time course. Right, averages traces of 10 consecutive EPSCs before application of CP 55,940 (black, control) and after (CP, red). B, same as A but in nociceptor-specific sns-CB1−/− mice. C, CP 55,940-induced depression of high-threshold EPSCs in wild-type mice was not reversed by AM 251 (50 μm). Left, time course. Right, averages traces of 10 consecutive EPSCs before application of CP 55,940 (black, control), after application of CP 55,940 (CP, red) and in the additional presence of AM 251 (CP AM, grey). D, statistics: normalised high-threshold EPSC amplitudes relative to pre-treatment values. Wild-type (wt, CB1fl/fl) are same as those shown in Fig. 4A. AM 251 was applied after inhibition by CP 55,940 had been established. ***P≤ 0.001; *P≤ 0.05 significantly different from wild-type, ANOVA followed by Dunnett's post hoc test.
Figure 3. CB1 receptor activation is required for induction but not for maintenance of high-threshold primary afferent LTD.
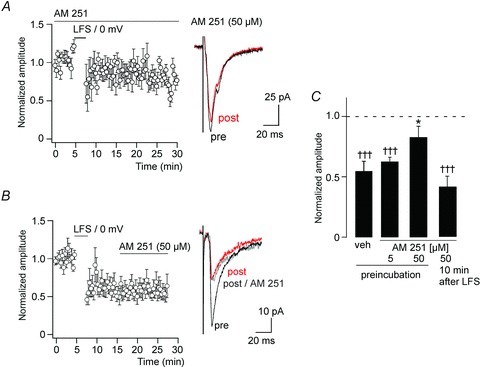
A, preincubation with the CB1 receptor antagonist AM 251 (50 μm) largely prevented high-threshold primary afferent LTD (n= 8). Left, time course. Right, EPSC traces averaged from 10 consecutive stimulations immediately before (pre, black) or 15 min after the conditioning stimulation (post, red). B, application of AM 251 (50 μm) after LTD had been established did not reverse LTD. Right, EPSC traces averaged from 10 consecutive stimulations immediately before (pre, black), 15 min after the conditioning stimulation (post, red), and 10 min after application of 50 μm AM 251 (post AM 251, grey). C, statistics. †††P≤ 0.001 significantly different from pre-conditioning values (paired Student's t test). *P≤ 0.05, significantly different from vehicle, F(3,29) = 4.46 (ANOVA followed by Dunnett's post hoc test).
Figure 7. Synaptic plasticity with NMDA receptor blocked.
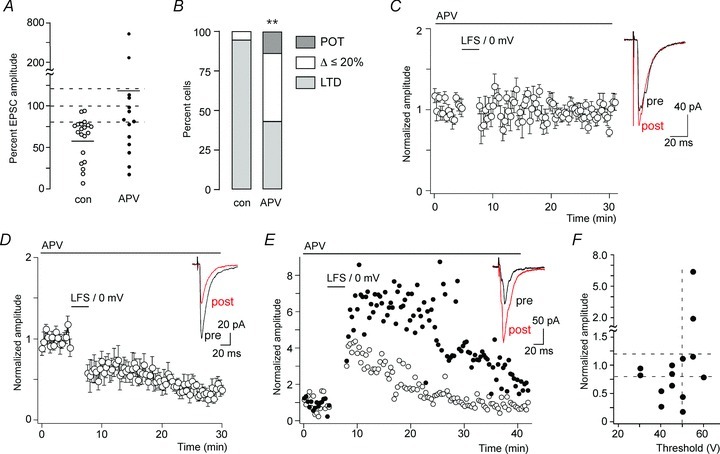
A, changes in EPSC amplitudes evoked by paired conditioning stimulation in the continuous presence of APV (50 μm) (n= 14 wild-type [CB1fl/fl] mice) compared with untreated slices (n= 20). Control cells are the same as those shown in Fig. 1C (CB1fl/fl mice). Horizontal bars indicate average amplitude changes. Dashed lines designate the range, which was considered as no change (80%≤ΔEPSC amplitude ≤ 120%). B, portion of cells showing LTD, no change or a potentiation by more than 20%. **P < 0.01. Pearson χ2(2,32) = 9.23. C–E, time course of normalised EPSC amplitudes in cells that responded to conditioning paired stimulation in the continuous presence of APV either with no change (n= 6) (C), LTD (n= 6) (D) or with a potentiation (n= 2) (E). Insets, averaged traces from 10 consecutively recorded EPSC immediately before (pre, black) and 15 min after (post, red) conditioning stimulation. F, averaged normalised EPSC amplitudes after paired conditioning stimulation plotted against EPSC thresholds.
Drugs and chemicals
(–)-Bicuculline methochloride, strychnine hydrochloride and d(−)-2-amino-5-phosphonopentanoic acid (APV) were dissolved in extracellular solution. Stock solutions of WIN 55-212-2 mesylate, AM 251 and CP 55,940 were prepared in DMSO and stored at −20°C. The DMSO concentration in the superfusate was ≤0.1%. All chemicals were purchased from Tocris.
Data analysis and statistics
For statistical analyses of LTD, average EPSCs were calculated from 10 consecutive current traces evoked during control conditions (i.e. before conditioning stimulation or before drug application), and when a steady-state effect was reached (typically 15 min after conditioning stimulation or 5 min after drug application). Statistical significance was analysed using ANOVA followed by appropriate post hoc tests or, in the case of pair-wise comparisons, with the paired Student's t test. For the comparison of the level of co-localization in CB1fl/fl and sns-CB1−/− mice, a χ2 test was used. All statistical analyses were made using SPSS Statistics 17.0.
Results
Low-frequency primary afferent stimulation paired with postsynaptic depolarisation induces LTD at dorsal horn nociceptor synapses
In a first set of experiments, we employed different conditioning stimulation protocols that are known to stimulate synaptic endocannabinoid production and to induce CB1 receptor-dependent plasticity in other parts of the CNS. We recorded high-threshold primary afferent-evoked EPSCs from lamina II dorsal horn neurons. Synaptic transmission was evoked by electrical stimulation of the afferent dorsal root at a frequency of 3 min−1. To identify such high-threshold EPSCs, the intensity of dorsal root stimulation was continuously increased from 1 V until a constant latency EPSC was reliably evoked. This was typically achieved with stimulation intensities 15–30% higher than the threshold intensity. EPSCs with activation thresholds ≥15 V were classified as high threshold and were selected for further investigation. To test whether primary nociceptor synapses exhibit DSE, we depolarised the recorded neuron to 0 mV for 3 s. Only 1 out of 15 cells exhibited a significant depression (by 23%). On average, the mean of the first three EPSC amplitudes following the conditioning depolarisation was 107.8 ± 7.3% (mean ± SEM) of the preconditioning value (n= 15, P= 0.59, paired Student's t test) (Fig. 1A).
Figure 1. Short-term and long-term depression of synaptic transmission between high-threshold primary afferent fibres and superficial dorsal horn neurons.
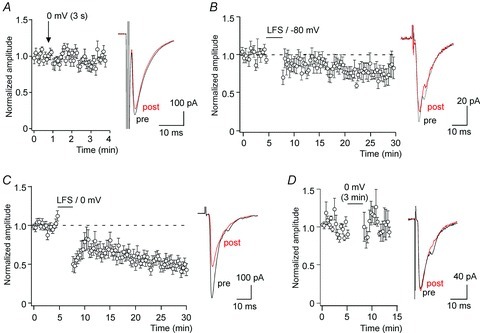
A, depolarisation-induced suppression of excitation (DSE). Primary afferent EPSCs were evoked at a frequency of 0.2 Hz. At the time indicated (arrow) the recorded cell was depolarised for 3 s from –80 mV to 0 mV. No suppression of EPSC amplitudes was observed following the depolarisation. Left, time course of averaged normalised EPSC amplitudes recorded from 15 cells. Right, superposition of representative EPSC traces of a single cell obtained immediately before (pre, black) and after the conditioning depolarisation (post, red). B, primary afferent EPSCs were elicited at a frequency of 3 min−1. Synaptic depression was induced by a conditioning low-frequency primary afferent fibre stimulation (200 pulses at 1.4 Hz). Left, time course of 8 cells averaged. Right, averages of 10 consecutive EPSC traces recorded in a single cell immediately before the conditioning stimulation (pre) and 15 min after (post). C, same as B but conditioning presynaptic stimulation was paired with postsynaptic depolarisation from −80 mV to 0 mV for 2.5 min (n= 20). D, same as C but without presynaptic stimulation. No depression of EPSC amplitudes was observed under this condition (n= 6).
In several brain areas, release of endocannabinoids induces LTD of excitatory synaptic transmission. In the spinal dorsal horn, LTD of synapses between primary afferent nociceptors and second order neurons can be elicited by brief high-frequency stimulation (Randic et al. 1993) or by prolonged (15 min) low-frequency synaptic stimulation (Sandkühler et al. 1997). In our experiments, we applied a short low-frequency stimulation protocol consisting of 200 pulses applied at a frequency of 1.4 Hz, which was chosen because both Aδ and C fibre nociceptors can sustain prolonged firing at this frequency (Torsney & MacDermott, 2006; see also Ikeda et al. 2006). This conditioning stimulation produced on average a minor but already statistically significant depression of EPSC amplitudes to 74.0 ± 6.4% (mean ± SEM, n= 8, P= 0.045, paired Student's t test, quantified at 15 min after conditioning stimulation) (Fig. 1B). We then combined this low-frequency stimulation with postsynaptic depolarisation to facilitate endocannabinoid production and release (Kreitzer & Regehr, 2001; Maejima et al. 2001; Wilson & Nicoll, 2001). This paired conditioning stimulation induced a pronounced, robust and long-lasting depression of EPSC amplitudes by at least 20% in 18 out of 20 cells. In the remaining two cells, EPSC amplitudes were reduced by 8.5 and 9.5%. On average, EPSC amplitudes were decreased to 57.5 ± 5.9% at 15 min after the conditioning paired stimulation protocol (n= 20; P < 0.0001 paired Student's t test) and remained depressed for the remaining experiment (Fig. 1C). We finally tested whether prolonged depolarisation (2.5 min) alone was sufficient to induce LTD. Only 1 out of 6 cells exhibited a transient depression of the EPSC amplitude. On average, this latter conditioning protocol had only a minor and statistically insignificant effect on the EPSC amplitudes (8.8 ± 11.7%, n= 6, P= 0.65, paired Student's t test) (Fig. 1D).
High-threshold primary afferent LTD in CB1 receptor-deficient mice
We next determined the contribution of the endo-cannabinoid system to this primary nociceptor LTD and tested whether LTD evoked by the conditioning paired stimulation protocol was altered in global CB1−/− mice. On average, conditioning paired stimulation had virtually no effect on EPSC amplitudes (99.8 ± 15.4% of the preconditioning values, n= 13, P= 0.35, paired Student's t test) in global CB1−/− mice (Fig. 2A and D), strongly suggesting that CB1 receptors contribute to LTD at nociceptor synapses. However, the lack of LTD in CB1−/− mice could also have been due to secondary changes in synaptic circuits caused by the continuous absence of CB1 receptors throughout development (Berghuis et al. 2007). Previous reports have shown that the CB1 receptors responsible for endocannabinoid-dependent LTD are located presynaptically (for recent reviews see e.g. Chevaleyre et al. 2006; Kano et al. 2009). We therefore went on to examine the expression of CB1 receptors on the presynaptic nociceptor terminals in more detail and then investigated mice lacking CB1 receptors specifically in nociceptors (sns-CB1−/− mice; Agarwal et al. 2007).
Figure 2. LTD of high-threshold primary afferent EPSCs in global CB1−/− and nociceptor-specific sns-CB1−/− mice.
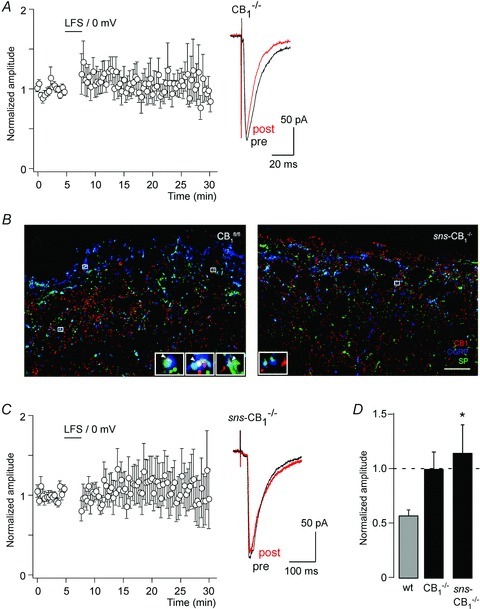
A, time course of EPSC amplitudes (normalised and averaged from 13 cells) following the paired conditioning stimulation protocol (same as protocol as in Fig. 1C) in slices obtained from global CB1−/− mice. Right, EPSC traces averaged from 10 consecutive stimulations (pre, black, preconditioning; post, red, 15 min after conditioning stimulation). B, confocal micrographs depicting CB1 (red), SP (green) and CGRP (blue) triple immunofluorescence staining in the superficial laminae of CB1fl/fl (left) and sns-CB1−/− (right) mouse spinal dorsal horns. Left, white arrowheads in enlarged boxed areas indicate co-localization of the three antigens, corresponding to CB1-containing nociceptive primary afferents in CB1fl/fl. Right, by contrast, only few SP+/CGRP+ puncta showed CB1 immunoreactivity in sns-CB1−/- samples (enlarged boxed area shows lack of CB1 immunoreactivity in SP+/CGRP+ boutons). Scale bar, 10 μm. C, same as A but in nociceptor-specific sns-CB1−/− mice (n= 13 cells). D, statistical analysis of A and C. *P < 0.05, sns-CB1−/− mice versus wild-type mice, F(2,43) = 4.23 (ANOVA, followed by Dunnett's post hoc test). Wild-type data shown for comparison in D are the same as shown in Fig. 1C.
Presynaptic CB1 receptors in high-threshold primary afferent LTD
CB1 receptors are expressed densely in the superficial dorsal horn, the termination area of primary nociceptors. However, the extent to which they are expressed by primary nociceptors or by intrinsic dorsal horn neurons has not fully been resolved. Previous work has shown that dorsal rhizotomy reduced dorsal horn CB1 receptor density only marginally (Farquhar-Smith et al. 2000). We therefore performed triple immunofluorescence confocal microscopy on spinal cord sections stained for CB1 along with substance P (SP) and calcitonin-gene related peptide (CGRP) to quantify the presence of CB1 receptors on a subpopulation of peptidergic primary afferent nociceptors. The co-occurrence of the two neuropeptides unequivocally identifies a subpopulation of peptidergic nociceptive primary afferents (Wiesenfeld-Hallin et al. 1984; Lawson et al. 1996). In the superficial dorsal horn of CB1fl/fl mice, on average at least 20% of all SP/CGRP double immunopositive axon terminals coexpressed CB1 receptors. This coexpression was strongly reduced in sns-CB1−/− mice (Fig. 2B): the ratio of CB1 containing SP+/CGRP+ axon terminals was 2.5- to 3-fold higher in the CB1fl/fl animals than in their sns-CB1−/− littermates (19% in CB1fl/fl versus 7% in sns-CB1−/−; n= 3 for each genotype). Pair-wise comparison also showed that in all three pairs of mice SP+/CGRP+ boutons immunoreactive for CB1 receptors were significantly more abundant in wild-type animals than in their corresponding sns-CB1−/- littermates (P < 0.0001, χ2 test). As in these mice, cre-mediated gene excision occurs only around birth (Agarwal et al. 2004), developmental effects should be less likely than in global CB1−/− mice.
In sns-CB1−/− mice, LTD evoked by conditioning paired stimulation was strongly impaired. On average, EPSC amplitudes even increased slightly by 13.6 ± 26.1% (n= 13, P= 0.61, paired Student's t test) (Fig. 2C and D). This result not only argues against a developmental deficit as the underlying cause of missing LTD in CB1−/− mice but also indicates that the CB1 receptors relevant for this form of LTD are residing on the presynaptic nociceptors terminals.
CB1 receptors are required for induction but not for maintenance for endocannabinoid-dependent LTD
In a subsequent series of experiments, we applied our LTD induction protocol in the presence of different concentrations of the CB1 receptor antagonist AM 251 (5 and 50 μm). AM 251 hindered the induction of LTD (quantified at 15 min) in a dose-dependent manner with a minor (statistically insignificant) reduction in LTD at 5 μm (ΔEPSC amplitudes from pre-conditioning: −41.7 ± 4.3%, n= 10, versus−46.5 ± 8.3%, n= 11 in the presence of vehicle (DMSO)), but with a significant reduction in LTD at 50 μm (ΔEPSC amplitude: −15.6 ± 10.2%, n= 8) (Fig. 3A and C). The use of AM 251 also allowed us to investigate whether CB1 receptor activation was required for induction of LTD only, or also for its maintenance. When AM 251 (50 μm) was applied 10 min after the conditioning stimulation (i.e. when LTD was fully established) AM 251 had no effect (ΔEPSC amplitude: −43.9 ± 11.1%versus−41.3 ± 8.9%, n= 5, P= 0.57, paired Student's t test) (Fig. 3B and C).
Pharmacological activation of CB1 receptors induces primary afferent LTD
We then tested whether pharmacological activation of CB1 receptors could mimic the LTD induced by the paired conditioning protocol. To this end, we investigated the effects of two mixed CB1 and CB2 receptor agonists, CP 55,940 and WIN 55,212-2 (both at 3 μm). Both compounds decreased high-threshold primary afferent-evoked EPSCs, by 40.2 ± 5.1% (n= 7, P= 0.008, paired Student's t test) and 35.1 ± 3.6 (n= 8, P= 0.01, paired Student's t test), respectively (Fig. 4A and D). As the difference between CP 55,940 and WIN 55,212-2 mediated inhibition was small and statistically insignificant, we pooled both data sets for further analyses. We also compared the effect of CB1 receptor activation on high-threshold and low-threshold primary afferent evoked EPSCs and plotted the degrees of inhibition by CP 55,940 or WIN 55,212-2 versus the stimulation threshold of the EPSC under study. Significant inhibition was only observed for EPSCs with stimulation thresholds ≥15 V (ΔEPSC amplitude: −37.9 ± 3.0%, n= 15), whereas EPSCs with thresholds <15 V were virtually insensitive to CP 55,940 and WIN 55,212-2 (ΔEPSC amplitude: −5.7 ± 2.4%, n= 11) (Fig. 4B–D).
To ensure that this inhibition occurred through activation of CB1 receptors, we subsequently analysed the effects of CP 55,940 in CB1−/− and sns-CB1−/− mice. As expected from the results obtained with the synaptic conditioning protocol, the inhibitory action of CP 55,940 (3 μm) on primary afferent evoked EPSCs was completely abolished in CB1−/− mice (ΔEPSC amplitude 10 min after drug application started: −5.4 ± 8.5%, n= 5, P= 0.88, paired Student's t test, Fig. 5A). In sns-CB1−/− mice, inhibition by CP 55,940 was also strongly reduced (ΔEPSC amplitude: −11.6 ± 4.4%, n= 6; P= 0.05; paired Student's t test; Fig. 5B). Like LTD evoked by the synaptic conditioning protocol, CP 55,940-induced depression of EPSC amplitudes was long-lasting and did not depend on continuous CB1 receptor activation, as indicated by its resistance to AM 251 (either 5 μm or 50 μm) (Fig. 5C). EPSC inhibition was 40.2 ± 5.1% before AM 251 versus 40.2 ± 11.8% (n= 7) during AM 251 (5 μm), and 24.1 ± 4.7%versus 30.1 ± 4.0% (n= 5) for AM 251 (50 μm) (for statistical analyses see Fig. 5D).
Heterogeneity of responses in CB1−/− and sns-CB1−/− mice
A close comparison of the consequences of the conditioning paired stimulation in wild-type mice and in CB1 receptor-deficient mice reveals not only differences in their average responses but also a much higher variability of post-conditioning EPSC amplitudes in CB1−/− and sns-CB1−/− mice as compared with wild-type mice (Fig. 6A, compare also Fig. 1C and Fig. 2A and C). In order to analyse this variability in more detail, we classified the responses of the recorded cells as ‘LTD’, if their EPSC amplitudes were reduced by more than 20% after the conditioning protocol, as ‘no change’ if EPSC amplitudes changed by less than 20%, and as potentiation (POT) if amplitudes increased by >20% (Fig. 6B). A threshold of 20% was chosen, because the average variability in EPSC amplitudes in control mice was close to 20% (19.3 ± 2.8%, n= 20). In 8 of 13 cells recorded in slices obtained from CB1−/− mice, and in 4 of 13 recordings made in slices from sns-CB1−/− mice, EPSC amplitudes remained virtually stable with changes ≤20%, suggesting that LTD in these synapses depended largely on CB1 receptors. However, 3 of the 13 recordings made in cells from CB1−/− mice, and 6 of the 13 recordings made from sns-CB1−/− mice still exhibited LTD. Furthermore, 2 of 13 recordings made in slices from CB1−/− mice and 3 of 13 made in slices from sns-CB1−/− mice, showed an LTP-like behaviour with increases in amplitude by between 90 and 240%. The distribution of the three response types in CB1−/− and sns-CB1−/− mice was significantly different from that in wild-type mice. However, the distributions in CB1−/− and sns-CB1−/- mice were not significantly different from each other (Fig. 6B, for details of the statistical analyses see figure legend).
Figure 6. Synaptic plasticity in CB1−/− and sns-CB1−/− mice.
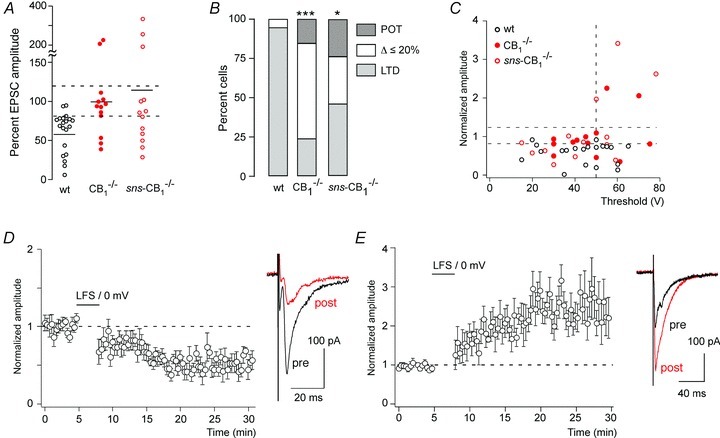
A, changes in EPSC amplitudes evoked by paired conditioning stimulation in CB1−/− (n= 13) and sns-CB1−/− mice (n= 13) compared with wild-type (CB1fl/fl) mice (n= 20). Same cells as shown in Fig. 3A and B (CB1−/− and sns-CB1−/− mice) and Fig. 2C (CB1fl/fl mice). Horizontal bars indicate average amplitude changes. Dashed lines designate the range, which was considered as no change (80%≤ΔEPSC amplitude ≤ 120%). B, portion of cells showing LTD, no change or potentiation (POT) in the three genotypes. *P≤ 0.05, ***P≤ 0.001. Pearson χ2(4,42) = 17.6 followed by pair-wise comparisons. The distribution of responses in sns-CB1−/− mice was not significantly different from that in CB1−/− mice (P= 0.28, χ2(2,24) = 2.53). C, post-conditioning EPSC amplitudes (normalised to pre-conditioning values) versus EPSC threshold in wild-type mice and in CB1−/− and sns-CB1−/− mice. D, time course of normalised EPSC amplitudes following the paired conditioning stimulation in the subset of CB1−/− and sns-CB1−/− mice (n= 9), which exhibited LTD. E, same as D but for cells exhibiting a potentiation by at least 90% (n= 5).
One likely factor contributing to this higher variability is the previously described presence of NMDA receptor-dependent LTD in the dorsal horn (Sandkühler et al. 1997; Ikeda et al. 2000), which would lead to the retention of LTD in a subset of nociceptor synapses of CB1−/− and sns-CB1−/− mice. A second contribution might come from the heterogeneity of nociceptor types (e.g. Aδ and C fibre nociceptors). We first addressed the latter possibility and plotted the post-conditioning EPSC amplitudes against their stimulation thresholds (Fig. 6C). This analysis revealed that LTP developed only in those cells from CB1−/− and sns-CB1−/− mice that had EPSC thresholds ≥50 V. These EPSCs most probably originated from C fibres.
We then performed separate analyses for those cells from CB1−/− and sns-CB1−/− mice that still developed LTD or LTP. Depression of EPSC amplitudes in these cells was very similar to that obtained in wild-type mice (49.6 ± 5.9%, n= 8, in CB1−/− and sns-CB1−/− mice, versus 46.2 ± 5.9%, n= 18, in wild-type mice, P= 0.73, unpaired Student's t test) (Fig. 6D).
Five cells recorded from either global CB1−/− or sns-CB1−/− mice exhibited a pronounced potentiation (by at least 90%) of their EPSC amplitudes following the conditioning paired stimulation protocol. In these cells, EPSC amplitudes showed a progressive increase to 245 ± 26% of the preconditioning value, which reached a plateau about 15 min after the conditioning stimulation and then remained stable until the end of the experiment (Fig. 6E). The time course of this potentiation was very much reminiscent of the NMDA receptor-dependent LTP reported by others at C fibre synapses in the dorsal horn (Ikeda et al. 2003, 2006). It should be added here that the paired conditioning stimulation protocol used in the present study did not evoke LTP in any of the 20 neurons studied in untreated slices of wild-type mice, despite the fact that the thresholds of the EPSCs recorded in wild-type mice covered a similar voltage range (15–65 V, average: 43 ± 3.2 V, n= 20 (wild-type mice) versus 15–78 V, average 45 ± 3.2, n= 26 (sns-CB1 and CB1−/− mice pooled); P= 0.62, unpaired Student's t test). Ablation of CB1 receptors from primary nociceptor terminals hence renders their synapses more susceptible to long-lasting facilitation of synaptic transmission. Preventing or limiting the development of LTP might thus be an important function of CB1 receptors residing on C fibre terminals.
LTD evoked in the presence of NMDA receptor blockers
As shown in Fig. 6D, a subset of neurons from CB1−/− and sns-CB1−/− still exhibited pronounced LTD. It has previously been reported that primary nociceptor (Aδ fibre) LTD is partially blocked by the NMDA receptor antagonist APV (Sandkühler et al. 1997; Ikeda et al. 2000). We therefore tested our paired conditioning protocol also in slices obtained from wild-type mice with NMDA receptors blocked. The responses obtained in the presence of APV (50 μm) were again more variable than those under control conditions (Fig. 7A and B). In 6 out of 14 cells, EPSC amplitudes remained virtually constant (97 ± 5.4%) (Fig. 7B and C). However, six cells exhibited NMDA receptor-independent LTD of >20%. The degree of this LTD was very similar to that measured in untreated wild-type slices (52.6 ± 9.2% (n= 6) versus 53.7 ± 5.9% (n= 18), in the presence of APV versus control conditions) (Fig. 7D). It should be added that we also observed a potentiation of EPSC amplitudes in two cells pretreated with APV (Fig. 7E). In these cells, potentiation reached about 400% and 600% of the preconditioning EPSC amplitude. This potentiation showed kinetic properties distinct from the LTP seen in CB1−/− and sns-CB1−/− mice, with an almost immediate onset after conditioning stimulation and relatively fast recovery to baseline amplitudes within about 30 min. Finally, we analysed again whether NMDA receptor-independent and presumed CB1 receptor-dependent synaptic plasticity correlated with the activation threshold of the EPSCs. As shown in Fig. 7F, LTD was observed across the whole range of stimulation thresholds (30–60 V), whereas the two cells that responded with pronounced potentiation had EPSC thresholds >50 V.
These results strongly suggest that NMDA receptor-dependent and CB1 receptor-dependent LTD exist in nociceptor synapses as distinct forms of synaptic plasticity. One might, however, argue that genetic ablation or blockade of CB1 receptors could interfere with NMDA receptor function. We therefore verified that NMDA receptor-mediated EPSCs were not altered in CB1−/− mice. Neither amplitudes (130.2 ± 13.4 pA, n= 10, versus 170.9 ± 39.8 pA, n= 7, P= 0.28, unpaired Student's t test) nor decay kinetics (108.6 ± 23.6 ms, n= 10, versus 74.0 ± 16.0, n= 7, P= 0.29) were significantly different between wild-type and CB1−/− mice.
Discussion
In the present study, we investigated CB1 receptor-dependent plasticity at spinal nociceptor synapses. A moderate depression of synaptic transmission was evoked when low-frequency (1.4 Hz) presynaptic stimulation was applied for 2 to 3 min, and a much more pronounced inhibition was obtained when synaptic stimulation was paired with postsynaptic depolarisation. This latter depression developed progressively over 5 to 10 min and then remained constant for the rest of the experiment.
NMDA receptor-dependent and CB1 receptor-dependent LTD in the spinal dorsal horn
LTD of high-threshold, presumed nociceptive, input to the dorsal horn has been reported previously (Randic et al. 1993; Sandkühler et al. 1997; Ikeda et al. 2000). This LTD was partially blocked by the NMDA receptor antagonist APV (Sandkühler et al. 1997) and subsequent work showed that, besides NMDA receptors, metabotropic glutamate receptors of the group I and group II families can evoke LTD in these synapses (Chen & Sandkühler, 2000). However, a contribution of endocannabinoids and CB1 receptors has not been demonstrated previously. In other areas of the CNS, there is clear evidence that NMDA receptor-dependent and endocannabinoid-dependent LTD exist in parallel (for reviews see Anwyl, 2006; Kano et al. 2009). In some cases, such as that of neocortical neurons, NMDA receptors do contribute to endocannabinoid-dependent LTD, but their contribution is presynaptic (Sjostrom et al. 2003; Bender et al. 2006; Nevian & Sakmann, 2006), while in the case of endocannabinoid-independent LTD, the relevant NMDA receptors are located postsynaptically (Anwyl, 2006). In the present study, neither genetic ablation of CB1 receptor nor blockade of NMDA receptors abolished LTD completely. Under both conditions, one fourth to one third of the cells still underwent LTD with no change in the degree of depression as compared with untreated slices from wild-type mice (compare Fig. 1C with Figs 6D and 7D), suggesting that endocannabinoid-dependent and NMDA receptor-dependent LTD occur independent of each other.
LTD in Aδ fibre synapses
Previous reports on LTD in the rat dorsal horn showed that it occurs mainly at Aδ fibre synapses (Sandkühler et al. 1997; Ikeda et al. 2000), while LTP is the typical response of C fibres to ongoing repetitive stimulation (Ikeda et al. 2003, 2006; for a review, see Sandkühler, 2007). In the present study, we found that pharmacological activation of CB1 receptors inhibited exclusively EPSCs with a high activation threshold corresponding to Aδ and/or C fibre EPSCs. This is consistent with a previous study by Nyilas et al. (2009), which provided compelling evidence for the expression of CB1 receptors on spinal terminals of both Aδ and C fibres. The same study also localised DGL-α to spines opposing dorsal horn nociceptor terminals indicating that primary nociceptor synapses express the molecular machinery required for endocannabinoid-dependent synaptic plasticity. In the present study we extend these findings and provide unambiguous evidence for the presence of CB1 receptors in peptidergic nociceptors of the superficial dorsal horn. In this context, it is important to note that both SP and CGRP are consistently found not only in C fibres but also in Aδ nociceptors (McCarthy & Lawson, 1989, 1990; Lawson et al. 1996). Furthermore, Aδ fibres also strongly express the sns (Nav1.8) gene (Djouhri et al. 2003), which was used in the present study to drive cre-mediated deletion of the CB1 receptor gene in nociceptors. Our finding that the portion of neurons exhibiting LTD was strongly reduced in sns-CB1−/− mice is therefore consistent with LTD being mainly expressed in Aδ fibres.
Possible prevention of C fibre LTP by presynaptic CB1 receptors
The morphological analyses in the present study and in previous studies (Hegyi et al. 2009; Nyilas et al. 2009) indicate that CB1 receptors are expressed on the presynaptic terminals of nociceptors in lamina II, although only few Aδ fibres terminate in this lamina. These CB1 receptors most probably reside on C fibre terminals, which extensively innervate lamina II. Our study reveals a possible function also for these receptors. The paired conditioning protocol used in this study did not induce significant potentiation in any of the wild-type neurons recorded. By contrast, LTP was evoked in about 20% of neurons from CB1−/− mice and sns-CB1−/− mice. All these neurons (5 out of 26) had EPSC thresholds of 50 mV or higher. Their EPSCs should therefore be considered as C fibre EPSCs. CB1 receptors on the spinal terminals of C fibre nociceptors may thus serve a specific function by hindering the development of LTP in C fibre synapses. Previous work by Wei et al. (2006) has demonstrated that dorsal horn nociceptor synapses can undergo LTP in the presence of intact cannabinoid signalling in response to a similar conditioning protocol (80 synaptic stimulations at 2 Hz paired with depolarization to +30 mV). In their study, GABAA and glycine receptors were blocked with bicuculline and strychnine in the entire spinal cord slice. In our experiments, we partially substituted chloride with fluoride in the internal recording solution in order to interfere with GABAergic and glycinergic inhibition of the recorded neuron only, while otherwise keeping fast synaptic inhibition intact. It is likely that the complete block of GABAergic and glycinergic inhibition greatly increases the level of excitation during the conditioning stimulation and thereby facilitates LTP induction.
Long-term and short-term CB1 receptor-dependent synaptic plasticity in the dorsal horn
The long-lasting and ‘irreversible’ inhibition of high-threshold EPSCs described here contrasts to the short-lasting and readily reversible depression of GABAergic and glycinergic synaptic transmission which we described previously in the same preparation (Pernía-Andrade et al. 2009). In this previous study, we have shown that GABAergic and glycinergic synapses in the dorsal horn are susceptible to transient DSI and that pharmacological activation of CB1 receptors or mGluR1/5 elicits a readily reversible reduction in GABAergic and glycinergic synaptic transmission. By contrast, the high-threshold primary afferent EPSCs studied here did not display transient DSE but underwent LTD after conditioning primary afferent stimulation paired with postsynaptic depolarisation. Such distinct forms of endocannabinoid-dependent plasticity have been repeatedly observed in many other CNS areas (for recent reviews see Heifets & Castillo, 2009; Kano et al. 2009). It is generally accepted that the short-term plasticity involves classical Gβγ-mediated inhibition of presynaptic voltage-gated Ca2+ channels, whereas LTD requires more intense conditioning stimulation, and additional intracellular transduction pathways such as reduced protein kinase A-dependent phosphorylation of the presynaptic scaffolding protein RIM1α (Chevaleyre et al. 2006, 2007). It is conceivable that the two pathways are also differentially active in inhibitory and excitatory synapses of the spinal dorsal horn.
Recent reports have suggested a role of CB1 receptors expressed on astrocytes in hippocampal or neocortical synaptic plasticity (Navarrete & Araque, 2010; Han et al. 2012; Min & Nevian, 2012). In our experiments, such a contribution would have become apparent as a difference in LTD expression between global CB1−/− mice, which lack CB1 receptors from both neurons and glia cells, and sns-CB1−/− mice, in which astrocytic CB1 receptors are retained. Such a difference was not observed.
Specific inhibition of high-threshold primary afferent EPSCs by CB1 receptor activation
The present experiments with CP 55,940 and WIN 55,212-2 indicate that only high-threshold but not low-threshold primary afferent synapses are susceptible to CB1 receptor-dependent inhibition. A similar preferential inhibition of high-threshold primary afferent evoked EPSCs has previously been observed by Liang et al. (2004). This specificity is unexpected given the fact that virtually all myelinated (low-threshold) primary sensory neurons express CB1 receptor mRNA and protein (Hohmann & Herkenham, 1999; Agarwal et al. 2007). A similar discrepancy of expression and functional activity is also found in the case of dorsal horn excitatory interneurons. More than one third of these terminals, identified by the presence of the vesicular glutamate transporter VGluT2 protein, carry CB1 receptors (Hegyi et al. 2009), but their activation does not affect synaptic glutamate release (Pernía-Andrade et al. 2009), suggesting that these CB1 receptors are uncoupled from synaptic release control. These receptors may serve functions different from synaptic inhibition or may become coupled to synaptic release only under certain conditions.
Implications for spinal nociception and pain plasticity
CB1 receptors and endocannabinoids exert complex effects on dorsal horn sensory processing. Previous work from our group (Pernía-Andrade et al. 2009) and from others (Jennings et al. 2001) has shown that activation of CB1 receptors on inhibitory interneuron terminals evokes a transient and readily reversible inhibition of synaptic GABA and glycine release. This CB1 receptor-mediated disinhibition contributes to a specific form of secondary hyperalgesia occurring in response to high-intensity C fibre stimulation (Pernía-Andrade et al. 2009). In the present study, we show that CB1 receptor activation does also reduce nociceptive transmission. This action occurs through induction of LTD at spinal nociceptor synapses. Which of these two apparently opposing actions occurring on distinct cellular elements of the dorsal horn circuit dominates may depend on the initial activity of the dorsal horn sensory network. Such state-dependent bidirectional modulation has recently been demonstrated in the rat and mouse cortex, where blockade of CB1 receptors increased network activity when basal activity was low, but decreased activity in the case of high basal activity (Piet et al. 2011). In the case of the spinal dorsal horn, similar state-dependent effects could explain the pronociceptive actions of endocannabinoids in the presence of very high nociceptive input, while analgesic actions would predominate at low or moderate activity levels.
Translational aspects
Our results suggest that spinal nociceptor synapses constitute an important site for the analgesic actions of endogenous and exogenous cannabinoids. They may thus have several implications for the discussion about the medicinal use of cannabis in pain patients. First, CB1 receptor-dependent LTD as well as the LTP preventing actions of dorsal horn CB1 receptors are likely to make significant contributions to the analgesic actions of cannabinoid receptor agonists in inflammatory or neuropathic pain states. These findings may be taken as support for the medical use of cannabinoids; it should, however, be kept in mind that the pharmacological targeting of these synapses will require drugs that are capable of penetrating into the CNS and which will probably exert significant psychotropic effects in addition to desired analgesia. Second, the processes described here should also be considered as a possible mechanism of conditioned analgesia, evoked e.g. by transcutaneous electrical nerve stimulation (TENS). TENS is most effective when applied at frequencies below 5 Hz and at intensities that cause mild to moderate nociceptor activation (Claydon et al. 2011). These conditions resemble quite closely the conditioning stimulation used here to induce cannabinoid-dependent LTD of primary nociceptor synapses. Finally, CB1 receptor-dependent LTD may constitute a previously unknown mechanism of stress-induced endogenous pain control (Hohmann et al. 2005), which has been suggested to depend at least partially on spinal endocannabinoid production (Nyilas et al. 2009). In summary, we are confident that the findings presented in this report will foster a rational debate on the use of cannabinoid receptor agonists, including medicinal marijuana, in pain patients.
Acknowledgments
The authors are very grateful to Louis Scheurer, Isabelle Camenisch and Erika Tischler for excellent technical assistance, to Drs Eszter Horváth, Zsolt Lele and Pascal Gratz for their contribution to the immunohistochemical experiments, to Drs Rohini Kuner, Heidelberg, Beat Lutz, Mainz, and Giovanni Marsicano, Bordeaux, for sns-cre and CB1fl/fl mice, to Dr Tamas Görcs, Budapest, and Dr Masahiko Watanabe, Hokkaido, for CGRP and CB1 receptor antisera, and to Dr Gonzalo E. Yévenes for critical reading of the manuscript. This work was partially supported by grants from the Swiss contribution (SH7/2/18) and by the Swiss National Science Foundation (31003AB_131093). R.N. and I.K. thank Mr László Barna, the Nikon Microscopy Center at IEM, Nikon Austria GmbH and Auro-Science Consulting Ltd for kindly providing microscopy support.
Glossary
- 2-AG
2-arachidonoyl glycerol
- APV
d(−)-2-amino-5-phosphonopentanoic acid
- BAC
bacterial artificial chromosome
- CB1
type 1 cannabinoid receptor
- CGRP
calcitonin gene-related peptide
- DGL-α
diacylglycerol lipase-α
- DSE
depolarisation-induced suppression of excitation
- DSI
depolarisation-induced suppression of inhibition
- GPCR
G protein-coupled receptor
- LTD
long-term depression
- LTP
long-term potentiation
- mGluR1/5
type 1/5 metabotropic glutamate receptor
- PB
phosphate buffer
- PFA
paraformaldehyde
- RIM1alpha
Rab3-interacting molecule 1alpha
- sns
sensory neuron specific sodium channel
- SP
substance P
- STD
short-term depression
- TBS
Tris-buffered saline
- TENS
transcutaneous electrical nerve stimulation
Author contributions
A.K., P.P., A.J.P.-A., C.v.S. and S.S. performed electrophysiological recordings. R.N. and I.K. performed and analysed the morphological experiments. H.U.Z. designed and analysed the electrophysiological experiments together with A.K. and P.P. H.U.Z wrote the manuscript. All authors made comments to the manuscript and all approved the final version.
Present addresses
A. K.: Department of Neurophysiology, Graduate School of Medicine, University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, 113-0033 Tokyo, Japan.
A.J.P.-A.: Institute of Science and Technology, Am Campus 1, A-3400 Klosterneuburg, Austria.
P.P.: Department of Biomedicine, Institute of Physiology, Pharmazentrum, University of Basel, Klingelbergstrasse 50/70, CH-4056 Basel, Switzerland.
S.S.: Institute of Physiology and Pathophysiology, University of Mainz, Duesbergweg 6, D-55128 Mainz, Germany.
References
- Agarwal N, Offermanns S, Kuner R. Conditional gene deletion in primary nociceptive neurons of trigeminal ganglia and dorsal root ganglia. Genesis. 2004;38:122–129. doi: 10.1002/gene.20010. [DOI] [PubMed] [Google Scholar]
- Agarwal N, Pacher P, Tegeder I, Amaya F, Constantin CE, Brenner GJ, et al. Cannabinoids mediate analgesia largely via peripheral type 1 cannabinoid receptors in nociceptors. Nat Neurosci. 2007;10:870–879. doi: 10.1038/nn1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmadi S, Lippross S, Neuhuber WL, Zeilhofer HU. PGE2 selectively blocks inhibitory glycinergic neurotransmission onto rat superficial dorsal horn neurons. Nat Neurosci. 2002;5:34–40. doi: 10.1038/nn778. [DOI] [PubMed] [Google Scholar]
- Anwyl R. Induction and expression mechanisms of postsynaptic NMDA receptor-independent homosynaptic long-term depression. Prog Neurobiol. 2006;78:17–37. doi: 10.1016/j.pneurobio.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Baffi J, Gorcs T, Slowik F, Horvath M, Lekka N, Pasztor E, Palkovits M. Neuropeptides in the human superior cervical ganglion. Brain Res. 1992;570:272–278. doi: 10.1016/0006-8993(92)90591-v. [DOI] [PubMed] [Google Scholar]
- Bender VA, Bender KJ, Brasier DJ, Feldman DE. Two coincidence detectors for spike timing-dependent plasticity in somatosensory cortex. J Neurosci. 2006;26:4166–4177. doi: 10.1523/JNEUROSCI.0176-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berghuis P, Rajnicek AM, Morozov YM, Ross RA, Mulder J, Urban GM, et al. Hardwiring the brain: endocannabinoids shape neuronal connectivity. Science. 2007;316:1212–1216. doi: 10.1126/science.1137406. [DOI] [PubMed] [Google Scholar]
- Bisogno T, Howell F, Williams G, Minassi A, Cascio MG, Ligresti A, et al. Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J Cell Biol. 2003;163:463–468. doi: 10.1083/jcb.200305129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Sandkühler J. Induction of homosynaptic long-term depression at spinal synapses of sensory Aδ-fibers requires activation of metabotropic glutamate receptors. Neuroscience. 2000;98:141–148. doi: 10.1016/s0306-4522(00)00080-4. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE. Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron. 2003;38:461–472. doi: 10.1016/s0896-6273(03)00235-6. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Heifets BD, Kaeser PS, Sudhof TC, Castillo PE. Endocannabinoid-mediated long-term plasticity requires cAMP/PKA signaling and RIM1a. Neuron. 2007;54:801–812. doi: 10.1016/j.neuron.2007.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaleyre V, Takahashi KA, Castillo PE. Endocannabinoid-mediated synaptic plasticity in the CNS. Annu Rev Neurosci. 2006;29:37–76. doi: 10.1146/annurev.neuro.29.051605.112834. [DOI] [PubMed] [Google Scholar]
- Claydon LS, Chesterton LS, Barlas P, Sim J. Dose-specific effects of transcutaneous electrical nerve stimulation (TENS) on experimental pain: a systematic review. Clin J Pain. 2011;27:635–647. doi: 10.1097/AJP.0b013e31821962b4. [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Lichtman AH. The endogenous cannabinoid system and its role in nociceptive behavior. J Neurobiol. 2004;61:149–160. doi: 10.1002/neu.20080. [DOI] [PubMed] [Google Scholar]
- Djouhri L, Fang X, Okuse K, Wood JN, Berry CM, Lawson SN. The TTX-resistant sodium channel Nav1.8 (SNS/PN3): expression and correlation with membrane properties in rat nociceptive primary afferent neurons. J Physiol. 2003;550:739–752. doi: 10.1113/jphysiol.2003.042127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodt HU, Zieglgänsberger W. Infrared videomicroscopy: a new look at neuronal structure and function. Trends Neurosci. 1994;17:453–458. doi: 10.1016/0166-2236(94)90130-9. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farquhar-Smith WP, Egertova M, Bradbury EJ, McMahon SB, Rice AS, Elphick MR. Cannabinoid CB1 receptor expression in rat spinal cord. Mol Cell Neurosci. 2000;15:510–521. doi: 10.1006/mcne.2000.0844. [DOI] [PubMed] [Google Scholar]
- Fukudome Y, Ohno-Shosaku T, Matsui M, Omori Y, Fukaya M, Tsubokawa H, Taketo MM, et al. Two distinct classes of muscarinic action on hippocampal inhibitory synapses: M2-mediated direct suppression and M1/M3-mediated indirect suppression through endocannabinoid signalling. Eur J Neurosci. 2004;19:2682–2692. doi: 10.1111/j.0953-816X.2004.03384.x. [DOI] [PubMed] [Google Scholar]
- Han J, Kesner P, Metna-Laurent M, Duan T, Xu L, Georges F, et al. Acute cannabinoids impair working memory through astroglial CB1 receptor modulation of hippocampal LTD. Cell. 2012;148:1039–1050. doi: 10.1016/j.cell.2012.01.037. [DOI] [PubMed] [Google Scholar]
- Hegyi Z, Kis G, Hollo K, Ledent C, Antal M. Neuronal and glial localization of the cannabinoid-1 receptor in the superficial spinal dorsal horn of the rodent spinal cord. Eur J Neurosci. 2009;30:251–262. doi: 10.1111/j.1460-9568.2009.06816.x. [DOI] [PubMed] [Google Scholar]
- Heifets BD, Castillo PE. Endocannabinoid signaling and long-term synaptic plasticity. Annu Rev Physiol. 2009;71:283–306. doi: 10.1146/annurev.physiol.010908.163149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann AG, Herkenham M. Localization of central cannabinoid CB1 receptor messenger RNA in neuronal subpopulations of rat dorsal root ganglia: a double-label in situ hybridization study. Neuroscience. 1999;90:923–931. doi: 10.1016/s0306-4522(98)00524-7. [DOI] [PubMed] [Google Scholar]
- Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, et al. An endocannabinoid mechanism for stress-induced analgesia. Nature. 2005;435:1108–1112. doi: 10.1038/nature03658. [DOI] [PubMed] [Google Scholar]
- Ikeda H, Asai T, Murase K. Robust changes of afferent-induced excitation in the rat spinal dorsal horn after conditioning high-frequency stimulation. J Neurophysiol. 2000;83:2412–2420. doi: 10.1152/jn.2000.83.4.2412. [DOI] [PubMed] [Google Scholar]
- Ikeda H, Heinke B, Ruscheweyh R, Sandkühler J. Synaptic plasticity in spinal lamina I projection neurons that mediate hyperalgesia. Science. 2003;299:1237–1240. doi: 10.1126/science.1080659. [DOI] [PubMed] [Google Scholar]
- Ikeda H, Stark J, Fischer H, Wagner M, Drdla R, Jager T, Sandkühler J. Synaptic amplifier of inflammatory pain in the spinal dorsal horn. Science. 2006;312:1659–1662. doi: 10.1126/science.1127233. [DOI] [PubMed] [Google Scholar]
- Jennings EA, Vaughan CW, Christie MJ. Cannabinoid actions on rat superficial medullary dorsal horn neurons in vitro. J Physiol. 2001;534:805–812. doi: 10.1111/j.1469-7793.2001.00805.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. Endocannabinoid-mediated control of synaptic transmission. Physiol Rev. 2009;89:309–380. doi: 10.1152/physrev.00019.2008. [DOI] [PubMed] [Google Scholar]
- Katona I, Freund TF. Multiple functions of endocannabinoid signaling in the brain. Annu Rev Neurosci. 2012;35:529–558. doi: 10.1146/annurev-neuro-062111-150420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Dopamine modulation of state-dependent endocannabinoid release and long-term depression in the striatum. J Neurosci. 2005;25:10537–10545. doi: 10.1523/JNEUROSCI.2959-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG. Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron. 2001;29:717–727. doi: 10.1016/s0896-6273(01)00246-x. [DOI] [PubMed] [Google Scholar]
- Lawson SN, McCarthy PW, Prabhakar E. Electrophysiological properties of neurones with CGRP-like immunoreactivity in rat dorsal root ganglia. J Comp Neurol. 1996;365:355–366. doi: 10.1002/(SICI)1096-9861(19960212)365:3<355::AID-CNE2>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Lever IJ, Rice ASC. Cannabinoids and pain. Handb Exp Pharmacol. 2006;177:265–306. doi: 10.1007/978-3-540-33823-9_10. [DOI] [PubMed] [Google Scholar]
- Lewis JW, Cannon JT, Liebeskind JC. Opioid and nonopioid mechanisms of stress analgesia. Science. 1980;208:623–625. doi: 10.1126/science.7367889. [DOI] [PubMed] [Google Scholar]
- Liang YC, Huang CC, Hsu KS, Takahashi T. Cannabinoid-induced presynaptic inhibition at the primary afferent trigeminal synapse of juvenile rat brainstem slices. J Physiol. 2004;555:85–96. doi: 10.1113/jphysiol.2003.056986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy PW, Lawson SN. Cell type and conduction velocity of rat primary sensory neurons with substance P-like immunoreactivity. Neuroscience. 1989;28:745–753. doi: 10.1016/0306-4522(89)90019-5. [DOI] [PubMed] [Google Scholar]
- McCarthy PW, Lawson SN. Cell type and conduction velocity of rat primary sensory neurons with calcitonin gene-related peptide-like immunoreactivity. Neuroscience. 1990;34:623–632. doi: 10.1016/0306-4522(90)90169-5. [DOI] [PubMed] [Google Scholar]
- Maejima T, Hashimoto K, Yoshida T, Aiba A, Kano M. Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron. 2001;31:463–475. doi: 10.1016/s0896-6273(01)00375-0. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Goodenough S, Monory K, Hermann H, Eder M, Cannich A, et al. CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science. 2003;302:84–88. doi: 10.1126/science.1088208. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, et al. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- Meng ID, Manning BH, Martin WJ, Fields HL. An analgesia circuit activated by cannabinoids. Nature. 1998;395:381–383. doi: 10.1038/26481. [DOI] [PubMed] [Google Scholar]
- Min R, Nevian T. Astrocyte signaling controls spike timing-dependent depression at neocortical synapses. Nat Neurosci. 2012;15:746–753. doi: 10.1038/nn.3075. [DOI] [PubMed] [Google Scholar]
- Navarrete M, Araque A. Endocannabinoids potentiate synaptic transmission through stimulation of astrocytes. Neuron. 2010;68:113–126. doi: 10.1016/j.neuron.2010.08.043. [DOI] [PubMed] [Google Scholar]
- Nevian T, Sakmann B. Spine Ca2+ signaling in spike-timing-dependent plasticity. J Neurosci. 2006;26:11001–11013. doi: 10.1523/JNEUROSCI.1749-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyilas R, Gregg LC, Mackie K, Watanabe M, Zimmer A, Hohmann AG, Katona I. Molecular architecture of endocannabinoid signaling at nociceptive synapses mediating analgesia. Eur J Neurosci. 2009;29:1964–1978. doi: 10.1111/j.1460-9568.2009.06751.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Maejima T, Kano M. Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron. 2001;29:729–738. doi: 10.1016/s0896-6273(01)00247-1. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Tsubokawa H, Mizushima I, Yoneda N, Zimmer A, Kano M. Presynaptic cannabinoid sensitivity is a major determinant of depolarization-induced retrograde suppression at hippocampal synapses. J Neurosci. 2002;22:3864–3872. doi: 10.1523/JNEUROSCI.22-10-03864.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Batkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58:389–462. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernía-Andrade AJ, Kato A, Witschi R, Nyilas R, Katona I, Freund TF, et al. Spinal endocannabinoids and CB1 receptors mediate C-fiber-induced heterosynaptic pain sensitization. Science. 2009;325:760–764. doi: 10.1126/science.1171870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrosino S, Palazzo E, de Novellis V, Bisogno T, Rossi F, Maione S, Di Marzo V. Changes in spinal and supraspinal endocannabinoid levels in neuropathic rats. Neuropharmacology. 2007;52:415–422. doi: 10.1016/j.neuropharm.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Piet R, Garenne A, Farrugia F, Le Masson G, Marsicano G, Chavis P, Manzoni OJ. State-dependent, bidirectional modulation of neural network activity by endocannabinoids. J Neurosci. 2011;31:16591–16596. doi: 10.1523/JNEUROSCI.4297-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randic M, Jiang MC, Cerne R. Long-term potentiation and long-term depression of primary afferent neurotransmission in the rat spinal cord. J Neurosci. 1993;13:5228–5241. doi: 10.1523/JNEUROSCI.13-12-05228.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbe D, Kopf M, Remaury A, Bockaert J, Manzoni OJ. Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proc Natl Acad Sci U S A. 2002;99:8384–8388. doi: 10.1073/pnas.122149199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandkühler J. Understanding LTP in pain pathways. Mol Pain. 2007;3:9. doi: 10.1186/1744-8069-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandkühler J, Chen JG, Cheng G, Randic M. Low-frequency stimulation of afferent Aδ-fibers induces long-term depression at primary afferent synapses with substantia gelatinosa neurons in the rat. J Neurosci. 1997;17:6483–6491. doi: 10.1523/JNEUROSCI.17-16-06483.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singla S, Kreitzer AC, Malenka RC. Mechanisms for synapse specificity during striatal long-term depression. J Neurosci. 2007;27:5260–5264. doi: 10.1523/JNEUROSCI.0018-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjostrom PJ, Turrigiano GG, Nelson SB. Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron. 2003;39:641–654. doi: 10.1016/s0896-6273(03)00476-8. [DOI] [PubMed] [Google Scholar]
- Tanimura A, Yamazaki M, Hashimotodani Y, Uchigashima M, Kawata S, Abe M, et al. The endocannabinoid 2-arachidonoylglycerol produced by diacylglycerol lipase alpha mediates retrograde suppression of synaptic transmission. Neuron. 2010;65:320–327. doi: 10.1016/j.neuron.2010.01.021. [DOI] [PubMed] [Google Scholar]
- Torsney C, MacDermott AB. Disinhibition opens the gate to pathological pain signaling in superficial neurokinin 1 receptor-expressing neurons in rat spinal cord. J Neurosci. 2006;26:1833–1843. doi: 10.1523/JNEUROSCI.4584-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JM, Hohmann AG. Cannabinoid mechanisms of pain suppression. Handb Exp Pharmacol. 2005;168:509–554. doi: 10.1007/3-540-26573-2_17. [DOI] [PubMed] [Google Scholar]
- Wei F, Vadakkan KI, Toyoda H, Wu LJ, Zhao MG, Xu H, Shum FW, Jia YH, Zhuo M. Calcium calmodulin-stimulated adenylyl cyclases contribute to activation of extracellular signal-regulated kinase in spinal dorsal horn neurons in adult rats and mice. J Neurosci. 2006;26:851–861. doi: 10.1523/JNEUROSCI.3292-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesenfeld-Hallin Z, Hokfelt T, Lundberg JM, Forssmann WG, Reinecke M, Tschopp FA, Fischer JA. Immunoreactive calcitonin gene-related peptide and substance P coexist in sensory neurons to the spinal cord and interact in spinal behavioral responses of the rat. Neurosci Lett. 1984;52:199–204. doi: 10.1016/0304-3940(84)90374-4. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature. 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]