

Abstract
Hypoglossal (XII) motoneurons (MNs) innervate the genioglossus muscle of the tongue, which plays an important role in maintaining upper airway patency, particularly during sleep, and modulating upper airway resistance. Discovering methods for inducing long-term increases in genioglossal motoneuronal excitability to AMPA-mediated drive may help in the development of therapeutics for upper airway motor disorders such as obstructive sleep apnoea. We show that the diuretic, anti-hypertensive, AMPA receptor modulator cyclothiazide (CTZ) induces a profound and long-lasting increase in the amplitude of respiratory-related XII nerve activity in rhythmically active neonatal rat medullary slices. Treatment of the slice with CTZ (90 μm) for 1 h increased the integrated XII (∫XII) nerve burst amplitude to 262 ± 23% of pre-treatment control at 1 h post-treatment; much of this increase lasted at least 12 h. The amount of CTZ-induced facilitation (CIF) was dependent upon both CTZ dose and exposure time and was accompanied by a long-lasting increase in endogenous AMPA-mediated drive currents to XII MNs. CIF, however, is not a form of plasticity and does not depend on AMPA or NMDA receptor activation for its induction. Nor does it depend on coincident protein kinase A or C activity. Rather, measurement of mEPSCs along with mass spectrometric analysis of CTZ-treated slices indicates that the cause is prolonged bioavailability of CTZ. These results illustrate a latent residual capacity for potentiating AMPA-mediated inspiratory drive to XII MNs that might be applied to the treatment of upper airway motor deficits.
Key points
Hypoglossal (XII) motoneurons (MNs) innervate the genioglossus muscle of the tongue, which plays an important role in maintaining upper airway patency and modulating upper airway resistance.
Cyclothiazide (CTZ) is a diuretic, anti-hypertensive, AMPA receptor modulator.
We show that CTZ induces a profound and long-lasting increase in the amplitude of respiratory-related XII nerve activity in rhythmically active neonatal rat medullary slices, with integrated XII nerve burst amplitude rising to 262 ± 23% of pre-treatment control at 1 h post-treatment and much of this increase lasting at least 12 h.
This phenomenon is not plasticity and does not depend on AMPA or NMDA receptor activation or on coincident protein kinase A or C activity for its induction.
Electrophysiological and mass spectrometric analyses indicate that the cause is prolonged bioavailability of CTZ.
Discovering methods for inducing long-term increases in genioglossal motoneuronal excitability to AMPA-mediated drive may help in the development of therapeutics for upper airway motor disorders such as obstructive sleep apnoea (OSA). These results illustrate a latent residual capacity for potentiating AMPA-mediated inspiratory drive to XII MNs that might be applied to the treatment of upper airway motor deficits such as OSA.
Introduction
Motor pools innervating muscles of the upper airway maintain proper upper airway alignment and patency against collapse from subatmospheric pressures during inspiration. In pathological conditions, such as obstructive sleep apnoea (OSA), often in combination with craniofacial abnormalities that narrow the upper airway, reduction or loss of motoneuron (MN) activity innervating genioglossus (tongue retractor) and other upper airway muscles during sleep leads to upper airway collapse. This results in repeated apnoeic and hypopnoeic events and often, severe disruption of sleep. In particular, collapse of the base of the tongue contributes to apnoeas in 25–44% of OSA cases (Ryan CM & Bradley TD, 2005). Occurring in 15–20% of adults (Young et al. 2002, 2009), OSA leads to increased daytime drowsiness, risk of workplace or car accidents and increased long-term risks of cardiovascular disease, stroke and hypertension (Young et al. 2002, 2009), as well as increased psychological and physiological stress for the bed partners of OSA sufferers (Smith et al. 2009). Therapies that increase excitability of these MNs to maintain airway patency have the potential to ameliorate OSA.
Hypoglossal (XII) MNs innervate the genioglossus muscle. In vitro (Greer et al. 1991; Funk et al. 1995) and under anaesthesia in vivo (Steenland et al. 2006; 2008), phasic respiratory drive to these MNs is mediated primarily by glutamatergic signalling through AMPA and NMDA receptors, suggesting that the excitability of XII MNs can be modulated by drugs that change AMPA receptor kinetics. One class of drugs, which impedes AMPA receptor deactivation and, to a lesser extent, desensitization is ampakines (Arai & Kessler, 2007). Ampakines have therapeutic potential for respiratory disorders: they have been used in rodents to successfully treat central depression of breathing due to anaesthetics (Ren et al. 2006; 2009) and knockout of the Rett syndrome-related gene MeCP2 (Ogier et al. 2007). They also facilitate respiratory-related activity in XII MNs in vitro (Lorier et al. 2010). Another class of drugs that can upregulate AMPA receptor-mediated excitability is benzothiadiazide diuretics (Bertolino et al. 1993; Arai & Kessler, 2007), of which cyclothiazide (CTZ) is the most potent (Bertolino et al. 1993; Yamada & Tang, 1993). CTZ affects the amplitude and rate of respiratory-related activity in vitro measured on the XII nerve (Funk et al. 1995). Interestingly, its effects last for at least 1–2 h post-treatment (Funk et al. 1995). What underlies this long-lasting facilitation is unclear, with the possibility that a novel form of plasticity, induced by CTZ, may be the source (Funk et al. 1995).
We studied the mechanisms underlying the persistence of CTZ-induced facilitation (CIF) of inspiratory-related XII nerve activity. CTZ profoundly increased the amplitude of inspiratory activity; the effects lasted up to 12 h post-treatment. Surprisingly, the source of this persistent facilitation was prolonged bioavailability of CTZ, rather than a novel form of plasticity.
Methods
Ethical approval
All animal procedures were performed according to National Institutes of Health guidelines and approved by the Office for the Protection of Research Subjects, University of California Research Committee. In addition, experiments complied with the policies and regulations documented by Drummond (2009), which the authors have read.
Preparation
Neonatal (P0–P4) Sprague–Dawley rats (n= 94; Charles River Laboratories International Inc., Wilmington, MA, USA) of both sexes were anaesthetized in a small chamber by inhalation of isoflurane (5 ml for ∼15 min). A lack of pedal withdrawal reflex assured that the level of anaesthesia was sufficient. The anaesthetized rat was placed ventral side up and rapidly decerebrated with a scalpel. A second cut caudal to the cervical backbone was made, separating the skull and vertebrae containing the brainstem and cervical spinal cord from the rest of the body. This reduced preparation was pinned in a dish and submerged in chilled (4–8°C) artificial cerebral spinal fluid (ACSF, in mm, 120 NaCl, 3 KCl, 1.5 CaCl2, 1 MgSO4, 23.5 NaHCO3, 0.5 NaH2PO4, 30 d-glucose, pH 7.4) that was saturated with 95% O2–5% CO2. Via a ventral entry, the brainstem and cervical spinal cord were exposed and removed with care to preserve the XII nerve rootlets. The dura mater was stripped away, the individual XII rootlets teased apart, and the cerebellum and choroid plexus removed, exposing the IVth ventricle. Finally, a scalpel cut was made near the pontomedullary border.
Still in oxygenated ACSF, the resulting en bloc preparation was pinned ventral surface up to a holder made of Sylgard 184 (Dow Corning Corp., Midland, MI, USA) backed with rigid plastic and placed in the chuck of a Vibratome 1000 (Vibratome, Bannockburn, IL, USA). Several cuts were made from the rostral end of the preparation. Once the facial nucleus was removed and the compact formation of the nucleus ambiguus exposed, a 700 μm slice was cut. This slice retained a sufficient proportion of the respiratory network to generate an inspiratory rhythm that could be measured in the activity of the XII nerve rootlets (Smith et al. 1991). The slice was transferred to a 1.5 ml recording chamber (Warner Instruments, Hamden, CT, USA) and held in place with a harp. The slice was superfused (≥5 ml min−1) with ACSF containing elevated K+ (9 mm) to sustain stable inspiratory activity (Smith et al. 1991). The slice was maintained at a constant temperature of 28°C and allowed to recover for ∼1 h before beginning experiments.
XII nerve recordings
XII nerve activity was recorded using a suction electrode and differential amplifier (A-M Systems, Carlsborg, WA, USA). The signal coming from the amplifier was split into two channels, one for direct data acquisition and a second that was rectified and integrated (Paynter filter, τ= 100 ms) using a custom-built integrator. Signals were sampled at 10–20 kHz and stored using a Digidata 1440A analog-to-digital converter and pCLAMP 10 software (Molecular Devices, Sunnyvale, CA, USA) running on a PC. The rhythmic burst discharges of the XII nerve defined the inspiratory period.
Whole-cell recordings
Inspiratory-active XII MNs were visualized with an infinity corrected 40× water immersion objective using differential interference contrast microscopy on an Axioskop FS1 microscope (Carl Zeiss, Thornwood, NY, USA). The image was displayed on a monitor using a CCD72 camera (Dage-MTI, Michigan City, IN, USA). Whole-cell voltage-clamp recordings (Vh=–70 mV for inspiratory drive currents, Vh=–90 mV for mEPSCs) were made using borosilicate glass electrodes (8250 glass, 3–5 MΩ). The internal solution contained (in mm): 135 potassium gluconate, 1.1 EGTA, 5 NaCl, 0.1 CaCl2, 10 Hepes, 2 ATP (Mg2+ salt), 0.3 GTP (Na+ salt), pH 7.3 adjusted using KOH. Caesium methanesulfonate and CsOH were substituted for potassium gluconate and KOH, respectively, for mEPSC recordings. Signals were recorded with an Axopatch-1D patch-clamp amplifier and CV-4 1/100 headstage (Molecular Devices). Signals were filtered in the patch-clamp amplifier with a low-pass Bessel filter (−3 dB at 5 kHz) and sampled at 20 kHz via Digidata 1440A both of which were controlled through pCLAMP software. Post hoc, currents were filtered further in pCLAMP using a low-pass 8-pole Bessel filter (−3 dB at 1 kHz). During recordings, access resistance was monitored for stability throughout the experiment. Whole-cell capacitance was compensated before break-in. For recording mEPSCs, XII MNs having inspiratory activity were silenced with tetrodotoxin (TTX, 1 μm) and non-NMDA glutamate receptor mEPSCs isolated using d-AP5 (50 μm), picrotoxin (100 μm) and strychnine (10 μm). Because of the requirement for inspiratory-modulated XII MNs, only one MN per slice was used.
Mass spectrometry
A liquid chromatography–tandem mass spectrometry–multiple reaction monitoring (LC–MSMS–MRM) assay was used to determine whether significant amounts of CTZ remained in tissue at different times during washout. Non-rhythmogenic brainstem slices (700 μm thick, as many as 4 from a single animal) from neonatal (P0–P4) Sprague–Dawley rats were exposed to ACSF only, 1 h of CTZ (90 μm), 1 h of CTZ plus 1 h wash with ACSF, or 1 h of CTZ plus 6 h of wash with ACSF. Excess surface ACSF was removed from the treated slices, which were then placed in pre-weighed microcentrifuge tubes, weighed again, and frozen at −20°C for later processing. For processing, 1 ml of methanol was added, and the tissue was disrupted with an ultrasonic cell disrupter. After centrifugation (13,000 g for 5 min), the supernatant was removed into a clean tube and taken to dryness in a vacuum centrifuge. To the dried residue water–acetonitrile–formic acid (100 μl, 95/5/0.1) was added and the samples were vigorously mixed, centrifuged again (13,000 g for 5 min) and the supernatant was transferred to LC injector vials. Aliquots (typically 20 μl) of each sample were injected to a reverse phase column (Agilent Eclipse Plus C18 RRHD, 2.1 × 50 mm, 1.8 μm particle size) equilibrated in Buffer A (0.01% acetic acid in water) and eluted (0.1 ml min−1) with an increasing concentration of Buffer B (0.01% acetic acid in acetonitrile: minutes/% B; 0/3; 5/3; 30/100; 33/3; 40/3). The effluent from the column was directed to an electrospray ion source (Agilent Jet Stream) attached to a triple quadrupole mass spectrometer (Agilent 6460) operating in the positive ion mode. Data were collected in the multiple reaction monitoring (MRM) mode in which the intensity of CTZ parent → fragment ion transitions were recorded under previously optimized conditions (m/z 392 → 326 and 392 → 105 with fragmentor and collision energy of 70 and 6, respectively; m/z stands for mass-to-charge ratio. This notation is fairly common, however. (http://en.wikipedia.org/wiki/Mass_spectrum) 390 → 324 and 390 → 105 with fragmentor and collision energy of 100 and 6, respectively). To determine the amount of CTZ present in samples, the peak areas from the liquid chromatography retention time corresponding to CTZ (19 min) were integrated with instrument manufacturer-supplied software (Agilent Mass Hunter Quantitative Analysis). Calculations were made using the mean of the results obtained for the two strongest fragment ion transitions (390/324, 392/326). To correlate peak areas measured in the tissue samples with known quantities of CTZ, standard solutions of pure CTZ diluted to 0, 2, 20, 200 and 2000 nm concentrations (0, 0.04, 0.4, 4 and 40 pmol per 20 μl) were run along with each batch of tissue samples. Using the peak areas run from the standard CTZ solutions, a curve of peak area vs. concentration was constructed. Then the amount of CTZ for a given tissue sample was calculated by using the peak area measured for that sample to interpolate the associated CTZ concentration from the standard curve calculated using the pure CTZ samples. Final values were reported as picomoles per milligram of tissue. A more detailed exploration of the limit of detection for the LC–MSMS–MRM assay found the limits of detection for CTZ to be approximately 2.7 nm (54 fmol per 20 μl). All tissue samples showing the presence of CTZ had amounts vastly greater than this detection threshold, confirming that the detection method used was of satisfactory sensitivity.
Drugs
6-Chloro-3,4-dihydro-3-(5-norbornen-2-yl)-2H-1,2,4-benzothiazidiazine-7-sulfonamide-1,1-dioxide (CTZ), N-[2-[[3-(4-bromophenyl)-2-propenyl]amino]ethyl]-5-isoquinolinesulfonamide (H89) dihydrochloride, 1,2-dimethoxy-12-methyl[1,3]benzodioxolo[5,6-c]phenanthridinium (chelerythrine) chloride, d-(–)-2-amino-5-phosphonopentanoic acid (d-AP5), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), and tetrodotoxin citrate were acquired from Tocris (Ellisville, MO, USA). 1-(1,4-Benzodioxan-6-ylcarbonyl)piperidine (CX546), strychnine and picrotoxin were acquired from Sigma-Aldrich (St Louis, MO, USA). Dimethyl sulfoxide (DMSO; Sigma-Aldrich) was used to dissolve CTZ, CX546, picrotoxin and CNQX into 100 mm stocks and chelerythrine into a 10 mm stock. Stocks were then diluted directly to their final concentrations as reported.
Electrophysiological data analysis
For recordings of ∫XII nerve activity, nerve bursts were identified using threshold detection in pCLAMP. The peak value for each burst was measured and averaged with the peak values from the other bursts in 5 min epochs. Averages were normalized to the first 25 min of a 30 min control period. The last 5 min of the control period, also normalized to the first 25 min of the control period, served as the control measurement. XII nerve burst rate was determined by taking the inverse of the average of the individual inter-burst intervals occurring over the same periods used to measure amplitude and normalized to the control rate.
For whole-cell recordings of XII MN inspiratory drive currents, charge transfer was calculated for individual inspiratory periods using DataView (Version 5.2.2, WJ Heitler, University of St Andrews) and averaged over 5 min periods. mEPSCs were identified using template matching in pCLAMP. Two- to four-hundred mEPSCs per cell were recorded and used in analysing amplitude and inter-event interval. The maximum amplitude of each individual event was averaged to determine the average amplitude, while individual inter-event intervals were averaged to determine the average interval. To calculate decay time constants, all mEPSC waveforms recorded from a given MN were averaged into a single average waveform from which the decay time constant was computed by fitting a single decaying exponential using data from the minimum amplitude of the average mEPSC waveform back to baseline.
Statistics
Data are summarized as mean ± SEM unless otherwise indicated. Differences associated with P values ≤ 0.05 were deemed significant. Although they were not considered significant, values of 0.05 < P≤ 0.10 are reported. Values for P > 0.10 are reported as n.s. (not significant).
For data containing more than two groups an ANOVA was first conducted. One-way ANOVAs were used for dose–response and exposure–response analyses. Two-way repeated-measures ANOVA (RMANOVA) was used for analysing longitudinal data from different treatment groups. Then individual difference tests (see below) were used for pairwise comparisons of interest. Individual P values are reported, but Holm–Bonferroni analysis for multiple comparisons ensured a familywise error rate ≤0.05 for all difference comparisons made within a data set. The similarity of cumulative distributions for mEPSC amplitudes and intervals for neurons exposed to different treatment conditions was assessed by piecewise comparison mEPSC amplitude and interval distributions in addition to group mean comparisons.
The normality and homoscedasticity of sample populations were assessed using the Shapiro–Wilk test and Levene's test, respectively. A number of the sample populations from this study violated one or both of these criteria. In addition, in some cases ANOVAs were carried out on slightly unequal data populations to take advantage of all available data. Since normality (t test and ANOVA) and homoscedasticity (pooled variance t test and ANOVA) are basic assumptions of parametric statistics (Cohen & Lea, 2004), bootstrapping statistical methods were used to analyse all data. Bootstrapping avoids making any underlying assumptions about parent data distributions, instead working solely with existing data distributions. Bootstrapping also allows the use of data values rather than ranks. Similar to regular t tests and ANOVA, the difference tests and ANOVAs assumed as a null hypothesis that data from all treatment groups were part of the same distribution. This null hypothesis was tested by sampling, with replacement, from the composite population to come up with differences in means or F ratios for comparison. This process was repeated 10,000 times to construct distributions of the mean differences or F ratios based on the null hypothesis. The actual two-sample difference in means or multi-sample F ratio for the treatment groups was compared with these distributions to assess the likelihood that they would have occurred if the null hypothesis were true. In repeated measures (RM) situations, resampling was done at the level of the subject, maintaining within-subject correlations across time. More detail on the rationale for and use of bootstrapping in analysis of biological data may be found in Manly (2006).
Bootstrapping was also employed to compare the similarity of mEPSC amplitude and interval distributions among treatment groups. The control distribution was binned into amplitude and interval histograms. Bin edges were chosen so that the number of samples in each bin was approximately equivalent to the number of samples in the other bins, maintaining similar levels of power for each individual bin comparison. Bins had >100 samples each, except for the highest bin, which had ∼50 samples in it. Then, the control distribution was resampled with replacement 10,000 times and binned into histograms using the same bin edges. Variations in the number of samples falling into each bin from resampling trial to resampling trial was used to develop confidence intervals. The number of data points selected during each resample depended upon the number of samples contained in the histogram for the treatment (either CTZ or CTZ plus 1 h wash) being compared with the control distribution. Then the non-control treatment distribution was binned into the same bins and compared against the control distribution confidence intervals. If the number of observations in a given bin was outside of confidence intervals for more than 20% of the bins consecutively, the distributions were deemed significantly different at the level of the confidence intervals. If confidence intervals were violated sporadically, e.g. the bin representing the largest amplitude mEPSCs violating the upper confidence interval by a factor of 2 without the preceding bin violating its confidence interval, those violations were evaluated instead at the Bonferroni level of confidence (α divided by the number of bins) to determine significance. Since this method generates bins whose widths are unequal, the resulting display of this histogram distorts the actual shape of the distribution events. To avoid this distortion, estimated probability density functions are displayed in Fig. 8. Probability density functions of event amplitudes were estimated by binning events in bins 1.5 pA in width, bin centres ranging from 6.25 to 80 pA. Probability density functions of event intervals were estimated by binning events in bins of 100 ms width from 0 to 2.5 s.
Figure 8. Estimated non-NMDA mEPSC probability density functions (PDFs).

PDFs for mEPSC peak amplitude (A) and interval (B) of untreated, CTZ-treated and CTZ-treated with 1 h wash slices. Insets show detail of PDF tail with enlarged y-axis. Comparison of distributions performed using piecewise bootstrap method (see Methods). *P < 0.05, ***P < 0.001.
Regressions
Single-variable linear regressions were performed using StatPlus (AnalystSoft Inc., Vancouver, BC, Canada). Only regressions having an F test with P≤ 0.05 and normally distributed residuals were considered significant.
Results
CIF
Previous investigation of the effects of CTZ on inspiratory activity in the medullary slice employed a cumulative dose–response protocol (Funk et al. 1995), where slices were exposed to five increasing concentrations of CTZ in the range 10–100 μm for ∼10 min. Here, bath application of 100 μm CTZ for ∼2 h caused ∫XII nerve burst amplitude to increase continuously for the entire duration of treatment, although the rate of rise slowed considerably within the first hour (Fig. 1A). The increase in ∫XII nerve burst amplitude, however, appeared to be much larger than previously reported. Also, there were sporadic increases in tonic activity after 1 h (Fig. 1A), as was previously reported (Funk et al. 1995).
Figure 1. Bath application of CTZ leads to long-lasting facilitation of endogenous inspiratory XII nerve bursts in neonatal rat medullary slice.

A, ∫XII nerve burst amplitude and rate continue to increase in the presence of bath-applied CTZ for entire duration (∼2 h) of application. Arrows show periods of increased tonicity. Asterisk denotes period of tonicity on expanded timescale (right of main trace). B, protocol for experiments in C and Fig. 2. C, example traces showing the impact of bath application of CTZ (top), CX546 (middle) and DMSO (bottom) on ∫XII nerve burst amplitude and rate. Traces on expanded timescales below main traces show samples of ∫XII nerve bursts before and 1, 6 and 12 h post-treatment.
To avoid these sporadic periods of increased nerve tonicity, the maximum CTZ concentration was limited to 90 μm and the treatment duration was no more than 1 h (Fig. 1B and C). ∫XII nerve burst amplitude increased to 236 ± 21% of (pre-treatment) control (P < 0.001, n= 5, paired difference test) at the conclusion of bath application and peaked at 262 ± 23% 1 h post-treatment (P < 0.001, n= 5, paired difference test), i.e. 1 h after start of washout. Given our interest in the persistent effects of CTZ, activity was followed for 12 h post-treatment. ∫XII nerve burst amplitude remained significantly elevated relative to pre-treatment for at least 12 h post-treatment (Figs 1C and 2A; Table 1).
Figure 2. CTZ, but not CX546 or DMSO, leads to long-lasting facilitation of endogenous inspiratory ∫XII nerve burst amplitude.

A–B, longitudinal data for effect of 1 h application of CTZ (90 μm, n= 5), CX546 (90 μm; n= 7) or DMSO (0.1%; n= 7) on normalized ∫XII nerve burst amplitude (A) and rate (B). Thick lines show group averages. Dotted lines show individual experiments. CTZ (squares), CX546 (diamonds), DMSO (triangles). Black bar above data shows timing and duration of treatment. C–D, comparisons for the effects of CTZ (squares) and CX546 (diamonds) vs. DMSO control (triangles) on normalized ∫XII nerve burst amplitude (C) and rate (D) at 1, 6 and 12 h post-treatment. Significance, assessed using difference tests following two-way RMANOVA, is as indicated in the figures. E–F, regressions assessing whether the effect of CTZ (90 μm; n= 11) on normalized ∫XII nerve burst amplitude at 1 h post-treatment vs. raw control ∫XII nerve burst amplitude (E) and normalized XII nerve burst rate at 1 h post-treatment vs. raw XII nerve burst rate (F) were significantly correlated.
Table 1.
Summary of statistical comparisons for medullary slices treated for 1 h with CTZ (90 μm), DMSO (0.1%) or CX546 (90 μm)
| ∫XII amplitude | ∫XII rate | |
|---|---|---|
| RMANOVA CTZ DMSO CX546 | ||
| Treatment | F(2,16) = 47.7*** | F(2,16) = 42.9*** |
| Time | F(13,208) = 6.37 | F(26,208) = 8.94*** |
| Treatment × Time | F(26,208) = 5.46*** | F(13,208) = 49.8* |
| Tests vs. pre-treatment control | ||
| CTZ 0 h post-treatment | 236 ± 21%*** | 147 ± 14%*** |
| CTZ 1 h post-treatment | 262 ± 23%*** | 151 ± 12%*** |
| CTZ 6 h post-treatment | 205 ± 4.8%*** | 108 ± 15% |
| CTZ 12 h post-treatment | 190 ± 9.4%*** | 62.9 ± 19% |
| DMSO 0 h post-treatment | 98.5 ± 3.8% | 82.8 ± 5.7%* |
| DMSO 1 h post-treatment | 90.2 ± 4.2%** | 59.7 ± 4.8%*** |
| DMSO 6 h post-treatment | 91.5 ± 6.6% | 36.0 ± 4.1%*** |
| DMSO 12 h post-treatment | 90.3 ± 13% | 22.3 ± 5.1%*** |
| CX546 0 h post-treatment | 160 ± 15%*** | 137 ± 6.6%*** |
| CX546 1 h post-treatment | 126 ± 11%*** | 74.9 ± 6.7%*** |
| CX546 2 h post-treatment | 114 ± 8.4% | n.t. |
| CX546 6 h post-treatment | 118 ± 18% | 40.9 ± 4.6%*** |
| CX546 12 h post-treatment | 119 ± 19% | 29.5 ± 3.3%*** |
Values are normalized relative to pre-treatment control (see Methods) and reported as mean ± SEM (n= 5 for CTZ-treated slices, n= 7 for DMSO and CX546-treated slices). Two-way RMANOVA included all slices. Measurements that were taken pre-treatment, immediately (0 h) and 1–12 h (hourly) post-treatment (14 data points in all per slice) were used in RMANOVA. Tests vs. pre-treatment control were RM difference tests where measurements of activity were compared at two time points: once immediately prior to treatment and once at the time point indicated. Family-wise error rate, which also includes statistical tests shown in Fig. 2, was protected to P≤ 0.05 using Holm–Bonferroni method. *P < 0.05, **P < 0.01, ***P < 0.001 n.t., not tested.
The effect of CTZ on the rate of XII nerve bursting (Fig. 2B) was significant but more modest. Respiratory rate elevated to 147 ± 14% of control at the conclusion of treatment (P < 0.001, n= 5, paired difference test) and rose to 151 ± 12% of control 1 h post-treatment (P < 0.001, n= 5, paired difference test). By 6 h, the rate returned to baseline 108 ± 15%, remaining there at 12 h (63 ± 19%, n= 5, paired difference test).
DMSO, the solvent used for CTZ, has excitatory effects on neurons of the lamprey locomotor CPG (Tsvyetlynska et al. 2005). To control for any effects DMSO might have on the neurons we studied, control slices were exposed to 0.1% DMSO alone for 1 h (Figs 1C and 2A and B). (This concentration was slightly greater than the 0.09% that slices were exposed to when 90 μm CTZ was applied.) DMSO did not increase ∫XII nerve burst amplitude or rate, which was depressed at all time points relative to pre-treatment (Table 1).
The response to CTZ was compared with DMSO and CX546 (see below) in a two-way RMANOVA to look for treatment effects as well as time effects followed by difference tests at specific time points (reported in Table 1 and Fig. 2). For ∫XII nerve burst amplitude, the effects of treatment and the interaction of time and treatment were both significant, while the effect of time was not significant (Table 1). The increase in ∫XII nerve burst amplitude for CTZ-treated slices was significantly larger than for DMSO-treated slices at all time points (Fig. 2C). Treatment and time effects on the XII nerve burst rate showed significant effects for treatment, the interaction of time and treatment, as well as a significant effect for time (Table 1). Similar to amplitude, the increase in the XII nerve burst rate of CTZ-treated slices was higher than in DMSO slices (Fig. 2D).
From slice to slice, absolute ∫XII nerve burst amplitude can vary by more than an order of magnitude, and the XII nerve burst rate can vary by a factor of 2–3. For this reason, normalized values are useful in evaluating data taken from groups of slices. Analysing normalized values, however, left the possibility that, for example, the effects of CTZ on XII nerve burst rate might be greater in slices whose pre-treatment rates were at the lower end of the expected range. Also, the effects of CTZ on a slice with burst amplitudes at the high end of the amplitude range during the pre-treatment period might be suppressed, because the level of nerve activity could already be saturated. To assess whether the pre-treatment raw values of amplitude or rate of inspiratory XII nerve bursts predicted the response to CTZ, the percentage effect on ∫XII nerve burst amplitude or rate at 1 h post-treatment was regressed against the raw burst amplitude or rate for that slice during the pre-treatment period. Pre-treatment amplitude ranged from 0.31 to 13.1 a.u., and pre-treatment control rate ranged from 6.34 to 20.4 bursts min−1. No significant relationships between amplitude effect size and raw pre-treatment amplitude or rate effect size and raw pre-treatment burst rate were seen (Fig. 2E and F).
Finally, to see if the long-lasting effects of CTZ on amplitude and rate were typical of agents that affect AMPA receptor desensitization and deactivation, CX546 (90 μm) was tested using the same protocol (Fig. 1C). In slices treated with CX546, ∫XII nerve burst amplitude increased immediately post-treatment, then declined greatly but was still elevated for 1 h post-treatment, returned to baseline by 2 h post-treatment and remained there for the rest of the experiment (Fig. 2A, Table 1). Relative to DMSO, ∫XII nerve burst amplitude was significantly different only immediately post-treatment (Fig. 2C).
Similar to ∫XII nerve burst amplitude, the rate of inspiratory ∫XII nerve bursts was significantly elevated in CX546-treated slices relative to pre-treatment control, although only in the immediate post-treatment period (Fig. 2B, Table 1). As in slices treated with DMSO to which they were statistically similar (Fig. 2D), slices treated with CX546 had a lower burst rate relative to pre-treatment (Table 1). Therefore, in the presence of CX546, inspiratory activity in the medullary slice responds in a way similar to CTZ, but the effects of CX546 abate during washout.
Dose–response
Next, the effects of CTZ dose and duration of exposure on slice inspiratory activity were characterized. Either 3, 9 or 30 μm was applied to separate groups of slices for 1 h or 90 μm CTZ was applied to separate groups of slices for 10 or 30 min. These data were then grouped with the data for slices exposed to 90 μm for 1 h in dose–response (Fig. 3A) or exposure–response (Fig. 3B) curves. The effect of CTZ on ∫XII nerve burst amplitude was significant at 1 h post-treatment for both dose and exposure time (Fig. 3). CTZ dose but not exposure time had a significant effect on the respiratory rate at 1 h post-treatment (Fig. 3), indicating the possibility that the effect of CTZ on ∫XII nerve burst amplitude and rate were separable. Since the effect of CTZ on ∫XII nerve burst amplitude was so much larger and longer lasting than its effect on the rate of bursting, subsequent experiments focused solely on the former.
Figure 3. Dose–response and exposure–response effects of CTZ on ∫XII nerve burst amplitude and rate 1 h post-treatment.

A, dose–response for 1 h bath application of 3 μm, 9 μm, 30 μm or 90 μm CTZ (n= 5 for each concentration) on ∫XII nerve burst amplitude and rate. Concentration had significant effect on both amplitude (F(3,16) = 6.38, P < 0.01) and rate (F(3,16) = 6.50, P < 0.01) as determined by one-way ANOVA. B, exposure–response curves for 90 μm CTZ applied for 10 min (n= 6), 30 min (n= 6) or 1 h (n= 5). Exposure time had significant effect on ∫XII nerve burst amplitude (F(2,14) = 7.68, P < 0.01) but not rate (F(2,14) = 2.31, n.s.) as assessed by one-way ANOVA. In both panels, squares represent amplitude responses and circles represent rate responses. Large symbols show group averages and small symbols individual experiments. All measurements were taken at 1 h post-treatment.
Long-term effects of CTZ on XII MN drive
Manipulation of the excitability of XII MNs affects the amplitude but not the rate of ∫XII nerve bursts in the medullary slice (Funk et al. 1993). Inspiratory drive to these MNs is primarily AMPA-receptor dependent in vitro (Funk et al. 1993) and is enhanced by acute CTZ exposure (Funk et al. 1995). To assess the long-term post-treatment effects of CTZ on inspiratory drive to XII MNs, currents were measured continuously from XII MNs before, during and up to 1 h post-treatment with 90 μm CTZ. CTZ was applied for only 10 min in order to reliably maintain stable whole-cell recordings for the duration of the experiment, while still getting reliable, though weaker, CIF, i.e. long-lasting facilitation of ∫XII nerve burst amplitude (Figs 3B and 4A). At 1 h post-treatment, endogenously generated drive to XII MNs was facilitated (Fig. 4A). Charge transfer was 153 ± 8.1% of pre-treatment (P < 0.001, n= 5, paired difference test) while ∫XII nerve burst amplitude increased to 146 ± 19% (P < 0.001, n= 5, paired difference test; Fig. 4B). Furthermore, the size of increase of ∫XII nerve burst amplitude correlated well with the increase in charge transfer (Fig. 4C).
Figure 4. Bath application of CTZ induces long-lasting increases in endogenous inspiratory drive to XII MNs.
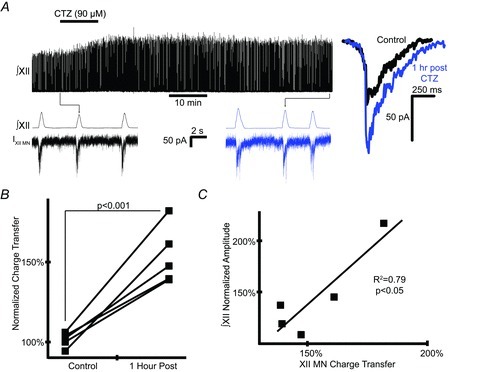
A, top trace shows effect of treating medullary slice with CTZ (90 μm) for 10 min. Expanded traces below show sample ∫XII nerve bursts and accompanying XII MN drive currents immediately prior to and 1 h post-treatment. Overlaid current traces to the right show average of 25 consecutive drive currents for each time point. B, comparison of normalized charge transfer of inspiratory drive currents in XII MNs before and 1 h post-treatment. Lines connect measurements from the same cell before and 1 h post-treatment. Significance tested using RM difference test (n= 5). C, regression showing high correlation between increases in XII MN drive currents and ∫XII nerve burst amplitude 1 h post-treatment.
Investigation of intracellular signalling as the mechanism underlying CIF
Since CTZ led to a profound and long-lasting facilitation of ∫XII nerve burst amplitude and AMPA receptor-mediated synaptic drive to XII MNs, identifying the mechanism underlying this long-lasting component of CIF was investigated next. Increased internal Ca2+ is a signal critical for inducing a variety of long-term plasticity phenomena (Malenka & Bear, 2004; Vogt & Canepari, 2010). NMDA receptors, which are highly permeable to Ca2+, require depolarization (to remove Mg2+ block) in addition to glutamate to open. NMDA receptors are colocalized with AMPA receptors at excitatory synapses in XII MNs (O’Brien et al. 1997). Therefore, a reasonable possibility existed that enhanced depolarization, resulting from facilitated AMPA receptor-mediated synaptic currents, could raise the level of intracellular Ca2+ through a commensurate increase in activation of NMDA receptors. To investigate this possibility AMPA (10 μm CNQX) and NMDA (50 μm d-APV) receptors were blocked for 30 min before and continuing through 1 h of CTZ application and 1 h of washout. Eliminating the main source of XII MN excitability in the slice, probably also reduced activation of another major source of Ca2+, voltage-gated Ca2+ channels.
Blockade of AMPA and NMDA receptors abolished inspiratory activity (Fig. 5A). To guard against the possibility that any facilitation seen during recovery from CNQX and d-APV was due to rebound excitation from silencing neurons rather than from the effects of CTZ, another set of slices was exposed to the same protocol but without applying CTZ (Fig. 5A). Treatment with CTZ in the presence of these glutamate receptor antagonists significantly increased ∫XII nerve burst amplitude relative to the antagonists alone (Fig. 5B). Slices treated with CTZ in the presence of CNQX and d-APV saw their activity start to return within 30 min of starting antagonist washout (85.6 ± 22% of pre-treatment control ∫XII nerve burst amplitude, n= 6). ∫XII nerve burst amplitude after CTZ was significantly greater than pre-treatment from 1 h after the start of CNQX and d-APV washout to the end of the experiment 5 h after start of washout of CNQX and d-APV (Fig. 5B). In contrast, only 1 of 5 slices treated with the antagonists alone saw activity return within 30 min of starting antagonist washout. Activity in all five slices returned by 1 h after the start of antagonist washout (80.8 ± 16% of pre-treatment control ∫XII nerve burst amplitude, n= 5) but, on average, ∫XII nerve burst amplitude in these slices was neither facilitated nor depressed relative to pre-treatment control through the rest of the experiment (Fig. 5B). At both 1 and 5 h post CNQX and d-APV, ∫XII nerve burst amplitude was larger in slices treated with CTZ relative to those not treated with CTZ (Fig. 5C). We considered whether a component of the facilitation seen when slices were treated with CTZ without glutamate receptor antagonists (Figs 1 and 2) was missing when treatments were done in the presence of antagonists (Fig. 5), i.e. activity still may have been facilitated but not as much as when fast glutamatergic signalling had not been blocked. To explore this, the amplitude of ∫XII nerve burst activity was compared at equivalent time points for CTZ-treated slices in these glutamate antagonist experiments (Fig. 5B) with the level of activity in CTZ-treated slices from the initial characterization (Fig. 2A). There was no significant difference in amplitude of response between slices treated with CTZ and slices treated with CTZ in the presence of CNQX and d-APV (F(1,9) = 3.04, n.s., two-way RMANOVA). Together these data indicated that it was unlikely CIF depended, in whole or in part, on activation of AMPA or NMDA receptors during treatment with CTZ.
Figure 5. CIF does not depend upon activation of AMPA or NMDA receptors during treatment with CTZ.

A, sample traces showing the effects on ∫XII nerve activity after bath application of CNQX (10 μm) and APV (50 μm) (control slices, top trace) or CTZ (90 μm) in the presence of CNQX and APV (bottom trace). CNQX and APV were applied for 2.5 h. When CTZ was applied, it was applied for 1 h, 30 min after the start of CNQX and APV. This allowed CNQX and APV to take effect before CTZ and 1 h for the slices to be washed after CTZ before removing CNQX and APV. Black bars above traces illustrate the timing and duration of application. Transients resulting from electrostatic discharge during the slice silent periods have been removed. B, longitudinal data for all experiments run according to the protocols in A. CTZ had a significant effect (F(1,9) = 12.8, RMANOVA) on ∫XII nerve burst amplitude. In slices treated with CTZ (n= 6) ∫XII nerve burst amplitude was significantly greater than pre-treatment from 1 h post CNQX and d-APV (182 ± 6.8%, P < 0.001, RM difference test) to the end of the experiment 5 h post CNQX and d-APV (180 ± 25%, P < 0.001, RM difference test). ∫XII nerve burst amplitude in slices not treated with CTZ (n= 5) was neither facilitated nor depressed relative to pre-treatment (115 ± 21% 5 h post CNQX and d-APV, n.s., RM difference test). Slices receiving treatment with CTZ are marked by squares. Control slices (n= 5) are marked with triangles. Thick traces represent group means. Thin traces represent individual experiments. C, comparison of activity 1 h and 5 h post CNQX and d-APV in slices treated and not treated with CTZ. Significance was computed using difference tests that followed a two-way RMANOVA.
Next, the possibility that CTZ affected the activity of PKA and PKC, both of which have a role in increasing XII MN excitability (Bocchiaro et al. 2003; Neverova et al. 2007; DuBord et al. 2010), was investigated by blocking PKA (10 μm H89) and PKC (10 μm chelerythrine) activity 30 min prior to applying CTZ for 1 h. The kinase antagonists were maintained throughout CTZ treatment and for 1 h post-treatment. The long-term effects of H89 and chelerythrine, alone, on ∫XII nerve burst amplitude in the slice were controlled for by applying these antagonists without applying CTZ. Long-term exposure to H89 and chelerythrine greatly reduced ∫XII nerve burst amplitude in slices, which only partially recovered after 30 min of washout (Fig. 6A). Some slices were allowed more than an hour of washout but never showed complete recovery (data not shown). Despite the effect of these kinase antagonists on activity, CTZ still facilitated the ∫XII nerve burst amplitude, which was significant when compared with slices treated only with H89 and chelerythrine alone at 30 min following washout of the kinase antagonists (Fig. 6B). These experiments demonstrated that CTZ most probably was acting in a manner independent of PKA and PKC.
Figure 6. CIF is not PKA- or PKC-dependent.

A, sample trace showing effects of bath application of chelerythrine (10 μm) and H89 (10 μm) on inspiratory ∫XII nerve activity. Last half hour of trace (during washout of H89 and chelerythrine) demonstrates failure of some slices not treated with CTZ to recover pre-treatment activity levels. (Transients resulting from perfusion noise have been removed.) B, comparison of ∫XII nerve burst amplitude (relative to pre-treatment control) between CTZ-treated (CTZ) and untreated (No CTZ) slices (n= 6 for both groups) 30 min post washout of H89 and chelerythrine, which was 1 h post the start of washout of CTZ, when CTZ was used. Symbols represent individual experiments and lines represent group averages. Significance assessed using difference test.
Does CTZ wash out?
Having eliminated several of the intracellular signalling mechanisms that would most probably underlie the long-lasting component of CIF, we considered the alternative hypothesis that CTZ did not wash out. While some studies report that CTZ washes out from neural tissue (Ballerini et al. 1995; Funk et al. 1995; Qi et al. 2006), other studies maintain that CTZ lingers, sequestered in the lipid bilayer of the neuron plasma membrane (Patneau et al. 1993; Larson et al. 1994), where it continues to affect AMPA receptor desensitization. We addressed this issue in two ways. First, a functional assay, measuring non-NMDA, presumably AMPA, mEPSCs, was used to identify telltale signatures of the continued presence of CTZ. Specifically, CTZ increases the amplitude, decay time constant and frequency of non-NMDA mEPSCs (Diamond & Jahr, 1995). mEPSCs were measured in XII MNs from slices that were bathed in ACSF only, bathed in CTZ for 1 h, or bathed in CTZ for 1 h and then washed for 1 h (Fig. 7A). Following a given treatment regimen, XII MNs were patched and verified to have inspiratory-related drive. Then action potentials (TTX 1 μm) and receptors for NMDA (50 μm d-APV), GABAA (100 μm picrotoxin) and glycine (10 μm strychnine) were blocked. Average mEPSC peak amplitude was significantly larger in neurons treated with CTZ and washed for 1 h relative to neurons treated with CTZ for 1 h and to neurons bathed in ACSF alone, which was consistent with results from comparing mEPSC peak amplitude distributions in a piecewise mannerh (Figs 7B and 8A). mEPSC decay time, as assessed by measuring the decay time constant, was greater for neurons treated with CTZ and washed for 1 h relative to neurons bathed in ACSF alone, although less than for neurons treated with CTZ but not washed (Fig. 7C).
Figure 7. CTZ treatment of medullary slices leads to long-lasting increases in XII MN non-NMDA mEPSC amplitude and decay.
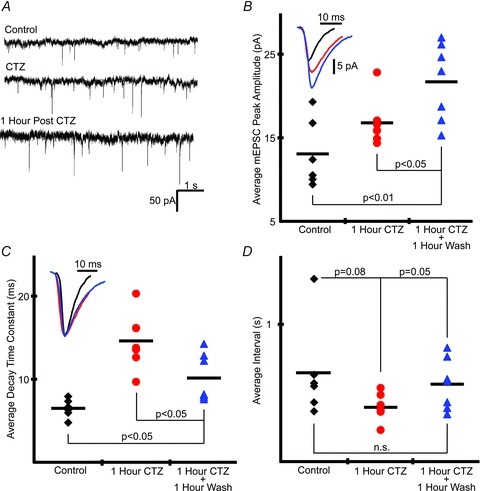
A, sample mEPSCs from control cell (top), cell treated for 1 h with CTZ (90 μm; middle) and a cell treated with CTZ that was then washed for 1 h before recording (bottom). B–D, group data comparing average mEPSC peak amplitude (B), mEPSC decay time constant (C) and average mEPSC interval (D) for cells treated under 1 of the 3 conditions described in A (n= 6 for control cells and n= 7 for cells treated with CTZ and cells treated with CTZ and then washed for 1 h). Inset in B shows average of the average waveforms for each experiment under a given condition. Inset in C shows waveforms in B scaled to same peak value. Significance in B–D assessed using difference test. Individual symbols in B–D represent values for single experiments. Lines represent group averages.
CTZ had at most a marginally significant effect on the average interval between mEPSCs (Fig. 7D). Use of the more sensitive piecewise comparison of interval distributions, however, showed a significant difference between the interval distribution of CTZ-treated slices compared with both ACSF-treated neurons and CTZ-treated neurons that were washed for 1 h (Fig. 8B). There was also a marginally significant difference between the group of slices treated with ACSF and the group of slices treated with CTZ and then washed for 1 h that indicated XII MNs treated with CTZ and then washed had fewer large (>1.25 s) intervals between mEPSC events (Fig. 8B).
The mEPSC amplitude and decay data provided indirect evidence that CTZ remained in the tissue. A second, more direct measure for the residual presence of CTZ in treated but washed tissue, i.e. liquid chromatography–tandem mass spectrometry, was pursued. Slices were analysed from four different treatment groups: (1) exposed to ACSF only, (2) treated with CTZ (90 μm) for 1 h and removed immediately, (3) treated with CTZ (90 μm) for 1 h and then washed with ACSF for 1 h before removal, (4) treated with CTZ (90 μm) for 1 h and then washed with ACSF for 6 h before removal. Slices exposed only to ACSF showed no evidence of CTZ at the level of assay sensitivity (∼50 fmol), while all other treatment groups showed large quantities of residual CTZ (10 ± 2.8 pmol mg−1 for CTZ-treated slices, 5.3 ± 0.9 pmol mg−1 for slices treated with CTZ and washed for 1 h, and 5.7 ± 0.6 pmol mg−1 for slices treated with CTZ and washed for 6 h; Fig. 9). Although there was a trend towards more CTZ remaining in tissue immediately post-treatment compared with 1 or 6 h of washout, the difference was not significant (F(2,12) = 2.92, n.s., n= 5 for each group, one-way ANOVA), suggesting most of the CTZ remained in the slice despite hours of washing (Fig. 9). Assuming the slices have the approximate density of water and that CTZ could go anywhere in the slice, which is conservative, since CTZ probably does not easily cross the plasma membrane (Patneau et al. 1993), the measurements indicate the residual concentration of CTZ in the tissue following treatment and wash was on average >5 μm and not statistically significantly different from unwashed CTZ-treated slices.
Figure 9. Large quantities of CTZ remain trapped in medullary slice following wash with ACSF.
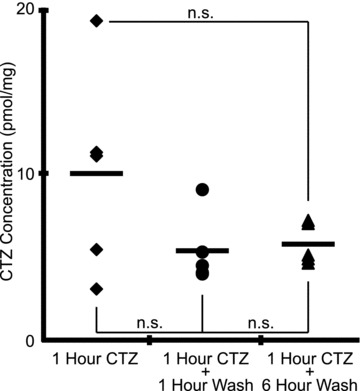
Measurements made using liquid chromatography–tandem mass spectrometry. Slices treated with CTZ (90 μm) in 1 of 3 ways: 1 h, 1 h and washed for 1 h, 1 h and washed for 6 h (n= 5 for all groups). No significant difference among the three groups using one-way ANOVA or difference tests between groups. Individual symbols represent individual experiments. Lines represent group averages.
Discussion
CTZ profoundly increased inspiratory ∫XII nerve burst amplitude in the respiratory rhythmic medullary slice preparation and modestly increased the burst rate. The ∼150% average increase in amplitude is about 3 times the maximum facilitation previously reported for similar concentrations of CTZ (Funk et al. 1995), with much of this facilitation remaining for at least 12 h post-treatment, and also about 3 times that of in vitro long-term facilitation (ivLTF), another form of long-lasting facilitation of inspiratory activity (Bocchiaro & Feldman, 2004; Neverova et al. 2007).
As a neuromodulator, CTZ is best known for its ability to limit or abolish desensitization in primarily flip splice variants of AMPA receptors (Yamada & Tang, 1993; Patneau et al. 1993; Partin et al. 1994). CTZ, however, has a variety of other activity-independent effects at AMPA receptors, including dramatically lowering agonist EC50 (Patneau et al. 1993; Partin et al. 1994; Dzubay & Jahr, 1999; Fucile et al. 2006), lengthening rate and length of channel open time (Yamada & Tang, 1993; Fucile et al. 2006), increasing the preference for larger conductance states (Fucile et al. 2006) and increasing deactivation time (Patneau et al. 1993). Another activity-independent property of CTZ, enhancement of presynaptic release, can combine with these postsynaptic AMPA-receptor effects to increase the rate, peak currents and decay time constants of spontaneous and mEPSC activity simultaneously at synapses of CA1 neurons in hippocampal culture (Diamond & Jahr, 1995).
The increase in inspiratory ∫XII nerve burst amplitude that we observed was associated with an increase in XII MNs of AMPA-mediated inspiratory drive currents and the amplitude of AMPA mEPSCs, while the decay time constant of AMPA mEPSCs increased. As previously discussed, this combination of signatures is a hallmark of the presence of CTZ. Given the continued presence of CTZ, which was directly measured to be at physiologically relevant concentrations (Patneau et al. 1993) in tissue for at least 6 h following treatment of slices with CTZ, the most parsimonious explanation for the large and persistent facilitation of respiratory activity we observed in the medullary slice is continued presence of CTZ.
Based on the lipophilic nature of CTZ, some might conclude a priori that CTZ does not wash out of tissue. Yet, we failed to find other published data that present direct evidence of the washout characteristics of CTZ in in vitro preparations, despite some previous speculation based on indirect evidence (Patneau et al. 1993; Larson et al. 1994; Funk et al. 1995). Furthermore, reversible enhancement of AMPA receptor-mediated signalling with another lipophilic drug, CX546, in part by blockade of AMPA receptor desensitization, did not lead to long-lasting increases in respiratory activity, consistent with the idea that the extended bioavailability particular to CTZ underlies its persistent effects, rather than some general properties of temporary desensitization/deactivation blockade. Therefore, these data provide additional perspective for interpreting results acquired when CTZ is used in vitro. These results include reports of persistence of enhanced motor activity (Ballerini et al. 1995; Funk et al. 1995) and the induction of epileptiform activity (Qi et al. 2006) following treatment with CTZ.
In addition to its effects on AMPA receptors, CTZ generates a variety of effects not involving AMPA receptors. CTZ was developed as a diuretic (Martz et al. 1962), acting on the thiazide-sensitive Na+–Cl− cotransporter in the distal loop of the kidney (Martinez-Maldonado & Cordova, 1990). This mechanism does not affect the medullary respiratory control circuit, however, since this transporter is not found in the mammalian brain (Gamba, 2005). CTZ can inhibit metabotropic glutamate type 1 (Sharp et al. 1994; Surin et al. 2007) and α3- but not α7-containing acetylcholine receptors (Nooney & Feltz, 1995). Most probably blockade of these receptors would decrease XII MN excitability, since both the effects of acetylcholine and activation of metabotropic glutamate receptor type 1 tend to be excitatory in the preBötzinger complex and XII motor nucleus in vitro (Chamberlin et al. 2002; Sharifullina et al. 2004; Shao & Feldman, 2005; Shao et al. 2008).
Perhaps the most important non-AMPA-related property of CTZ, when considering enhancement of inspiratory activity, is antagonism of GABAA (Deng & Chen, 2003), GABAC (Xie et al. 2008) and α2-subunit-containing glycine (Zhang et al. 2008) receptors. Whereas blockade of inhibitory neurotransmitters has little effect on respiratory rhythm generation in vitro, such blockade can change the amplitude of XII nerve bursts (Feldman & Smith, 1989; Shao & Feldman, 1997; Saywell & Feldman, 2004). Together with enhancement of AMPA-mediated respiratory drive currents, antagonism of inhibitory transmission by CTZ probably contributes in a meaningful way to the observed increase in nerve burst amplitude that we observed. This effect of CTZ, however, also relies upon its continued presence.
Blockade of fast GABA currents, however, could further contribute by fostering activity-dependent changes in transcription. A 15 min blockade of GABAA currents in rat hippocampal slice culture increases network activity that drives early- and late-phase long-term potentiation (LTP) of AMPA currents mediated by enhanced presynaptic release, which is evidenced by increases in mEPSC frequency at pyramidal cell synapses for as long as 10 h (Wiegert et al. 2009). While CTZ is thought not to affect early-phase LTP (Rammes et al. 1999), its effects on late-phase LTP are unknown. Increasing activity directly at AMPA receptors in rat neuronal cultures using CTZ (Qi et al. 2006) or S18986 (Lockhart et al. 2007), another AMPA receptor modulator, enhances brain-derived neurotrophic factor (BDNF) production, which enhances mEPSC amplitude and frequency (Nagano et al. 2003; Abidin et al. 2006). These changes appear to be important in one model of epilepsy, where high enough concentrations of CTZ can kindle epileptiform activity (Wang et al. 2009). Enhancement of excitatory synaptic efficacy by activity-dependent enhancement of BDNF signalling, however, may not be universal to all synapses; in dorsal root ganglion, neuron identity is critical to determining BDNF effects, with enhanced BDNF signalling either increasing or decreasing mEPSC frequency and amplitude depending upon neuron type (Lu et al. 2007).
Although the aforementioned mechanisms do not depend upon continued presence of CTZ, we highlight that they do depend upon continued, enhanced network activity to induce the observed transcription-dependent excitatory synaptic plasticity. To this point, we interpret our results where AMPA and NMDA receptor activity was blocked from 30 min before to 1 h after completion of treatment with CTZ as indicating that the phenomenon we observed is activity independent, especially since we did not observe even a partial loss of the CTZ-driven activity enhancement once activity recovered. We acknowledge, though, that these experiments are not equivalent to completely blocking all network-driven activity, e.g. using TTX, and that the abolition of observable XII nerve activity does not necessarily indicate the abolition of a reduced level of network activity that might support plasticity. In addition, the direct effects of CTZ on an unidentified second messenger substrate that mediates plasticity cannot be ruled out.
Physiological significance
Our observations point to the importance of endogenous regulation of AMPA receptor kinetics for proper functioning of neural circuits. These findings should be applicable to older or more intact preparations, despite the immaturity and lower temperature of the medullary slice preparation. Despite being prepared from neonatal animals, the slice preparation has provided important information and predictions about the neural control of breathing that subsequently have been verified in highly intact preparations (Feldman & Del Negro, 2006). The effect of temperature in particular on synaptic transmission is largely presynaptic, with lower temperatures leading to increased synaptic failure (Schiff & Somjen, 1985; Hardingham & Larkman, 1998). In addition, the peak amplitude of miniature EPSPs is unaffected by raising temperatures to physiological levels (Hardingham & Larkman, 1998). Furthermore, AMPA receptors desensitize more rapidly with increases in temperature (Raman & Trussell, 1992). Finally, all AMPA receptor subunit types behave similarly with respect to activation, deactivation and desensitization (Gouaux, 2004), making reported developmental shifts in AMPA receptor subtypes within the respiratory control circuit (Liu & Wong-Riley, 2005) unlikely to influence our results.
Proper levels of AMPA receptor desensitization are important for neuronal function. Knock-in of a desensitization inhibiting L483Y mutation to the gene encoding the GluA2 receptor subunit is homozygous lethal, with most heterozygous mice suffering from runted development and seizures developing at around P16, before premature death, usually in the third postnatal week. Interestingly, no respiratory distress is reported (Christie et al. 2010). Also, Conus snails paralyse and kill their prey with a toxin that blocks AMPA receptor desensitization (Walker et al. 2009). In addition, mammals have numerous endogenous proteins, including TARPs, cornichons and CKAMP44, to regulate desensitization, deactivation and activation kinetics of AMPA receptors, suggesting the necessity for the careful management of these phenomena (Milstein & Nicoll, 2008; Brockie & Maricq, 2010; Kato et al. 2010; von Engelhardt et al. 2010).
Remarkably, induction of large increases in the efficacy of AMPA receptor signalling and reductions in inhibition greatly enhanced the amplitude of inspiratory nerve discharge while not grossly distorting rhythmogenic behaviour. Only the highest concentrations of CTZ at the longest durations of application caused bouts of tonic activity that distorted respiratory rhythm. Thus, there is enormous residual capacity to increase inspiratory motor output, at least under in vitro conditions, with minimal disruption to rhythmogenic behaviour. We posit that a similar capacity and effect for CTZ, where the amplitude of activity rather than the pattern of activity is differentially affected, might exist in other motor pools and systems, e.g. the locomotor system, although study of the effects of CTZ in neonatal rat spinal cord preparations suggest that the effects of CTZ on the balance of excitation and inhibition in such networks must be considered carefully (Ballerini et al. 1995).
We suggest that CTZ, or related drugs that more readily cross the blood–brain barrier, e.g. IDRA-21 of S18986, may be useful as therapeutics for enhancing motor output that is reduced due to disease or dysfunction. Whereas some data exist for the safe use of compounds such as IDRA-21 and S18986 in the nervous system of in vivo models for enhancing neurological function (Black, 2005), similar data related to dose and half-life in the nervous system do not exist for CTZ. Potential drawbacks include negative consequences at higher CTZ doses (Qi et al. 2006; Wang et al. 2009) and effective delivery of CTZ to the nervous system, as it does not cross the blood–brain barrier. Nevertheless, the potency and prolonged nature of the effects of CTZ might be advantageous. One such therapeutic application would be in the treatment of OSA, where loss of tone in upper airway muscles but not respiratory rhythmogenesis (as evidenced by continued diaphragmatic efforts) during sleep is associated with repetitive upper airway collapse leading to periods of apnoea or hypopnoea, disrupted sleep, and increased risk for a variety long-term pathological cardiovascular and neurological consequences (Young et al. 2002; Young et al. 2009).
Acknowledgments
The authors wish to acknowledge the late Shane Saywell, who participated in some of the initial experiments presented in this paper, Alan Garfinkel, who helped us in applying the bootstrap statistical techniques, and Tom Otis, who made many invaluable suggestions as this project progressed. W.E. Babiec is now affiliated with the Department of Physiology at the David Geffen School of Medicine, UCLA. This work was performed with funding from the NIH (NS24742, NS67933, NS58280). The authors are aware of no conflicts of interest.
Translational perspective
Obstructive sleep apnoea (OSA), a disease of upper airway collapse during sleep whose cause is unknown, generates frequent, repetitive cycles of hypoxaemic hypoxia and compensatory sympathetic facilitation that cause disrupted sleep, with consequent neurocognitive impairment and increased risk for automobile and workplace accidents. Untreated OSA raises the risk of hypertension, cardiovascular disease, type 2 diabetes, and stroke. Estimated to affect as many as 20% of adult Americans, OSA represents a significant public health problem.
OSA is most commonly treated by splinting open the airway with continuous positive airway pressure or surgical reconstruction of the soft palate, tongue, and/or jaw to remove or relocate tissue likely to cause constriction of the upper airway: the former effective but with low patient compliance, the benefits of the latter eventually diminishing. There are no pharmacological treatments.
Respiratory drive to the upper airway (and diaphragm) is mediated largely via AMPA receptor signalling. In vitro, we tested the extent to which cyclothiazide, a clinically approved diuretic/antihypertensive that abolishes AMPA receptor desensitization, enhanced respiratory drive to hypoglossal motoneurons, which control the genioglossus muscle of the tongue (loss of genioglossus tone can precipitate OSA). Treatment with cyclothiazide greatly enhanced the peak amplitude of respiratory drive to hypoglossal motoneurons with limited facilitation of respiratory rate. These effects persisted for hours due to the slow washoff kinetics of cyclothiazide. Our data provide proof of principle for using AMPA receptor modulators in treating motor dysfunctions like OSA, warranting follow-up in intact animal and potentially human studies for development of pharmacological treatments.
Glossary
- ACSF
artificial cerebral spinal fluid
- BDNF
brain-derived neurotrophic factor
- CIF
cyclothiazide-induced facilitation
- CTZ
cyclothiazide
- ivLTF
in vitro long-term facilitation
- mEPSC
miniature excitatory postsynaptic current
- MN
motoneuron
- MRM
multiple reaction monitoring
- OSA
obstructive sleep apnoea
probability density function
- RM
repeated measures
- TARP
transmembrane AMPA receptor regulatory proteins
- XII
hypoglossal
- ∫XII
integrated hypoglossal
Author contributions
W.E.B.: Overall study design and design of experiments. Collection and analysis of all electrophysiological data. Statistical analysis for all data. Preparation/review of manuscript. K.F.F.: Design, conduct, collection and analysis of data for mass spectrometry experiments. Preparation/review of manuscript. J.L.F.: Overall study design and design of experiments. Guidance of data analysis/review. Preparation/review of manuscript. All authors approved the final version.
References
- Abidin I, Köhler T, Weiler E, Zoidl G, Eysel UT, Lessmann V, Mittmann T. Reduced presynaptic efficiency of excitatory synaptic transmission impairs LTP in the visual cortex of BDNF-heterozygous mice. Eur J Neurosci. 2006;24:3519–3531. doi: 10.1111/j.1460-9568.2006.05242.x. [DOI] [PubMed] [Google Scholar]
- Arai AC, Kessler M. Pharmacology of ampakine modulators: from AMPA receptors to synapses and behavior. Curr Drug Targets. 2007;8:583–602. doi: 10.2174/138945007780618490. [DOI] [PubMed] [Google Scholar]
- Ballerini L, Bracci E, Nistri A. Desensitization of AMPA receptors limits the amplitude of EPSPs and the excitability of motoneurons of the rat isolated spinal cord. Eur J Neurosci. 1995;7:1229–1234. doi: 10.1111/j.1460-9568.1995.tb01113.x. [DOI] [PubMed] [Google Scholar]
- Bertolino M, Bareli M, Parenti C, Braghiroli D, DiBella M, Vicini S, Costa E. Modulation of AMPA/kainate receptors by analogues of diazoxide and cyclothiazide in thin slices of rat hippocampus. Receptors Channels. 1993;1:267–278. [PubMed] [Google Scholar]
- Black MD. Therapeutic potential of positive AMPA modulators and their relationship to AMPA receptor subunits. A review of preclinical data. Psychopharmacology (Berl) 2005;179:154–163. doi: 10.1007/s00213-004-2065-6. [DOI] [PubMed] [Google Scholar]
- Bocchiaro CM, Feldman JL. Synaptic activity-independent persistent plasticity in endogenously active mammalian motoneurons. Proc Natl Acad Sci U S A. 2004;101:4292–4295. doi: 10.1073/pnas.0305712101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocchiaro CM, Saywell SA, Feldman JL. Dynamic modulation of inspiratory drive currents by protein kinase A and protein phosphatases in functionally active motoneurons. J Neurosci. 2003;23:1099–1103. doi: 10.1523/JNEUROSCI.23-04-01099.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockie PJ, Maricq AV. In a pickle: is cornichon just relish or part of the main dish? Neuron. 2010;68:1017–1019. doi: 10.1016/j.neuron.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlin NL, Bocchiaro CM, Greene RW, Feldman JL. Nicotinic excitation of rat hypoglossal motoneurons. Neuroscience. 2002;115:861–870. doi: 10.1016/s0306-4522(02)00454-2. [DOI] [PubMed] [Google Scholar]
- Christie LA, Russell TA, Xu J, Wood L, Shepherd GM, Contractor A. AMPA receptor desensitization mutation results in severe developmental phenotypes and early postnatal lethality. Proc Natl Acad Sci U S A. 2010;107:9412–9417. doi: 10.1073/pnas.0908206107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen BH, Lea RB. Essentials of Statistics for the Social and Behavioral Sciences. Hoboken: John Wiley & Sons, Inc; 2004. [Google Scholar]
- Deng L, Chen G. Cyclothiazide potently inhibits γ-aminobutyric acid type A receptors in addition to enhancing glutamate responses. Proc Natl Acad Sci U S A. 2003;100:13025–13029. doi: 10.1073/pnas.2133370100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS, Jahr CE. Asynchronous release of synaptic vesicles determines the time course of the AMPA receptor-mediated EPSC. Neuron. 1995;15:1097–1107. doi: 10.1016/0896-6273(95)90098-5. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuBord MA, Liu H, Horner RL. Protein kinase A activators produce a short-term, but not long-term, increase in respiratory-drive transmission at the hypoglossal motor nucleus in vivo. Neurosci Lett. 2010;486:14–18. doi: 10.1016/j.neulet.2010.09.034. [DOI] [PubMed] [Google Scholar]
- Dzubay JA, Jahr CE. The concentration of synaptically released glutamate outside of the climbing fiber-Purkinje cell synaptic cleft. J Neurosci. 1999;19:5265–5274. doi: 10.1523/JNEUROSCI.19-13-05265.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman JL, Del Negro CA. Looking for inspiration: new perspectives on respiratory rhythm. Nat Rev Neurosci. 2006;7:232–242. doi: 10.1038/nrn1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman JL, Smith JC. Cellular mechanisms underlying modulation of breathing pattern in mammals. Ann N Y Acad Sci. 1989;563:114–130. doi: 10.1111/j.1749-6632.1989.tb42194.x. [DOI] [PubMed] [Google Scholar]
- Fucile S, Miledi R, Eusebi F. Effects of cyclothiazide on GluR1/AMPA receptors. Proc Natl Acad Sci U S A. 2006;103:2943–2947. doi: 10.1073/pnas.0511063103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk GD, Smith JC, Feldman JL. Generation and transmission of respiratory oscillations in medullary slices: role of excitatory amino acids. J Neurophysiol. 1993;70:1497–1515. doi: 10.1152/jn.1993.70.4.1497. [DOI] [PubMed] [Google Scholar]
- Funk GD, Smith JC, Feldman JL. Modulation of neural network activity in vitro by cyclothiazide, a drug that blocks desensitization of AMPA receptors. J Neurosci. 1995;15:4046–4056. doi: 10.1523/JNEUROSCI.15-05-04046.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamba G. Molecular pathophysiology of electroneutral cation-chloride cotransporters. Physiol Rev. 2005;85:423–493. doi: 10.1152/physrev.00011.2004. [DOI] [PubMed] [Google Scholar]
- Gouaux E. Structure and function of AMPA receptors. J Physiol. 2004;554:249–253. doi: 10.1113/jphysiol.2003.054320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer JJ, Smith JC, Feldman JL. Role of excitatory amino acids in the generation and transmission of respiratory drive in neonatal rat. J Physiol. 1991;437:727–749. doi: 10.1113/jphysiol.1991.sp018622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham NR, Larkman AU. The reliability of excitatory synaptic transmission in slices of rat visual cortex in vitro is temperature dependent. J Physiol. 1998;507:249–256. doi: 10.1111/j.1469-7793.1998.249bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato AS, Gill MB, Yu H, Nisenbaum ES, Bredt DS. TARPs differentially decorate AMPA receptors to specify neuropharmacology. Trends Neurosci. 2010;33:241–248. doi: 10.1016/j.tins.2010.02.004. [DOI] [PubMed] [Google Scholar]
- Larson J, Le T-T, Hall RA, Lynch G. Effects of cyclothiazide on synaptic responses in slices of adult and neonatal rat hippocampus. Neuroreport. 1994;5:389–392. doi: 10.1097/00001756-199401120-00004. [DOI] [PubMed] [Google Scholar]
- Liu Q, Wong-Riley MT. Postnatal developmental expressions of neurotransmitters and receptors in various brain stem nuclei of rats. J Appl Physiol. 2005;98:1442–1457. doi: 10.1152/japplphysiol.01301.2004. [DOI] [PubMed] [Google Scholar]
- Lockhart BP, Rodriguez M, Mourlevat S, Peron P, Catesson S, Villain N, Galizzi JP, Boutin JA, Lestage P. S18986: a positive modulator of AMPA-receptors enhances (S)-AMPA-mediated BDNF mRNA and protein expression in rat primary cortical neuronal cultures. Eur J Pharmacol. 2007;561:23–31. doi: 10.1016/j.ejphar.2007.01.030. [DOI] [PubMed] [Google Scholar]
- Lorier AR, Funk GD, Greer JJ. Opiate-induced suppression of rat hypoglossal motoneuron activity and its reversal by ampakine therapy. PLoS One. 2010;5:e8766. doi: 10.1371/journal.pone.0008766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu VB, Ballanyi K, Colmers WF, Smith PA. Neuron type-specific effects of brain-derived neurotrophic factor in rat superficial dorsal horn and their relevance to ‘central sensitization’. J Physiol. 2007;584:543–563. doi: 10.1113/jphysiol.2007.141267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Manly BFJ. Randomization, Bootstrap and Monte Carlo Methods in Biology. 3rd ed. Boca Raton: Chapman & Hall/CRC; 2006. [Google Scholar]
- Martinez-Maldonado M, Cordova HR. Cellular and molecular aspects of the renal effects of diuretic agents. Kidney Int. 1990;38:632–641. doi: 10.1038/ki.1990.253. [DOI] [PubMed] [Google Scholar]
- Martz BL, Steinmetz E, Kraner JC. Studies of a new diuretic, cyclothiazide. J Indiana State Med Assoc. 1962;55:173–176. [PubMed] [Google Scholar]
- Milstein AD, Nicoll RA. Regulation of AMPA receptor gating and pharmacology by TARP auxiliary subunits. Trends Pharmacol Sci. 2008;29:333–339. doi: 10.1016/j.tips.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano T, Yanagawa Y, Obata K, Narisawa-Saito M, Namba H, Otsu Y, Takei N, Nawa H. Brain-derived neurotrophic factor upregulates and maintains AMPA receptor currents in neocortical GABAergic neurons. Mol Cell Neurosci. 2003;24:340–356. doi: 10.1016/s1044-7431(03)00172-6. [DOI] [PubMed] [Google Scholar]
- Neverova NV, Saywell SA, Nashold LJ, Mitchell GS, Feldman JL. Episodic stimulation of α1-adrenoreceptors induces protein kinase C-dependent persistent changes in motoneuronal excitability. J Neurosci. 2007;27:4435–4442. doi: 10.1523/JNEUROSCI.2803-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nooney JM, Feltz A. Inhibition by cyclothiazide of neuronal nicotinic responses in bovine chromaf-fin cells. Br J Pharmacol. 1995;114:648–655. doi: 10.1111/j.1476-5381.1995.tb17188.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien JA, Isaacson JS, Berger AJ. NMDA and non-NMDA receptors are co-localized at excitatory synapses of rat hypoglossal motoneurons. Neurosci Lett. 1997;227:5–8. doi: 10.1016/s0304-3940(97)00293-0. [DOI] [PubMed] [Google Scholar]
- Ogier M, Wang H, Hong E, Wang Q, Greenberg ME, Katz DM. Brain-derived neurotrophic factor expression and respiratory function improve after ampakine treatment in a mouse model of Rett syndrome. J Neurosci. 2007;27:10912–10917. doi: 10.1523/JNEUROSCI.1869-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partin KM, Patneau DK, Mayer ML. Cyclothiazide differentially modulates desensitization of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor splice variants. Mol Pharmacol. 1994;46:129–138. [PubMed] [Google Scholar]
- Patneau DK, Vyklicky L, Mayer ML. Hippocampal neurons exhibit cyclothiazide-sensitive rapidly desensitizing responses to kainate. J Neurosci. 1993;13:3496–3509. doi: 10.1523/JNEUROSCI.13-08-03496.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi J, Wang Y, Jiang M, Warren P, Chen G. Cyclothiazide induces robust epileptiform activity in rat hippocampal neurons both in vitro and in vivo. J Physiol. 2006;571:605–618. doi: 10.1113/jphysiol.2005.103812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Trussell LO. The kinetics of the response to glutamate and kainate in neurons of the avian cochlear nucleus. Neuron. 1992;9:173–186. doi: 10.1016/0896-6273(92)90232-3. [DOI] [PubMed] [Google Scholar]
- Rammes G, Zeilhofer HU, Collingridge GL, Parsons CG, Swandulla D. Expression of early hippocampal CA1 LTP does not lead to changes in AMPA-EPSC kinetics or sensitivity to cyclothiazide. Pflugers Arch. 1999;437:191–196. doi: 10.1007/s004240050768. [DOI] [PubMed] [Google Scholar]
- Ren J, Ding X, Funk GD, Greer JJ. Ampakine CX717 protects against fentanyl-induced respiratory depression and lethal apnea in rats. Anesthesiology. 2009;110:1364–1370. doi: 10.1097/ALN.0b013e31819faa2a. [DOI] [PubMed] [Google Scholar]
- Ren J, Poon BY, Tang Y, Funk GD, Greer JJ. Ampakines alleviate respiratory depression in rats. Am J Respir Crit Care Med. 2006;174:1384–1391. doi: 10.1164/rccm.200606-778OC. [DOI] [PubMed] [Google Scholar]
- Ryan CM, Bradley TD. Pathogenesis of obstructive sleep apnea. J Appl Physiol. 2005;99:2440–2450. doi: 10.1152/japplphysiol.00772.2005. [DOI] [PubMed] [Google Scholar]
- Saywell SA, Feldman JL. Dynamic interactions of excitatory and inhibitory inputs in hypoglossal motoneurones: respiratory phasing and modulation by PKA. J Physiol. 2004;554:879–889. doi: 10.1113/jphysiol.2003.054528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiff SJ, Somjen GG. The effects of temperature on synaptic transmission in hippocampal tissue slices. Brain Res. 1985;345:279–284. doi: 10.1016/0006-8993(85)91004-2. [DOI] [PubMed] [Google Scholar]
- Shao XM, Feldman JL. Respiratory rhythm generation and synaptic inhibition of expiratory neurons in pre-Bötzinger complex: differential roles of glycinergic and GABAergic neural transmission. J Neurophysiol. 1997;77:1853–1860. doi: 10.1152/jn.1997.77.4.1853. [DOI] [PubMed] [Google Scholar]
- Shao XM, Feldman JL. Cholinergic neurotransmission in the preBötzinger Complex modulates excitability of inspiratory neurons and regulates respiratory rhythm. Neuroscience. 2005;130:1069–1081. doi: 10.1016/j.neuroscience.2004.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao XM, Tan W, Xiu J, Puskar N, Fonck C, Lester HA, Feldman JL. Alpha4* nicotinic receptors in preBotzinger complex mediate cholinergic/nicotinic modulation of respiratory rhythm. J Neurosci. 2008;28:519–528. doi: 10.1523/JNEUROSCI.3666-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharifullina E, Ostroumov K, Nistri A. Activation of group I metabotropic glutamate receptors enhances efficacy of glutamatergic inputs to neonatal rat hypoglossal motoneurons in vitro. Eur J Neurosci. 2004;20:1245–1254. doi: 10.1111/j.1460-9568.2004.03590.x. [DOI] [PubMed] [Google Scholar]
- Sharp RL, Mayne NG, Burnett JP. Cyclothiazide differentially modulates human metabotropic glutamate receptors linked to phosphoinositide hydrolysis stimulation in oocytes. Eur J Pharmacol. 1994;269:R5–R7. doi: 10.1016/0922-4106(94)90049-3. [DOI] [PubMed] [Google Scholar]
- Smith AK, Togeiro SM, Tufik S, Roizenblatt S. Disturbed sleep and musculoskeletal pain in the bed partner of patients with obstructive sleep apnea. Sleep Med. 2009;10:904–912. doi: 10.1016/j.sleep.2008.08.013. [DOI] [PubMed] [Google Scholar]
- Smith JC, Ellenberger HH, Ballanyi K, Richter DW, Feldman JL. Pre-Bötzinger complex: a brainstem region that may generate respiratory rhythm in mammals. Science. 1991;254:726–729. doi: 10.1126/science.1683005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenland HW, Liu H, Horner RL. Endogenous glutamatergic control of rhythmically active mammalian respiratory motoneurons in vivo. J Neurosci. 2008;28:6826–6835. doi: 10.1523/JNEUROSCI.1019-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenland HW, Liu H, Sood S, Liu X, Horner RL. Respiratory activation of the genioglossus muscle involves both non-NMDA and NMDA glutamate receptors at the hypoglossal motor nucleus in vivo. Neuroscience. 2006;138:1407–1424. doi: 10.1016/j.neuroscience.2005.12.040. [DOI] [PubMed] [Google Scholar]
- Surin A, Pshenichkin S, Grajkowska E, Surina E, Wroblewski JT. Cyclothiazide selectively in-hibits mGluR1 receptors interacting with a common allosteric site for non-competitive antagonists. Neuropharmacol. 2007;52:744–754. doi: 10.1016/j.neuropharm.2006.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsvyetlynska NA, Hill RH, Grillner S. Role of AMPA receptor desensitization and the side effects of a DMSO vehicle on reticulospinal EPSPs and locomotor activity. J Neurophysiol. 2005;94:3951–3960. doi: 10.1152/jn.00201.2005. [DOI] [PubMed] [Google Scholar]
- Vogt KE, Canepari M. On the induction of postsynaptic granule cell-Purkinje neuron LTP and LTD. Cerebellum. 2010;9:284–290. doi: 10.1007/s12311-010-0174-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Engelhardt J, Mack V, Sprengel R, Kavenstock N, Li KW, Stern-Bach Y, Smit AB, Seeburg PH, Monyer H. CKAMP44: a brain-specific protein attenuating short-term synaptic plasticity in the dentate gyrus. Science. 2010;327:1518–1522. doi: 10.1126/science.1184178. [DOI] [PubMed] [Google Scholar]
- Walker CS, Jensen S, Ellison M, Matta JA, Lee WY, Imperial JS, Duclos N, Brockie PJ, Madsen DM, Isaac JTR, Olivera B, Maricq AV. A novel Conus snail polypeptide causes excitotoxicity by blocking desensitization of AMPA receptors. Curr Biol. 2009;19:900–908. doi: 10.1016/j.cub.2009.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Qi JS, Kong S, Sun Y, Fan J, Jiang M, Chen G. BDNF-TrkB signaling pathway mediates the induction of epileptiform activity induced by a convulsant drug cyclothiazide. Neuropharmacology. 2009;57:49–59. doi: 10.1016/j.neuropharm.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegert JS, Hofmann F, Bading H, Bengtson CP. A transcription-dependent increase in miniature EPSC frequency accompanies late-phase plasticity in cultured hippocampal neurons. BMC Neurosci. 2009;10:124. doi: 10.1186/1471-2202-10-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie A, Song X, Ripps H, Qian H. Cyclothiazide: a subunit-specific inhibitor of GABAC receptors. J Physiol. 2008;586:2743–2752. doi: 10.1113/jphysiol.2008.153346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada KA, Tang C-M. Benzothiadiazides inhibit rapid glutamate receptor desensitization and enhance glutamatergic synaptic currents. J Neurosci. 1993;13:3904–3915. doi: 10.1523/JNEUROSCI.13-09-03904.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young T, Palta M, Dempsey J, Peppard PE, Nieto FJ, Hla KM. Burden of sleep apnea: rationale, design, and major findings of the Wisconsin Sleep Cohort study. WMJ. 2009;108:246–249. [PMC free article] [PubMed] [Google Scholar]
- Young T, Peppard PE, Gottlieb DJ. Epidemiology of obstructive sleep apnea: a population health perspective. Am J Respir Crit Care Med. 2002;165:1217–1239. doi: 10.1164/rccm.2109080. [DOI] [PubMed] [Google Scholar]
- Zhang XB, Sun GC, Liu LY, Yu F, Xu TL. Alpha2 subunit specificity of cyclothiazide inhibition on glycine receptors. Mol Pharmacol. 2008;73:1195–1202. doi: 10.1124/mol.107.042655. [DOI] [PubMed] [Google Scholar]